 Mi Zhang1,2
Mi Zhang1,2 Zexi Chen
Zexi Chen Jiang Chen
Jiang Chen- 1School and Hospital of Stomatology, Fujian Medical University, Fuzhou, China
- 2Fujian Key Laboratory of Oral Diseases, School and Hospital of Stomatology, Fujian Medical University, Fuzhou, China
- 3Research Center of Dental and Craniofacial Implants, School and Hospital of Stomatology, Fujian Medical University, Fuzhou, China
- 4Department of Oral and Maxillofacial Surgery, The First Affiliated Hospital of Fujian Medical University, Fuzhou, China
Accumulating evidence indicates that long non-coding RNAs (lncRNAs) play crucial roles in tongue squamous cell carcinoma (TSCC) tumorigenesis. However, the comprehensive regulation of lncRNAs-transcription factors (TFs)-messenger RNAs (mRNAs) in TSCC remains largely unknown. The purpose of this study was to identify aberrantly expressed lncRNAs and the associated TF-mRNA network in TSCC. To explore lncRNA and mRNA expression profiles and their biological functions in TSCC, we surveyed the lncRNA and mRNA expression profiles of TSCC and adjacent tissues using next-generation RNA sequencing in six patients. Thousands of significantly differentially expressed lncRNAs (DELs) and mRNAs (DEGs) were identified. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed to demonstrate the principal functions of the significantly dysregulated lncRNAs and genes. A total of 40 DELs were screened between TSCC and adjacent non-cancerous tissues. Results obtained from GEPIA and StarBase confirmed the expression levels of nine pivotal DELs obtained in our study. Three of the nine deregulated DELs were identified to have a significant impact on the overall survival of TSCC patients, which were evaluated with GEPIA and StarBase. LncMAP was used to predict the lncRNA-TF-mRNA triplets in TSCC. Furthermore, based on these results, we established lncRNA-TF-mRNA coexpression networks for the up- and downregulated lncRNAs using Cytoscape. We also found that among the nine pivotal lncRNAs, there is limited research on the abnormally expressed lncRNAs, including RP11-54H7.4, CTD-2545M3.8, RP11-760H22.2, RP4-791M13.3, and LINC01405, in TSCC pathogenesis. This is the first study to show that RP11-54H7.4, LINC00152, and LINC01405 can be acted as a prognostic target for TSCC. Our findings provide a novel perspective and lay the foundation for future research on the potential roles of lncRNAs, TFs, and mRNAs in TSCC. Verification of the potential lncRNA-TF-mRNA regulatory networks will provide a more comprehensive understanding of the pathogenesis of TSCC.
Introduction
As the most lethal and commonly occurring oral malignancy, oral squamous cell carcinoma (OSCC) accounts for 95% of all oral cancer diagnoses and causes more than 500,000 deaths per year (1). Approximately 25–40% of OSCCs are diagnosed as tongue squamous cell carcinoma (TSCC) (2). TSCC is characterized by frequent lymphoid metastasis, a high rate of regional recurrence, and a poor prognosis. Moreover, despite great progress in surgical techniques, diagnostic procedures, chemotherapy, and radiotherapy, the 5-year overall survival rate of TSCC patients has not improved significantly over the past decades. Therefore, a better understanding of the molecular mechanisms behind the initiation and progression of TSCC may facilitate the diagnosis and treatment of this malignant tumor.
Long non-coding RNA (lncRNA) represents a class of RNA species that are transcribed predominantly by RNA polymerase II, with a length exceeding 200 nucleotides and no apparent protein-coding potential, as indicated by the lack of strongly translated open reading frames (ORFs) (3). lncRNA plays a crucial role in diverse biological systems through genomic imprinting, cell cycle regulation, and cell differentiation and has been linked to a number of human diseases, especially cancer (4, 5). Recently, growing evidence has demonstrated that lncRNAs may also have essential roles in TSCC. For example, overexpression of the lncRNA FALEC represses malignant behaviors in TSCC (6). Upregulation of the lncRNA HOTTIP in TSCC patients indicates a poor clinical prognosis (7). In TSCC cell lines, cell growth, invasion, and migration abilities are associated with the overexpression of MALAT-1 and AFAP1-AS1 via the Wnt signaling pathway, while the knockdown of MALAT-1 in TSCC cells leads to the upregulation of certain SPRR proteins (8–10). High expression of the lncRNA UCA1 has been linked to the migratory ability of epithelial cancer cells and regional lymph node metastasis in TSCC (11). Moreover, the lncRNAs KCNQ1OT1 and SNHG17 act as competing endogenous RNAs (ceRNAs) to regulate cell proliferation and cancer progression in TSCC (12, 13).
In our study, we analyzed the expression profiles of lncRNAs and mRNAs in TSCC tissue through high-throughput sequencing. In addition, we identified molecular function, cellular component, biological process, and pathway analyses enriched by mRNA-associated lncRNAs and differentially expressed mRNAs in TSCC. Furthermore, our work revealed several pivotal lncRNAs related to the overall survival of TSCC patients. Finally, to optimize the regulatory mechanism of lncRNAs, we further explored several reliable transcription factors (TFs) in the regulatory regions of lncRNAs and constructed a lncRNA-TF-mRNA coexpression network.
Materials and Methods
Clinical Samples and Data Processing
Tumors and adjacent tongue tissues were obtained from patients with TSCC who underwent surgery at The First Affiliated Hospital of Fujian Medical University. Following surgical resection, six pairs of tissue samples were immediately immersed in liquid nitrogen and then stored at −80°C. Written informed consent was obtained from all participants in accordance with the Declaration of Helsinki, and the study protocol was approved by the Ethics Committee of The First Affiliated Hospital of Fujian Medical University (Fuzhou, China).
Next-generation RNA sequencing assays were performed to detect the mRNA and lncRNA expression profiles by KangChen Biotech (Shanghai, China) using an Illumina HiSeq 4,000 system (Illumina, San Diego, CA, USA). Solexa pipeline version 1.8 was used to align the reads to the genome, generate raw counts corresponding to each known gene (a total of 17,242 genes), and calculate the reads per kilobase per million (RPKM) values. The differentially expressed lncRNAs (DELs) and mRNAs (DEGs) were identified through fold change/p-value/FDR filtering [fold change ≥1.5, p < 0.05, and FDR (adjusted p-value) < 0.05].
Then, the data associated with TSCC were also retrieved from The Cancer Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov/) and were downloaded through GDC data transfer tool, including the RNA sequencing (RNA-Seq) of transcriptome profiling and clinical data. Then, in the predictive survival related model, we excluded 27 samples because they lacked complete clinical data. Finally, 146 TSCC samples and 15 normal control samples were collected in our study. EdgeR package in R (version 3.6.3) was used to identify the DELs. After that, we used the annotation file in GTF format (Homo_sapiens.GRCh38.100.chr.gtf) to identify and annotate DELs with the thresholds of |log2FC|>2.0 and FDR < 0.05.
Cell Lines
Three human TSCC cell lines, CAL-27, SCC-9, and SCC-25 were purchased from the American Type Culture Collection (ATCC; Manassas, VA, USA), and the human normal oral keratinocyte (hNOK) cell line was obtained from the Institute of Biochemistry and Cell Biology of the Chinese Academy of Sciences (Shanghai, China). CAL-27 cells and hNOK cells were grown in DMEM (Gibco) supplemented with 10% FBS (Invitrogen, Carlsbad, CA, USA). SCC-9 and SCC-25 cells were cultured in DMEM/F-12 (Gibco) supplemented with 10% FBS and 0.4 μg/ml hydrocortisone. All cells were maintained at 37°C in an incubator supplied with 5% CO2.
RNA Extraction and Quantitative Real-Time PCR Analysis
Total RNA was extracted from tissues or cells using TRIzol reagent (Invitrogen, CA, USA) according to the manufacturer's instructions. Total RNA was reverse transcribed into cDNA using a PrimeScript RT reagent kit (TaKaRa). RT-qPCR analyses were performed using SYBR Green Master Mix (TaKaRa). This process was performed using an Applied Biosystems 7,500 Real-Time PCR System. The results were normalized to the expression of glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The relative expression level was calculated by the 2−ΔΔ Ct method. The primers were provided by the SunYa Company.
Filtering DELs and DEGs
DELs and DEGs were selected using the following cut-off criteria: for DELs: the top 20 upregulated and downregulated lncRNAs, for DEGs: |log2-fold change| >2 and p < 0.05. Heatmap charts and volcano plots were established the gplots package in R software.
Verification of DEL and DEG Expression in GEPIA and StarBase
To identify the DELs, we used GEPIA (http://gepia.cancer-pku.cn/detail.php) (14) and StarBase (http://starbase.sysu.edu.cn/) (15), publicly available online databases, to explore gene expression levels in tumors and normal tissues.
Gene Set Enrichment Analysis (GSEA)
GSEA is a calculation method used to determine whether a given gene set is significantly different between different groups. The genes in these gene sets have a certain degree of correlation (e.g., the same biological function, located close to each other on the chromosome or defined by themselves according to a certain standard). However, LncRNA can not be directly used for GSEA as mRNA, but the correlation between each DEL and all mRNAs can be calculated. Then, GSEA of DELs can be performed using GSEAPreranked in GSEA software.
The Gene Ontology (GO) project provides a controlled vocabulary to describe gene and gene products attributed in any organism (http://www.geneontology.org). GO covers three domains: biological process, cellular component, and molecular function. The lower the p-value is, the more significant the GO term enrichment among the differentially expressed genes (a p ≤ 0.05 is recommended). Pathway analysis is a functional analysis that maps genes to Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (http://www.genome.jp/kegg/). The p-value (EASE score, Fisher's p-value, or hypergeometric p-value) denotes the significance of the pathway correlated to the condition. The GO and KEGG pathway analyses were performed by KangChen Biotech (Shanghai, China).
Survival Analysis of the Candidate lncRNAs
The GEPIA and StarBase databases were used to evaluate the effects of DELs on the overall survival of patients with TSCC. The final lncRNA correlated with overall survival was thus regarded as the pivotal lncRNA. p < 0.05 was considered statistically significant using the log-rank test. The cut-off value between two groups was set as the ‘‘median.''
Establishing the lncRNA-TF-mRNA Network
LncMAP (http://bio-bigdata.hrbmu.edu.cn/LncMAP/index.jsp) was used to analyze the lncRNA-TF-mRNA triplets in TSCC. A Venn diagram was drawn to determine the intersection between the mRNA in triplets and DEGs. The expression levels of these overlapping mRNAs were validated with GEPIA, and the correlation of the TF and mRNA was analyzed with StarBase. On the basis of these findings, we constructed a lncRNA-TF-mRNA regulatory network using Cytoscape (version 3.7.2) software to visualize their interactions. lncRNA-TF-mRNA triplets that showed no correlations with other triplets were excluded from the network.
Identification of the Prognostic Biomarkers
After analyzing the effects of pivotal DELs on the overall survival of patients with TSCC, we verified our findings in the TCGA difference analysis results, and it confirmed that LINC00152, LINC01405, RP11-54H7.4, and RP11-760H22.2 were simultaneously identified to be differentially expressed in TSCC. Thus, we also made the expression scatter and pairing plots of this four pivotal lncRNA by R software.
Statistical Analysis
Data are presented as the mean ± standard error (SE). Measured data were compared between groups using Student's paired t-tests or independent t-test. All statistical analyses were performed using SPSS and R software (version 22; IBM, Armonk, NY, USA) and presented graphically in GraphPad Prism 5.0 (GraphPad, La Jolla, CA, USA). Making the scatter diagram and paired plot by wilcoxon test in R software. All tests were two-tailed, and p < 0.05 was considered statistically significant.
Results
Identification of the Candidate DELs and DEGs in TSCC
Tumor and adjacent tissue samples were collected from six patients with TSCC. The main clinical characteristics of the enrolled patients are summarized in Supplementary Table 1. After sequencing, a total of 51,349 transcripts were presented, of which 38,567 are involved in protein coding. Among these transcripts, 4,224 mRNAs and 1,470 lncRNAs were differentially expressed between TSCC tumor and adjacent tissues. Among the mRNAs, 2,792 were upregulated and 1,432 were downregulated. Among the lncRNAs identified, 947 were upregulated and in TSCC and 523 were downregulated in TSCC (p < 0.05).
We further screened the 40 DELs and 450 DEGs that were aberrantly expressed between all TSCC tumor and adjacent tissues. As shown in Figure 1, the heatmap plot of (A) the top 20 upregulated and downregulated DELs and (B) all significantly expressed mRNAs were shown. Moreover, the volcano plot of (C) the top 20 upregulated and downregulated DELs and (D) all significantly expressed mRNAs were constructed. The annotation of these DELs is summarized in Table 1. Information on the 450 DEGs is presented in Table 2.
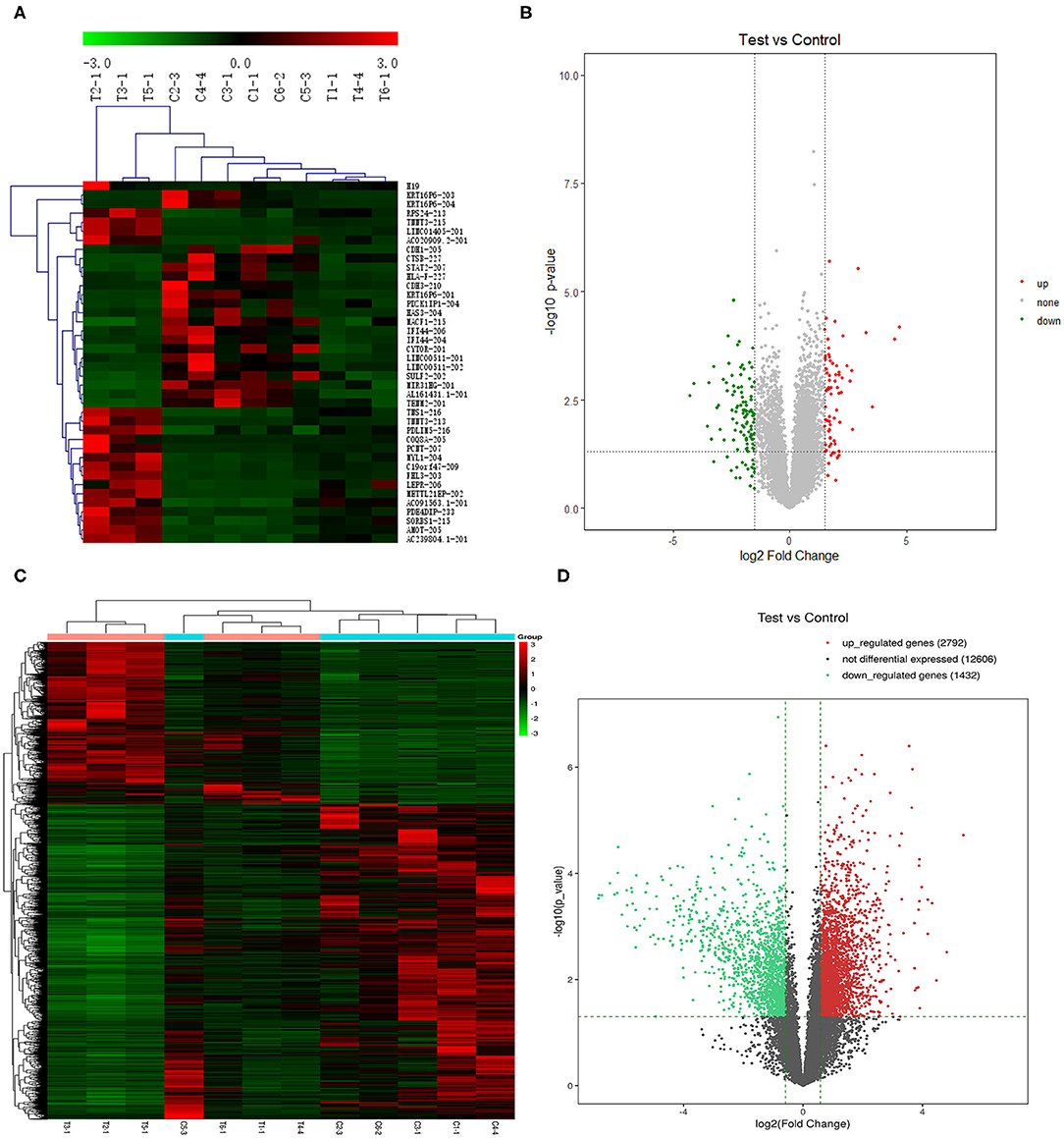
Figure 1. Heatmap plot of (A) 20 upregulated lncRNAs and 20 downregulated DELs and (C) the significantly expressed mRNAs. Volcano plot of (B) the significantly expressed lncRNAs and (D) mRNAs (p < 0.05, fold change >1.5). Synonyms: RP11-54H7.4 (AL161431.1-201), LINC00152 (CYTOR-201), RP11-760H22.2 (AC091563.1-201), CTD-2545M3.8 (AC020909.2-201), and RP4-791M13.3 (AC239804.1-201).
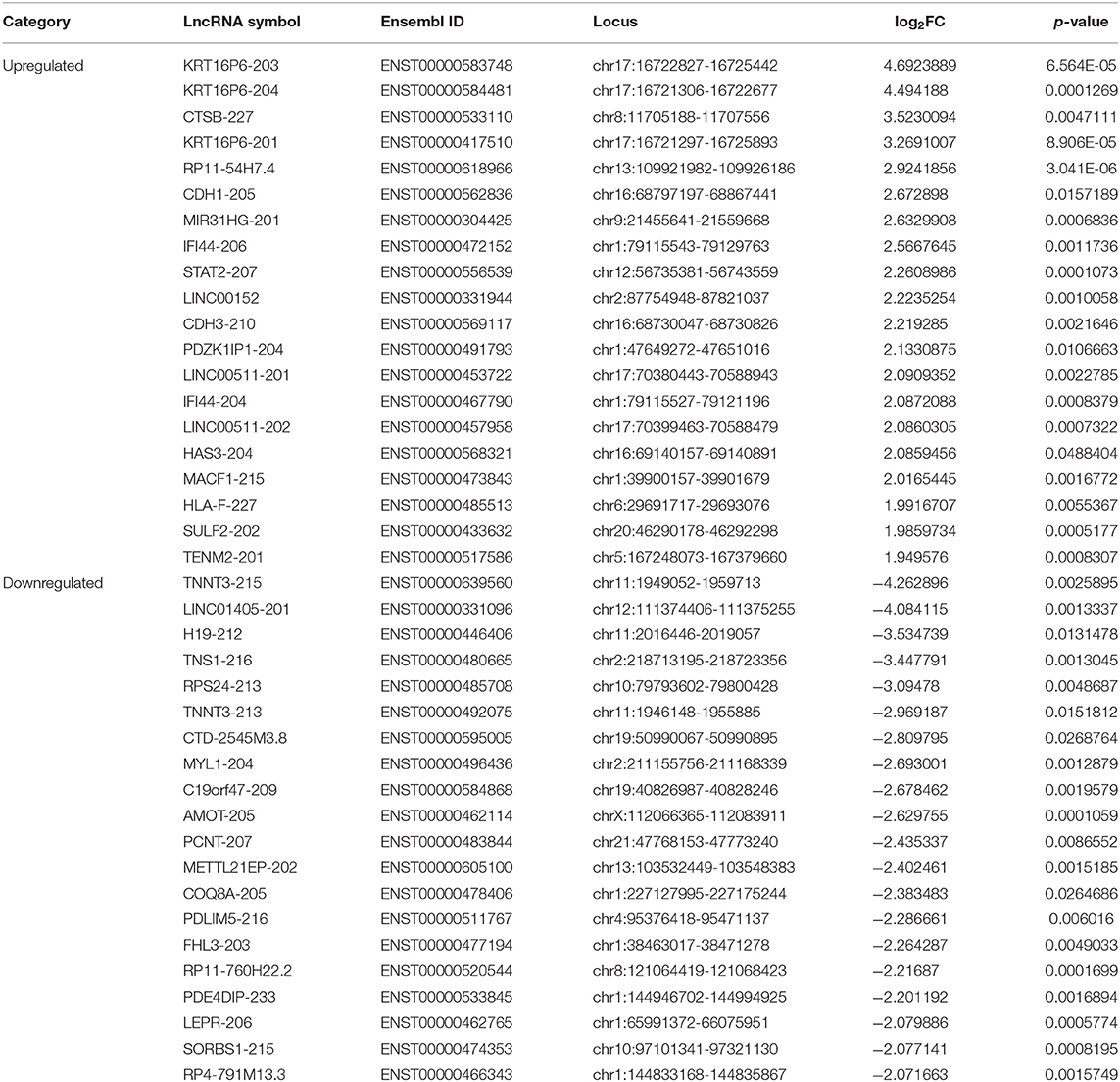
Table 1. Information on 40 DELs in TSCC.

Table 2. Information on DEGs in TSCC.
Verification of the Expression of DELs and DEGs in Available Databases
After screening, the expression profiles of the candidate DELs and DEGs were further validated with GEPIA and StarBase. We confirmed that nine keyDELs were aberrantly expressed between TSCC tumor and normal tissues. In GEPIA, with RP11-54H7.4, MIR31HG, LINC00152, and LINC00511 being upregulated and H19, CTD-2545M3.8, RP11-760H22.2, and RP4-791M13.3 being consistently downregulated in head and neck squamous cell carcinoma (HNSC) (Figure 2). Moreover, the results acquired in StarBase verified that the expression of the candidate DELs, such as RP11-54H7.4, MIR31HG, LINC00152, and LINC00511, was upregulated while the expression of H19, RP11-760H22.2, and LINC01405 was downregulated in HNSC (Figure 3).
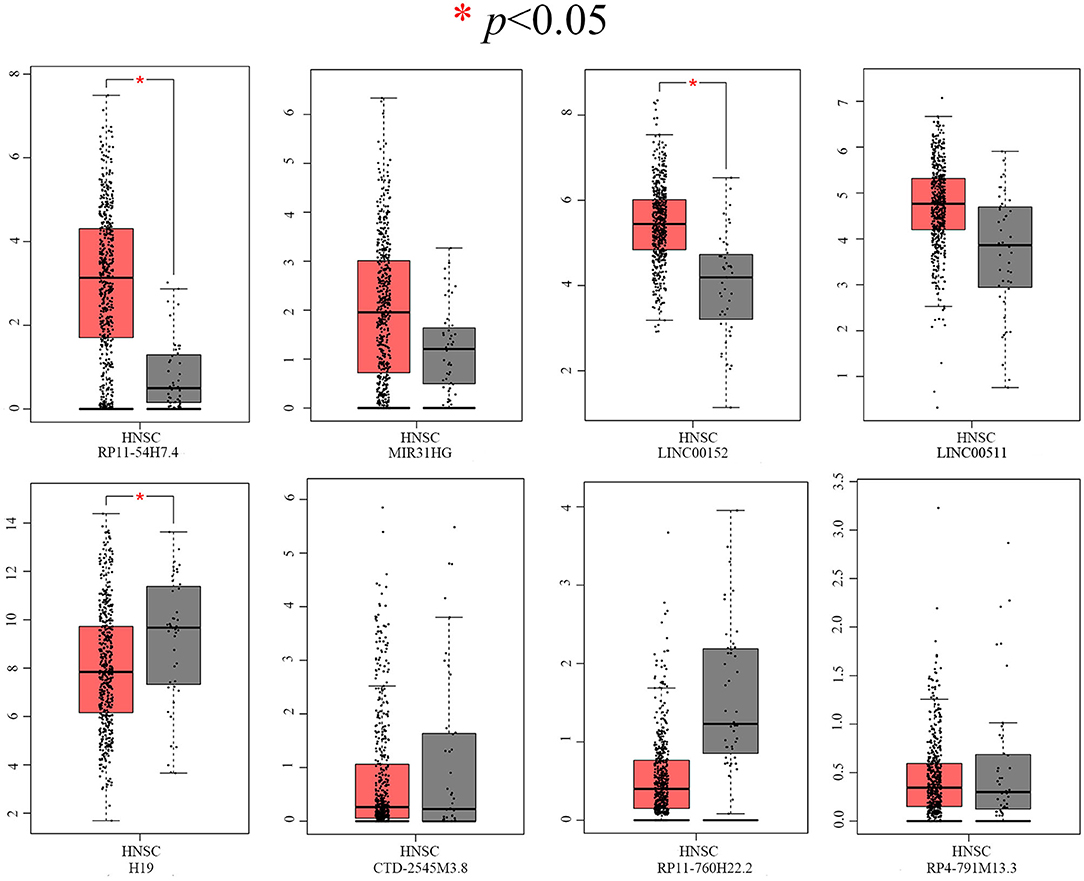
Figure 2. Verification of RP11-54H7.4, MIR31HG, LINC00152, LINC00511, H19, CTD-2545M3.8, RP11-760H22.2, and RP4-791M13.3 expression in HNSC by GEPIA. The red box represents expression in tumor tissue, and the gray box represents expression in normal tissue. HNSC, head, and neck squamous cell carcinoma (p < 0.05).
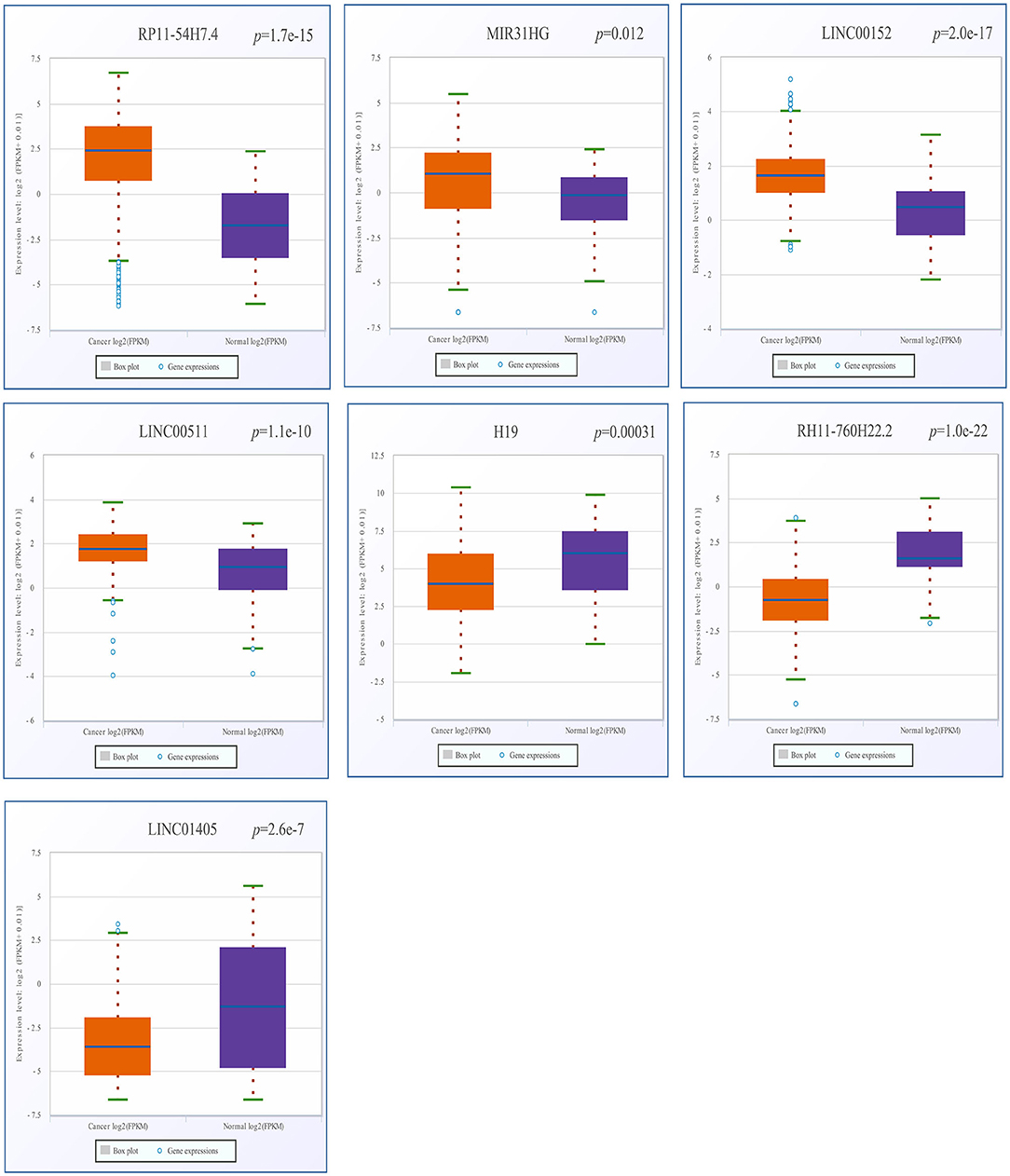
Figure 3. Verification of RP11-54H7.4, MIR31HG, LINC00152, LINC00511, H19, RP11-760H22.2, and LINC01405 expression in HNSC by StarBase. The orange box represents expression in tumor tissue, and the purple box represents expression in normal tissue. HNSC, head, and neck squamous cell carcinoma.
The GSEA Results of DELs
We also calculated the correlation between each DEL and all mRNAs so that GSEA could be performed using GSEAPreranked in GSEA software. The biological process (Figure 4A), cellular component (Figure 4B), molecular function (Figure 4C), and pathway (Figure 4D) analyses of the pivotal DELs, including KRT16P6-203, CTSB-227, RP11-54H7.4-201, CDH1-205, MIR31HG-201, IFI44-206, STAT2-207, LINC00152-201, CDH3-210, TNNT3-215, LINC01405-201, H19-212, TNS1-216, RPS24-213, and CTD-2545M3.8, were performed. Our data showed that the biological processes associated with the aberrantly regulated lncRNAs were cellular respiration, oxidative phosphorylation, and ATP synthesis coupled electron transport. The cellular components associated with the aberrantly regulated lncRNAs were the oxidoreductase complex mitochondrial respiratory chain and respiratory chain. The molecular functions associated with the aberrantly regulated lncRNAs were NADH dehydrogenase activity and oxidoreductase activity. KEGG pathway enrichment analysis of the significant DELs was performed to understand gene-related pathways and molecular interactions. Our results showed that oxidative phosphorylation, RNA transport, and mRNA surveillance pathway were associated with the dysregulated lncRNAs. Meanwhile, the GSEA results of LINC00152, LINC01405, RP11-54H7.4, and RP11-760H22.2 were showed independently in Supplementary Table 2.
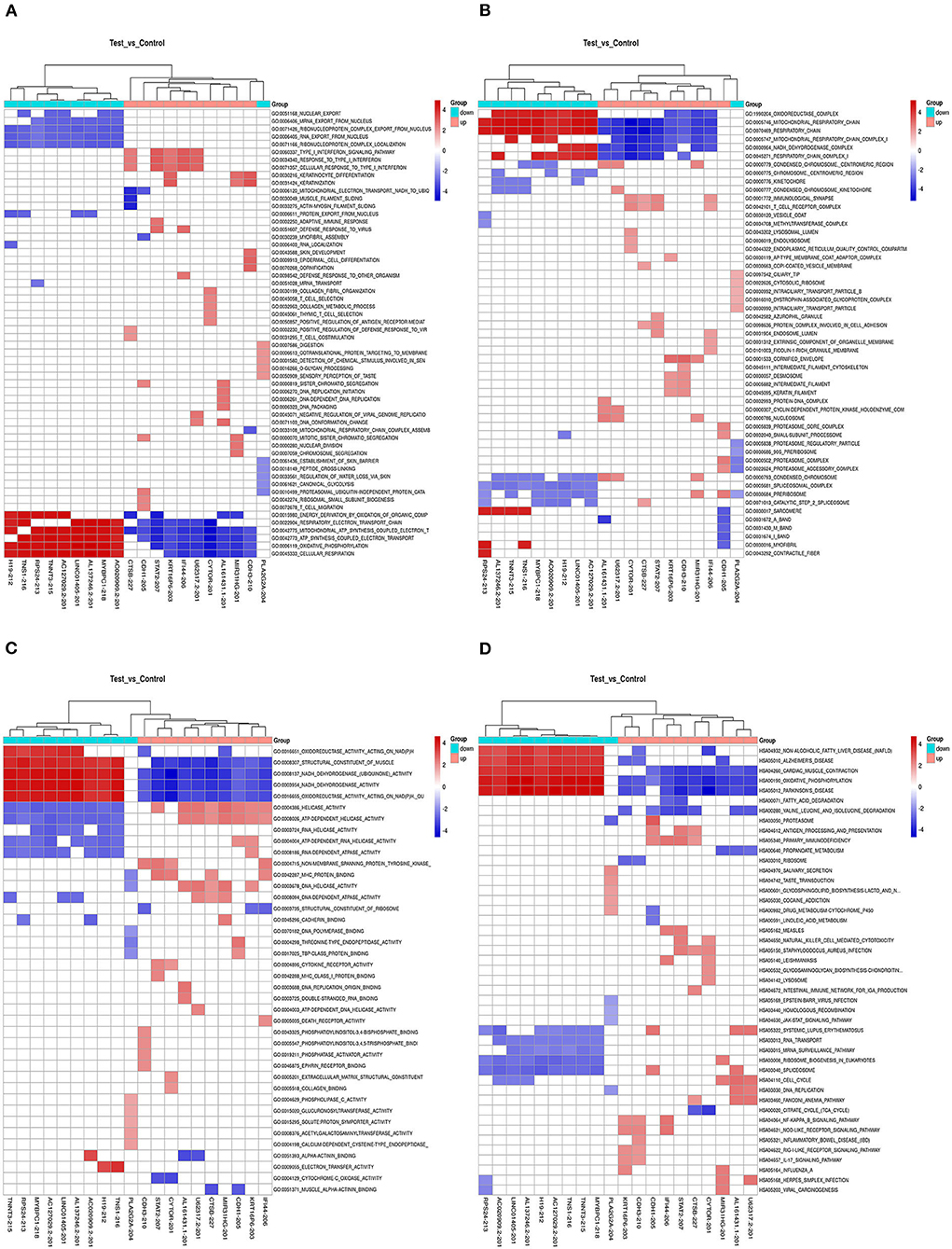
Figure 4. The GSEA results the pivotal DELs. (A) Biological processes of the DELs. (B) Cellular component of the DELs. (C) Molecular functions of the DELs. (D) Pathway analysis of the DELs.
Delineation of GO and KEGG Pathway Analyses of DEGs
GO enrichment analysis of the DEGs may reveal the roles of the significantly differentially regulated lncRNAs. Our data also showed that the biological processes associated with the upregulated mRNAs were immune system processes and the immune response (Figure 5A). The downregulated mRNAs were mostly enriched in the generation of precursor metabolites and energy, energy derivation by oxidation of organic compounds, and muscle system processes (Figure 5B). Regarding the cellular components, most of the upregulated mRNAs were related to the cytoplasm, intracellular and cytosol, and the downregulated mRNAs were mostly related to the contractile fiber, myofibril and sarcomere. Moreover, the molecular functions mainly associated with the upregulated mRNAs were protein binding, peptide binding, and amide binding, while those associated with the downregulated mRNAs were oxidoreductase activity, the structural constituent of muscle and actin binding. Importantly, KEGG pathway analysis of the DEGs revealed 20 pathways that could play pivotal roles in the mechanism of TSCC tumorigenesis, including the cell cycle, metabolic and NOD-like receptor signaling pathway (Figures 5C,D).
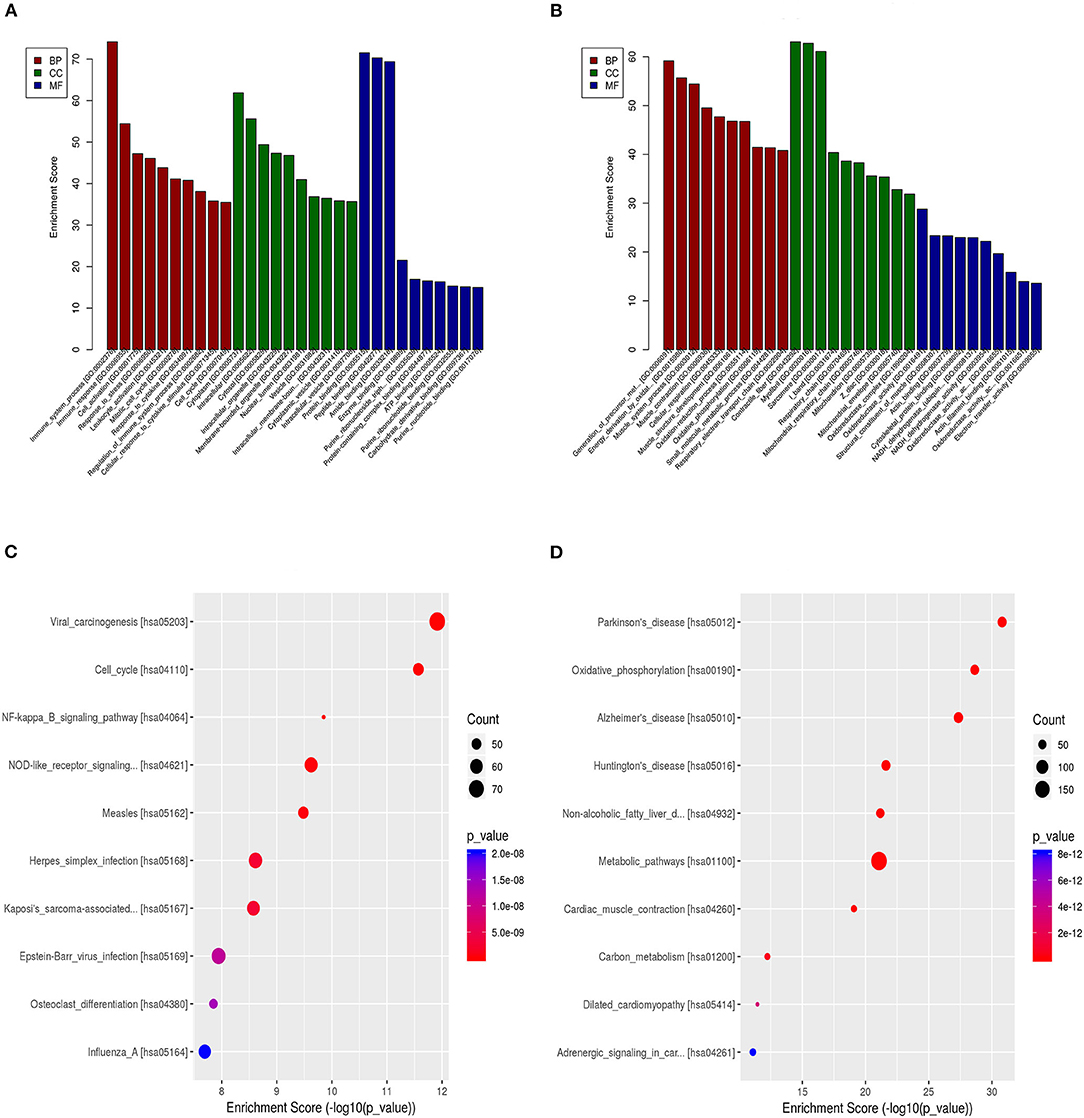
Figure 5. GO and pathway analyses of the DEGs. (A) GO annotation of upregulated mRNAs with the top 10 enrichment scores covering biological process, cellular component, and molecular function terms. (B) GO annotation of downregulated mRNAs with the top 10 enrichment scores covering biological process, cellular component, and molecular function terms. (C) Pathway analysis of upregulated DEGs. (D) Pathway analysis of downregulated DEGs.
Clinical Prognostic Significance of the Pivotal DELs for TSCC Patients
Then, the significance of the nine key DELs on the overall survival of patients with TSCC was evaluated by GEPIA and StarBase independently. According to the results obtained from GEPIA (Figure 6), RP11-54H7.4, and LINC00152 are related to poor overall survival in patients with HNSC, while according to StarBase (Figure 7), RP11-54H7.4 and LINC01405 are associated with a poor prognosis for patients with HNSC. Clinical prognostic information on LINC01405 was unavailable in GEPIA, and details on CTD-2545M3.8 were not accessible in StarBase.
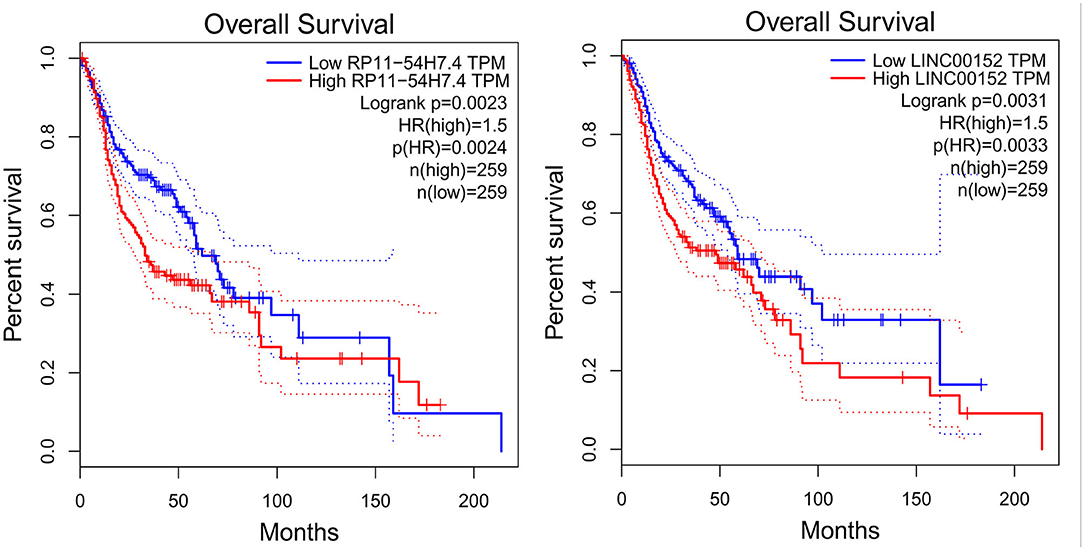
Figure 6. Clinical prognostic significance of RP11-54H7.4 and LINC00152 in HNSC by GEPIA. HNSC, head, and neck squamous cell carcinoma.
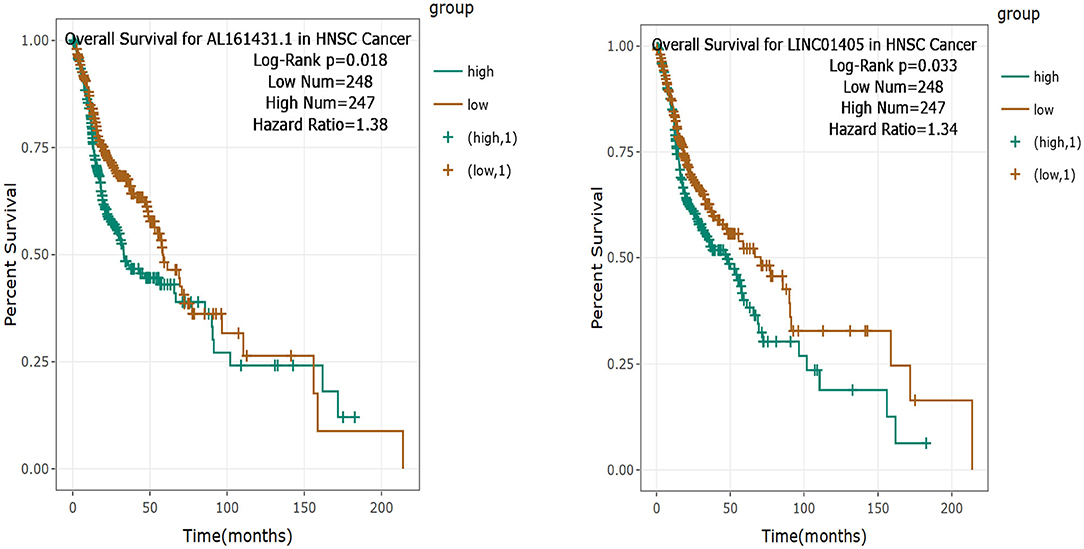
Figure 7. Clinical prognostic significance of RP11-54H7.4 and LINC01405 in HNSC by StarBase. HNSC, head, and neck squamous cell carcinoma.
Construction and Analysis of the lncRNA-TF-mRNA Network
Next, LncMAP was applied to determine lncRNA-TF-mRNA triplets in TSCC. Seven of the nine pivotal DELs associated with TFs were traced. Venn diagrams were used to identify 33 overlapping mRNAs (i.e., overlapping mRNA in triplets and DEGs; Table 3). The expression of the intersecting mRNAs was further confirmed by GEPIA. The expression of S100A12, HSPB3, GAMT, FABP5, ASB16, CPA4, LIMCH1, MLXIPL, PDZK1IP1, TRIM55, USP13, XIRP1, and ZNF106 was not significantly different between HNSC and normal tissues (Supplementary Figures 1, 2). Moreover, the coexpression of TFs and overlapping mRNAs was analyzed with StarBase (Supplementary Figures 3, 4). Pairs of closely related TFs-mRNAs were used to build a regulatory network. Ultimately, we successfully constructed and visualized the lncRNA-TF-mRNA network, including the upregulated (Figure 8A) and downregulated (Figure 8B) DELs, with Cytoscape.
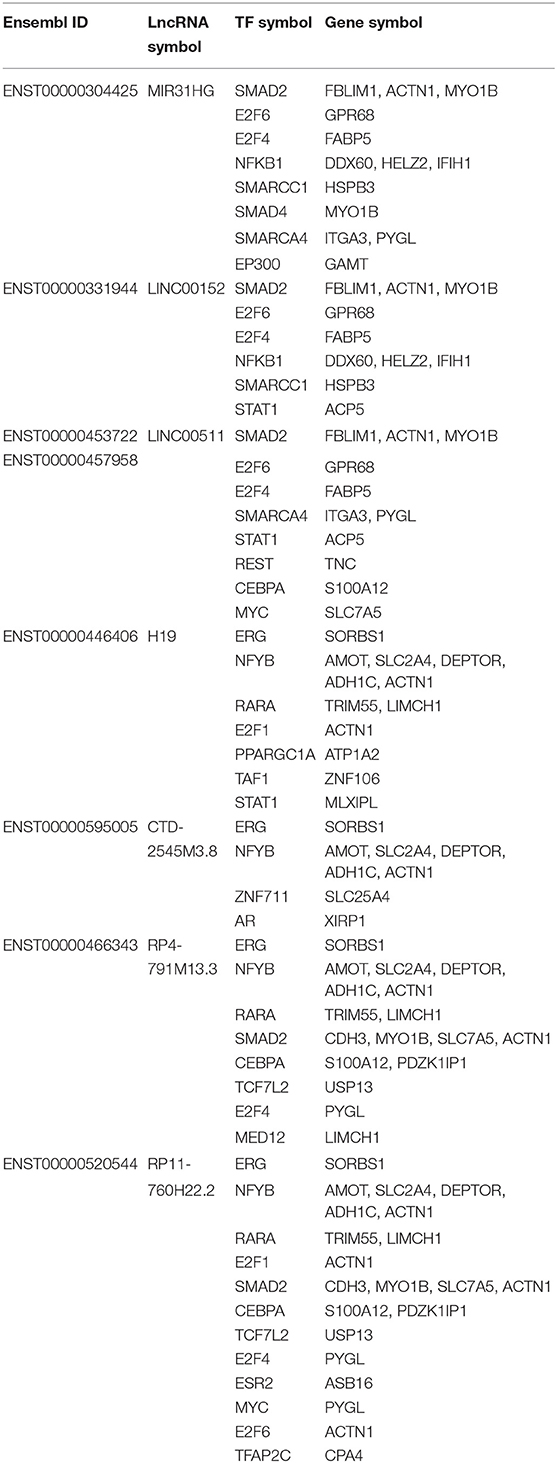
Table 3. Intersecting mRNAs between mRNA in triplets and DEGs.
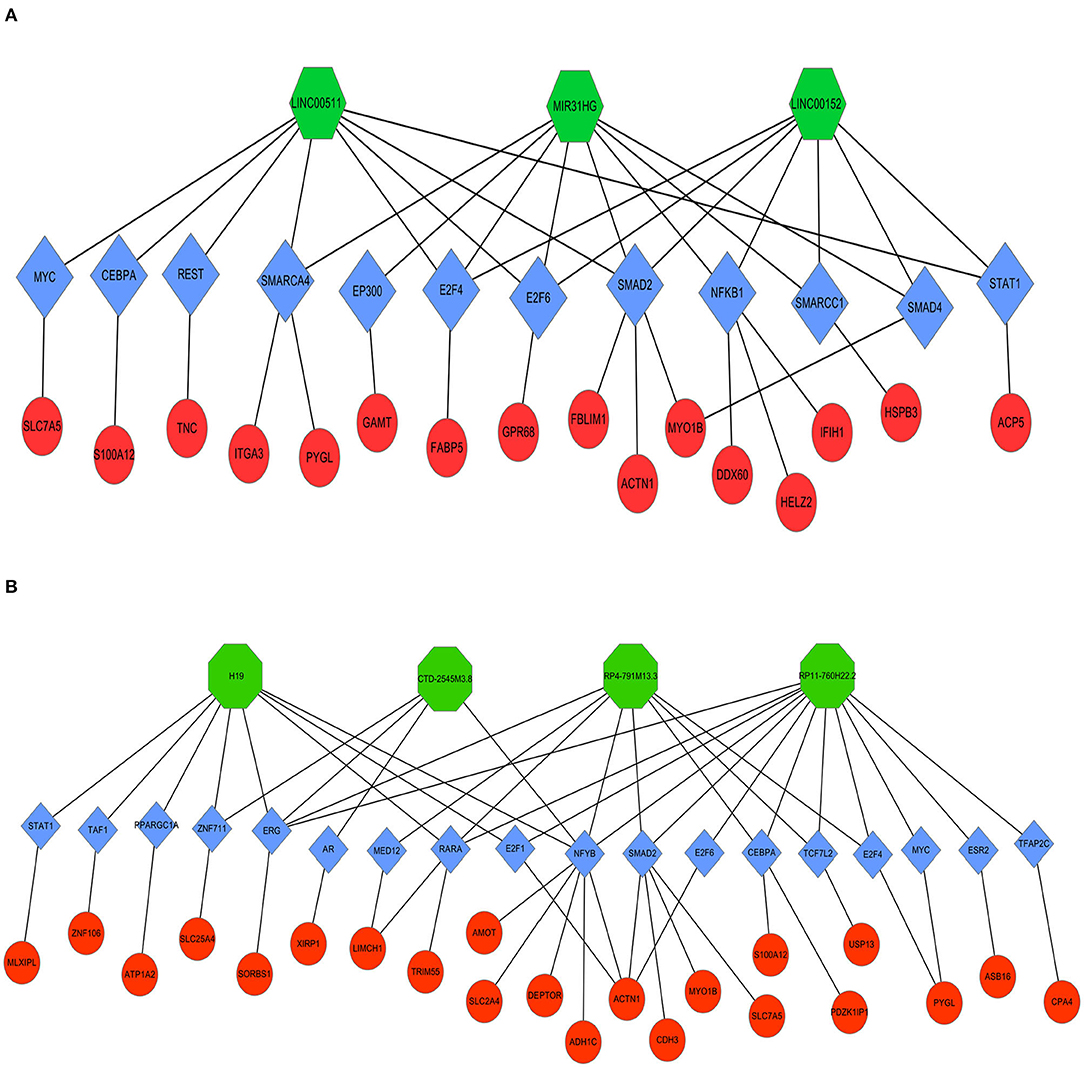
Figure 8. Coexpression regulatory network of lncRNAs-TFs-mRNAs in TSCC. Octagons represent lncRNAs. Diamonds represent TFs. Ellipses represent mRNAs. (A) Upregulated DELs and (B) Downregulated DELs.
Screening Biomarkers
Furthermore, we also compared our findings with the DELs screened by TCGA database. As expected, several biomarker expression levels in our results were consistent with TCGA. A few DELs worth noting are RP11-54H7.4, LINC00152, LINC01405, and RP11-760H22.2. The expression of RP11-54H7.4, LINC00152, LINC01405, and RP11-760H22.2 were then analyzed in the TSCC and normal tissues, and their expression in the paired groups (Figure 9). Moreover, several pivotal GSEA results of them were performed by GSEA software independently (Figure 10).
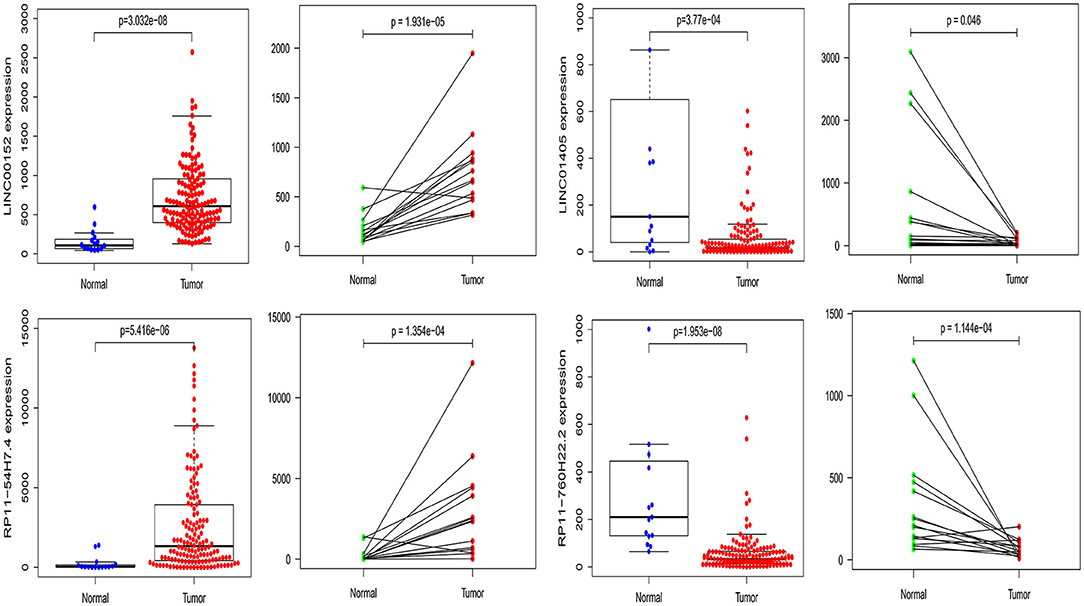
Figure 9. The expression of RP11-54H7.4, LINC00152, LINC01405, and RP11-760H22.2 were then analyzed in the TSCC and normal tissues, and their expression in the paired groups.
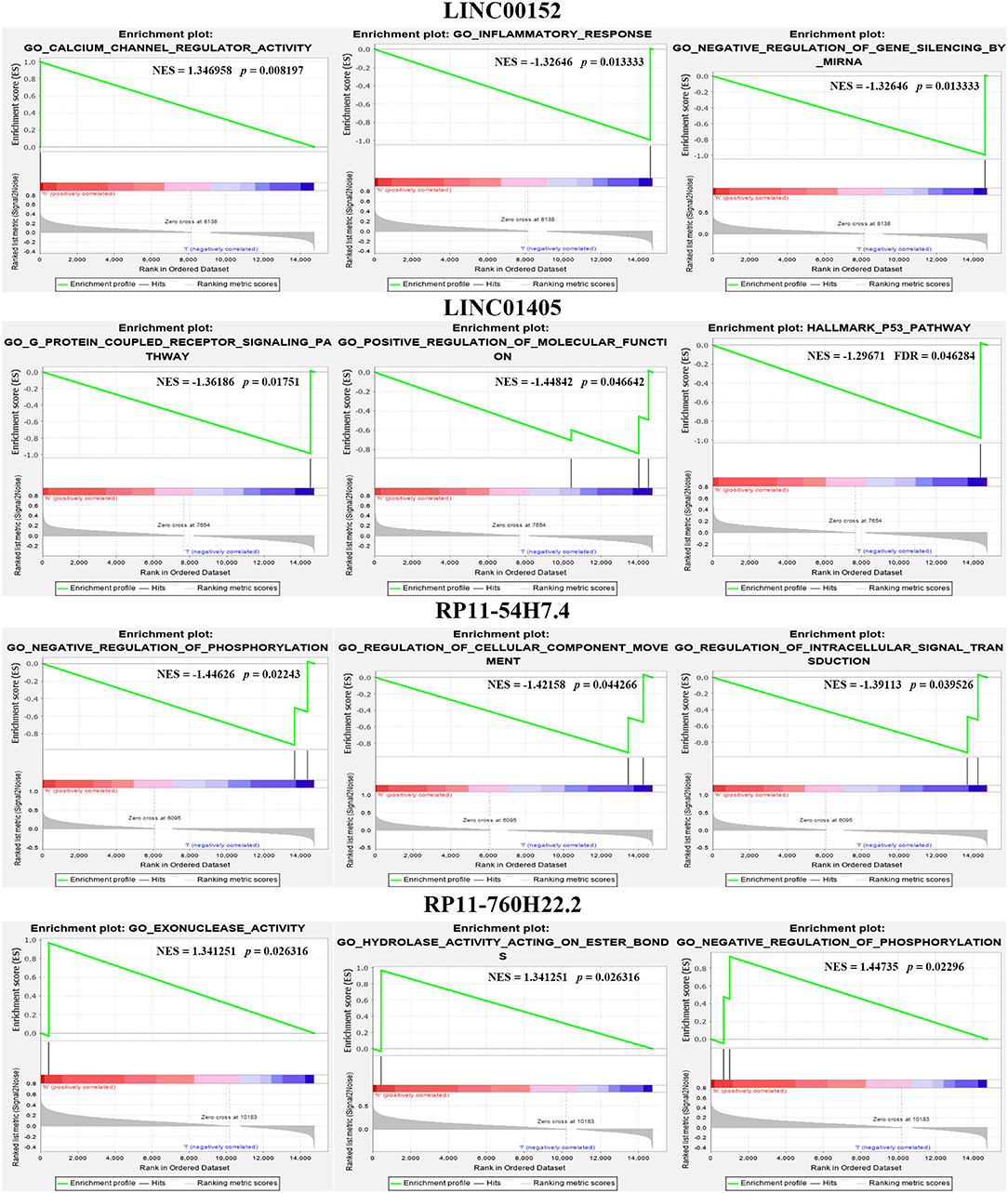
Figure 10. Several pivotal GSEA results of RP11-54H7.4, LINC00152, LINC01405, and RP11-760H22.2 were performed by GSEA software independently.
Validation of the Candidate Biomarkers
qRT-PCR was performed to verify the expression levels of the candidate DELs in TSCC cell lines. The expression of CDH1-205, MIR31HG, IFI44-204, IFI44-206, RP11-54H7.4, CTSB-227, HAS3-204, CDH3-210, LEPR-206, AMOT-205, CTD-2545M3.8, LINC01405, RP11-760H22.2, LINC00152, and LINC00511 was consistent with the sequencing results (Figure 11). All primers information are listed in Supplementary Table 3.
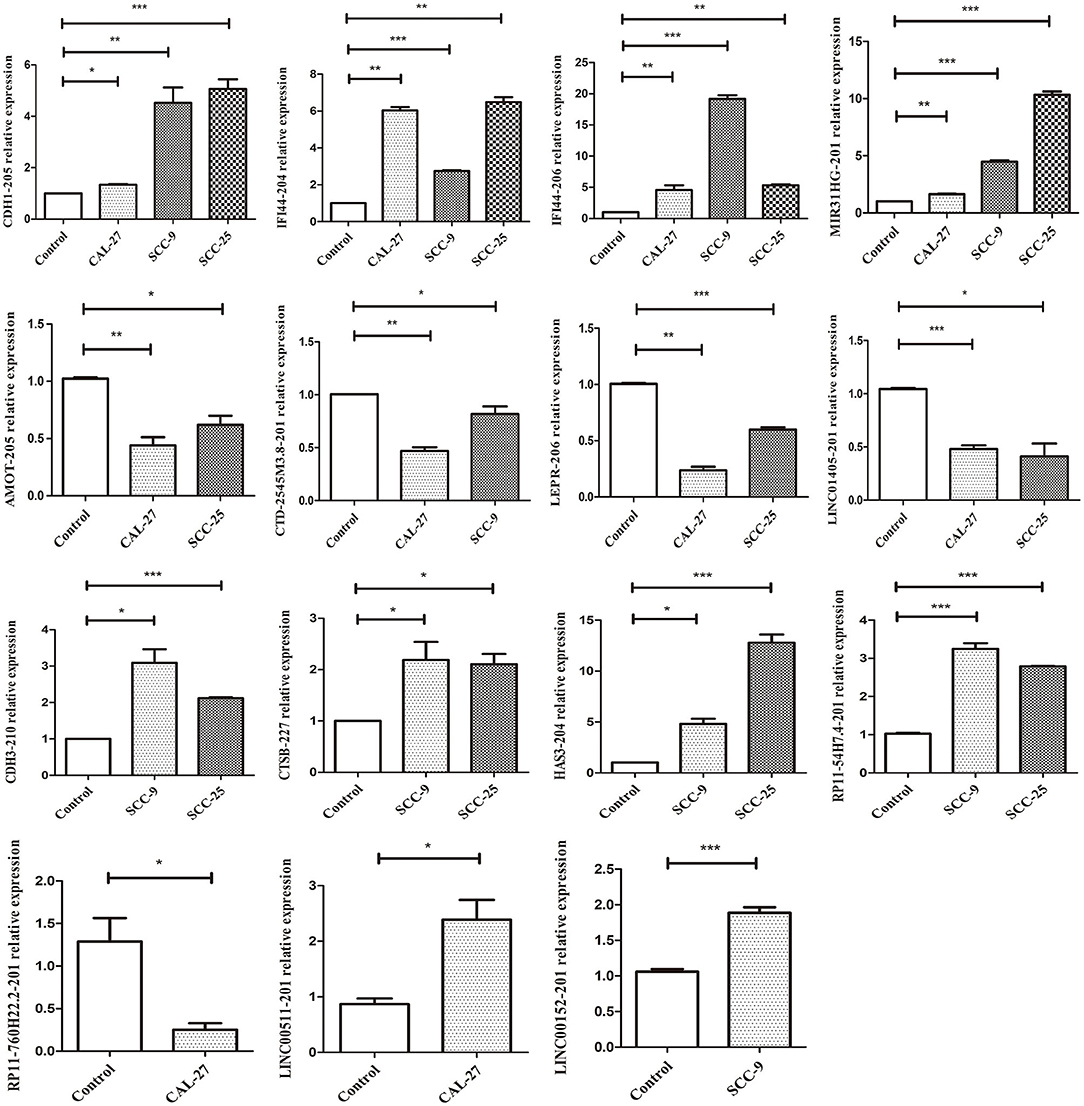
Figure 11. qRT-PCR validation of significant DELs from sequencing (***p < 0.001, **p < 0.01, and *p < 0.05).
Discussion
Various studies have demonstrated that lncRNAs are abnormally expressed in diverse tumor types and have broad application prospects in cancer diagnosis, monitoring, prognosis, and treatment response prediction (7, 16, 17).
TSCC is the most common type of OSCC, and its pathogenesis involves the dysregulation of gene networks, including both protein-coding genes and non-coding RNAs (6–8). To date, multiple lncRNAs have been identified in TSCC, providing new perspectives for exploring the molecular mechanism of TSCC pathogenesis (18). In the current study, we sought to screen aberrantly expressed lncRNAs by performing RNA-seq analysis of TSCC tumor tissues. These data indicate that 51,349 transcripts, 38,567 of which are protein coding, 4,224 mRNAs and 1,470 lncRNAs are differentially expressed between TSCC tumor and adjacent tissues. Through data preprocessing and limma package analysis, we identified 40 candidate DELs in TSCC. Then, 15 dysregulated lncRNAs were further verified using qRT-PCR, the results may be related to the heterogeneity between different TSCC cell lines. We also selected nine pivotal lncRNAs through an online public database. Among them, in TSCC, RP11-54H7.4, MIR31HG, LINC00152, and LINC00511 were upregulated, while H19, CTD-2545M3.8, RP11-760H22.2, RP4-791M13.3, and LINC01405 were consistently downregulated. The most well-known lncRNA, MIR31HG, can act as a HIF-1α coactivator to modulate hypoxia signaling pathways in oral cancer (17). It has also been reported that the lncRNA MIR31HG facilitates HNSC cell proliferation and tumorigenesis via HIF1A and p21 by promoting cell cycle progression and inhibiting cell apoptosis (19). A previous study showed that LINC00152 might promote the growth and invasion of OSCC by regulating miR-139-5p (20). The same study proved that LINC00511 could modulate TSCC progression (21). Various findings have revealed that H19 plays a critical role in the regulation of TSCC migration and invasion (22, 23). Our results are consistent with those of previous studies, and we also found that there is limited research on RP11-54H7.4, CTD-2545M3.8, RP11-760H22.2, RP4-791M13.3, and LINC01405 in TSCC pathogenesis.
GO analysis of the DELs revealed that some biological processes and molecular functions, such as cellular respiration, and oxidative phosphorylation could be involved in the progression of TSCC. Annotation of the most significant KEGG pathways associated with the DELs manifested the important pathways that could play pivotal roles in the mechanism of TSCC tumorigenesis, including oxidative phosphorylation, RNA transport, and mRNA surveillance pathway, suggesting that dysregulated lncRNAs may have a great influence on these targets by regulating associated pathways in TSCC. However, the functions of most lncRNAs are not well-understood.
The most significant GO terms of the DEGs were immune system processes, the immune response, and protein binding. KEGG pathway analysis of the DEGs revealed 20 pathways that could play pivotal roles in the mechanism of TSCC tumorigenesis, including the cell cycle, metabolic, and NOD-like receptor signaling pathway. The results suggest that these pathways might contribute significantly to the pathogenesis and progression of TSCC.
This study found that RP11-54H7.4, LINC00152, and LINC01405 are significantly aberrantly expressed based on the RNA-seq expression profile and associated with poor clinical outcomes in TSCC patients, the results are consistent with prior studies (24, 25). The expression of these lncRNAs was further confirmed in GEPIA and StarBase. Besides, the expression of RP11-54H7.4, LINC00152, LINC01405, and RP11-760H22.2 were also verified with TSCC samples in TCGA. Thus, RP11-54H7.4, LINC00152, and LINC01405 could represent prognostic biomarkers of TSCC.
Research shows that lncRNAs might play a role in trans-acting regulation via TFs, while the interactions between lncRNAs and TFs may ameliorate the expression levels of their target genes (26). The expression of target genes could also be regulated by TFs combining cis-elements at promoter locations (27). It is known that some TFs are involved in TSCC pathogenesis. To further investigate the functions of DELs in TSCC, seven of nine pivotal DELs associated TFs, namely, SMAD2, E2F6, E2F4, CEBPA, STAT1, MYC, ERG, and NFYB, were further analyzed. Research has demonstrated that SMAD2 acts as a predictor of overall survival in OSCC patients (28). Additionally, SMAD2 directly binds to the lncRNA MACC1-AS1 promoter and thus increases MACC1-AS1 expression in nasopharyngeal carcinoma (29). Studies have also shown that silencing the lncRNA CEBPA-AS1 can regulate OSCC cell proliferation, cell apoptosis, migration, and invasion by targeting CEBPA and via a novel pathway, CEBPA/Bcl2 (30). Similarly, abundant studies demonstrated that E2F4, STAT1, MYC, ERG, and NFYB may play an important role in the pathogenesis and progression of various cancers, including OSCC, and the underlying mechanism may be related to dysregulated lncRNA (31–37). All of these studies confirm that the TFs screened in our research play a critical role in tumor pathogenesis. However, the underlying mechanisms remain unknown. Therefore, we hypothesized that these TFs play vital roles in the tumorigenesis of TSCC by regulating lncRNAs and mRNAs. In addition, the relationship between lncRNAs and TFs requires further investigation.
The overlapping genes were screened by comparing the mRNA in triplets and DEGs. Some vital mRNAs, including MYO1B, ACTN1, PYGL, S100A12, and SLC7A5, are associated with TSCC (38–40). We also found that many TF expression levels were significantly correlated with the expression of multiple protein-coding genes. We thus deduce that these TFs might be correlated with the carcinogenesis of TSCC by regulating coexpressing genes. Therefore, a coexpression network was built to further investigate the relationship of the dysregulated TFs and mRNAs. We were able to successfully build the lncRNA-TF-mRNA network by combining all the results. The lncRNA-TF-mRNA coexpression network was constructed to predict lncRNA function. More critically, the interaction network of the significantly dysregulated lncRNAs with their TFs and coding genes was delineated, which might provide new clues for elucidating the underlying mechanism of TSCC.
Conclusion
To conclude, we found a profile of dysregulated lncRNAs, TFs, and mRNAs that could serve as prospective clinical biomarkers because of their tissue specificity and association with the tumorigenesis and progression of TSCC. Our study might lay a foundation for further functional research on lncRNAs-TFs-mRNAs in TSCC. The composite analysis of lncRNAs, TFs, and differentially coexpressed genes may provide a more comprehensive understanding of the pathogenesis of TSCC. Our results also suggest that specific lncRNAs, TFs, and mRNAs could be valuable for the diagnosis and treatment of tongue cancer, contribute to the application of lncRNAs in cancer and provide deep insights into the biological mechanism of TSCC. Intriguingly, this is the first study to show that RP11-54H7.4, LINC00152, and LINC01405 can be acted as a prognostic target for TSCC.
Similar to other approaches, our study also had certain limitations. First, in the survival analysis, because of the lack of clinical data that matched our data, it was difficult to further validate some prognosis-related lncRNAs. Furthermore, the interaction between lncRNAs, TFs, and mRNAs still lacks experimental verification. These deficiencies will be improved by complementing these data in the future.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/geo/, GSE149008/b.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee of the First Affiliated Hospital of Fujian Medical University. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
MZ: research design and implementation. ZC, YL, and YH: data analysis and interpretation. SZ, YJ, and LW: statistical analysis. MZ: writing of the manuscript. JC and BS: revision of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 81771126) and Startup Fund for Scientific Research, Fujian Medical University (Grant No. 2016QH075).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to express our sincere thanks to all those who have helped us in the course of our writing this paper.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.01421/full#supplementary-material
Abbreviations
DEL, differentially expressed lncRNA; DEG, differentially expressed mRNA; LncRNA, long non-coding RNA; TF, transcription factor; TSCC, tongue squamous cells carcinomas; HNSC, head and neck squamous cell carcinoma; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes.
References
1. Pannone G, Santoro A, Papagerakis S, Lo ML, De Rosa G, Bufo P. The role of human papillomavirus in the pathogenesis of head & neck squamous cell carcinoma: an overview. Infect Agents Cancer. (2011) 6:4. doi: 10.1186/1750-9378-6-4
3. Prensner JR, Chinnaiyan AM. The emergence of lncRNAs in cancer biology. Cancer Discov. (2011) 1:391–407. doi: 10.1158/2159-8290.CD-11-0209
4. Guo X, Gao L, Wang Y, Chiu DK, Wang T, Deng Y. Advances in long noncoding RNAs: identification, structure prediction and function annotation. Brief Funct Genomics. (2016) 15:38–46. doi: 10.1093/bfgp/elv022
5. Zhang Y, Liao G, Bai J, Zhang X, Xu L, Deng C, et al. Identifying cancer driver lncRNAs bridged by functional effectors through integrating multi-omics data in human cancers. Mol Ther Nucleic Acids. (2019) 17:362–73. doi: 10.1016/j.omtn.2019.05.030
6. Jia B, Xie T, Qiu X, Sun X, Chen J, Huang Z, et al. Long noncoding RNA FALEC inhibits proliferation and metastasis of tongue squamous cell carcinoma by epigenetically silencing ECM1 through EZH2. Aging. (2019) 11:4990–5007. doi: 10.18632/aging.102094
7. Zhang H, Zhao L, Wang YX, Xi M, Liu SL, Luo LL. Long non-coding RNA HOTTIP is correlated with progression and prognosis in tongue squamous cell carcinoma. Tumour Biol. (2015) 36:8805–9. doi: 10.1007/s13277-015-3645-2
8. Fang Z, Zhang S, Wang Y, Shen S, Wang F, Hao Y, et al. Long non-coding RNA MALAT-1 modulates metastatic potential of tongue squamous cell carcinomas partially through the regulation of small proline rich proteins. BMC Cancer. (2016) 16:706. doi: 10.1186/s12885-016-2735-x
9. Liang J, Liang L, Ouyang K, Li Z, Yi X. MALAT1 induces tongue cancer cells' EMT and inhibits apoptosis through Wnt/beta-catenin signaling pathway. J Oral Pathol Med. (2017) 46:98–105. doi: 10.1111/jop.12466
10. Wang ZY, Hu M, Dai M, Xiong J, Zhang S, Wu H, et al. Upregulation of the long non-coding RNA AFAP1-AS1 affects the proliferation, invasion and survival of tongue squamous cell carcinoma via the Wnt/β-catenin signaling pathway. Mol Cancer. (2018) 17:3. doi: 10.1186/s12943-017-0752-2
11. Fang Z, Wu L, Wang L, Yang Y, Meng Y, Yang H. Increased expression of the long non-coding RNA UCA1 in tongue squamous cell carcinomas: a possible correlation with cancer metastasis. Oral Surg Oral Med Oral Pathol Oral Radiol. (2014) 117:89–95. doi: 10.1016/j.oooo.2013.09.007
12. Zhang S, Ma H, Zhang D, Xie S, Wang W, Li Q, et al. LncRNA KCNQ1OT1 regulates proliferation and cisplatin resistance in tongue cancer via miR-211-5p mediated Ezrin/Fak/Src signaling. Cell Death Dis. (2018) 9:742. doi: 10.1038/s41419-018-0793-5
13. Liu X, Zhang B, Jia Y, Fu M. SNHG17 enhances the malignant characteristics of tongue squamous cell carcinoma by acting as a competing endogenous RNA on microRNA-876 and thereby increasing specificity protein 1 expression. Cell Cycle. (2020) 19:711–25. doi: 10.1080/15384101.2020.1727399
14. Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. (2017) 45:W98–102. doi: 10.1093/nar/gkx247
15. Li JH, Liu S, Zhou H, Qu LH, Yang JH. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. (2014) 42:D92–7. doi: 10.1093/nar/gkt1248
16. Arunkumar G, Murugan AK, Prasanna SRH, Subbiah S, Rajaraman R, Munirajan AK. Long non-coding RNA CCAT1 is overexpressed in oral squamous cell carcinomas and predicts poor prognosis. Biomed Rep. (2017) 6:455–62. doi: 10.3892/br.2017.876
17. Shih JW, Chiang WF, Wu A, Wu MH, Wang LY, Yu YL, et al. Long noncoding RNA LncHIFCAR/MIR31HG is a HIF-1alpha co-activator driving oral cancer progression. Nat Commun. (2017) 8:15874. doi: 10.1038/ncomms15874
18. Zhang L, Meng X, Zhu XW, Yang DC, Chen R, Jiang Y, et al. Long non-coding RNAs in Oral squamous cell carcinoma: biologic function, mechanisms and clinical implications. Mol Cancer. (2019) 18:102. doi: 10.1186/s12943-019-1021-3
19. Wang R, Ma Z, Feng L, Yang Y, Tan C, Shi Q, et al. LncRNA MIR31HG targets HIF1A and P21 to facilitate head and neck cancer cell proliferation and tumorigenesis by promoting cell-cycle progression. Mol Cancer. (2018) 17:162. doi: 10.1186/s12943-018-0916-8
20. Li M, Ning J, Li Z, Wang J, Zhao C, Wang L. LINC00152 promotes the growth and invasion of oral squamous cell carcinoma by regulating miR-139-5p. Onco Targets Ther. (2018) 11:6295–304. doi: 10.2147/OTT.S168807
21. Ding J, Yang C, Yang S. LINC00511 interacts with miR-765 and modulates tongue squamous cell carcinoma progression by targeting LAMC2. J Oral Pathol Med. (2018) 47:468–76. doi: 10.1111/jop.12677
22. Zhang DM, Lin ZY, Yang ZH, Wang YY, Wan D, Zhong JL, et al. IncRNA H19 promotes tongue squamous cell carcinoma progression through beta-catenin/GSK3beta/EMT signaling via association with EZH2. Am J Transl Res. (2017) 9:3474–86.
23. Kou N, Liu S, Li X, Li W, Zhong W, Gui L, et al. H19 facilitates tongue squamous cell carcinoma migration and invasion via sponging miR-let-7. Oncol Res. (2019) 27:173–82. doi: 10.3727/096504018X15202945197589
24. Yu J, Liu Y, Guo C, Zhang S, Gong Z, Tang Y, et al. Upregulated long non-coding RNA LINC00152 expression is associated with progression and poor prognosis of tongue squamous cell carcinoma. J Cancer. (2017) 8:523–30. doi: 10.7150/jca.17510
25. Pentenero M, Bowers LM, Jayasinghe R, Yap T, Cheong SC, Kerr AR, et al. World workshop on oral medicine VII: clinical evidence of differential expression of lncRNAs in oral squamous cell carcinoma: a scoping review. Oral Dis. (2019) 25 (Suppl. 1):88–101. doi: 10.1111/odi.13076
26. Vance KW, Sansom SN, Lee S, Chalei V, Kong L, Cooper SE, et al. The long non-coding RNA paupar regulates the expression of both local and distal genes. EMBO J. (2014) 33:296–311. doi: 10.1002/embj.201386225
27. Tian L, Cao J, Deng X, Zhang C, Qian T, Song X, et al. Unveiling transcription factor regulation and differential co-expression genes in duchenne muscular dystrophy. Diagn Pathol. (2014) 9:210. doi: 10.1186/s13000-014-0210-z
28. Mangone FR, Walder F, Maistro S, Pasini FS, Lehn CN, Carvalho MB, et al. Smad2 and Smad6 as predictors of overall survival in oral squamous cell carcinoma patients. Mol Cancer. (2010) 9:106. doi: 10.1186/1476-4598-9-106
29. Chen S, Luo X, Wu W, Li Y, Yu H, Wang Y, et al. The long non-coding RNA MACC1-AS1 promotes nasopharyngeal carcinoma cell stemness via suppressing miR-145-mediated inhibition on SMAD2/MACC1-AS1 axis. Biomed Pharmacother. (2020) 125:109986. doi: 10.1016/j.biopha.2020.109986
30. Guo Y, Ma Y, Hu X, Song R, Zhu L, Zhong M. Long non-coding RNA CEBPA-AS1 correlates with poor prognosis and promotes tumorigenesis via CEBPA/Bcl2 in oral squamous cell carcinoma. Cancer Biol Ther. (2018) 19:205–13. doi: 10.1080/15384047.2017.1416276
31. Russo G, Zamparelli A, Howard CM, Minimo C, Bellan C, Carillo G, et al. Expression of cell cycle-regulated proteins pRB2/p130, p107, E2F4, p27, and pCNA in salivary gland tumors: prognostic and diagnostic implications. Clin Cancer Res. (2005) 11:3265–73. doi: 10.1158/1078-0432.CCR-04-2508
32. Jung HM, Patel RS, Phillips BL, Wang H, Cohen DM, Reinhold WC, et al. Tumor suppressor miR-375 regulates MYC expression via repression of CIP2A coding sequence through multiple miRNA-mRNA interactions. Mol Biol Cell. (2013) 24:1638–48, S1–7. doi: 10.1091/mbc.e12-12-0891
33. Wu L, Zhao JC, Kim J, Jin HJ, Wang CY, Yu J. ERG is a critical regulator of Wnt/LEF1 signaling in prostate cancer. Cancer Res. (2013) 73:6068–79. doi: 10.1158/0008-5472.CAN-13-0882
34. Bing F, Zhao Y. Screening of biomarkers for prediction of response to and prognosis after chemotherapy for breast cancers. Onco Targets Ther. (2016) 9:2593–600. doi: 10.2147/OTT.S92350
35. Arunkumar G, Anand S, Raksha P, Dhamodharan S, Prasanna SRH, Subbiah S, et al. LncRNA OIP5-AS1 is overexpressed in undifferentiated oral tumors and integrated analysis identifies as a downstream effector of stemness-associated transcription factors. Sci Rep. (2018) 8:7018. doi: 10.1038/s41598-018-25451-3
36. Xu H, Jiang Y, Xu X, Su X, Liu Y, Ma Y, et al. Inducible degradation of lncRNA Sros1 promotes IFN-γ-mediated activation of innate immune responses by stabilizing Stat1 mRNA. Nat Immunol. (2019) 20:1621–30. doi: 10.1038/s41590-019-0542-7
37. Ryan N, Anderson K, Volpedo G, Hamza O, Varikuti S, Satoskar AR, et al. STAT1 inhibits T-cell exhaustion and myeloid derived suppressor cell accumulation to promote antitumor immune responses in head and neck squamous cell carcinoma. Int J Cancer. (2020) 146:1717–29. doi: 10.1002/ijc.32781
38. Ohmura G, Tsujikawa T, Yaguchi T, Kawamura N, Mikami S, Sugiyama J, et al. Aberrant myosin 1b expression promotes cell migration and lymph node metastasis of HNSCC. Mol Cancer Res. (2015) 13:721–31. doi: 10.1158/1541-7786.MCR-14-0410
39. Gao W, Li JZ, Chen SQ, Chu CY, Chan JY, Wong TS. Decreased brain-expressed X-linked 4 (BEX4) expression promotes growth of oral squamous cell carcinoma. J Exp Clin Cancer Res. (2016) 35:92. doi: 10.1186/s13046-016-0355-6
Keywords: tongue squamous cell carcinomas, lncRNA, mRNA, transcription factor, network
Citation: Zhang M, Chen Z, Zhang S, Wu L, Jie Y, Liao Y, Huang Y, Chen J and Shi B (2020) Analysis of Differentially Expressed Long Non-coding RNAs and the Associated TF-mRNA Network in Tongue Squamous Cell Carcinoma. Front. Oncol. 10:1421. doi: 10.3389/fonc.2020.01421
Received: 13 April 2020; Accepted: 06 July 2020;
Published: 14 August 2020.
Edited by:
Wei Cao, Shanghai Jiao Tong University, ChinaReviewed by:
Zheqi Liu, Shanghai Jiao Tong University, ChinaYanjie Zhang, Shanghai Jiao Tong University, China
Copyright © 2020 Zhang, Chen, Zhang, Wu, Jie, Liao, Huang, Chen and Shi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiang Chen, amlhbmdjaGVuQGZqbXUuZWR1LmNu; Bin Shi, c2hpYmluQGZqbXUuZWR1LmNu