 Linyuan Xue
Linyuan Xue Hongyu Qi
Hongyu Qi He Zhang
He Zhang Lu Ding
Lu Ding Qingxia Huang
Qingxia Huang Daqing Zhao2
Daqing Zhao2 Boyang Jason Wu
Boyang Jason Wu Xiangyan Li
Xiangyan Li- 1Research Center of Traditional Chinese Medicine, College of Traditional Chinese Medicine, Changchun University of Chinese Medicine, Changchun, China
- 2Key Laboratory of Active Substances and Biological Mechanisms of Ginseng Efficacy, Ministry of Education, Jilin Provincial Key Laboratory of Bio-Macromolecules of Chinese Medicine, Jilin Ginseng Academy, Changchun University of Chinese Medicine, Changchun, China
- 3College of Traditional Chinese Medicine, Changchun University of Chinese Medicine, Changchun, China
- 4Department of Pharmaceutical Sciences, College of Pharmacy and Pharmaceutical Sciences, Washington State University, Spokane, WA, United States
Recently, targeting metabolic reprogramming has emerged as a potential therapeutic approach for fighting cancer. Sterol regulatory element binding protein-2 (SREBP-2), a basic helix-loop-helix leucine zipper transcription factor, mainly regulates genes involved in cholesterol biosynthesis and homeostasis. SREBP-2 binds to the sterol regulatory elements (SREs) in the promoters of its target genes and activates the transcription of mevalonate pathway genes, such as HMG-CoA reductase (HMGCR), mevalonate kinase and other key enzymes. In this review, we first summarized the structure of SREBP-2 and its activation and regulation by multiple signaling pathways. We then found that SREBP-2 and its regulated enzymes, including HMGCR, FPPS, SQS, and DHCR4 from the mevalonate pathway, participate in the progression of various cancers, including prostate, breast, lung, and hepatocellular cancer, as potential targets. Importantly, preclinical and clinical research demonstrated that fatostatin, statins, and N-BPs targeting SREBP-2, HMGCR, and FPPS, respectively, alone or in combination with other drugs, have been used for the treatment of different cancers. This review summarizes new insights into the critical role of the SREBP-2-regulated mevalonate pathway for cancer and its potential for targeted cancer therapy.
Introduction
Sterol regulatory-element binding proteins (SREBPs) were first identified as a subclass of membrane-bound, basic helix-loop-helix leucine zipper (bHLH-LZ) transcription factors which regulate the promoters of genes involved in lipid synthesis and uptake pathways (1–3). In mammals, two genes, SREBF1 and SREBF2, express three major SREBP proteins (SREBP-1a, SREBP-1c, and SREBP-2) with distinct but overlapping lipogenic transcriptional programs (3, 4). Most studies report that SREBP-1a and SREBP-1c primarily regulate fatty acid metabolism and that SREBP-2 is a main regulator of cholesterol metabolism (5–8). Over the past 30 years, the functions of SREBPs have been identified to participate in numerous crucial physiologic processes (9), highlighting metabolic integrators in cellular homeostasis (10, 11). Accumulating evidence has revealed that SREBPs integrate multiple cell signals to control lipogenesis as well as unexpected pathways in type II diabetes, atherosclerosis, and a series of cancers (12, 13).
In particular, multiple SREBP-2-mediated pathways have been extensively studied as attractive potential targets for cancer therapy (14–16). As reported, SREBP-2 binds to the sterol regulatory elements (SREs) in the promoters of its target genes and activates the transcription of mevalonate pathway genes, such as 3-hydroxy-3-methyl-glutaryl-CoA (HMG-CoA) reductase (HMGCR), mevalonate kinase (MVK), and other key enzymes (1). Recent reports found that the mevalonate pathway and its metabolites are essential for cancer growth and malignant progression in a series of cancers, including prostate, breast, lung, and liver cancer (17, 18). Moreover, multiple key pathways, such as the p53 and phosphatidylinositol-3-kinase (PI3K)/Akt signaling pathways, lead to the activation of SREBP-2 to promote tumorigenesis (19–21). Based on the findings above, targeting SREBP-2 and mevalonate pathways has emerged as an encouraging strategy for cancer therapy.
In this review, we first summarized recent advances in the study of SREBP-2 structure, activation, and regulation, followed by SREBP-2, key enzymes of mevalonate pathway, their regulation by various signal pathways or metabolites, and their roles in different cancers. Finally, we focused on the inhibition of the SREBP-2-regulated mevalonate pathway by fatostatin, natural products, statins, or amino-bisphosphonates (N-BPs), alone or in combination with other drugs, as potential therapeutic strategies for various cancers. This review provides new insights into the critical role of SREBP-2-regulated mevalonate metabolism in cancer and its potential as a target for cancer therapy.
SREBP-2 Structure, Activation, and Regulation
SREBP-2 Structure
Human SREBP-2, identified by cDNA cloning in 1993, is produced from one gene, SREBF-2, 72kb, human chromosome 22q13, as the result of alternative promoter usage and transcription start sites (1). Similar to SREBP-1 structure, SREBP-2 contains 1,141 amino acids and includes an NH2-terminal transcription factor domain, a middle hydrophobic region and a COOH-terminal regulatory domain (3). The NH2-terminal domain with ~480 amino acids contains the bHLH-Zip motif (DNA binding) and an acidic transcriptional motif (transcriptional activity), which binds co-activator specificity protein 1 (SP1) or nuclear transcription factor Y (NF-Y) to regulate gene expression (22, 23). A middle hydrophobic region of SREBP-2 with approximately 80 amino acids, a membrane-binding region, consists of two hydrophobic membrane-spanning segments separated by a hydrophilic loop, which extends into the lumen of the endoplasmic reticulum (ER). The COOH-terminal regulatory domain contains approximately 590 amino acids responsible for SREBP-2 subcellular localization and translocation (Figure 1A) (24).
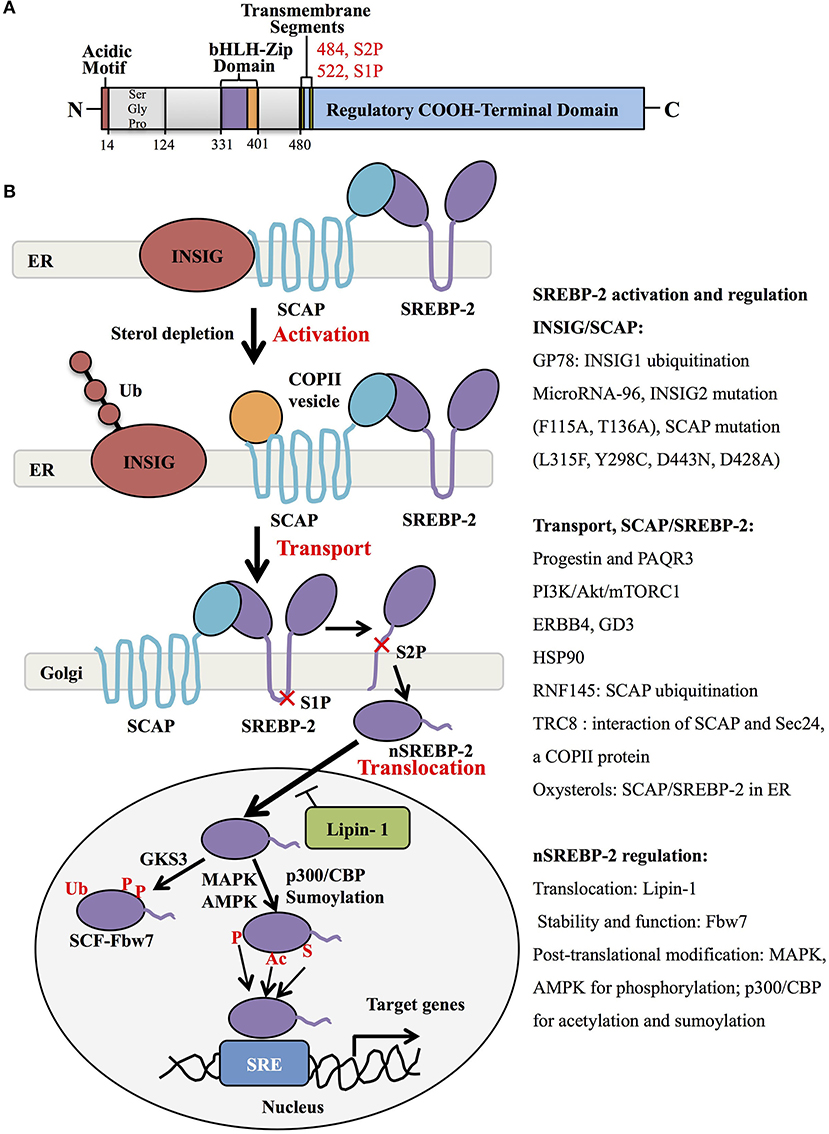
Figure 1. SREBP-2 structure, activation, and regulation. (A) SREBP-2 protein consists of three domains, including an NH2-terminal regulatory domain, a middle hydrophilic region, and a COOH-terminal regulatory domain. The NH2-terminal domain contains the bHLH-Zip motif and an acidic transcriptional motif. (B) SREBP-2 activation, transport, and translocation. After INSIG dissociation from SCAP by sterol depletion, SREBP-2 translocates to the Golgi apparatus and is cleaved by S1P and S2P proteases to release the NH2-terminal fragment of SREBP-2 (nSREBP-2). nSREBP-2 translocation and stability are regulated by multiple signaling pathways at different levels. ER, endoplasmic reticulum; INSIG, insulin-induced gene protein; SCAP, SREBP cleavage-activation protein; COPII, coatomer protein II; S1P, site-1 protease; S2P, site-2 protease; Ub, ubiquitination; GSK3, glycogen synthase kinase 3; P, phosphorylation; SCF-Fbw7, SKP1-cullin-F-Box protein-F-box and WD repeat domain-containing 7; AMPK, adenosine monophosphate-activated protein kinase; MAPK, mitogen-activated protein kinase; p300/CBP, p300 and cyclic AMP response element-binding protein; Ac, acetylation; S, Sumoylation; SRE, sterol regulatory element; GP78, a membrane-anchored ubiquitin ligase; PAQR3, progestin and adipoQ receptors member 3; ERBB4, Erb-b2 receptor tyrosine kinase 4; GD3, a dominant melanoma ganglioside; HSP90, heat shock protein 90; TRC8, translocation in renal cancer from chromosome 8; RNF145, RNF finger protein 145.
SREBP-2 Activation
Generally, SREBP-2 is synthesized as 125kDa inactive precursors in the ER (9). The COOH-terminal domain of SREBP-2 binds to the WD-repeat domain of SREBP cleavage-activation protein (SCAP), while the NH2-terminal domain of SCAP binds to the ER-resident insulin-induced gene proteins (INSIG), including INSIG1 and INSIG2, to form a complex of INSIG/SCAP/SREBP-2 for maintaining SREBP-2 in the ER (25, 26). When sterol level decreases, SCAP dissociates from INSIGs and facilitates the incorporation of SCAP/SREBP into coatomer protein II (COPII)-coated vesicles, which then transports the complex from the ER to the Golgi apparatus (27, 28). In the Golgi, SREBP-2 is sequentially cleaved by two membrane-bound proteases, site-1 protease (S1P) (29) and site-2 protease (S2P) (30) to release the NH2-terminal form of this transcription factor (nuclear SREBP-2, nSREBP-2) (28). This form translocates to the nucleus and binds to the sterol regulatory element (SRE) of target genes, including key enzymes of cholesterol biosynthesis and uptake (4).
Many studies have shown that the INSIG/SCAP/SREBP-2 complex and the transport of SREBP-2 from ER to Golgi are regulated by multiple signaling proteins. When sterols in the ER membrane are high, they bind to loop 1 of SCAP and switch the conformation of SCAP to interact with INSIG protein, which blocks COPII binding and causes the maintenance of the SCAP/SREBP-2 complex in the ER (31) (Figure 1B, upper). Three different mutants within the sterol-sensing domain of SCAP (L315F, Y298C, and D443N) disrupt the interaction of SCAP/INSIG to abolish the sterol-mediated feedback regulation of SREBP processing (32). A recent report showed that heat shock protein 90 (HSP90) stabilized the SCAP/SREBP complex to facilitate SREBP activation (33). Another intrinsic protein encoding an E3 ubiquitin ligase in ER, TRC8 (translocation in renal cancer from chromosome 8), is capable of binding both SREP-2 and SCAP to form a TRC8/SREBP-2/SCAP complex, which hampers the interaction between SCAP and Sec24, a COPII protein, to reduce the cleavage of SREBP-2 (34). Meanwhile, INSIG-1 binds to GP78, a membrane-bound ubiquitin ligase with high affinity, and is then ubiquitinated and rapidly degraded in sterol-depleted cells. However, INSIG-2 lacks interaction with Gp78, which may be related to its slower degradation than INSIG-1 (35, 36). In addition, oxysterols such as 25-hydroxycholesterol bind directly to INSIGs to trigger ER retention of the SCAP/SREBP-2 complex. Mutations at F115A and T136A of the transmembrane helices of INSIG-2 are important for binding to oxysterols and SCAP (37).
For the transporting process, the mutant SCAP with aspartic acid replacement by alanine at 428 (D428A) fails to dissociate from INSIGs and impairs the transportation of SREBP-2 to the Golgi (38). Similar to INSIG-1, INSIG-2 binds SCAP to block the export of SREBPs in the absence of exogenous sterols (25), which is inhibited by microRNA-96 to increase the abundance of active SREBP-2 (39). Furthermore, several signaling proteins were reported to control the transport of SREBP-2. One study showed that Golgi-localized transmembrane protein progestin and adipoQ receptor 3 (PAQR3) interacted with the SCAP/SREBP-2 complex to remain in the Golgi, which was disrupted to reduce cholesterol biosynthesis (40). Another report demonstrated that a RING-finger ubiquitin ligase, RNF finger protein 145, triggered the ubiquitination of SCAP on lysine residues within a cytoplasmic loop, potentially inhibiting the transport of SREBP-2 to Golgi and subsequent SREBP-2 processing (41). Additionally, the PI3K/Akt/mTORC1 pathway is involved in SREBP-2 transport to the Golgi, contributing to SREBP-2 activation (42, 43), which can be activated by neuregulin-activated ERBB4 and melanoma antigen ganglioside GD3 (19, 44). Collectively, the INSIG/SCAP/SREBP-2 complex and SREBP-2 transportation from ER to Golgi are regulated by multiple signaling molecules, as summarized in Figure 1B.
SREBP-2 Regulation at Different Levels
After the cleavage of full-length SREBP-2 by S1P and S2P in the Golgi, nSREBP-2 can translocate to the nucleus and be regulated at protein expression and transcription levels. The nutrient and growth factor-responsive kinase mTOR complex 1 (mTORC1) causes Lipin-1, a phosphatidic acid phosphatase, to reside in the cytoplasm, which increases the expression of nSREBP-2 protein (45, 46). mTORC1 can also suppress cholesterol delivery to lysosomes through the inhibition of autophagy and the maintenance of endosomal recycling, which reduces the level of cholesterol in ER to activate SREBP-2 (47). A nuclear receptor protein, peroxisome proliferator-activated receptor (PPAR) α in in rat liver cells, leads to a decrease of nSREBP-2 to lower cholesterol concentration (48). In addition, the stability and function of nuclear SREBP-2 are negatively regulated by a substrate receptor of the SCF ubiquitin ligase complex, Fbw7, through ubiquitination and proteasome-mediated degradation in a phosphorylation-dependent manner (49).
Importantly, the transcriptional activity of nSREBP-2 is also modulated by various post-translational modifications, including phosphorylation, acetylation, and sumoylation. For the phosphorylated regulation, insulin-activated Erk-mitogen-activated protein kinase (MAPK) increases SREBP-2 activity by phosphorylation at serine 432 and 455 (50). Glycogen synthase kinase 3 directly phosphorylates Ser443 on SREBP-2 to mediate Fbw7-induced ubiquitination and degradation of nSREBP-2 (49). A synthetic polyphenol, S17834, can promote AMP-activated protein kinase (AMPK) activation to decrease SREBP-2 transcription via its phosphorylation site on SREBP-2 (51). Aside from phosphorylation, histone acetyltransferase p300/CREB-binding protein (CBP) can bind and acetylate the N-terminus of SREBP-2 to enhance its expression and transcriptional activity (52), while sirtuin-1 (SIRT1) deacetylates SREBP-2 to decrease the abundance of SREBP-2 in the nucleus (53). Additionally, nSREBP-2 at Lys464 is also modified by sumoylation to decrease transcriptional activity (54). Taken together, SREBP-2 stability and activation are regulated by a series of key molecules and signaling pathways, which hold promise for understanding the role of SREBP-2 in physiological and pathological procedures.
The SREBP-2-Regulated Mevalonate Pathway
In the nucleus, nSREBP-2 binds to SREs in the promoter of target genes to activate the gene expression of most of the enzymes involved in the mevalonate pathway, including HMGCR, MVK, squalene synthase (SQS) (55), and 24-dihydrocholesterol reductase (DHCR24) (56), as well as increasing the expression of low-density lipoprotein receptors (LDLR) for exogenous cholesterol uptake (8, 57). For the mevalonate pathway, two molecules of acetyl-CoA from glucose metabolism or fatty acid degradation form acetoacetyl-CoA by acetoacetyl-CoA thiolase. In the presence of HMG-CoA synthase (HMGCS), acetyl-CoA and acetoacetyl-CoA form HMG-CoA, which is converted to mevalonate by HMGCR (58). Then, the mevalonate is phosphorylated sequentially to 5-phosphomevalonate by mevalonate kinase (MK) and to 5-pyrophosphomevalonate by phosphomevalonate kinase (PMK), which is further synthesized to isopentenylpyrophosphate (IPP) by mevalonate diphosphate decarboxylase (59). Furthermore, IPP and its isomer, dimethylallyl pyrophosphate (DMPP), can form geranyl pyrophosphate (GPP) by farnesylpyrophosphate synthase (FPPS, FDPS), which is condensed with another IPP to yield farnesylpyrophosphate (FPP). By the action of SQS, FPP is converted to squalene (60), which is converted sequentially to monooxidaosqualene (MOS) and lanosterol by squalene monooxygenase (SM) and lanosterol synthase, respectively (18). Lastly, lanosterol is further metabolized to cholesterol by 19 enzymes, including CYP51A (lanosterol-14α demethylase), TM7SF2 (steroid 14 reductase), SC4MOL (4 methyl sterol oxidase), NSDHL (C3 sterol dehydrogenase), HSD17B7 (3-ketoreductase), EBP (phenylalkylamine Ca2+ antagonist binding protein), SC5D (sterol-C5-desaturase), 7-dehydrocholesterol reductase (DHCR7), and DHCR24 (17, 56, 61). As reported, cholesterol plays a crucial role in maintaining the structure and function of cellular membranes and is also a precursor of steroid hormones and vitamin D (62). Collectively, SREBP-2 controls cholesterol biosynthesis by regulating mevalonate metabolism enzymes (Figure 2).
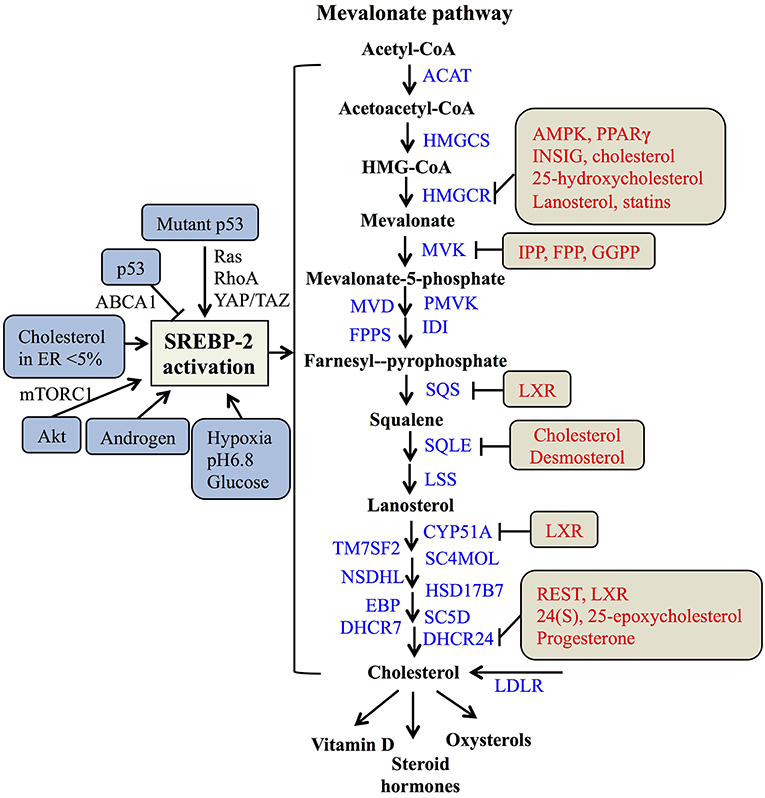
Figure 2. The SREBP-regulated mevalonate pathway and its regulation. Schematic representation summarizing the SREBP-2-regulated mevalonate pathway and key enzymes for synthesis from acetyl-CoA to cholesterol and its products. Multiple signaling pathways such as p53, Akt, and androgen can regulate SREBP-2 activation. Several regulatory feedback mechanisms exist for different enzymes by various signals and mevalonate metabolites, such as cholesterol, IPP (Isopentenylpyrophosphate) and FPP (farnesylpyrophosphate). ACAT, acyl CoA-cholesterol acyltransferase; HMGCSm HMG-CoA synthase; HMGCR, HMG-CoA reductase; MVK, mevalonate kinase; MVD, HMG-CoA synthase; PMVK, phosphomevalonate kinase; IDI, isopentenyl diphosphate isomerase; FPPS, farnesylpyrophosphate synthase; SQS, squalene synthase; SQLE, squalene epoxidase; LSS, lanosterol synthase; CYP51A, lanosterol-14α demethylase; TM7SF2, steroid 14 reductase; SC4MOL, 4 methyl sterol oxidase; NSDHL, C3 sterol dehydrogenase; HSD17B7, 3-ketoreductase, EBP, phenylalkylamine Ca2+ antagonist binding protein; SC5D, sterol-C5-desaturase; DHCR7, 7-dehydrocholesterol reductase; DHCR24, 24-dihydrocholesterol reductase; LDLR, low-density lipoprotein receptors; GGPP, geranylgeranylpyrophosphate; AMPK, adenosine monophosphate-activated protein kinase; PPARγ, peroxisome proliferators-activated receptor γ; INSIG, insulin-induced gene protein; LXR, Liver X receptor; REST, RE1-silencing transcription factor.
As many studies reported, SREBP-2 activation and pathways are regulated by multiple signals. A major tumor suppressor, p53, can block activation of SREBP-2 to decrease the transcription of mevalonate pathway genes through transcriptional up-regulation of the ATP-binding cassette (ABC) transporter A1 (ABCA1) gene, which mediates tumor suppression (21). On the other hand, SREBP-2 can increase the generation of oxysterol ligands for liver X receptors (LXRs) to positively regulate ABCA1 gene transcription (63). LXRs play a potential role in maintaining cholesterol homeostasis through promoting cholesterol efflux and suppressing de novo synthesis and uptake (64, 65). Mutant p53 is recruited to the promoters of genes encoding mevalonate pathway enzymes by binding to the SREBP-2, which subsequently increases the activities of oncogenic pathways such as Ras, RhoA (66), and YAP/TAZ (67) to promote cancer progression (20, 68). Apart from p53, protein kinase B (Akt) acutely activates SREBP-2 (43) to induce the expression of genes involved in cholesterol synthesis, which contributes to tumor development (19, 69). In addition, tumor microenvironments such hypoxia, extracellular pH, and nutrient levels also play critical roles in the regulation of SREBP-2 activation. Hypoxia inducible factor-1α is able to increase the activity of HMGCR by the translocation of SREBP-2 to the nucleus (70). Acidic extracellular pH (pH 6.8) triggers nuclear translocation of SREBP-2 to target acyl-CoA synthase short-chain family member 2 for maintaining overall survival of cancer patients (71). As a kind of nutrient, glucose promotes SCAP/SREBP complex trafficking from the ER to the Golgi and subsequent SREBP activation via N-glycosylation of SCAP (72). When cholesterol in the ER falls below 5% of total ER lipids, the cleavage of SREBP-2 is activated (31). A steroid hormone, androgen, can induce SREBP-2 activation in normal physiological or pathological conditions, such as prostate cancer (73, 74). Taken together, either multiple signaling pathways or cellular nutrient levels can regulate SREBP-2 activation to control the mevalonate pathway (Figure 2).
Meanwhile, the enzymes participating in the mevalonate pathway, such as HMGCR, MVK, SQS, and DHCR24, are regulated by various molecules or the metabolites from mevalonate metabolism. Both the phosphorylation by AMP-activated protein kinase (AMPK) and dephosphorylation by protein phosphatase 2A regulate HMGCR activity (75, 76). The binding of INSIG on the sterol-sensing domain can lead to the ubiquitination and degradation of HMGCR (77, 78). Interestingly, PPARγ can regulate multiple pathways, including decreasing the expressions of SREBP-2 and HMGCR and increasing the expression of LXRα to reduce cholesterol levels. Considering the role of LXRα on cholesterol efflux, the expression of ABC transporter G5 or G8 is increased by the PPARγ-LXRα pathway or their individual dependence, which needs to be further clarified (79). Two key enzymes of the post-squalene pathway, SQS and CYP51A are directly repressed by LXRα via negative binding with LXR DNA response elements (80). Moreover, DHCR24 as a final enzyme for cholesterol biosynthesis is regulated by RE1-silencing transcription factor, REST and LXRα through the binding of its promoter at the transcriptional level (56, 81). These findings suggest that LXRα plays an important role in regulating several enzymes of the mevalonate pathway, such as SQS, CYP51A, and DHCR24.
Additionally, key metabolites also can modulate metabolic enzymes of the SREBP-2-reguated mevalonate pathway. Cholesterol and 25-hydroxycholesterol can regulate HMGCR by increasing its alternative splicing (82). Mevalonate and certain of its derivatives such as dioxidolanosterol and geraniol regulate HMGCR mRNA translation or polysome distribution to reduce its synthesis and translation (83). Lanosterol and other C4-dimethylated sterol intermediates may regulate both HMGCR degradation and SREBP-2 cleavage (84). For geranylgeranyle diphosphate (GGPP), FPP and IPP, these intermediates post-transcriptionally inhibit MVK activity by negative feedback responses (85). Other metabolites, such as phytosterols, 24(S), 25-epoxycholesterol (24,25-EC) and steroid hormones (progesterone) can directly inhibit DHCR24 activity at the post-translational level (86–88). Overall, SREBP-2 and the enzymes for cholesterol biosynthesis, such as HMGCR, MVK, SQS, and DHCR24, can be regulated by various signaling pathways and mevalonate pathway metabolites at the transcriptional and post-translational levels (Figure 2).
SREBP-2 Signaling and the Enzymes From the Mevalonate Pathway in the Cancer Context
Reprogramming of lipid metabolism occurs in a variety of cancers and contributes to rapid tumor growth, which is regulated by SREBPs (89). SREBP-2 is markedly upregulated in various cancers, including prostate (14, 90), breast (15), and hepatocellular cancer (91). Moreover, SREBP-2-mediated mevalonate metabolism drives epithelial to mesenchymal transition (EMT) and supports cancer stemness, and has been suggested as a potential target for cancer treatment (17, 18, 92).
Prostate Cancer
Lipid synthesis and uptake are significantly elevated in prostate cancer (PCa) as important energy resources to support tumor growth and progression (93, 94). As is well-known, androgens bind to and activate the androgen receptor (AR) to maintain the survival and proliferation of PCa (95). Androgen-induced activation of SREBPs occurs not only under normal physiological conditions but also in the setting of steroid-regulated cancers (74, 96). Androgens markedly stimulate the expression of SCAP (97) and cause a switch in the isoform expression of INSIG, which play a pivotal role in the lipogenic effects of androgen in PCa (73). Meanwhile, dihydrotestosterone or R1881 marginally up-regulates the mRNA and protein levels of SREBP-2, which induces the expression of multiple genes encoding enzymes involved in cholesterol biosynthesis, including HMGCS, HMGCR, FPPS in PCa cells (98, 99). A recent report shows that an aberrant SREBP-dependent lipogenic program promotes PCa metastasis with double-null PML and PTEN (100). During the progression to androgen independence, nuclear SREBP-2 protein expression underwent a 3-fold increase in a PCa xenograft model (90). In addition, SREBP-2 expression is elevated in advanced pathologic grade and metastatic PCa and significantly associated with poor clinical outcomes. SREBP-2 promotes PCa cell growth, stemness and metastasis through transcriptional c-Myc activation mediated by direct interaction with a SREBP-2-binding element in the 5′-flanking c-Myc promoter region (14).
Key enzymes for mevalonate pathway such as HMGCS1, HMGCR, FPPS, and SQS also play important roles in PCa malignant progression. HMGCS1 and HMGCR are overexpressed in stroma of early stage PCa (101). Moreover, enzalutamide-resistant PCa cell lines express elevated HMGCR, and are more sensitive to statins, HMGCR inhibitors (102). FPPS is associated with increasing Gleason scores, PTEN functionally deficient status, and poor survival in PCa through modulation of the small GTPases/Akt axis (103, 104). SQS at rs2645429 is significantly associated with PCa risk and aggressive phenotypes (105). Taken together, SREBP-2 and key enzymes for the mevalonate pathway are potential targets for PCa treatment.
Breast Cancer
In breast cancer, CtBP expression negatively correlates with SREBP-2 and HMGCR expressions. CtBP can form a complex with ZEB1 to transcriptionally repress SREBP-2 expression and activate TGF-β signaling, which maintains intracellular cholesterol homeostasis in breast cancer (106). TP53 mutation correlates with elevated expression of a subset of mevalonate pathway genes in breast cancer patients. The levels of genes such as HMGCR, FPPS, SQS, and DHCR7 are positively associated with the risk of breast cancer. The functional interaction with SREBP-2 is critical for mutant p53-mediated up-regulation of the mevalonate pathway genes (20). Oncogenic PI3K (H1047R) or K-Ras (G12V) can induce de novo lipogenesis through convergent activation of mTORC1 to promote aberrant growth and proliferation of breast cancer, which is mediated by the activation of SREBP-2 or SREBP-1 (107). In addition, SREBP-2 is highly expressed in breast cancer tissues and correlated with a poor prognosis (15). SREBP-2 expression is increased during the early stages of osteoclast formation under the control of the RANKL/cAMP-CREB signaling cascade, which induces the expressions of NFATc1 and matrix metalloproteinase, thus contributing to breast cancer-induced osteolysis (15).
For patients with HER2+ metastatic breast cancer, dual targeted therapy with a tyrosine kinase inhibitor, lapatinib or its combination with an anti-HER2 monoclonal antibody, trastuzumab can significantly improve pathological complete response and overall survival (108). However, lapatinib and its combination with trastuzumab lead to the resistance of breast cancer cells to HER2-targeted therapy, which has been a clinical challenge (109). The mevalonate pathway has been considered as a new potential target for overcoming this acquired anti-HER2 treatment resistance, which may be mediated by activating the mTORC1-mediated YAP/TAZ pathway (110). Rate-limiting enzyme studies found that high levels of HMGCR are correlated with breast cancer risk (111) and poor survival (112, 113). Cholesterol is also implicated as a breast cancer risk factor and promotes breast tumor growth and metastasis (114). Another metabolite, 27-hydroxycholesterol, can increase the proliferation of estrogen receptor (ER)-positive breast cancer through the activation of ER and LXR (115). Therefore, inhibition of the SREBP-2-mediated mevalonate pathway has been recognized as a potential therapeutic approach for breast cancer.
Lung Cancer
The single-nucleotide polymorphism of HMGCR, rs12916, is associated with the subgroups of attained age for lung cancer (111) and the C allele of the SQS rs2645429 polymorphism gene can be a risk factor for non-small cell lung cancer (NSCLC) (116). Three key enzymes of the mevalonate pathway, FPPS, SQS and GGPPS, are also associated with stage and metastasis of NSCLC (117–119). Of these enzymes, SQS is increased in invasive lung cancer cells and in the tumor regions of lung cancer specimens, and significantly associated with metastasis and poor prognosis by enhancing NF-κB-mediated up-regulation of matrix metallopeptidase-1 (117) or modulating extracellular signal-regulated kinase (ERK) signaling (120). FPPS plays an important role in promoting cell invasion and EMT through the RhoA/ROCK1 pathway (118). Although GGPPS knockdown has no effect on lung adenocarcinoma cell proliferation and apoptosis, it significantly inhibits invasion and migration by regulating EMT (119). Overall, several enzymes from the mevalonate pathway as mentioned above have been identified as potential targets for treating lung cancer (121).
Hepatocellular Carcinoma
New studies reveal that several key molecules, such as p53 and fatty acid synthase (FASN), can activate SREBP-2 to promote cholesterol accumulation for maintaining the progression of hepatocellular carcinoma (HCC). In HCC, p53 tumor suppressor can induce the expression of MVA pathway enzymes through the accumulation and stabilization of mature SREBP-2 by transcriptionally inducing ABCA1, a cholesterol transporter gene. Like p53 loss, the ablation of ABCA1 promotes murine liver tumorigenesis and is associated with increased SREBP-2 maturation (21). In contrast to p53, a p53 activator, haplo-insufficient tumor suppressor ASPP2, can interact with SREBP-2 in the nucleus and negatively regulates the mevalonate pathway to mediate the inhibition of HCC tumor growth (122). Moreover, overexpression of Staphylococcal nuclease and tudor domain containing-1 (SND-1) in HCC results in the accumulation of cellular cholesteryl esters due to the altered activation of SREBP-2 (123). Interestingly, SREBP-2 also binds to specific sites in SND-1 promoter to induce its transcription, which contributes to lipid metabolism reprogramming in HCC (91). This suggests that there is a complex for the interaction of SND-1and SREBP-2 in the lipid reprogramming of HCC, which needs to be clarified. Another molecule, FASN, contributes to de novo fatty acid synthesis in a murine HCC model induced by Pten loss and c-Met overexpression. Compared with the control group, genes such as HMGCR involved in cholesterol biosynthesis were obviously upregulated in HCC in FASN knockout mice, related to the promotion of nuclear SREBP-2 (124). Reportedly, the inhibition of FASN ubiquitination and disruption of the SREBP-1/SREBP-2 degradation complexes may be potential molecular mechanisms of Akt-induced lipogenesis and HCC tumor development in mice (69). In addition, Forkhead Box M1 has a positive correlation with SREBP-2 or HMGCR in HCC tissues, which links the mevalonate pathway through protein geranylgeranylation as novel targets (125). Based on the findings above, targeting the SREBP-2-mediated mevalonate pathway seems to have potential as a strategy for HCC treatment.
Other Cancers
Similarly, SREBP-2 and its regulated mevalonate pathways also participate in other cancers. In esophageal squamous cell carcinoma, SREBP-2 is upregulated in clinical samples and promotes cell growth, migration and colony formation, which may be mediated by interaction with c-Myc to increase HMGCR expression (16). In renal carcinoma, kruppel-like factor 6 (KLF6) activates mTOR signaling and its downstream lipid metabolism regulator, SREBP-2 to enhance tumor growth (126). In pancreatic cancer, the novel small nucleolar RNA host gene 16 directly regulates the miR-195/SREBP-2 axis to promote lipogenesis and accelerate tumor progression (127). Furthermore, increasing cellular cholesterol can drive intestinal stem cell proliferation and tumorigenesis through the activation of nuclear SREBP-2 (128). Also, SQS is frequently mutated and dysregulated in the liver metastatic cohort of colorectal cancer (129). The final enzyme of the cholesterol pathway, DHCR24, is significantly elevated and associated with advanced clinical stage and overall survival in bladder and endometrial cancer, which is mediated by several oncogenesis-associated biological processes (130, 131). Collectively, these findings in different cancers indicate that the SREBP-2-regulated mevalonate pathway significantly participates in tumor growth and metastasis and may be an attractive target in a variety of malignancies (Table 1).
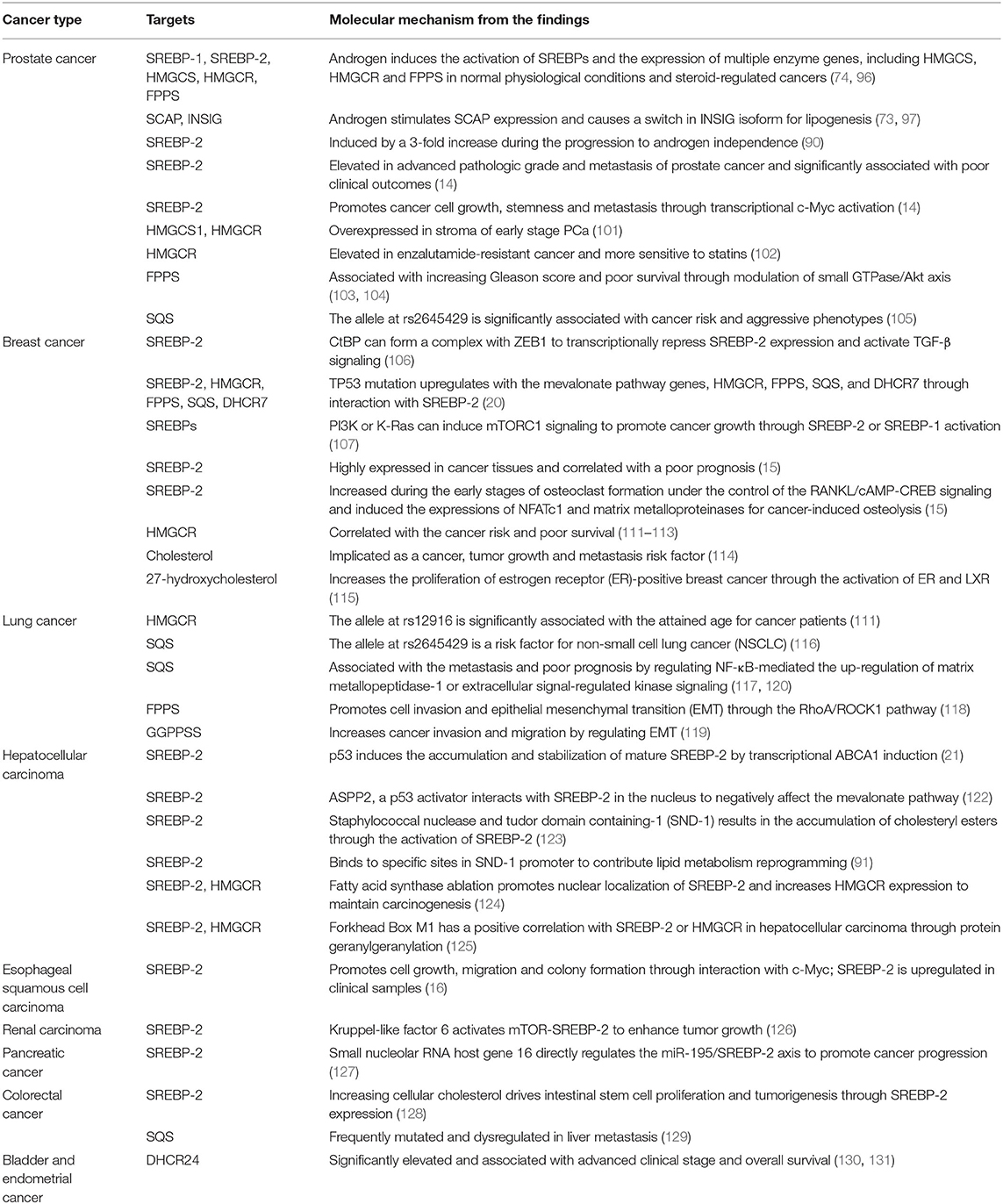
Table 1. The roles and molecular mechanisms of the SREBP-2-regulated mevalonate pathway in different cancers.
Targeting the SREBP-2-Regulated Mevalonate Pathway For Cancer Therapy
Based on the above reports, we choose SREBP-2, HMGCR, and FPPS as potential targets for cancer therapy and summarized the findings so far regarding several inhibitors or miRNAs used to address these targets in preclinical and clinical studies.
Targeting SREBP-2 for Cancer Therapy
As reported, SREBPs inhibition by small molecules such as fatostatin, natural products, and microRNAs such as miR-185, miR-342, and miR-33a have been extensively found to exert multiple anti-tumor effects in various cancers by reducing mevalonate metabolic dysfunction (132–136). Fatostatin, a non-sterol diarylthazole derivative, was first reported to inhibit insulin-induced adipogenesis and reduce body weight by blocking nuclear translocation of SREBPs in obese mice (137, 138). Fatostatin has been used for treating prostate (133), breast (139), and endometrial cancers (140). Mechanistically, fatostatin directly binds SCAP and blocks its transport from ER to Golgi apparatus, then inhibits the activation of SREBPs (138). A recent study also showed that fatostatin inhibits cell proliferation through a SCAP-independent mechanism (141). In PCa, in vitro and in vivo studies reveal that fatostatin suppresses cell proliferation and induces apoptosis through blockade of SREBP-regulated metabolic pathways (133), similar to the findings in endometrial carcinoma (140). The combination of fatostatin with docetaxel significantly increases proliferation inhibition and apoptosis induction in metastatic PCa harboring p53 mutations, compared with fatostatin alone (142). Moreover, fatostatin also inhibits mitotic microtubule spindle assembly and cell division in aggressive cancers in addition to the inhibition of SREBP activity (136). Fatostatin also causes lipid accumulation as a response to endoplasmic reticulum stress rather than the inhibition of SREBP-mediated lipogenesis in ER+ breast cancer cells (139). These studies suggest that the antitumor effects of fatostation are multiple and dependent on cancer type.
Recent studies indicate that natural products can directly target SREBP-2 to inhibit the expression of key enzymes for the mevalonate pathway, to reduce tumor growth. Tocotrienol, a minor form of vitamin E, can degrade mature SREBP-2 without affecting LXR activity to maintain cholesterol homoeostasis in PCa (143). In glioma, artesunate, initially developed as an anti-malaria drug, effectively inhibits cancer cell growth and distant metastasis, and further induces cell senescence by regulating the nuclear localization of SREBP-2 and the expression of HMGCR (144). As an anthraquinone derived from many plants, emodin inhibits SREBP-2 transcriptional activity to suppress cholesterol metabolism and Akt signaling, which sensitizes HCC cells to the anti-cancer effect of sorafenib in vitro and in xenograft models (145). Surprisingly, ursolic acid as a natural pentacyclic terpenoid activates SREBP-2 and increases the expression of cholesterol biosynthesis-related enzymes to induce cell cycle arrest and apoptosis in HCC cells (146). Additionally, archazolid B leads to the accumulation of free cholesterol and drastic disturbance in cholesterol homeostasis, which can activate nuclear SREBP-2 expression and up-regulate HMGCR for killing bladder cancer cells (147).
Some miRNAs, such as miR-98 and miR-33a, have been found to play critical roles in cholesterol metabolism by targeting SREBP-2 (134, 148, 149). Our previous study shows that miR-185 and miR-342 not only significantly block SREBP-2-mediated cholesterogenesis, but also inhibit SREBP-1-mediated lipogenesis in PCa (132). Another miRNA, miR-33a, an intronic miRNA located within the SREBP-2 gene, inhibits EMT targeting of Twist1 to block invasion and metastasis in NSCLC (135). According to present studies, searching for miRNAs directly and specifically targeting SREBP-2 could be a future direction for new cancer treatment strategies. Table 2 summarizes current SREBP-2 targeting by small molecules or miRNAs.
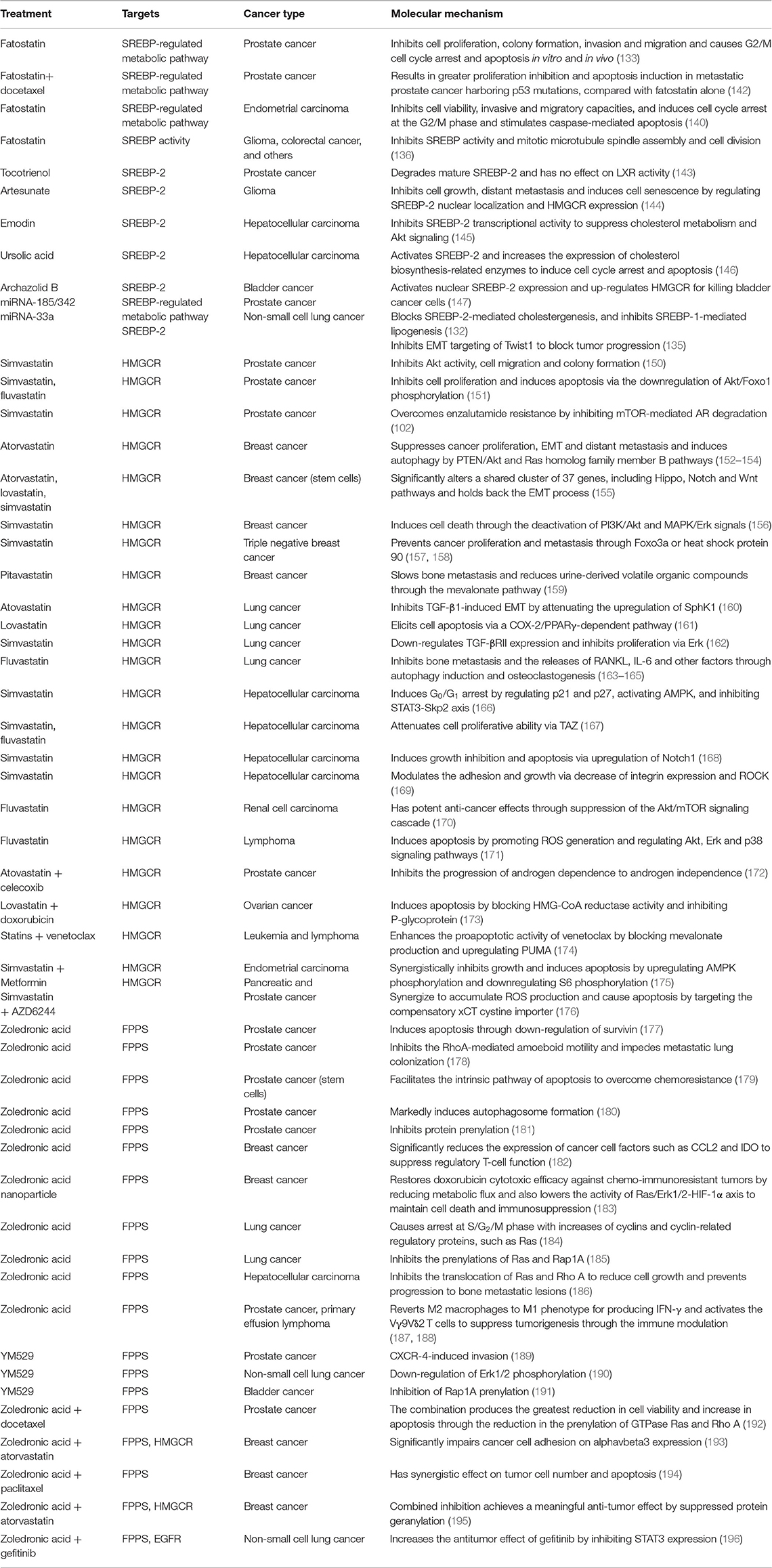
Table 2. Preclinical findings for targeting the SREBP-2-regulated mevalonate pathway in different cancers.
Targeting HMGCR for Cancer Therapy
Targeting HMGCR in Preclinical Cancer Therapy
Altered cholesterol metabolism is considered as a risk factor and driver of tumor growth, and is also associated with worse prognosis in a variety of cancers including breast, prostate, brain, and colorectal cancer (197, 198). Targeting HMGCR, a rate-limiting specific enzyme of cholesterol synthesis, has been identified as a potential therapeutic strategy for cancer treatment. Originally for treating cardiovascular diseases, statins like HMGCR inhibitors have become a standard of care for treating cancer patients with high cholesterol levels (199, 200) and also reduce the incidence and recurrence of various cancers, including colon (201), liver (202), and lung cancer (203). Statins can be divided mainly into two groups, depending on their origin by fungi fermentation or chemical syntheses, including type-1, mevastatin, lovastatin, simvastatin and type-2, fluvastatin, and atorvastatin (200). A number of studies have indicated that statins can inhibit cell proliferation, invasion and colony formation, and induce apoptosis to suppress tumorigenesis, tumor survival, angiogenesis and metastasis by regulating multiple signaling pathways (59, 199, 204).
In PCa xenograft mice models, simvastatin treatment at 25 μM inhibited serum-induced Akt activity, cell migration and colony formation (150). Both simvastatin and fluvastatin inhibit cell proliferation and induce apoptosis in a dose- and time-dependent manner via the downregulation of Akt/Foxo1 phosphorylation in PCa (151). Simvastatin treatment also overcomes enzalutamide-induced resistance through the inhibition of mTOR-mediated AR degradation (102).
In breast cancer, both statins and HMGCR transcriptional regulation can overcome statin resistance through the regulation of SREBP-2 cleavage (205). The findings in a 2D co-culture and a splenic mouse model demonstrate that atorvastatin suppresses breast cancer proliferation, EMT and distant metastasis (152) and also induces autophagy (153), which is related to regulating PTEN/Akt and Ras homolog family member B pathways (154). In breast cancer stem-like cells, statins at non-toxic doses significantly alter a shared cluster of 37 genes, including the Hippo, Notch, and Wnt pathways, to hold back EMT processes (155). Simvastatin induces breast cancer cell death through the deactivation of PI3K/Akt and MAPK/Erk signals (156) and also prevents triple-negative breast cancer proliferation and metastasis through Foxo3a phosphorylation (157) or HSP90 acetylation (158). Another statin, pitavastatin, can slow breast cancer-induced bone metastasis and reduce urine-derived volatile organic compounds through the mevalonate pathway (159).
Increasing evidence demonstrates the anticancer effects of statins including atorvastatin, lovastatin, and fluvastatin against lung cancer by decreasing proliferative and migratory capacity and inducing apoptosis, which is mediated by SphK1 (160), COX-2/PPARγ (161), TGF-β RII/Erk (162), and other key pathways (204). Lung cancer cells metastasize to the bone and release RANKL, IL-6, and other factors to stimulate osteoclasts, which can be inhibited by fluvastatin through autophagy induction and osteoclastogenesis (163–165).
In hepatocellular carcinoma, in vitro and in vivo studies reveal that simvastatin induces G0/G1 arrest by upregulating p21 and p27, activating AMPK and inhibiting the STAT3-Skp2 axis in HCC (166). Other studies report that TAZ, Notch1 or ROCK expression are also involved in the anti-proliferative effects of statins against HCC (167–169).
Fluvastatin has potent anti-cancer effects against renal cell carcinoma through the suppression of the Akt/mTOR signaling cascade (170) and induces lymphoma cell apoptosis by promoting ROS generation and regulating the Akt, Erk, and p38 signaling pathways via the inhibition of mevalonate metabolic products (171). Combination therapy studies demonstrated that statins combined with chemical molecules, including doxorubicin, celecoxib, venetoclax, metformin, or a MEK inhibitor, AZD6244, can synergistically suppress tumor growth in prostate, ovarian, endometrial, or pancreatic cancers, respectively (172–176). Overall, these findings suggest that statins alone or combined with other drugs inhibit the mevalonate pathway to achieve anti-cancer effects by a variety of molecular mechanisms (Table 2).
Targeting HMGCR in Clinical Cancer Therapy
Currently, statins are in use for preventing or treating cancer patients with prostate (206), breast (207), lung (208), liver (209), and other cancers (210, 211). The safety, efficacy and mortality benefits of statins have been assessed both alone and in combination therapy in clinical cancer patients studies (212–214).
In a 7.5 year follow-up of patients with PCa, statin use was associated with a decreased risk of death and delays in cancer progression, dependent on the increasing intensity of usage. However, statin use before diagnosis is not associated with PCa death risk (206). A meta-analysis of breast cancer patients indicates that statin can lower cancer-specific and all-cause mortality, which appears to be related to statin type (lipophilic or hydrophilic statin) and follow-up time (207). Seventeen studies in 98,445 patients indicate that statins potentially decrease cancer-specific mortality and promote the overall survival of patients with lung cancer in observational studies (215), which does not affect progression-free survival (208). In liver cancer, numerous studies have demonstrate decreased liver cancer mortality by statin treatment after adjusting for cholesterol level and body mass index, which is a novel approach for the prevention and treatment of HCC (209). In addition, post-diagnostic statin use is associated with improved survival of patients with other cancers, such as esophageal cancer (211) and ovarian cancer (210). Compared to statin alone, the combination of statins with therapeutic drugs such as thalidomide, idarubicin or tyrosine kinase inhibitor has synergistic effects for patients with refractory myeloma (216), acute myeloid leukemia (217) or NSCLC (218), respectively.
However, some contradictory studies indicate that statins have no protective effect on skin (219), colon (220), or other cancers in numerous clinical trials (221), which might be related to chemical nature, tumor stage and type, dose, use duration and patient characteristics. Therefore, well-defined patient information and clinical trial design need careful consideration in future studies of statins in cancer patients (222). Table 3 summarizes the detailed information about tumor type, the number of patients, and main findings from clinical studies of statins alone or combined with other therapeutic agents in patients with various cancers.
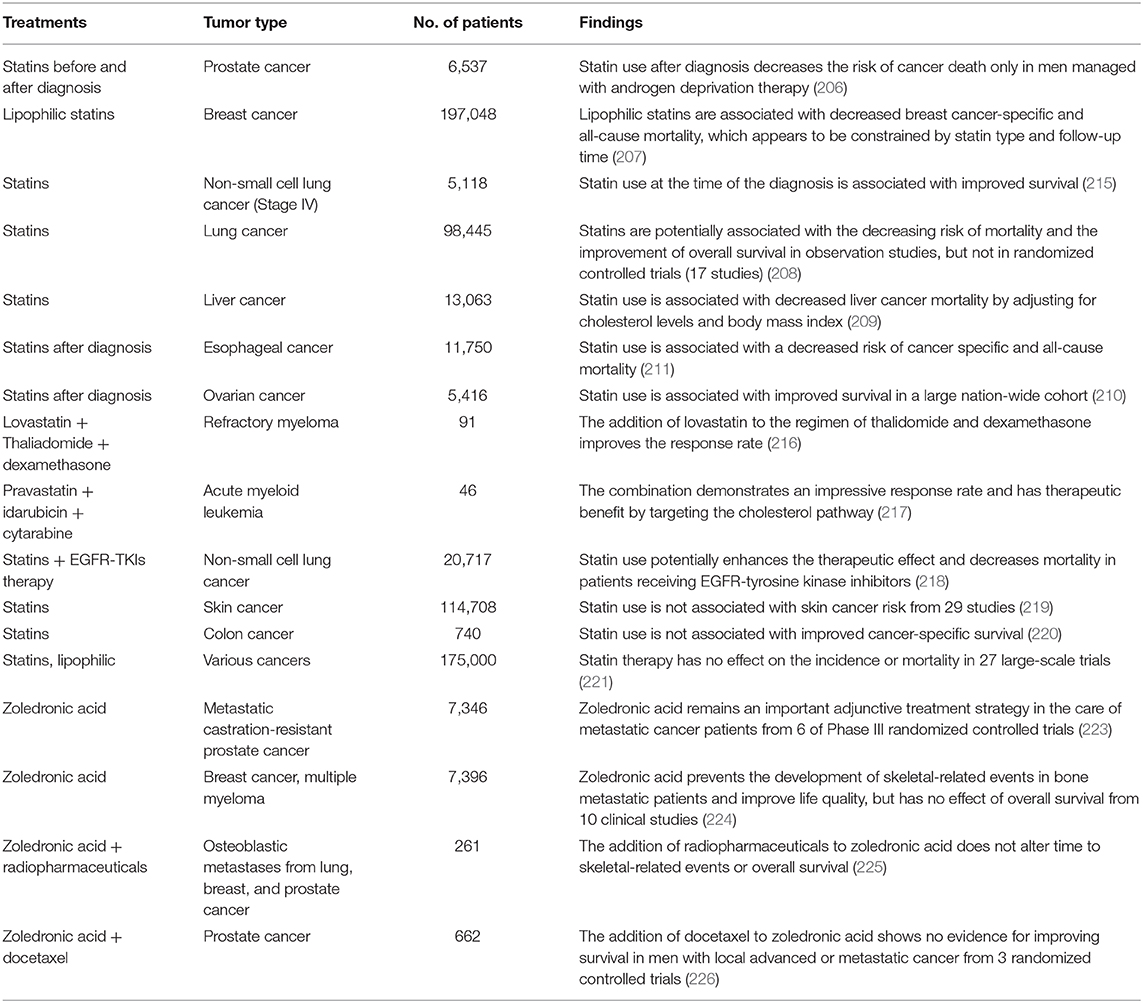
Table 3. Clinical findings for statins and N-BPs in different cancers.
Targeting FPPS for Cancer Therapy
Targeting FPPS in Preclinical Cancer Therapy
Amino-bisphosphonates (N-BPs), as FPPS inhibitors, represent another major class of inhibitors targeting the mevalonate pathway. Compared to original non-nitrogen containing bisphosphonates, N-BPs have an increased affinity to hydroxyapatite and interfere with FPPS in the mevalonate pathway (227), and are used for treating patients with osteoporosis (228) or osteolytic bone metastases (229). Several studies reveal anti-tumor effects of N-BPs apart from the inhibition of osteoclasts. Third-generation N-BPs, zoledronic acid (ZOL) and minodronate (YM529), are more potent inhibitors of FPPS than the first-generation bisphosphonates, and have been found to exhibit anti-tumor effects through inhibition of cell growth, induction of apoptosis, inhibition of angiogenesis, decrease in tumor cell adhesion to bone and other possible mechanisms in various cancers (230, 231).
In PCa, ZOL induces apoptosis through down-regulation of survivin (177), and inhibits RhoA-mediated amoeboid motility to impede metastatic lung colonization (178). In PCa stem cells, ZOL can facilitate the intrinsic apoptosis pathway to overcome chemoresistance (179). Moreover, ZOL exposure markedly induces autophagosome formation and inhibits protein prenylation for anti-prostate cancer activity (180, 181). In breast cancer, ZOL can significantly reduce the expression of cancer cell factors such as CCL2 and IDO to suppress regulatory T-cell function (182). Especially, a formed ZOL-nanoparticle restores doxorubicin cytotoxic efficacy against chemo-immunoresistant tumors by reducing metabolic flux and also lowers Ras/Erk1/2/HIF-1α axis activity to maintain cell death and immunosuppression (183). In lung cancer, in vitro and in vivo experiments demonstrate that ZOL-treated cells typically arrest the at S/G2/M phase with increases of cyclins and cyclin-related regulatory proteins such as Ras (184). ZOL can also inhibit Ras and Rap1A prenylation to target lung cancer (185). Similar findings in HCC demonstrate that ZOL inhibits the translocation of Ras and RhoA to reduce cell growth and prevent progression to bone metastatic lesions (186). Additionally, ZOL treatment reverts M2 macrophages to M1 phenotype for producing IFN-γ (188) or activating Vγ9Vδ2 T cells (187) to suppress tumorigenesis through the immune modulation.
Another N-BP, YM529, also exerts anti-tumor effects against various types of cancer cells, including PCa, NSCLC, and bladder cancer, by various mechanisms such as CXCR-4-induced invasion (189), down-regulation of Erk1/2 phosphorylation (190), and inhibition of Rap1A prenylation (191). In addition, N-BPs have been used in combination with chemotherapy, statins or enzyme inhibitors to achieve additive or synergistic effects by diverse mechanisms, including a reduction in protein prenylation, impairment of geranylgeranylation or inhibition of STAT3 in prostate (192), breast (193–195), and lung (196) cancers. The effects of FPPS inhibitors such as ZOL and YM529 alone and in combination with other drugs targeting multiple signaling pathways in cancer cell and xenograft models are summarized in Table 2.
Targeting FPPS in Clinical Cancer Therapy
Based on their strong inhibitory effect on osteoclasts, N-BPs are used to treat osteolytic bone metastases, which are frequent in advanced cancer, especially prostate and breast cancer. In PCa, ZOL has become an established first-line or adjunctive treatment in bone-targeted therapy for metastatic castration-resistant progression (223, 232). Though ZOL delays skeletal-related events (SREs), it reportedly has no effect on overall survival, other disease-oriented parameters, or radiographic progression improvement. It remains an important adjunctive treatment strategy in the care of metastatic castrate-resistant PCa patients (223). Findings in clinical trials indicate that the beneficial effect of ZOL on bone metastasis from advanced prostate cancer might be related to long-term therapy, generally for more than 2 years (226). Similarly, ZOL can prevent the development of SREs in bone metastatic patients with breast cancer and improve quality of life, although with no effect on overall survival (224, 225). However, long-term side effects of ZOL, such as impaired renal function and bone pain need to be taken into consideration for treatment decisions (233). Findings in clinical studies of ZOL treatment alone or in combination are summarized in Table 3.
Overall, the SREBP-2-regulated mevalonate pathway is a crucial regulator for tumor progression and a promising therapeutic target. Targeting SREBP-2, HMGCR or FPPS has become an attractive strategy for cancer therapy. Preclinical (Table 2) and clinical (Table 3) studies demonstrate that fatostatin, statins, ZOL, and YM529, alone or in combination with chemotherapy or other drugs, have anti-tumor effects through a variety of molecular mechanisms.
Conclusions
This review has summarized the structure, activation and regulation of SREBP-2 by multiple signaling pathways. SREBP-2 and its regulated enzymes from the mevalonate pathway, including HMGCR, FPPS, SQS, and DHCR4, participate in the progression of various cancers including prostate, breast, lung, and hepatocellular cancer, and thus are important potential therapeutic targets. Importantly, preclinical and clinical research has demonstrated that fatostatin, statins, and N-BPs targeting SREBP-2, HMGCR, and FPPS, respectively, alone or in combination with other drugs, are used for the treatment of different cancers. This review provides new insights into the critical role of the SREBP-2-regulated mevalonate pathway in cancer and its potential for targeted cancer therapy.
As a metabolic reprogramming process, the SREBP-2-regulated mevalonate pathway has a high-degree of similarity with glucose or glutamine metabolism and links them together to participate in cancer progression. Based on the function of SREBP-2 in cholesterol biosynthesis, it is necessary to develop new strategies specifically targeting SREBP-2 to treat various cancers with dysfunctional cholesterol metabolism. Combination treatments simultaneously targeting SREBP-2 and its regulated enzymes from the mevalonate pathway may achieve beneficial effects for cancer treatment and prevention, and represent important future directions in ongoing research.
Author Contributions
LX, XL, and BW: conceptualization. LX: writing—original draft preparation. HQ, HZ, LD, and QH: writing—review and editing of different sections. DZ, BW, and XL: supervision. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (81602257) and the Science and Technology Development Plan Project of Jilin Province (20190101010JH).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Hua X, Yokoyama C, Wu J, Briggs MR, Brown MS, Goldstein JL, et al. SREBP-2, a second basic-helix-loop-helix-leucine zipper protein that stimulates transcription by binding to a sterol regulatory element. Proc Natl Acad Sci USA. (1993) 90:11603–7. doi: 10.1073/pnas.90.24.11603
2. Yokoyama C, Wang X, Briggs MR, Admon A, Wu J, Hua X, et al. SREBP-1, a basic-helix-loop-helix-leucine zipper protein that controls transcription of the low density lipoprotein receptor gene. Cell. (1993) 75:187–97. doi: 10.1016/S0092-8674(05)80095-9
3. Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. (1997) 89:331–40. doi: 10.1016/S0092-8674(00)80213-5
4. Goldstein JL, DeBose-Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell. (2006) 124:35–46. doi: 10.1016/j.cell.2005.12.022
5. Horton JD, Shimomura I, Brown MS, Hammer RE, Goldstein JL, Shimano H. Activation of cholesterol synthesis in preference to fatty acid synthesis in liver and adipose tissue of transgenic mice overproducing sterol regulatory element-binding protein-2. J Clin Invest. (1998) 101:2331–9. doi: 10.1172/JCI2961
6. Shimano H, Yahagi N, Amemiya-Kudo M, Hasty AH, Osuga J, Tamura Y, et al. Sterol regulatory element-binding protein-1 as a key transcription factor for nutritional induction of lipogenic enzyme genes. J Biol Chem. (1999) 274:35832–9. doi: 10.1074/jbc.274.50.35832
7. Amemiya-Kudo M, Shimano H, Hasty AH, Yahagi N, Yoshikawa T, Matsuzaka T, et al. Transcriptional activities of nuclear SREBP-1a,−1c, and−2 to different target promoters of lipogenic and cholesterogenic genes. J Lipid Res. (2002) 43:1220–35. doi: 10.1194/jlr.M100417-JLR200
8. Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. (2002) 109:1125–31. doi: 10.1172/JCI0215593
9. Osborne TF, Espenshade PJ. Evolutionary conservation and adaptation in the mechanism that regulates SREBP action: what a long, strange tRIP it's been. Genes Dev. (2009) 23:2578–91. doi: 10.1101/gad.1854309
10. Jeon TI, Osborne TF. SREBPs: metabolic integrators in physiology and metabolism. Trends Endocrinol Metab. (2012) 23:65–72. doi: 10.1016/j.tem.2011.10.004
11. Shao W, Espenshade PJ. Expanding roles for SREBP in metabolism. Cell Metab. (2012) 16:414–9. doi: 10.1016/j.cmet.2012.09.002
12. Moon YA, Liang G, Xie X, Frank-Kamenetsky M, Fitzgerald K, Koteliansky V, et al. The Scap/SREBP pathway is essential for developing diabetic fatty liver and carbohydrate-induced hypertriglyceridemia in animals. Cell Metab. (2012) 15:240–6. doi: 10.1016/j.cmet.2011.12.017
13. Griffiths B, Lewis CA, Bensaad K, Ros S, Zhang Q, Ferber EC, et al. Sterol regulatory element binding protein-dependent regulation of lipid synthesis supports cell survival and tumor growth. Cancer Metab. (2013) 1:3. doi: 10.1186/2049-3002-1-3
14. Li X, Wu JB, Li Q, Shigemura K, Chung LW, Huang WC. SREBP-2 promotes stem cell-like properties and metastasis by transcriptional activation of c-Myc in prostate cancer. Oncotarget. (2016) 7:12869–84. doi: 10.18632/oncotarget.7331
15. Jie Z, Xie Z, Xu W, Zhao X, Jin G, Sun X, et al. SREBP-2 aggravates breast cancer associated osteolysis by promoting osteoclastogenesis and breast cancer metastasis. Biochim Biophys Acta Mol Basis Dis. (2019) 1865:115–25. doi: 10.1016/j.bbadis.2018.10.026
16. Zhong C, Fan L, Li Z, Yao F, Zhao H. SREBP2 is upregulated in esophageal squamous cell carcinoma and cooperates with cMyc to regulate HMGCR expression. Mol Med Rep. (2019) 20:3003–10. doi: 10.3892/mmr.2019.10577
17. Gruenbacher G, Thurnher M. Mevalonate metabolism in cancer. Cancer Lett. (2015) 356(Pt. A):192–6. doi: 10.1016/j.canlet.2014.01.013
18. Bathaie SZ, Ashrafi M, Azizian M, Tamanoi F. Mevalonate pathway and human cancers. Curr Mol Pharmacol. (2017) 10:77–85. doi: 10.2174/1874467209666160112123205
19. Yamauchi Y, Furukawa K, Hamamura K, Furukawa K. Positive feedback loop between PI3K-Akt-mTORC1 signaling and the lipogenic pathway boosts Akt signaling: induction of the lipogenic pathway by a melanoma antigen. Cancer Res. (2011) 71:4989–97. doi: 10.1158/0008-5472.CAN-10-4108
20. Freed-Pastor WA, Mizuno H, Zhao X, Langerod A, Moon SH, Rodriguez-Barrueco R, et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell. (2012) 148:244–58. doi: 10.1016/j.cell.2011.12.017
21. Moon SH, Huang CH, Houlihan SL, Regunath K, Freed-Pastor WA, Morris JP, et al. p53 represses the mevalonate pathway to mediate tumor suppression. Cell. (2019) 176:564–80 e519. doi: 10.1016/j.cell.2018.11.011
22. Sato R, Yang J, Wang X, Evans MJ, Ho YK, Goldstein JL, et al. Assignment of the membrane attachment, DNA binding, and transcriptional activation domains of sterol regulatory element-binding protein-1 (SREBP-1). J Biol Chem. (1994) 269:17267–73.
23. Shimano H, Horton JD, Shimomura I, Hammer RE, Brown MS, Goldstein JL. Isoform 1c of sterol regulatory element binding protein is less active than isoform 1a in livers of transgenic mice and in cultured cells. J Clin Invest. (1997) 99:846–54. doi: 10.1172/JCI119248
24. Weber LW, Boll M, Stampfl A. Maintaining cholesterol homeostasis: sterol regulatory element-binding proteins. World J Gastroenterol. (2004) 10:3081–7. doi: 10.3748/wjg.v10.i21.3081
25. Yabe D, Brown MS, Goldstein JL. Insig-2, a second endoplasmic reticulum protein that binds SCAP and blocks export of sterol regulatory element-binding proteins. Proc Natl Acad Sci USA. (2002) 99:12753–8. doi: 10.1073/pnas.162488899
26. Yang T, Espenshade PJ, Wright ME, Yabe D, Gong Y, Aebersold R, et al. Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell. (2002) 110:489–500. doi: 10.1016/S0092-8674(02)00872-3
27. Sun LP, Li L, Goldstein JL, Brown MS. Insig required for sterol-mediated inhibition of Scap/SREBP binding to COPII proteins in vitro. J Biol Chem. (2005) 280:26483–90. doi: 10.1074/jbc.M504041200
28. Brown MS, Radhakrishnan A, Goldstein JL. Retrospective on cholesterol homeostasis: the central role of scap. Annu Rev Biochem. (2018) 87:783–807. doi: 10.1146/annurev-biochem-062917-011852
29. Sakai J, Rawson RB, Espenshade PJ, Cheng D, Seegmiller AC, Goldstein JL, et al. Molecular identification of the sterol-regulated luminal protease that cleaves SREBPs and controls lipid composition of animal cells. Mol Cell. (1998) 2:505–14. doi: 10.1016/S1097-2765(00)80150-1
30. Rawson RB, Zelenski NG, Nijhawan D, Ye J, Sakai J, Hasan MT, et al. Complementation cloning of S2P, a gene encoding a putative metalloprotease required for intramembrane cleavage of SREBPs. Mol Cell. (1997) 1:47–57. doi: 10.1016/S1097-2765(00)80006-4
31. Radhakrishnan A, Goldstein JL, McDonald JG, Brown MS. Switch-like control of SREBP-2 transport triggered by small changes in ER cholesterol: a delicate balance. Cell Metab. (2008) 8:512–21. doi: 10.1016/j.cmet.2008.10.008
32. Yabe D, Xia ZP, Adams CM, Rawson RB. Three mutations in sterol-sensing domain of SCAP block interaction with insig and render SREBP cleavage insensitive to sterols. Proc Natl Acad Sci USA. (2002) 99:16672–7. doi: 10.1073/pnas.262669399
33. Kuan YC, Hashidume T, Shibata T, Uchida K, Shimizu M, Inoue J, et al. Heat shock protein 90 modulates lipid homeostasis by regulating the stability and function of Sterol Regulatory Element-binding Protein (SREBP) and SREBP cleavage-activating protein. J Biol Chem. (2017) 292:3016–28. doi: 10.1074/jbc.M116.767277
34. Irisawa M, Inoue J, Ozawa N, Mori K, Sato R. The sterol-sensing endoplasmic reticulum (ER) membrane protein TRC8 hampers ER to Golgi transport of sterol regulatory element-binding protein-2. (SREBP-2)/SREBP cleavage-activated protein and reduces SREBP-2 cleavage. J Biol Chem. (2009) 284:28995–9004. doi: 10.1074/jbc.M109.041376
35. Gong Y, Lee JN, Lee PC, Goldstein JL, Brown MS, Ye J. Sterol-regulated ubiquitination and degradation of Insig-1 creates a convergent mechanism for feedback control of cholesterol synthesis and uptake. Cell Metab. (2006) 3:15–24. doi: 10.1016/j.cmet.2005.11.014
36. Lee JN, Song B, DeBose-Boyd RA, Ye J. Sterol-regulated degradation of Insig-1 mediated by the membrane-bound ubiquitin ligase gp78. J Biol Chem. (2006) 281:39308–15. doi: 10.1074/jbc.M608999200
37. Radhakrishnan A, Ikeda Y, Kwon HJ, Brown MS, Goldstein JL. Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: oxysterols block transport by binding to Insig. Proc Natl Acad Sci USA. (2007) 104:6511–8. doi: 10.1073/pnas.0700899104
38. Feramisco JD, Radhakrishnan A, Ikeda Y, Reitz J, Brown MS, Goldstein JL. Intramembrane aspartic acid in SCAP protein governs cholesterol-induced conformational change. Proc Natl Acad Sci USA. (2005) 102:3242–7. doi: 10.1073/pnas.0500206102
39. Jo Y, Cha JY, Moon YA. Regulation of INSIG2 by microRNA-96. Anim Cells Syst. (2017) 21:263–8. doi: 10.1080/19768354.2017.1336483
40. Xu D, Wang Z, Zhang Y, Jiang W, Pan Y, Song BL, et al. PAQR3 modulates cholesterol homeostasis by anchoring Scap/SREBP complex to the Golgi apparatus. Nat Commun. (2015) 6:8100. doi: 10.1038/ncomms9100
41. Zhang L, Rajbhandari P, Priest C, Sandhu J, Wu X, Temel R, et al. Inhibition of cholesterol biosynthesis through RNF145-dependent ubiquitination of SCAP. Elife. (2017) 6:e28766. doi: 10.7554/eLife.28766
42. Du X, Kristiana I, Wong J, Brown AJ. Involvement of Akt in ER-to-Golgi transport of SCAP/SREBP: a link between a key cell proliferative pathway and membrane synthesis. Mol Biol Cell. (2006) 17:2735–45. doi: 10.1091/mbc.e05-11-1094
43. Luu W, Sharpe LJ, Stevenson J, Brown AJ. Akt acutely activates the cholesterogenic transcription factor SREBP-2. Biochim Biophys Acta. (2012) 1823:458–64. doi: 10.1016/j.bbamcr.2011.09.017
44. Haskins JW, Zhang S, Means RE, Kelleher JK, Cline GW, Canfran-Duque A, et al. Neuregulin-activated ERBB4 induces the SREBP-2 cholesterol biosynthetic pathway and increases low-density lipoprotein uptake. Sci Signal. (2015) 8:ra111. doi: 10.1126/scisignal.aac5124
45. Lewis CA, Griffiths B, Santos CR, Pende M, Schulze A. Regulation of the SREBP transcription factors by mTORC1. Biochem Soc Trans. (2011) 39:495–9. doi: 10.1042/BST0390495
46. Peterson TR, Sengupta SS, Harris TE, Carmack AE, Kang SA, Balderas E, et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell. (2011) 146:408–20. doi: 10.1016/j.cell.2011.06.034
47. Eid W, Dauner K, Courtney KC, Gagnon A, Parks RJ, Sorisky A, et al. mTORC1 activates SREBP-2 by suppressing cholesterol trafficking to lysosomes in mammalian cells. Proc Natl Acad Sci USA. (2017) 114:7999–8004. doi: 10.1073/pnas.1705304114
48. Konig B, Koch A, Spielmann J, Hilgenfeld C, Stangl GI, Eder K. Activation of PPARalpha lowers synthesis and concentration of cholesterol by reduction of nuclear SREBP-2. Biochem Pharmacol. (2007) 73:574–85. doi: 10.1016/j.bcp.2006.10.027
49. Sundqvist A, Bengoechea-Alonso MT, Ye X, Lukiyanchuk V, Jin J, Harper JW, et al. Control of lipid metabolism by phosphorylation-dependent degradation of the SREBP family of transcription factors by SCF(Fbw7). Cell Metab. (2005) 1:379–91. doi: 10.1016/j.cmet.2005.04.010
50. Kotzka J, Lehr S, Roth G, Avci H, Knebel B, Muller-Wieland D. Insulin-activated Erk-mitogen-activated protein kinases phosphorylate sterol regulatory element-binding Protein-2 at serine residues 432 and 455 in vivo. J Biol Chem. (2004) 279:22404–11. doi: 10.1074/jbc.M401198200
51. Li Y, Xu S, Mihaylova MM, Zheng B, Hou X, Jiang B, et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. (2011) 13:376–88. doi: 10.1016/j.cmet.2011.03.009
52. Giandomenico V, Simonsson M, Gronroos E, Ericsson J. Coactivator-dependent acetylation stabilizes members of the SREBP family of transcription factors. Mol Cell Biol. (2003) 23:2587–99. doi: 10.1128/MCB.23.7.2587-2599.2003
53. Walker AK, Yang F, Jiang K, Ji JY, Watts JL, Purushotham A, et al. Conserved role of SIRT1 orthologs in fasting-dependent inhibition of the lipid/cholesterol regulator SREBP. Genes Dev. (2010) 24:1403–17. doi: 10.1101/gad.1901210
54. Hirano Y, Murata S, Tanaka K, Shimizu M, Sato R. Sterol regulatory element-binding proteins are negatively regulated through SUMO-1 modification independent of the ubiquitin/26 S proteasome pathway. J Biol Chem. (2003) 278:16809–19. doi: 10.1074/jbc.M212448200
55. Inoue J, Sato R, Maeda M. Multiple DNA elements for sterol regulatory element-binding protein and NF-Y are responsible for sterol-regulated transcription of the genes for human 3-hydroxy-3-methylglutaryl coenzyme A synthase and squalene synthase. J Biochem. (1998) 123:1191–8. doi: 10.1093/oxfordjournals.jbchem.a022060
56. Zerenturk EJ, Sharpe LJ, Brown AJ. Sterols regulate 3beta-hydroxysterol Delta24-reductase (DHCR24) via dual sterol regulatory elements: cooperative induction of key enzymes in lipid synthesis by Sterol Regulatory Element Binding Proteins. Biochim Biophys Acta. (2012) 1821:1350–60. doi: 10.1016/j.bbalip.2012.07.006
57. Horton JD, Shah NA, Warrington JA, Anderson NN, Park SW, Brown MS, et al. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc Natl Acad Sci USA. (2003) 100:12027–32. doi: 10.1073/pnas.1534923100
58. Miziorko HM. Enzymes of the mevalonate pathway of isoprenoid biosynthesis. Arch Biochem Biophys. (2011) 505:131–43. doi: 10.1016/j.abb.2010.09.028
59. Gobel A, Rauner M, Hofbauer LC, Rachner TD. Cholesterol and beyond - the role of the mevalonate pathway in cancer biology. Biochim Biophys Acta Rev Cancer. (2020) 1873:188351. doi: 10.1016/j.bbcan.2020.188351
60. Do R, Kiss RS, Gaudet D, Engert JC. Squalene synthase: a critical enzyme in the cholesterol biosynthesis pathway. Clin Genet. (2009) 75:19–29. doi: 10.1111/j.1399-0004.2008.01099.x
61. Luo J, Yang H, Song BL. Mechanisms and regulation of cholesterol homeostasis. Nat Rev Mol Cell Biol. (2019) 21:225–45. doi: 10.1038/s41580-019-0190-7
62. Narwal V, Deswal R, Batra B, Kalra V, Hooda R, Sharma M, et al. Cholesterol biosensors: a review. Steroids. (2019) 143:6–17. doi: 10.1016/j.steroids.2018.12.003
63. Tamehiro N, Shigemoto-Mogami Y, Kakeya T, Okuhira K, Suzuki K, Sato R, et al. Sterol regulatory element-binding protein-2- and liver X receptor-driven dual promoter regulation of hepatic ABC transporter A1 gene expression: mechanism underlying the unique response to cellular cholesterol status. J Biol Chem. (2007) 282:21090–9. doi: 10.1074/jbc.M701228200
64. Wong J, Quinn CM, Brown AJ. SREBP-2 positively regulates transcription of the cholesterol efflux gene, ABCA1, by generating oxysterol ligands for LXR. Biochem J. (2006) 400:485–91. doi: 10.1042/BJ20060914
65. Hu YW, Zheng L, Wang Q. Regulation of cholesterol homeostasis by liver X receptors. Clin Chim Acta. (2010) 411:617–25. doi: 10.1016/j.cca.2009.12.027
66. Ingallina E, Sorrentino G, Bertolio R, Lisek K, Zannini A, Azzolin L, et al. Mechanical cues control mutant p53 stability through a mevalonate-RhoA axis. Nat Cell Biol. (2018) 20:28–35. doi: 10.1038/s41556-017-0009-8
67. Sorrentino G, Ruggeri N, Specchia V, Cordenonsi M, Mano M, Dupont S, et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat Cell Biol. (2014) 16:357–66. doi: 10.1038/ncb2936
68. Parrales A, Thoenen E, Iwakuma T. The interplay between mutant p53 and the mevalonate pathway. Cell Death Differ. (2018) 25:460–70. doi: 10.1038/s41418-017-0026-y
69. Calvisi DF, Wang C, Ho C, Ladu S, Lee SA, Mattu S, et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology. (2011) 140:1071–83. doi: 10.1053/j.gastro.2010.12.006
70. Pallottini V, Guantario B, Martini C, Totta P, Filippi I, Carraro F, et al. Regulation of HMG-CoA reductase expression by hypoxia. J Cell Biochem. (2008) 104:701–9. doi: 10.1002/jcb.21757
71. Kondo A, Yamamoto S, Nakaki R, Shimamura T, Hamakubo T, Sakai J, et al. Extracellular acidic pH activates the sterol regulatory element-binding protein 2 to promote tumor progression. Cell Rep. (2017) 18:2228–42. doi: 10.1016/j.celrep.2017.02.006
72. Guo D. SCAP links glucose to lipid metabolism in cancer cells. Mol Cell Oncol. (2016) 3:e1132120. doi: 10.1080/23723556.2015.1132120
73. Heemers H, Maes B, Foufelle F, Heyns W, Verhoeven G, Swinnen JV. Androgens stimulate lipogenic gene expression in prostate cancer cells by activation of the sterol regulatory element-binding protein cleavage activating protein/sterol regulatory element-binding protein pathway. Mol Endocrinol. (2001) 15:1817–28. doi: 10.1210/mend.15.10.0703
74. Chen M, Chen LM, Chai KX. Androgen regulation of prostasin gene expression is mediated by sterol-regulatory element-binding proteins and SLUG. Prostate. (2006) 66:911–20. doi: 10.1002/pros.20325
75. Ching YP, Davies SP, Hardie DG. Analysis of the specificity of the AMP-activated protein kinase by site-directed mutagenesis of bacterially expressed 3-hydroxy 3-methylglutaryl-CoA reductase, using a single primer variant of the unique-site-elimination method. Eur J Biochem. (1996) 237:800–8. doi: 10.1111/j.1432-1033.1996.0800p.x
76. Gaussin V, Skarlas P, Ching YP, Hardie DG, Hue L. Distinct type-2A protein phosphatases activate HMGCoA reductase and acetyl-CoA carboxylase in liver. FEBS Lett. (1997) 413:115–8. doi: 10.1016/S0014-5793(97)00890-9
77. Sever N, Yang T, Brown MS, Goldstein JL, DeBose-Boyd RA. Accelerated degradation of HMG CoA reductase mediated by binding of insig-1 to its sterol-sensing domain. Mol Cell. (2003) 11:25–33. doi: 10.1016/S1097-2765(02)00822-5
78. Tsai YC, Leichner GS, Pearce MM, Wilson GL, Wojcikiewicz RJ, Roitelman J, et al. Differential regulation of HMG-CoA reductase and Insig-1 by enzymes of the ubiquitin-proteasome system. Mol Biol Cell. (2012) 23:4484–94. doi: 10.1091/mbc.e12-08-0631
79. Han T, Lv Y, Wang S, Hu T, Hong H, Fu Z. PPARgamma overexpression regulates cholesterol metabolism in human L02 hepatocytes. J Pharmacol Sci. (2019) 139:1–8. doi: 10.1016/j.jphs.2018.09.013
80. Wang Y, Rogers PM, Su C, Varga G, Stayrook KR, Burris TP. Regulation of cholesterologenesis by the oxysterol receptor, LXRalpha. J Biol Chem. (2008) 283:26332–9. doi: 10.1074/jbc.M804808200
81. Tint GS, Pan L, Shang Q, Sharpe LJ, Brown AJ, Li M, et al. Desmosterol in brain is elevated because DHCR24 needs REST for Robust Expression but REST is poorly expressed. Dev Neurosci. (2014) 36:132–42. doi: 10.1159/000362363
82. Medina MW, Gao F, Naidoo D, Rudel LL, Temel RE, McDaniel AL, et al. Coordinately regulated alternative splicing of genes involved in cholesterol biosynthesis and uptake. PLoS ONE. (2011) 6:e19420. doi: 10.1371/journal.pone.0019420
83. Peffley DM, Gayen AK. Mevalonate regulates polysome distribution and blocks translation-dependent suppression of 3-hydroxy-3-methylglutaryl coenzyme A reductase mRNA: relationship to translational control. Somat Cell Mol Genet. (1995) 21:189–204. doi: 10.1007/BF02254770
84. Chen L, Ma MY, Sun M, Jiang LY, Zhao XT, Fang XX, et al. Endogenous sterol intermediates of the mevalonate pathway regulate HMGCR degradation and SREBP-2 processing. J Lipid Res. (2019) 60:1765–75. doi: 10.1194/jlr.RA119000201
85. Hinson DD, Chambliss KL, Toth MJ, Tanaka RD, Gibson KM. Post-translational regulation of mevalonate kinase by intermediates of the cholesterol and nonsterol isoprene biosynthetic pathways. J Lipid Res. (1997) 38:2216–23.
86. Metherall JE, Waugh K, Li H. Progesterone inhibits cholesterol biosynthesis in cultured cells. Accumulation of cholesterol precursors. J Biol Chem. (1996) 271:2627–33. doi: 10.1074/jbc.271.5.2627
87. Fernandez C, Suarez Y, Ferruelo AJ, Gomez-Coronado D, Lasuncion MA. Inhibition of cholesterol biosynthesis by Delta22-unsaturated phytosterols via competitive inhibition of sterol Delta24-reductase in mammalian cells. Biochem J. (2002) 366(Pt. 1):109–19. doi: 10.1042/bj20011777
88. Zerenturk EJ, Kristiana I, Gill S, Brown AJ. The endogenous regulator 24(S),25-epoxycholesterol inhibits cholesterol synthesis at DHCR24 (Seladin-1). Biochim Biophys Acta. (2012) 1821:1269–77. doi: 10.1016/j.bbalip.2011.11.009
89. Cheng C, Geng F, Cheng X, Guo D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun. (2018) 38:27. doi: 10.1186/s40880-018-0301-4
90. Ettinger SL, Sobel R, Whitmore TG, Akbari M, Bradley DR, Gleave ME, et al. Dysregulation of sterol response element-binding proteins and downstream effectors in prostate cancer during progression to androgen independence. Cancer Res. (2004) 64:2212–21. doi: 10.1158/0008-5472.CAN-2148-2
91. Armengol S, Arretxe E, Enzunza L, Llorente I, Mendibil U, Navarro-Imaz H, et al. SREBP-2-driven transcriptional activation of human SND1 oncogene. Oncotarget. (2017) 8:108181–94. doi: 10.18632/oncotarget.22569
92. Gruenbacher G, Thurnher M. Mevalonate metabolism in cancer stemness and trained immunity. Front Oncol. (2018) 8:394. doi: 10.3389/fonc.2018.00394
93. Suburu J, Chen YQ. Lipids and prostate cancer. Prostaglandins Other Lipid Mediat. (2012) 98:1–10. doi: 10.1016/j.prostaglandins.2012.03.003
94. Bull CJ, Bonilla C, Holly JM, Perks CM, Davies N, Haycock P, et al. Blood lipids and prostate cancer: a Mendelian randomization analysis. Cancer Med. (2016) 5:1125–36. doi: 10.1002/cam4.695
95. Fujita K, Nonomura N. Role of androgen receptor in prostate cancer: a review. World J Mens Health. (2019) 37:288–95. doi: 10.5534/wjmh.180040
96. Heemers HV, Verhoeven G, Swinnen JV. Androgen activation of the sterol regulatory element-binding protein pathway: current insights. Mol Endocrinol. (2006) 20:2265–77. doi: 10.1210/me.2005-0479
97. Heemers H, Verrijdt G, Organe S, Claessens F, Heyns W, Verhoeven G, et al. Identification of an androgen response element in intron 8 of the sterol regulatory element-binding protein cleavage-activating protein gene allowing direct regulation by the androgen receptor. J Biol Chem. (2004) 279:30880–7. doi: 10.1074/jbc.M401615200
98. Swinnen JV, Van Veldhoven PP, Esquenet M, Heyns W, Verhoeven G. Androgens markedly stimulate the accumulation of neutral lipids in the human prostatic adenocarcinoma cell line LNCaP. Endocrinology. (1996) 137:4468–74. doi: 10.1210/endo.137.10.8828509
99. Swinnen JV, Ulrix W, Heyns W, Verhoeven G. Coordinate regulation of lipogenic gene expression by androgens: evidence for a cascade mechanism involving sterol regulatory element binding proteins. Proc Natl Acad Sci USA. (1997) 94:12975–80. doi: 10.1073/pnas.94.24.12975
100. Chen M, Zhang J, Sampieri K, Clohessy JG, Mendez L, Gonzalez-Billalabeitia E, et al. An aberrant SREBP-dependent lipogenic program promotes metastatic prostate cancer. Nat Genet. (2018) 50:206–18. doi: 10.1038/s41588-017-0027-2
101. Ashida S, Kawada C, Inoue K. Stromal regulation of prostate cancer cell growth by mevalonate pathway enzymes HMGCS1 and HMGCR. Oncol Lett. (2017) 14:6533–42. doi: 10.3892/ol.2017.7025
102. Kong Y, Cheng L, Mao F, Zhang Z, Zhang Y, Farah E, et al. Inhibition of cholesterol biosynthesis overcomes enzalutamide resistance in castration-resistant prostate cancer (CRPC). J Biol Chem. (2018) 293:14328–41. doi: 10.1074/jbc.RA118.004442
103. Todenhofer T, Hennenlotter J, Kuhs U, Gerber V, Gakis G, Vogel U, et al. Altered expression of farnesyl pyrophosphate synthase in prostate cancer: evidence for a role of the mevalonate pathway in disease progression? World J Urol. (2013) 31:345–50. doi: 10.1007/s00345-012-0844-y
104. Seshacharyulu P, Rachagani S, Muniyan S, Siddiqui JA, Cruz E, Sharma S, et al. FDPS cooperates with PTEN loss to promote prostate cancer progression through modulation of small GTPases/AKT axis. Oncogene. (2019) 38:5265–80. doi: 10.1038/s41388-019-0791-9
105. Fukuma Y, Matsui H, Koike H, Sekine Y, Shechter I, Ohtake N, et al. Role of squalene synthase in prostate cancer risk and the biological aggressiveness of human prostate cancer. Prostate Cancer Prostatic Dis. (2012) 15:339–45. doi: 10.1038/pcan.2012.14
106. Zhao Z, Hao D, Wang L, Li J, Meng Y, Li P, et al. CtBP promotes metastasis of breast cancer through repressing cholesterol and activating TGF-beta signaling. Oncogene. (2019) 38:2076–91. doi: 10.1038/s41388-018-0570-z
107. Ricoult SJ, Yecies JL, Ben-Sahra I, Manning BD. Oncogenic PI3K and K-Ras stimulate de novo lipid synthesis through mTORC1 and SREBP. Oncogene. (2016) 35:1250–60. doi: 10.1038/onc.2015.179
108. Xu ZQ, Zhang Y, Li N, Liu PJ, Gao L, Gao X, et al. Efficacy and safety of lapatinib and trastuzumab for HER2-positive breast cancer: a systematic review and meta-analysis of randomised controlled trials. BMJ Open. (2017) 7:e013053. doi: 10.1136/bmjopen-2016-013053
109. Wang YC, Morrison G, Gillihan R, Guo J, Ward RM, Fu X, et al. Different mechanisms for resistance to trastuzumab versus lapatinib in HER2-positive breast cancers–role of estrogen receptor and HER2 reactivation. Breast Cancer Res. (2011) 13:R121. doi: 10.1186/bcr3067
110. Sethunath V, Hu H, De Angelis C, Veeraraghavan J, Qin L, Wang N, et al. Targeting the mevalonate pathway to overcome acquired anti-HER2 treatment resistance in breast cancer. Mol Cancer Res. (2019) 17:2318–30. doi: 10.1158/1541-7786.MCR-19-0756
111. Orho-Melander M, Hindy G, Borgquist S, Schulz CA, Manjer J, Melander O, et al. Blood lipid genetic scores, the HMGCR gene and cancer risk: a Mendelian randomization study. Int J Epidemiol. (2018) 47:495–505. doi: 10.1093/ije/dyx237
112. Clendening JW, Pandyra A, Boutros PC, El Ghamrasni S, Khosravi F, Trentin GA, et al. Dysregulation of the mevalonate pathway promotes transformation. Proc Natl Acad Sci USA. (2010) 107:15051–6. doi: 10.1073/pnas.0910258107
113. Di Benedetto A, Mottolese M, Sperati F, Ercolani C, Di Lauro L, Pizzuti L, et al. HMG-CoAR expression in male breast cancer: relationship with hormone receptors, Hippo transducers and survival outcomes. Sci Rep. (2016) 6:35121. doi: 10.1038/srep35121
114. Baek AE, Nelson ER. The contribution of cholesterol and its metabolites to the pathophysiology of breast cancer. Horm Cancer. (2016) 7:219–28. doi: 10.1007/s12672-016-0262-5
115. Nelson ER, Chang CY, McDonnell DP. Cholesterol and breast cancer pathophysiology. Trends Endocrinol Metab. (2014) 25:649–55. doi: 10.1016/j.tem.2014.10.001
116. Dehghani M, Samani Z, Abidi H, Manzouri L, Mahmoudi R, Hosseini Teshnizi S, et al. Relationship of SNP rs2645429 in farnesyl-diphosphate farnesyltransferase 1 gene promoter with susceptibility to lung cancer. Int J Genomics. (2018) 2018:4863757. doi: 10.1155/2018/4863757
117. Yang YF, Jan YH, Liu YP, Yang CJ, Su CY, Chang YC, et al. Squalene synthase induces tumor necrosis factor receptor 1 enrichment in lipid rafts to promote lung cancer metastasis. Am J Respir Crit Care Med. (2014) 190:675–87. doi: 10.1164/rccm.201404-0714OC
118. Lin L, Li M, Lin L, Xu X, Jiang G, Wu L. FPPS mediates TGF-beta1-induced non-small cell lung cancer cell invasion and the EMT process via the RhoA/Rock1 pathway. Biochem Biophys Res Commun. (2018) 496:536–41. doi: 10.1016/j.bbrc.2018.01.066
119. Wang X, Xu W, Zhan P, Xu T, Jin J, Miu Y, et al. Overexpression of geranylgeranyl diphosphate synthase contributes to tumour metastasis and correlates with poor prognosis of lung adenocarcinoma. J Cell Mol Med. (2018) 22:2177–89. doi: 10.1111/jcmm.13493
120. Ge H, Zhao Y, Shi X, Tan Z, Chi X, He M, et al. Squalene epoxidase promotes the proliferation and metastasis of lung squamous cell carcinoma cells though extracellular signal-regulated kinase signaling. Thorac Cancer. (2019) 10:428–36. doi: 10.1111/1759-7714.12944
121. Fatehi Hassanabad A, Mina F. Targeting the mevalonate pathway for treating lung cancer. Am J Clin Oncol. (2020) 43:69–70. doi: 10.1097/COC.0000000000000630
122. Liang B, Chen R, Song S, Wang H, Sun G, Yang H, et al. ASPP2 inhibits tumor growth by repressing the mevalonate pathway in hepatocellular carcinoma. Cell Death Dis. (2019) 10:830. doi: 10.1038/s41419-019-2054-7
123. Navarro-Imaz H, Chico Y, Rueda Y, Fresnedo O. Channeling of newly synthesized fatty acids to cholesterol esterification limits triglyceride synthesis in SND1-overexpressing hepatoma cells. Biochim Biophys Acta Mol Cell Biol Lipids. (2019) 1864:137–46. doi: 10.1016/j.bbalip.2018.11.004
124. Che L, Chi W, Qiao Y, Zhang J, Song X, Liu Y, et al. Cholesterol biosynthesis supports the growth of hepatocarcinoma lesions depleted of fatty acid synthase in mice and humans. Gut. (2019) 69:177–86. doi: 10.1136/gutjnl-2018-317581
125. Ogura S, Yoshida Y, Kurahashi T, Egawa M, Furuta K, Kiso S, et al. Targeting the mevalonate pathway is a novel therapeutic approach to inhibit oncogenic FoxM1 transcription factor in human hepatocellular carcinoma. Oncotarget. (2018) 9:21022–35. doi: 10.18632/oncotarget.24781
126. Syafruddin SE, Rodrigues P, Vojtasova E, Patel SA, Zaini MN, Burge J, et al. A KLF6-driven transcriptional network links lipid homeostasis and tumour growth in renal carcinoma. Nat Commun. (2019) 10:1152. doi: 10.1038/s41467-019-09116-x
127. Yu Y, Dong JT, He B, Zou YF, Li XS, Xi CH, et al. LncRNA SNHG16 induces the SREBP2 to promote lipogenesis and enhance the progression of pancreatic cancer. Future Oncol. (2019) 15:3831–44. doi: 10.2217/fon-2019-0321
128. Wang B, Rong X, Palladino END, Wang J, Fogelman AM, Martin MG, et al. Phospholipid remodeling and cholesterol availability regulate intestinal stemness and tumorigenesis. Cell Stem Cell. (2018) 22:206–20. e204. doi: 10.1016/j.stem.2017.12.017
129. Ma YS, Wu ZJ, Zhang HW, Cai B, Huang T, Long HD, et al. Dual regulatory mechanisms of expression and mutation involving metabolism-related genes FDFT1 and UQCR5 during CLM. Mol Ther Oncolytics. (2019) 14:172–8. doi: 10.1016/j.omto.2019.04.008
130. Dai M, Zhu XL, Liu F, Xu QY, Ge QL, Jiang SH, et al. Cholesterol synthetase DHCR24 induced by insulin aggravates cancer invasion and progesterone resistance in endometrial carcinoma. Sci Rep. (2017) 7:41404. doi: 10.1038/srep41404
131. Liu XP, Yin XH, Meng XY, Yan XH, Cao Y, Zeng XT, et al. DHCR24 predicts poor clinicopathological features of patients with bladder cancer: a STROBE-compliant study. Medicine. (2018) 97:e11830. doi: 10.1097/MD.0000000000011830
132. Li X, Chen YT, Josson S, Mukhopadhyay NK, Kim J, Freeman MR, et al. MicroRNA-185 and 342 inhibit tumorigenicity and induce apoptosis through blockade of the SREBP metabolic pathway in prostate cancer cells. PLoS ONE. (2013) 8:e70987. doi: 10.1371/journal.pone.0070987
133. Li X, Chen YT, Hu P, Huang WC. Fatostatin displays high antitumor activity in prostate cancer by blocking SREBP-regulated metabolic pathways and androgen receptor signaling. Mol Cancer Ther. (2014) 13:855–66. doi: 10.1158/1535-7163.MCT-13-0797
134. Yang M, Liu W, Pellicane C, Sahyoun C, Joseph BK, Gallo-Ebert C, et al. Identification of miR-185 as a regulator of de novo cholesterol biosynthesis and low density lipoprotein uptake. J Lipid Res. (2014) 55:226–38. doi: 10.1194/jlr.M041335
135. Yang L, Yang J, Li J, Shen X, Le Y, Zhou C, et al. MircoRNA-33a inhibits epithelial-to-mesenchymal transition and metastasis and could be a prognostic marker in non-small cell lung cancer. Sci Rep. (2015) 5:13677. doi: 10.1038/srep13677
136. Gholkar AA, Cheung K, Williams KJ, Lo YC, Hamideh SA, Nnebe C, et al. Fatostatin inhibits cancer cell proliferation by affecting mitotic microtubule spindle assembly and cell division. J Biol Chem. (2016) 291:17001–8. doi: 10.1074/jbc.C116.737346
137. Choi Y, Kawazoe Y, Murakami K, Misawa H, Uesugi M. Identification of bioactive molecules by adipogenesis profiling of organic compounds. J Biol Chem. (2003) 278:7320–4. doi: 10.1074/jbc.M210283200
138. Kamisuki S, Mao Q, Abu-Elheiga L, Gu Z, Kugimiya A, Kwon Y, et al. A small molecule that blocks fat synthesis by inhibiting the activation of SREBP. Chem Biol. (2009) 16:882–92. doi: 10.1016/j.chembiol.2009.07.007
139. Brovkovych V, Izhar Y, Danes JM, Dubrovskyi O, Sakallioglu IT, Morrow LM. Fatostatin induces pro- and anti-apoptotic lipid accumulation in breast cancer. Oncogenesis. (2018) 7:66. doi: 10.1038/s41389-018-0076-0
140. Gao S, Shi Z, Li X, Li W, Wang Y, Liu Z, et al. Fatostatin suppresses growth and enhances apoptosis by blocking SREBP-regulated metabolic pathways in endometrial carcinoma. Oncol Rep. (2018) 39:1919–29. doi: 10.3892/or.2018.6265
141. Shao W, Machamer CE, Espenshade PJ. Fatostatin blocks ER exit of SCAP but inhibits cell growth in a SCAP-independent manner. J Lipid Res. (2016) 57:1564–73. doi: 10.1194/jlr.M069583
142. Li X, Wu JB, Chung LW, Huang WC. Anti-cancer efficacy of SREBP inhibitor, alone or in combination with docetaxel, in prostate cancer harboring p53 mutations. Oncotarget. (2015) 6:41018–32. doi: 10.18632/oncotarget.5879
143. Krycer JR, Phan L, Brown AJ. A key regulator of cholesterol homoeostasis, SREBP-2, can be targeted in prostate cancer cells with natural products. Biochem J. (2012) 446:191–201. doi: 10.1042/BJ20120545
144. Wei S, Liu L, Chen Z, Yin W, Liu Y, Ouyang Q, et al. Artesunate inhibits the mevalonate pathway and promotes glioma cell senescence. J Cell Mol Med. (2019). doi: 10.1111/jcmm.14717
145. Kim YS, Lee YM, Oh TI, Shin DH, Kim GH, Kan SY, et al. Emodin sensitizes hepatocellular carcinoma cells to the anti-cancer effect of sorafenib through suppression of cholesterol metabolism. Int J Mol Sci. (2018) 19:3127. doi: 10.3390/ijms19103127
146. Kim GH, Kan SY, Kang H, Lee S, Ko HM, Kim JH, et al. Ursolic acid suppresses cholesterol biosynthesis and exerts anti-cancer effects in hepatocellular carcinoma cells. Int J Mol Sci. (2019) 20:4767. doi: 10.3390/ijms20194767
147. Hamm R, Chen YR, Seo EJ, Zeino M, Wu CF, Muller R, et al. Induction of cholesterol biosynthesis by archazolid B in T24 bladder cancer cells. Biochem Pharmacol. (2014) 91:18–30. doi: 10.1016/j.bcp.2014.06.018
148. Horie T, Ono K, Horiguchi M, Nishi H, Nakamura T, Nagao K, et al. MicroRNA-33 encoded by an intron of sterol regulatory element-binding protein 2 (Srebp2) regulates HDL in vivo. Proc Natl Acad Sci USA. (2010) 107:17321–6. doi: 10.1073/pnas.1008499107
149. Geng C, Dong T, Jin W, Yu B, Yin F, Peng F, et al. MicroRNA-98 regulates hepatic cholesterol metabolism via targeting sterol regulatory element-binding protein 2. Biochem Biophys Res Commun. (2018) 504:422–6. doi: 10.1016/j.bbrc.2018.08.205
150. Kochuparambil ST, Al-Husein B, Goc A, Soliman S, Somanath PR. Anticancer efficacy of simvastatin on prostate cancer cells and tumor xenografts is associated with inhibition of Akt and reduced prostate-specific antigen expression. J Pharmacol Exp Ther. (2011) 336:496–505. doi: 10.1124/jpet.110.174870
151. Deng JL, Zhang R, Zeng Y, Zhu YS, Wang G. Statins induce cell apoptosis through a modulation of AKT/FOXO1 pathway in prostate cancer cells. Cancer Manag Res. (2019) 11:7231–42. doi: 10.2147/CMAR.S212643
152. Beckwitt CH, Clark AM, Ma B, Whaley D, Oltvai ZN, Wells A. Statins attenuate outgrowth of breast cancer metastases. Br J Cancer. (2018) 119:1094–105. doi: 10.1038/s41416-018-0267-7
153. Hu MB, Zhang JW, Gao JB, Qi YW, Gao Y, Xu L, et al. Atorvastatin induces autophagy in MDA-MB-231 breast cancer cells. Ultrastruct Pathol. (2018) 42:409–15. doi: 10.1080/01913123.2018.1522406
154. Ma Q, Gao Y, Xu P, Li K, Xu X, Gao J, et al. Atorvastatin inhibits breast cancer cells by downregulating PTEN/AKT pathway via promoting ras homolog family member B (RhoB). Biomed Res Int. (2019) 2019:3235021. doi: 10.1155/2019/3235021
155. Koohestanimobarhan S, Salami S, Imeni V, Mohammadi Z, Bayat O. Lipophilic statins antagonistically alter the major epithelial-to-mesenchymal transition signaling pathways in breast cancer stem-like cells via inhibition of the mevalonate pathway. J Cell Biochem. (2018) 120:2515–31. doi: 10.1002/jcb.27544
156. Wang T, Seah S, Loh X, Chan CW, Hartman M, Goh BC, et al. Simvastatin-induced breast cancer cell death and deactivation of PI3K/Akt and MAPK/ERK signalling are reversed by metabolic products of the mevalonate pathway. Oncotarget. (2016) 7:2532–44. doi: 10.18632/oncotarget.6304
157. Wolfe AR, Debeb BG, Lacerda L, Larson R, Bambhroliya A, Huang X, et al. Simvastatin prevents triple-negative breast cancer metastasis in pre-clinical models through regulation of FOXO3a. Breast Cancer Res Treat. (2015) 154:495–508. doi: 10.1007/s10549-015-3645-3
158. Kou X, Jiang X, Liu H, Wang X, Sun F, Han J, et al. Simvastatin functions as a heat shock protein 90 inhibitor against triple-negative breast cancer. Cancer Sci. (2018) 109:3272–84. doi: 10.1111/cas.13748
159. Wang L, Wang Y, Chen A, Teli M, Kondo R, Jalali A, et al. Pitavastatin slows tumor progression and alters urine-derived volatile organic compounds through the mevalonate pathway. FASEB J. (2019) 33:13710–21. doi: 10.1096/fj.201901388R
160. Fan Z, Jiang H, Wang Z, Qu J. Atorvastatin partially inhibits the epithelial-mesenchymal transition in A549 cells induced by TGF-beta1 by attenuating the upregulation of SphK1. Oncol Rep. (2016) 36:1016–22. doi: 10.3892/or.2016.4897
161. Walther U, Emmrich K, Ramer R, Mittag N, Hinz B. Lovastatin lactone elicits human lung cancer cell apoptosis via a COX-2/PPARgamma-dependent pathway. Oncotarget. (2016) 7:10345–62. doi: 10.18632/oncotarget.7213
162. Shang L, Jia SS, Jiang HM, Wang H, Xu WH, Lv CJ. Simvastatin downregulates expression of TGF-betaRII and inhibits proliferation of A549 cells via ERK. Tumour Biol. (2015) 36:4819–24. doi: 10.1007/s13277-015-3134-7
163. Nakashima Y, Haneji T. Stimulation of osteoclast formation by RANKL requires interferon regulatory factor-4 and is inhibited by simvastatin in a mouse model of bone loss. PLoS ONE. (2013) 8:e72033. doi: 10.1371/journal.pone.0072033
164. Sarkar D. Statins as inhibitors of lung cancer bone metastasis. EBioMedicine. (2017) 19:6–7. doi: 10.1016/j.ebiom.2017.04.028
165. Yang Z, Su Z, DeWitt JP, Xie L, Chen Y, Li X, et al. Fluvastatin prevents lung adenocarcinoma bone metastasis by triggering autophagy. EBioMedicine. (2017) 19:49–59. doi: 10.1016/j.ebiom.2017.04.017
166. Wang ST, Ho HJ, Lin JT, Shieh JJ, Wu CY. Simvastatin-induced cell cycle arrest through inhibition of STAT3/SKP2 axis and activation of AMPK to promote p27 and p21 accumulation in hepatocellular carcinoma cells. Cell Death Dis. (2017) 8:e2626. doi: 10.1038/cddis.2016.472
167. Higashi T, Hayashi H, Kitano Y, Yamamura K, Kaida T, Arima K, et al. Statin attenuates cell proliferative ability via TAZ (WWTR1) in hepatocellular carcinoma. Med Oncol. (2016) 33:123. doi: 10.1007/s12032-016-0845-6
168. Huang X, Ma J, Xu J, Su Q, Zhao J. Simvastatin induces growth inhibition and apoptosis in HepG2 and Huh7 hepatocellular carcinoma cells via upregulation of Notch1 expression. Mol Med Rep. (2015) 11:2334–40. doi: 10.3892/mmr.2014.2976
169. Relja B, Meder F, Wang M, Blaheta R, Henrich D, Marzi I, et al. Simvastatin modulates the adhesion and growth of hepatocellular carcinoma cells via decrease of integrin expression and ROCK. Int J Oncol. (2011) 38:879–85. doi: 10.3892/ijo.2010.892
170. Woodard J, Sassano A, Hay N, Platanias LC. Statin-dependent suppression of the Akt/mammalian target of rapamycin signaling cascade and programmed cell death 4 up-regulation in renal cell carcinoma. Clin Cancer Res. (2008) 14:4640–9. doi: 10.1158/1078-0432.CCR-07-5232
171. Qi XF, Zheng L, Lee KJ, Kim DH, Kim CS, Cai DQ, et al. HMG-CoA reductase inhibitors induce apoptosis of lymphoma cells by promoting ROS generation and regulating Akt, Erk and p38 signals via suppression of mevalonate pathway. Cell Death Dis. (2013) 4:e518. doi: 10.1038/cddis.2013.44
172. Zheng X, Cui XX, Gao Z, Zhao Y, Lin Y, Shih WJ, et al. Atorvastatin and celecoxib in combination inhibits the progression of androgen-dependent LNCaP xenograft prostate tumors to androgen independence. Cancer Prev Res. (2010) 3:114–24. doi: 10.1158/1940-6207.CAPR-09-0059
173. Martirosyan A, Clendening JW, Goard CA, Penn LZ. Lovastatin induces apoptosis of ovarian cancer cells and synergizes with doxorubicin: potential therapeutic relevance. BMC Cancer. (2010) 10:103. doi: 10.1186/1471-2407-10-103
174. Lee JS, Roberts A, Juarez D, Vo TT, Bhatt S, Herzog LO, et al. Statins enhance efficacy of venetoclax in blood cancers. Sci Transl Med. (2018) 10:eaaq1240. doi: 10.1126/scitranslmed.aaq1240
175. Kim JS, Turbov J, Rosales R, Thaete LG, Rodriguez GC. Combination simvastatin and metformin synergistically inhibits endometrial cancer cell growth. Gynecol Oncol. (2019) 154:432–40. doi: 10.1016/j.ygyno.2019.05.022
176. McGregor GH, Campbell AD, Fey SK, Tumanov S, Sumpton D, Blanco GR, et al. Targeting the metabolic response to statin-mediated oxidative stress produces a synergistic antitumor response. Cancer Res. (2020) 80:175–88. doi: 10.1158/0008-5472.CAN-19-0644
177. Fragni M, Bonini SA, Stabile A, Bodei S, Cristinelli L, Simeone C, et al. Inhibition of survivin is associated with zoledronic acid-induced apoptosis of prostate cancer cells. Anticancer Res. (2016) 36:913–20.
178. Pietrovito L, Comito G, Parri M, Giannoni E, Chiarugi P, Taddei ML. Zoledronic Acid inhibits the RhoA-mediated amoeboid motility of prostate cancer cells. Curr Cancer Drug Targets. (2019) 19:807–16. doi: 10.2174/1568009619666190115142858
179. Rouhrazi H, Turgan N, Oktem G. Zoledronic acid overcomes chemoresistance by sensitizing cancer stem cells to apoptosis. Biotech Histochem. (2018) 93:77–88. doi: 10.1080/10520295.2017.1387286
180. Lin JF, Lin YC, Lin YH, Tsai TF, Chou KY, Chen HE, et al. Zoledronic acid induces autophagic cell death in human prostate cancer cells. J Urol. (2011) 185:1490–6. doi: 10.1016/j.juro.2010.11.045
181. Coxon JP, Oades GM, Kirby RS, Colston KW. Zoledronic acid induces apoptosis and inhibits adhesion to mineralized matrix in prostate cancer cells via inhibition of protein prenylation. BJU Int. (2004) 94:164–70. doi: 10.1111/j.1464-4096.2004.04831.x
182. Liu H, Wang SH, Chen SC, Chen CY, Lin TM. Zoledronic acid blocks the interaction between breast cancer cells and regulatory T-cells. BMC Cancer. (2019) 19:176. doi: 10.1186/s12885-019-5379-9
183. Kopecka J, Porto S, Lusa S, Gazzano E, Salzano G, Pinzon-Daza ML, et al. Zoledronic acid-encapsulating self-assembling nanoparticles and doxorubicin: a combinatorial approach to overcome simultaneously chemoresistance and immunoresistance in breast tumors. Oncotarget. (2016) 7:20753–72. doi: 10.18632/oncotarget.8012
184. Li YY, Chang JW, Liu YC, Wang CH, Chang HJ, Tsai MC, et al. Zoledronic acid induces cell-cycle prolongation in murine lung cancer cells by perturbing cyclin and Ras expression. Anticancer Drugs. (2011) 22:89–98. doi: 10.1097/CAD.0b013e3283400a05
185. Xie F, Li P, Gong J, Zhang J, Ma J. The bisphosphonate zoledronic acid effectively targets lung cancer cells by inhibition of protein prenylation. Biochem Biophys Res Commun. (2015) 467:664–9. doi: 10.1016/j.bbrc.2015.10.089
186. Honda Y, Takahashi S, Zhang Y, Ono A, Murakami E, Shi N, et al. Effects of bisphosphonate zoledronic acid in hepatocellular carcinoma, depending on mevalonate pathway. J Gastroenterol Hepatol. (2015) 30:619–27. doi: 10.1111/jgh.12715
187. Goto H, Matsuda K, Srikoon P, Kariya R, Hattori S, Taura M, et al. Potent antitumor activity of zoledronic acid-induced Vgamma9Vdelta2 T cells against primary effusion lymphoma. Cancer Lett. (2013) 331:174–82. doi: 10.1016/j.canlet.2012.12.021
188. Comito G, Pons Segura C, Taddei ML, Lanciotti M, Serni S, Morandi A, et al. Zoledronic acid impairs stromal reactivity by inhibiting M2-macrophages polarization and prostate cancer-associated fibroblasts. Oncotarget. (2017) 8:118–32. doi: 10.18632/oncotarget.9497
189. Miwa S, Mizokami A, Keller ET, Taichman R, Zhang J, Namiki M. The bisphosphonate YM529 inhibits osteolytic and osteoblastic changes and CXCR-4-induced invasion in prostate cancer. Cancer Res. (2005) 65:8818–25. doi: 10.1158/0008-5472.CAN-05-0540
190. Koshimune R, Aoe M, Toyooka S, Hara F, Ouchida M, Tokumo M, et al. Anti-tumor effect of bisphosphonate (YM529) on non-small cell lung cancer cell lines. BMC Cancer. (2007) 7:8. doi: 10.1186/1471-2407-7-8
191. Sato K, Yuasa T, Nogawa M, Kimura S, Segawa H, Yokota A, et al. A third-generation bisphosphonate, minodronic acid (YM529), successfully prevented the growth of bladder cancer in vitro and in vivo. Br J Cancer. (2006) 95:1354–61. doi: 10.1038/sj.bjc.6603423
192. Jones RM, Morgan C, Bertelli G. Effects of zoledronic acid and docetaxel on small GTP-binding proteins in prostate cancer. Tumour Biol. (2015) 36:4861–9. doi: 10.1007/s13277-015-3140-9
193. Wilke M, Gobel A, Rauner M, Benad-Mehner P, Schutze N, Fussel S, et al. Zoledronic acid and atorvastatin inhibit alphavbeta3-mediated adhesion of breast cancer cells. J Bone Oncol. (2014) 3:10–7. doi: 10.1016/j.jbo.2014.02.001
194. Jagdev SP, Coleman RE, Shipman CM, Rostami HA, Croucher PI. The bisphosphonate, zoledronic acid, induces apoptosis of breast cancer cells: evidence for synergy with paclitaxel. Br J Cancer. (2001) 84:1126–34. doi: 10.1054/bjoc.2001.1727
195. Gobel A, Thiele S, Browne AJ, Rauner M, Zinna VM, Hofbauer LC, et al. Combined inhibition of the mevalonate pathway with statins and zoledronic acid potentiates their anti-tumor effects in human breast cancer cells. Cancer Lett. (2016) 375:162–71. doi: 10.1016/j.canlet.2016.03.004
196. Feng C, Liu X, Li X, Guo F, Huang C, Qin Q, et al. Zoledronic acid increases the antitumor effect of gefitinib treatment for non-small cell lung cancer with EGFR mutations. Oncol Rep. (2016) 35:3460–70. doi: 10.3892/or.2016.4741
197. Kuzu OF, Noory MA, Robertson GP. The role of cholesterol in cancer. Cancer Res. (2016) 76:2063–70. doi: 10.1158/0008-5472.CAN-15-2613
198. Ding X, Zhang W, Li S, Yang H. The role of cholesterol metabolism in cancer. Am J Cancer Res. (2019) 9:219–27.
199. Corcos L, Le Jossic-Corcos C. Statins: perspectives in cancer therapeutics. Dig Liver Dis. (2013) 45:795–802. doi: 10.1016/j.dld.2013.02.002
200. Iannelli F, Lombardi R, Milone MR, Pucci B, De Rienzo S, Budillon A, et al. Targeting mevalonate pathway in cancer treatment: repurposing of statins. Recent Pat Anticancer Drug Discov. (2018) 13:184–200. doi: 10.2174/1574892812666171129141211
201. Singh H, Mahmud SM, Turner D, Xue L, Demers AA, Bernstein CN. Long-term use of statins and risk of colorectal cancer: a population-based study. Am J Gastroenterol. (2009) 104:3015–23. doi: 10.1038/ajg.2009.574
202. Chiu HF, Ho SC, Chen CC, Yang CY. Statin use and the risk of liver cancer: a population-based case-control study. Am J Gastroenterol. (2011) 106:894–8. doi: 10.1038/ajg.2010.475
203. Khurana V, Bejjanki HR, Caldito G, Owens MW. Statins reduce the risk of lung cancer in humans: a large case-control study of US veterans. Chest. (2007) 131:1282–8. doi: 10.1378/chest.06-0931
204. Fatehi Hassanabad A, McBride SA. Statins as potential therapeutics for lung cancer: molecular mechanisms and clinical outcomes. Am J Clin Oncol. (2019) 42:732–6. doi: 10.1097/COC.0000000000000579
205. Gobel A, Breining D, Rauner M, Hofbauer LC, Rachner TD. Induction of 3-hydroxy-3-methylglutaryl-CoA reductase mediates statin resistance in breast cancer cells. Cell Death Dis. (2019) 10:91. doi: 10.1038/s41419-019-1322-x
206. Murtola TJ, Peltomaa AI, Talala K, Maattanen L, Taari K, Tammela TLJ, et al. statin use and prostate cancer survival in the finnish randomized study of screening for prostate cancer. Eur Urol Focus. (2017) 3:212–20. doi: 10.1016/j.euf.2016.05.004
207. Liu B, Yi Z, Guan X, Zeng YX, Ma F. The relationship between statins and breast cancer prognosis varies by statin type and exposure time: a meta-analysis. Breast Cancer Res Treat. (2017) 164:1–11. doi: 10.1007/s10549-017-4246-0
208. Xia DK, Hu ZG, Tian YF, Zeng FJ. Statin use and prognosis of lung cancer: a systematic review and meta-analysis of observational studies and randomized controlled trials. Drug Des Devel Ther. (2019) 13:405–22. doi: 10.2147/DDDT.S187690
209. Kim GA, Shim JJ, Lee JS, Kim BH, Kim JW, Oh CH, et al. Effect of statin use on liver cancer mortality considering hypercholesterolemia and obesity in patients with non-cirrhotic chronic hepatitis B. Yonsei Med J. (2019) 60:1203–8. doi: 10.3349/ymj.2019.60.12.1203
210. Couttenier A, Lacroix O, Vaes E, Cardwell CR, De Schutter H, Robert A. Statin use is associated with improved survival in ovarian cancer: a retrospective population-based study. PLoS ONE. (2017) 12:e0189233. doi: 10.1371/journal.pone.0189233
211. Nguyen T, Khan A, Liu Y, El-Serag HB, Thrift AP. The association between statin use after diagnosis and mortality risk in patients with esophageal cancer: a retrospective cohort study of United States veterans. Am J Gastroenterol. (2018) 113:1310. doi: 10.1038/s41395-018-0169-6
212. Mei Z, Liang M, Li L, Zhang Y, Wang Q, Yang W. Effects of statins on cancer mortality and progression: a systematic review and meta-analysis of 95 cohorts including 1,111,407 individuals. Int J Cancer. (2017) 140:1068–81. doi: 10.1002/ijc.30526
213. Farooqi MAM, Malhotra N, Mukherjee SD, Sanger S, Dhesy-Thind SK, Ellis P, et al. Statin therapy in the treatment of active cancer: a systematic review and meta-analysis of randomized controlled trials. PLoS ONE. (2018) 13:e0209486. doi: 10.1371/journal.pone.0209486
214. Zaleska M, Mozenska O, Bil J. Statins use and cancer: an update. Future Oncol. (2018) 14:1497–509. doi: 10.2217/fon-2017-0543
215. Lin JJ, Ezer N, Sigel K, Mhango G, Wisnivesky JP. The effect of statins on survival in patients with stage IV lung cancer. Lung Cancer. (2016) 99:137–42. doi: 10.1016/j.lungcan.2016.07.006
216. Hus M, Grzasko N, Szostek M, Pluta A, Helbig G, Woszczyk D, et al. Thalidomide, dexamethasone and lovastatin with autologous stem cell transplantation as a salvage immunomodulatory therapy in patients with relapsed and refractory multiple myeloma. Ann Hematol. (2011) 90:1161–6. doi: 10.1007/s00277-011-1276-2
217. Advani AS, Li H, Michaelis LC, Medeiros BC, Liedtke M, List AF, et al. Report of the relapsed/refractory cohort of SWOG S0919: A phase 2 study of idarubicin and cytarabine in combination with pravastatin for acute myelogenous leukemia (AML). Leuk Res. (2018) 67:17–20. doi: 10.1016/j.leukres.2018.01.021
218. Hung MS, Chen IC, Lee CP, Huang RJ, Chen PC, Tsai YH, et al. Statin improves survival in patients with EGFR-TKI lung cancer: a nationwide population-based study. PLoS ONE. (2017) 12:e0171137. doi: 10.1371/journal.pone.0171137
219. Li X, Wu XB, Chen Q. Statin use is not associated with reduced risk of skin cancer: a meta-analysis. Br J Cancer. (2014) 110:802–7. doi: 10.1038/bjc.2013.762
220. Gray RT, Loughrey MB, Bankhead P, Cardwell CR, McQuaid S, O'Neill RF, et al. Statin use, candidate mevalonate pathway biomarkers, and colon cancer survival in a population-based cohort study. Br J Cancer. (2017) 116:1652–9. doi: 10.1038/bjc.2017.139
221. Cholesterol Treatment Trialists Collaboration, Emberson JR, Kearney PM, Blackwell L, Newman C, Reith C, et al. Lack of effect of lowering LDL cholesterol on cancer: meta-analysis of individual data from 175,000 people in 27 randomised trials of statin therapy. PLoS ONE. (2012) 7:e29849. doi: 10.1371/journal.pone.0029849
222. Abdullah MI, de Wolf E, Jawad MJ, Richardson A. The poor design of clinical trials of statins in oncology may explain their failure - lessons for drug repurposing. Cancer Treat Rev. (2018) 69:84–9. doi: 10.1016/j.ctrv.2018.06.010
223. Finianos A, Aragon-Ching JB. Zoledronic acid for the treatment of prostate cancer. Expert Opin Pharmacother. (2019) 20:657–66. doi: 10.1080/14656566.2019.1574754
224. Lluch A, Cueva J, Ruiz-Borrego M, Ponce J, Perez-Fidalgo JA. Zoledronic acid in the treatment of metastatic breast cancer. Anticancer Drugs. (2014) 25:1–7. doi: 10.1097/CAD.0000000000000020
225. Seider MJ, Pugh SL, Langer C, Wyatt G, Demas W, Rashtian A, et al. Randomized phase III trial to evaluate radiopharmaceuticals and zoledronic acid in the palliation of osteoblastic metastases from lung, breast, and prostate cancer: report of the NRG Oncology RTOG 0517 trial. Ann Nucl Med. (2018) 32:553–60. doi: 10.1007/s12149-018-1278-4
226. Vale CL, Burdett S, Rydzewska LHM, Albiges L, Clarke NW, Fisher D, et al. Addition of docetaxel or bisphosphonates to standard of care in men with localised or metastatic, hormone-sensitive prostate cancer: a systematic review and meta-analyses of aggregate data. Lancet Oncol. (2016) 17:243–56. doi: 10.1016/S1470-2045(15)00489-1
227. van Beek E, Pieterman E, Cohen L, Lowik C, Papapoulos S. Farnesyl pyrophosphate synthase is the molecular target of nitrogen-containing bisphosphonates. Biochem Biophys Res Commun. (1999) 264:108–11. doi: 10.1006/bbrc.1999.1499
228. Rakel A, Boucher A, Ste-Marie LG. Role of zoledronic acid in the prevention and treatment of osteoporosis. Clin Interv Aging. (2011) 6:89–99. doi: 10.2147/CIA.S7282
229. Van Acker HH, Anguille S, Willemen Y, Smits EL, Van Tendeloo VF. Bisphosphonates for cancer treatment: mechanisms of action and lessons from clinical trials. Pharmacol Ther. (2016) 158:24–40. doi: 10.1016/j.pharmthera.2015.11.008
230. Green JR. Antitumor effects of bisphosphonates. Cancer. (2003) 97:840–7. doi: 10.1002/cncr.11128
231. Zekri J, Mansour M, Karim SM. The anti-tumour effects of zoledronic acid. J Bone Oncol. (2014) 3:25–35. doi: 10.1016/j.jbo.2013.12.001
232. Pinkawa M. Zoledronic acid in first-line treatment of prostate cancer. Int J Radiat Oncol Biol Phys. (2017) 97:6–8. doi: 10.1016/j.ijrobp.2016.06.2453
Keywords: SREBP-2, HMG-CoA reductase, mevalonate, cholesterol, cancer therapy
Citation: Xue L, Qi H, Zhang H, Ding L, Huang Q, Zhao D, Wu BJ and Li X (2020) Targeting SREBP-2-Regulated Mevalonate Metabolism for Cancer Therapy. Front. Oncol. 10:1510. doi: 10.3389/fonc.2020.01510
Received: 24 April 2020; Accepted: 14 July 2020;
Published: 21 August 2020.
Edited by:
Miriam Martini, University of Turin, ItalyReviewed by:
Cinzia Domenicotti, University of Genoa, ItalyPaola Defilippi, University of Turin, Italy
Copyright © 2020 Xue, Qi, Zhang, Ding, Huang, Zhao, Wu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Boyang Jason Wu, Ym95YW5nLnd1QHdzdS5lZHU=; Xiangyan Li, eGlhbmd5YW5fbGkxOTgxQDE2My5jb20=