 Ken Asada1,2*†
Ken Asada1,2*† Syuzo Kaneko1,2†
Syuzo Kaneko1,2† Ken Takasawa1,2Hidenori Machino1,2Satoshi Takahashi1,2Norio Shinkai1,2,3
Ken Takasawa1,2Hidenori Machino1,2Satoshi Takahashi1,2Norio Shinkai1,2,3 Ryo Shimoyama1,2
Ryo Shimoyama1,2 Masaaki Komatsu1,2
Masaaki Komatsu1,2 Ryuji Hamamoto1,2,3*
Ryuji Hamamoto1,2,3*- 1Cancer Translational Research Team, RIKEN Center for Advanced Intelligence Project, Tokyo, Japan
- 2Division of Medical AI Research and Development, National Cancer Center Research Institute, Tokyo, Japan
- 3Department of NCC Cancer Science, Graduate School of Medical and Dental Sciences, Tokyo Medical and Dental University, Tokyo, Japan
With the completion of the International Human Genome Project, we have entered what is known as the post-genome era, and efforts to apply genomic information to medicine have become more active. In particular, with the announcement of the Precision Medicine Initiative by U.S. President Barack Obama in his State of the Union address at the beginning of 2015, “precision medicine,” which aims to divide patients and potential patients into subgroups with respect to disease susceptibility, has become the focus of worldwide attention. The field of oncology is also actively adopting the precision oncology approach, which is based on molecular profiling, such as genomic information, to select the appropriate treatment. However, the current precision oncology is dominated by a method called targeted-gene panel (TGP), which uses next-generation sequencing (NGS) to analyze a limited number of specific cancer-related genes and suggest optimal treatments, but this method causes the problem that the number of patients who benefit from it is limited. In order to steadily develop precision oncology, it is necessary to integrate and analyze more detailed omics data, such as whole genome data and epigenome data. On the other hand, with the advancement of analysis technologies such as NGS, the amount of data obtained by omics analysis has become enormous, and artificial intelligence (AI) technologies, mainly machine learning (ML) technologies, are being actively used to make more efficient and accurate predictions. In this review, we will focus on whole genome sequencing (WGS) analysis and epigenome analysis, introduce the latest results of omics analysis using ML technologies for the development of precision oncology, and discuss the future prospects.
Introduction
The structure of DNA was first reported by Watson and Crick in 1953 (1). Following this, the first sequencing technique known as the Sanger sequencing method was developed in 1977 (2). In 1987, the first automatic sequencing machine (AB370) was introduced by Applied Biosystems, which uses capillary electrophoresis without the need for a gel, which enabled the sequencing process to be more convenient in terms of accuracy and time (3). This technology truly accelerated the completion of the International Human Genome Project, which was aimed at decoding three billion human nucleotide base pairs (4). With the completion of the International Human Genome Project, the era known as the post-genome era began, and attempts to apply genomic information to medicine began to be actively pursued. Consequently, the concept of personalized medicine has also come to attract attention (5–7). Under such circumstances, the advent of a new analysis method called next-generation sequencing (NGS) technology has rapidly accelerated the speed of nucleotide sequence analysis and dramatically lowered the cost of performing whole genome analysis (8, 9). As a result, genome-wide analysis can now be performed routinely. In addition to DNA sequence analysis, various analysis methods using NGS technology have emerged, such as RNA sequencing (RNA-seq) for gene expression analysis, chromatin immunoprecipitation sequencing (ChIP-seq) for histone modification analysis and identification of transcription factor binding sites, Assay for Transposase-Accessible Chromatin using sequencing (ATAC-seq) and Hi-C for chromatin structure analysis (10, 11) (Figure 1). Along with technological innovation, there have also been attempts to apply genomic information to actual clinical practice. Targeted-gene panels (TGPs), which use NGS to examine the mutation status of a limited number of cancer-related genes, are actively being used to select the optimal treatment (12–14). On the other hand, one of the major problems in promoting precision oncology using the TGP method is that the number of patients who will benefit from the information obtained by the TGP method alone is limited (15–17). In order to increase the number of patients who will benefit from the promotion of precision oncology in the future, it is necessary to add more detailed omics data, such as whole genome analysis data and epigenome data, for integrated analysis. In recent years, it has been reported that epigenomic abnormalities play an important role in the development and progression of cancer (10, 18–25), and it is important to take into account information on epigenomic abnormalities when genomic mutations alone cannot elucidate the molecular mechanisms. In fact, the concept of epigenetic driver (epi-driver) is currently being used to describe the phenomenon of cancer development and progression based on epigenomic abnormalities (26, 27).
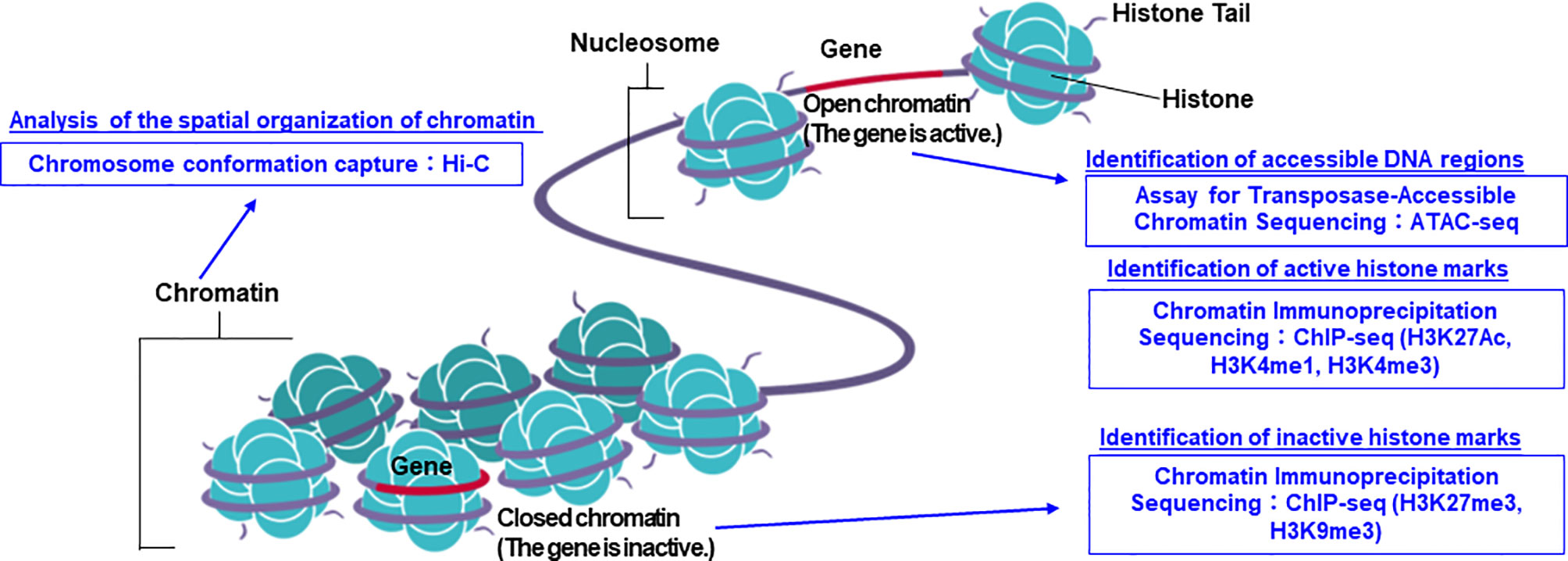
Figure 1 The summarized figure of chromatin structure and epigenomic analysis methods. ChIP-seq, ATAC-seq, and Hi-C methods can be used to predict the state of transcriptional activation or inactivation, and chromatin structure. Image credit: Shutterstock.com/ellepigrafica.
Another important issue is that the amount of data that researchers have to deal with has become enormous due to the emergence of various new methods with NGS analysis at their core as a result of technological innovation. For example, the amount of data generated by a single NGS run can be up to a million times larger than the data generated by a single Sanger sequencing run (28). In addition, there is a growing need for multimodal analysis, such as integrated analysis of genomic and epigenomic data, not just data from one modality. This kind of advanced analysis using a large amount of data is difficult to perform using conventional statistical methods, but nowadays, by proactively introducing artificial intelligence (AI) with machine learning (ML) and deep learning (DL) technologies at its core, good results can be obtained (29–31). In our view, there are four properties of ML and DL that are of particular importance. First, multimodal learning, which allows us to integrate multiple omics data as input (32–35). Second, multitask learning, which allows us to learn multiple different tasks simultaneously by sharing parts of the model (36, 37). Third, representation learning and semi-supervised learning, which allows us to acquire representations of data from large amounts of unlabeled data and thereby obtain small amounts of labels (38–41). The fourth is the ability to automatically acquire hierarchical features to capture higher-order correlations in the input (10, 42). More importantly, AI has already become one of the key technologies in the medical field, with a number of AI-powered medical devices approved by the US FDA (43). Under these circumstances, the active introduction of AI in the field of precision oncology seems to be an inevitable trend in the future.
Therefore, this review introduces the current status of efforts to establish precision oncology, focusing on whole genome sequencing (WGS) analysis and epigenome analysis, with particular emphasis on the results obtained through the use of ML and DL technologies.
Whole Genome Analysis
In this section, we introduce the recently published up to date WGS analyses using ML and DL. The cost of WGS dropped from 100 million US dollars in 2001 to 1,000 dollars in 2020 (NIH National Human Genome Research Institute; https://www.genome.gov/about-genomics/fact-sheets/Sequencing-Human-Genome-cost; Cost per genome data - 2020). In 2020, an international collaboration to identify common mutation patterns in more than 2,600 cancer whole genomes was performed by the Cancer Genome Atlas Research Network as The Cancer Genome Atlas Pan-Cancer Analysis of Whole Genomes (PCAWG) project (44). The results described in the flagship paper were accompanied with related papers that focused on specific analysis, such as peak calls, structural variations (SV), and non-coding variants.
As summarized in Table 1, we categorized WGS analyses into five groups based on the purpose of their use. The first type of analysis considered is peak calling. Finding an accurate peak calling is one of the most important and difficult parts of WGS analysis. Aligning several hundred bps to the whole genome (three billion bps in length) while considering sequencing errors is technically challenging (65, 66). Thus, reports comparing the benchmarks and new pipelines, particularly deep neural networks (DNNs), have been published for both peak calling and the identification of variants (45–51) in Table 1. In general, DNN models were first trained with publicly available datasets followed by the evaluation of their performance with the test dataset. Validation is performed with the validation dataset either using publicly available data or their in-house dataset. For example, the WGS dataset obtained from the PCAWG was used for training and testing the model. To independently validate the DNN model, the authors assembled several datasets outside the PCAWG (67).
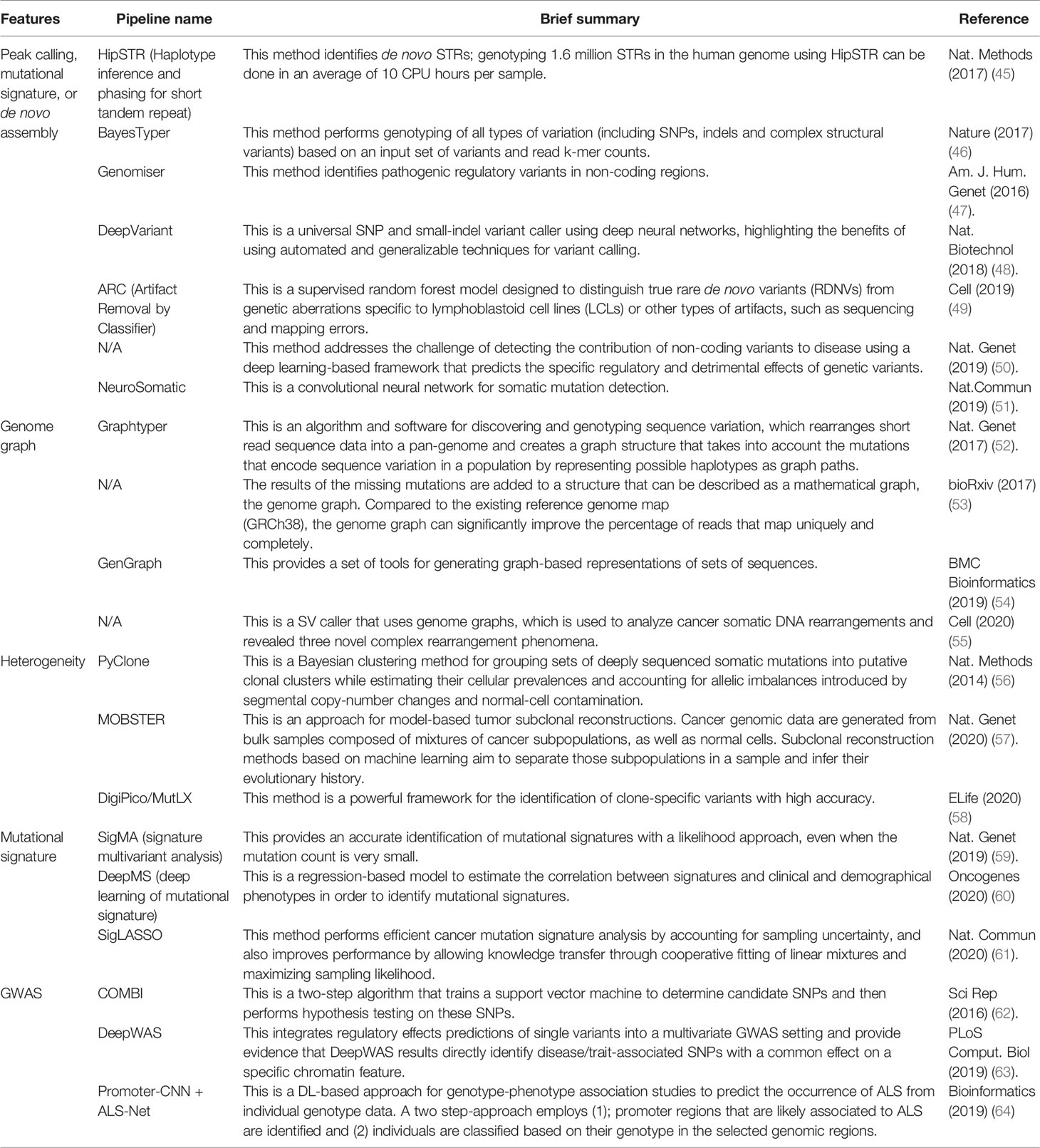
Table 1 Overview of whole genome analysis using machine learning.
The second analysis type is a genome graph or graph-based genome alignment. This approach has been recently reported and summarized (68). The advantage of using genome graphs is that they can accurately map (genotype) the polymorphisms of genomes with a good visualization, as well as perform fast and memory-efficient alignments (52–55) in Table 1. There is increasing recognition that a single, linear, monoploid reference genome is not always the best reference structure for human genetics, because they represent only a small fraction of existing human variations, particularly when they span SV breakpoints.
Third, heterogeneity in samples can be analyzed. Cancers are often observed to have various morphologies. These types of results are inconsistent with peak calls because they reflect where tissue samples are dissected. However, it is also true that tumors are composed of subpopulations of cells, and some cancer cells can migrate to other tissues. This heterogeneity results in a variety of features that can affect cancer phenotypes. To handle this, some published papers specifically focused on and investigated these phenotypes (56–58) in Table 1.
The fourth category is mutational signatures. The patterns of mutation or substitution signatures in cancer genome are discernible. Therefore, to categorize them, mutational signatures have been reported. Mutational signature analysis algorithms produce a decomposition matrix by using ML, a non-negative matrix factorization (NMF) approach, to extract mutational signatures (69–72). Additionally, other pipelines have been reported to perform mutational signature analyses to classify the samples (59–61) in Table 1.
The last is ML in a genome-wide association study (GWAS). GWAS has been used to discover genetic variants that are associated with diseases (73). To improve the analysis of GWAS, a combination of ML and DL analyses was reported (62) in Table1. However, how to improve mapping of regulatory variants (non-coding regions) identified by GWAS is still on going. Therefore, Arloth et al. developed DL-based approach and showed SNPs identified by DL were nominally significant in classical univariate GWAS analysis (63) in Table 1. They also identified disease/trait-relevant transcriptionally active genomic loci by integrating gene expression and DNA methylation quantitative trait loci (eQTL and meQTL) information of multiple resources and tissues. Although this is not a cancer research, another ML- and DL-based approach using GWAS data showed a good classification of amyotrophic lateral sclerosis (ALS) patient, and this approach can identify potentially ALS-associated promoter regions (64) in Table 1.
By integrating other omics data and analyzing single nucleotide variants (SNVs), indels, SV, and copy number alterations in non-coding regions, researchers can address the question of how pan-negative cancers developed, which we introduce in the following sections.
DNA Methylation
DNA methylation is an epigenetic modification that can discriminate specific patterns between in normal tissue cells and in cancer cells (74, 75). These epigenetic alterations affect gene expression, and thus, cell-specific DNA methylation patterns are used in the diagnosis and treatment selection of cancer by identifying cancer-specific DNA methylation patterns in biopsy specimens and blood samples (76, 77). A few diagnostic measures utilizing cancer-specific DNA methylation patterns have already received FDA approval (78, 79). Moreover, ML and DL analyses have been increasingly used to identify novel disease-specific DNA methylation patterns; they have also been used in research that aims to utilize the DNA methylation data from cancer patients for diagnosis, staging, and prognosis predictions (80–83).
Cell-free DNA (cfDNA) is circulating DNA found in plasma, and is known to be elevated in cancer patients (84). The clinical significance of analyzing cfDNA is that (1) it is noninvasive (2), it can be applied for monitoring, and (3) it can detect a more global signature compared to the data obtained from a biopsy on a single metastatic site. Therefore, ML can be applied for DNA methylation analyses using cfDNA. The DNA methylation levels of plasma cfDNA in renal cell carcinoma (RCC) patients have been assessed by cell-free methylated DNA immunoprecipitation and high-throughput sequencing (cfMeDIP-seq), and RCC detection was performed using the elastic net regularized generalized linear model method (80). In this aforementioned study, DNA methylation data obtained from blood and urine samples were used for validation, and the area under the receiver operating characteristic (AUROC) curve was found to be of 0.99 for blood samples and 0.86 for urine samples, respectively. In another study, cfDNA methylation data from blood samples of patients with intracranial tumors were obtained with cfMeDIP-seq and successfully used to generate a cancer detection model using the Random Forest algorithm (81). This model was also shown to have high discriminative capacity among the five tumor types (isocitrate dehydrogenase (IDH) wild-type glioma, IDH mutant glioma, low-grade glial-neuronal, hemangiopericytoma, and meningioma).
Next, we review DNA methylation analyses that use solid tumor samples. First, to distinguish metastatic head and neck squamous cell carcinoma (HNSC) from primary squamous cell carcinoma of the lung (LUSC), DNA methylation data were extracted from surgical specimens of lung cancer patients and artificial neural networks (NN), and a support vector machine (SVM) and a random forest (RF) classifier was constructed because current diagnostics show no possibility to distinguish metastatic HNSC from primary LUSC. Authors developed models that classified 96.4% of the cases by NN, 95.7% by SVM, and 87.8% by RF (82). The DL-based approach is also used to detect DNA methylation patterns related to breast cancer metastases and predict recurrence by conducting feature selection using an autoencoder with a single hidden layer followed by ML techniques for classification, or enrichment analysis for finding a biological relevance, genomic context, and functional annotation of best genes (83).
Cancer Epigenetics With A Focus On Enhancer Function
As mentioned earlier, since the advent of NGS technology and analyses based on ML, remarkable progress has been made in understanding the genetic basis of cancer. These studies have mainly defined genetic alterations as either causal (driver mutations), which confer a selective advantage to cancer cells, or consequential (passenger mutations, not directly causal), which do not have a selective advantage (26). Furthermore, genomic sequencing of tumor samples has revealed that different patients share a unique combination of one or two strong driver mutations such as gain-of-function EGFR and loss-of-function TP53 mutations typically detected in lung cancer and less frequent driver mutations (85, 86). On the other hand, the genetic component of the general disease risk is distributed mainly in the non-coding regions, which seem to be particularly rich in enhancers specific to the cell types associated with the disease (87, 88). Therefore, this has led to a growing interest in the annotation and understanding of human enhancers.
Measurable genome-wide biochemical annotations for enhancer regions include ChIP-seq or cleavage under targeted and release using nucleases (CUT&RUN) assays (89) for histone modifications or transcription factor (TF) binding, DNase I hypersensitivity sequencing (DNase-seq) for open chromatin (90), and ATAC-seq (91). On the other hand, it has long been hypothesized that enhancers loop in 3D space to access their target promoters. In recent years, the more powerful chromosome conformation capture (3C) method has yielded a series of high-resolution 3D conformation maps of the human genome in several cell types. In the 3C method, genomic DNA fragments are ligated to other genomic DNA fragments in physical proximity in the nucleus (92). These results have led to the identification of large compartments related to genomic organization, including enhancer-promoter loops (93), topologically associating domains (TADs) (94), and A/B compartments (92). In addition, 3C methods have been integrated with biochemical assays to annotate potentially functional interactions. For example, paired-end tag sequencing (ChIA-PET) (95), HiChIP (96), and proximity ligation-assisted ChIP-seq (PLAC-seq) (97) provide an overview of genome structures with a focus on proteins. Despite the development of various epigenomic methods as described above, and the obvious importance of human enhancers in both basic and disease biology, we still do not understand the repertoire of enhancers, including where they reside, how they act, and through which genes they mediate their effects.
In addition, it has recently been reported that super-enhancers are involved in abnormal gene expression in cancer cells (98). A super-enhancer is a region of the mammalian genome consisting of multiple enhancers, which are joined by a sequence of transcription factor proteins to drive the transcription of genes involved in cell identity (Figure 2) (99). An interesting finding is that disease-associated genetic mutations are particularly prevalent in super-enhancers of disease-associated cell types (100). Furthermore, cancer cells have been found to produce super-enhancers for oncogenes and other genes important in cancer development, suggesting that super-enhancers play an important role in human cell health and disease identity (100, 101). Importantly, super-enhancers are enriched in active chromatin marks such as H3K27ac and H3K4me3, while they are depleted in posed marks such as H3K27me3 (102). Therefore, epigenetic dysregulation may be involved in the production of super-enhancers in cancer cells. Since many disease-specific genetic variants are observed in super-enhancers, it seems to be pretty important to combine the information on genetic variants in non-coding regions obtained by WGS with the information on super enhancers based on epigenome data and analyze them in an integrated manner. As an example of super-enhancer analysis using ML, Gong et al. used two-dimensional lasso to improve the reproducibility of the Hi-C contact matrix and then classified the TAD boundaries based on the insulation score (103). The results showed that a higher TAD boundary insulation score was associated with higher CTCF levels, which may vary by cell type. They also showed that strong TAD boundaries and super-enhancer elements frequently overlap in cancer patients, suggesting that super-enhancer insulated by strong TAD boundaries may be used by cancer cells as a functional unit to promote tumorigenesis (103). Furthermore, Bu et al. proposed a new computational method, DEEPSEN, for super-enhancer prediction using a convolutional neural network, which is a DL algorithm (104). The proposed method integrates 36 different features and shows that it is capable of genome-wide prediction of super enhancers compared to existing methods.
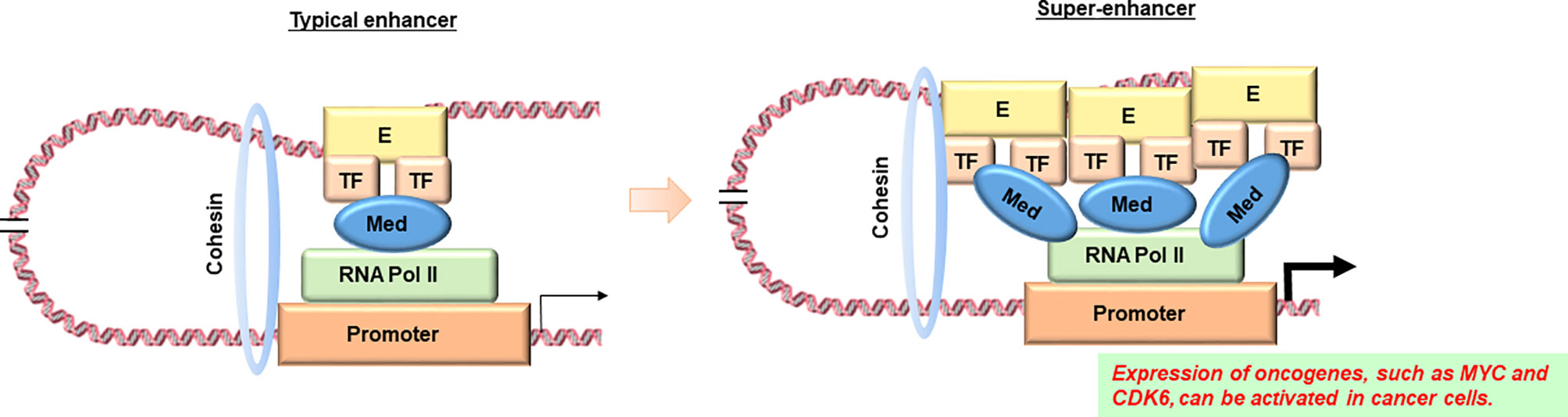
Figure 2 Diagram of comparison between a typical enhancer and a super-enhancer. According to reference 87, super enhancers are observed in the transcriptional regulatory regions of oncogenes such as MYC in cancer cells, but not in their counterparts in normal tissues. E, enhancer; TF, transcription factor; Med, Mediator complex; RNA pol II, RNA polymerase II. Image credit: Shutterstock.com/ellepigrafica.
In transcriptome and epigenome profiling, one of the conservative ML approaches of cluster analysis often yields reproducible regulatory subtypes. In this way, somatic mutations in cancer, although chaotic, often converge in a regulatory manner. These events suggest that cancer cells follow the same rules of transcriptional regulation as normal cells, despite the presence of aberrant combinations of transcription factors and genomic enhancers (105). Furthermore, a major unresolved question is how primary cancer cells metastasize and what the molecular events underlying this process are. However, extensive sequencing studies have shown that mutations may not be the causative factors in the transition from primary to metastasis (106). On the other hand, epigenetic changes are dynamic in nature and may play an important role in determining the metastatic phenotype, and research in this area is only beginning to be evaluated (107, 108). Unlike genetic studies, the current limitations in studying epigenetic events in cancer metastasis are the lack of conceptual understanding and the lack of an analytical framework to identify the putative driver and passenger epigenetic changes. We would therefore like to introduce an ML analysis that has the potential to address these issues.
Challenges That Machine Learning Can Overcome
Genomic and epigenetic data-driven science operates by comprehensively exploring genome-wide data to discover new properties, rather than testing existing models and hypotheses (109). These data-driven approaches include finding relationships between genotypes and phenotypes, searching for biomarkers for personalized medicine, discovering driver genes and predicting their functions, and tracking genomic regions with biochemical activities such as transcriptional enhancers, as mentioned in the previous section. Due to the large scale and complexity of genomic and epigenetic data, it is often not sufficient to check pairwise correlations to make predictions. Therefore, analytical tools are needed to support the discovery of new relationships, the derivation of new hypotheses and models, and to make predictions. ML is designed to automatically detect patterns in data, unlike algorithms that have predetermined assumptions and expertise. Therefore, ML is well suited for data-driven science, especially genomics and epigenomics (110). However, the performance of ML is highly dependent on how the data are represented and how each variable or a feature is extracted. Epigenetic information and various modalities are known to be interrelated events, which are thought to interact with each other to change gene activity patterns. Based on these hypotheses, Wang et al. predicted the DNA methylation state of a specific region using a deterministic ML model [stacked denoising autoencoders (SdAs)] based on the 3D genome topology and DNA sequence obtained from Hi-C experiments (111). Against the backdrop of the high cost and difficulty of experimental techniques, which is the bottleneck of Hi-C data acquisition, inference from 1D information such as ChIP-seq, ATAC-seq, and RNA-seq to 3D genome topology structure has been actively attempted using various ML methods (Table 2). However, the prediction accuracy may not be improved due to inaccurate extraction of the essential structures within the epigenetic dataset, such as the still unelucidated mechanism of gene transcription regulation by high-dimensional interactions between enhancer and promoter regions. To solve these issues, an integrated approach that combines not only the acquisition of multi-layered omics data over time but also the generation and selection of phenotypic features and ML, is necessary.
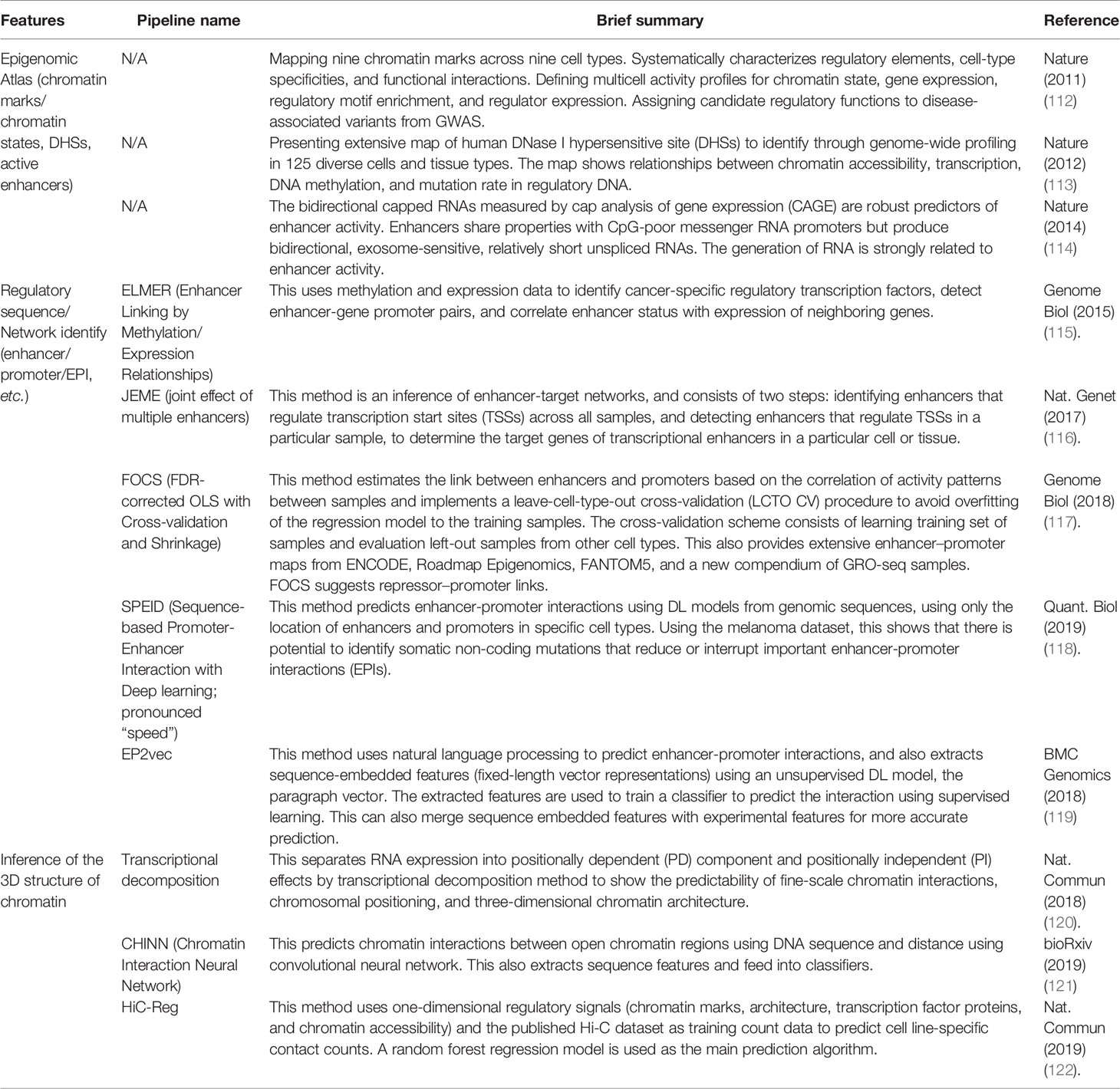
Table 2 Epigenetic analysis typically focusing on regulatory regions.
Integrated Analysis Of Whole Genome Sequencing And Epigenome Datasets
For decades, cancer genome research has made significant progresses in the identification of driver gene mutations, largely owing to the wide application of WES. However, we are now realizing that druggable gene mutations are limited, and the majority of cancer patients are left with unmet medical needs. Therefore, academic interest has gradually shifted to the analysis of mutations in non-coding genomes based on WGS analysis and the search for “epi-drivers”, which are mechanisms of cancer development and progression caused by epigenomic abnormalities. For this purpose, WGS and epigenetic sequence technologies such as ChIP-seq, ATAC-seq, and Hi-C are effective tools because they offer comprehensive information about the genome, epigenome, and crosstalk between these (Figure 1).
Integrated analysis of genome and epigenetic data can be applied to predict the functional significance of single nucleotide polymorphisms (SNPs) and germline/somatic mutations. In order to analyze the function of DNA mutations in non-coding genomes, it is important to focus on eQTLs, which are genomic sites involved in the variation of expression levels of target genes. It is known that most functionally active SNPs and mutations fall within the open chromatin region, especially at inferred transcription factor binding sites. Indeed, approximately 55% of eQTLs SNPs are reported to coincide with those of open chromatin-associated SNPs and mutations (123). An impressive study on integrated analyses of WGS, ATAC-seq, and RNA-seq datasets has been posted (124). In a case of bladder cancer, they found that a single base mutation in enhancer region of the FGD4 gene generated a putative de novo binding site for an NKX transcription factor, associated with an increase in chromatin accessibility and FGD4 gene expression (124). Since high expression of the FGD4 gene correlates with worse clinical outcomes in bladder cancer patients, this non-coding mutation might contribute to the malignant transformation of the cells by altering chromatin structure, thereby upregulating FGD4 gene expression.
However, it should be noted that the majority of non-coding mutations might not exert an active function. In general, the regional mutation rates of human cancer cells tend to be higher in repressive chromatin states than in active chromatin states, which may reflect differing efficiencies of DNA repair signals or mutagen exposure (125). Thus, from a probabilistic view, most of mutations in the heterochromatin region occur only because of their closed chromatin states; that is, they are less likely to have any selective advantages or active functions. Intriguingly, this tendency toward higher mutational occurrences in heterochromatin states offers potentially useful information. By applying the ML model, genome-wide mutation data can be utilized to infer the cell-of-origin of cancer cells. For example, the mutational landscape of melanoma is best correlated with the epigenetic profile of skin melanocytes than skin fibroblasts or skin keratinocytes, suggesting the true cell-of-origin of melanoma (126). This approach can be clinically applicable to predict the cell-of-origin for cancer of unknown primary origin and may yield a better phenotypic understanding of them. WGS can resolve non-coding SVs and CNVs. RNA-seq detects the expression levels of driver genes and aberrantly expressed genes caused by alternative promoter usage and exon skipping (127–130). The utility of an integrative, comprehensive approach, with WGS, RNA-seq, and DNA methylation, independently and in combination, has been reported (130). Comprehensive molecular tumor profiling comprising WGS, RNA-seq, and DNA methylation analyses identified pathogenic variants and provided therapy recommendations, which could accelerate the development of precision medications.
Overall, the genomic and epigenetic data of non-coding regions contain enormous, complex and interdependent information, and we believe that integrated analysis, effectively utilizing ML and DL technologies, is important to discover new drivers of human cancer.
Discussion
The genetic variants or SNPs were refined by the international haplotype map (HapMap) project to create a haplotype map of genes and genetic variants that affect health and disease (131–133). This project was attempted to genotype one common SNP in every 5,000 bps. At that time, it was believed that more than 99.9% of DNA sequences between any two people were identical, suggesting that only less than 0.1% of the genetic variants affect health and disease (https://www.genome.gov/11511175/about-the-international-hapmap-project-fact-sheet). Nowadays, analyzing WGS data has identified a considerable number of the genomic variants. The international consortium embarked on the 1000 Genomes Project to find common human genetic variations by applying WGS to a diverse set of individuals from multiple populations. High-throughput sequencing technologies do facilitate WGS in terms of accuracy, cost, and time. Almost two decades after the completion of the Human Genome Project, we have already entered a new era of sequencing, which led to individual genomic information becoming analyzable data. In practical terms, WGS analysis is becoming cost-effective. In addition, there is a trend to apply WGS routinely in both basic sciences and clinical cancer care to help us better understand and identify potential therapeutic targets or predictive biomarkers.
Epigenetics analyses were also drastically and positively affected by NGS. Chromatin conformations analyzed by ChIP-seq, ATAC-seq, or Hi-C are known to be related to cancer phenotypes (124, 134). Epigenetic alterations of DNA methylation at promoter and enhancer regions that induce chromatin dysregulation are found in cancer (135, 136). NGS analysis can help resolve both genetic and epigenetic alterations, and we expect to reveal the mechanism of pan-negative cancers using these data. From this point of view, we further introduced enhancers as an important concept in precision oncology. The current understanding is that enhancers bind to cell type-specific transcription factors, associate with regions of open chromatin, and are flanked by histones with H3K27ac and/or H3K4me1 modifications. These enhancers interact with promoters in 3D space and are either potentially primed or activated. Despite their obvious importance in both basic biology and disease biology, much remains to be learned about the relationship between enhancers and chromatin higher-order structure, including the identification of enhancer regions, how enhancers work, and through which genes they mediate their effects. In the future, we hope that multimodal analysis of multidimensional omics data by effective use of ML and DL techniques may contribute to precision oncology by providing an integrated understanding of more detailed molecular mechanisms.
Concluding Remarks
In this review, we first summarized the importance of genomic and epigenetic data and introduced the importance of omics data of interest in each section. Cancer is one of the leading causes of death worldwide, and molecular mechanisms remain unknown in certain cancers, which are categorized as pan-negative cancers. Multi-omics analyses by simply integrating omics data may encounter difficulties in identifying the mechanism causing cancer because none of the methodologies can address the comprehensive understanding underlying pan-negative cancers. Therefore, as we reviewed here, integrating multi-omics analysis with the assistance of ML is required for future cancer studies because each omics data is tightly linked to each other, and all omics data are associated with patient outcomes. Currently, there are high expectations for the development of medical AI, and it is expected that AI technology will be actively introduced in actual clinical practice in the future. On the other hand, medical AI research for clinical applications is currently focused on medical image analysis (137–144), and research on the introduction of AI to omics analysis such as whole genome analysis and epigenome analysis, as well as its clinical application, has not progressed sufficiently yet. In this regard, one of the problems associated with the widespread adoption of AI-based methodologies in omics analysis is that even though sequencing technology and other advanced analytics are increasingly being used in research and clinical practice, there is still a lot of confusion about the best protocols to adopt for analysis. For example, the RNA-seq pipeline is not sufficiently standardized, and the methodology relies heavily on the expertise and experience of a single research group/bioinformatics. As a result, in areas where uncertainty remains, the spread of AI-specific technologies may be delayed. We hope that this review will trigger the interest of more researchers in this field, and that the standardization of omics analysis will actively promote the adoption of AI and contribute to the establishment of the field of precision oncology in the future.
Author Contributions
KA and RH contributed to the study concept, design, and are guarantor of integrity of the entire study. KA, SK, KT, HM, ST, NS, RS, MK, and RH all contributed to literature search, manuscript preparation, and manuscript editing. All authors contributed to the article and submitted version.
Funding
This work was supported by JST CREST (Grant Number JPMJCR1689), JST AIP-PRISM (Grant Number JPMJCR18Y4), JSPS Grant-in-Aid for Scientific Research on Innovative Areas (Grant Number JP18H04908), and JSPS KAKENHI (Grant Number JP20K17982).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Watson JD, Crick FH. Molecular Structure of Nucleic Acids; a Structure for Deoxyribose Nucleic Acid. Nature (1953) 171(4356):737–8. doi: 10.1038/171737a0
2. Sanger F, Nicklen S, Coulson AR. DNA Sequencing With Chain-Terminating Inhibitors. Proc Natl Acad Sci USA (1977) 74(12):5463–7. doi: 10.1073/pnas.74.12.5463
3. Watson JD, Cook-Deegan RM. Origins of the Human Genome Project. FASEB J (1991) 5(1):8–11. doi: 10.1096/fasebj.5.1.1991595
4. Collins FS, Morgan M, Patrinos A. the Human Genome Project: Lessons From Large-Scale Biology. Science (2003) 300(5617):286–90. doi: 10.1126/science.1084564
5. Katsnelson A. Momentum Grows to Make ‘Personalized’ Medicine More ‘Precise’. Nat Med (2013) 19(3):249. doi: 10.1038/nm0313-249
6. Tran B, Dancey JE, Kamel-Reid S, McPherson JD, Bedard PL, Brown AM, et al. Cancer Genomics: Technology, Discovery, and Translation. J Clin Oncol (2012) 30(6):647–60. doi: 10.1200/JCO.2011.39.2316
7. Roychowdhury S, Chinnaiyan AM. Translating Genomics for Precision Cancer Medicine. Annu Rev Genomics Hum Genet (2014) 15:395–415. doi: 10.1146/annurev-genom-090413-025552
8. Levy SE, Myers RM. Advancements in Next-Generation Sequencing. Annu Rev Genomics Hum Genet (2016) 15:95–115. doi: 10.1146/annurev-genom-083115-022413
9. Slatko BE, Gardner AF, Ausubel FM. Overview of Next-Generation Sequencing Technologies. Curr Protoc Mol Biol (2018) 122(1):e59. doi: 10.1002/cpmb.59
10. Hamamoto R, Komatsu M, Takasawa K, Asada K, Kaneko S. Epigenetics Analysis and Integrated Analysis of Multiomics Data, Including Epigenetic Data, Using Artificial Intelligence in the Era of Precision Medicine. Biomolecules (2020) 10(1):62. doi: 10.3390/biom10010062
11. Wang Z, Gerstein M, Snyder M. RNA-Seq: A Revolutionary Tool for Transcriptomics. Nat Rev Genet (2009) 10(1):57–63. doi: 10.1038/nrg2484
12. Cimmino F, Lasorsa VA, Vetrella S, Iolascon A, Capasso MA. Targeted Gene Panel for Circulating Tumor DNA Sequencing in Neuroblastoma. Front Oncol (2020) 10:596191. doi: 10.3389/fonc.2020.596191
13. Fernandes MGO, Jacob M, Martins N, Moura CS, Guimaraes S, Reis JP, et al. Targeted Gene Next-Generation Sequencing Panel in Patients With Advanced Lung Adenocarcinoma: Paving the Way for Clinical Implementation. Cancers (Basel) (2019) 11(9):1229. doi: 10.3390/cancers11091229
14. Surrey LF, MacFarland SP, Chang F, Cao K, Rathi KS, Akgumus GT, et al. Clinical Utility of Custom-Designed NGS Panel Testing in Pediatric Tumors. Genome Med (2019) 11(1):32. doi: 10.1186/s13073-019-0644-8
15. Zhang X, Yang H, Zhang R. Challenges and Future of Precision Medicine Strategies for Breast Cancer Based on a Database on Drug Reactions. Biosci Rep (2019) 39(9):BSR20190230. doi: 10.1042/BSR20190230
16. Prasad V. Perspective: The Precision-Oncology Illusion. Nature (2016) 537(7619):S63. doi: 10.1038/537S63a
17. Meric-Bernstam F, Brusco L, Shaw K, Horombe C, Kopetz S, Davies MA, et al. Feasibility of Large-Scale Genomic Testing to Facilitate Enrollment Onto Genomically Matched Clinical Trials. J Clin Oncol (2015) 33(25):2753–62. doi: 10.1200/JCO.2014.60.4165
18. Oki S, Sone K, Oda K, Hamamoto R, Ikemura M, Maeda D, et al. Oncogenic Histone Methyltransferase EZH2: A Novel Prognostic Marker With Therapeutic Potential in Endometrial Cancer. Oncotarget (2017) 8(25):40402–11. doi: 10.18632/oncotarget.16316
19. Kogure M, Takawa M, Saloura V, Sone K, Piao L, Ueda K, et al. The Oncogenic Polycomb Histone Methyltransferase EZH2 Methylates Lysine 120 on Histone H2B and Competes Ubiquitination. Neoplasia (2013) 15(11):1251–61.
20. Asada K, Bolatkan A, Takasawa K, Komatsu M, Kaneko S, Hamamoto R. Critical Roles of N(6)-Methyladenosine (M(6)a) in Cancer and Virus Infection. Biomolecules (2020) 10(7):1071. doi: 10.3390/biom10071071
21. Hayami S, Kelly JD, Cho HS, Yoshimatsu M, Unoki M, Tsunoda T, et al. Overexpression of LSD1 Contributes to Human Carcinogenesis Through Chromatin Regulation in Various Cancers. Int J Cancer (2011) 128(3):574–86. doi: 10.1002/ijc.25349
22. Kim S, Bolatkan A, Kaneko S, Ikawa N, Asada K, Komatsu M, et al. Deregulation of the Histone Lysine-Specific Demethylase 1 is Involved in Human Hepatocellular Carcinoma. Biomolecules (2019) 9(12):810. doi: 10.3390/biom9120810
23. Sone K, Piao L, Nakakido M, Ueda K, Jenuwein T, Nakamura Y, et al. Critical Role of Lysine 134 Methylation on Histone H2AX for Gamma-H2AX Production and DNA Repair. Nat Commun (2014) 5:5691. doi: 10.1038/ncomms6691
24. Saloura V, Cho HS, Kyiotani K, Alachkar H, Zuo Z, Nakakido M, et al. Whsc1 Promotes Oncogenesis Through Regulation of Nima-Related-Kinase-7 in Squamous Cell Carcinoma of the Head and Neck. Mol Cancer Res (2015) 13(2):293–304. doi: 10.1158/1541-7786.MCR-14-0292-T
25. Wada M, Kukita A, Sone K, Hamamoto R, Kaneko S, Komatsu M, et al. Epigenetic Modifier SETD8 as a Therapeutic Target for High-Grade Serous Ovarian Cancer. Biomolecules (2020) 10(12):1686. doi: 10.3390/biom10121686
26. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA,Jr., Kinzler KW. Cancer Genome Landscapes. Science (2013) 339(6127):1546–58. doi: 10.1126/science.1235122
27. Chatterjee A, Rodger EJ, Eccles MR. Epigenetic Drivers of Tumourigenesis and Cancer Metastasis. Semin Cancer Biol (2018) 51:149–59. doi: 10.1016/j.semcancer.2017.08.004
28. Eliseev A, Gibson KM, Avdeyev P, Novik D, Bendall ML, Perez-Losada M, et al. Evaluation of Haplotype Callers for Next-Generation Sequencing of Viruses. Infect Genet Evol (2020) 82:104277. doi: 10.1016/j.meegid.2020.104277
29. Asada K, Kobayashi K, Joutard S, Tubaki M, Takahashi S, Takasawa K, et al. Uncovering Prognosis-Related Genes and Pathways by Multi-Omics Analysis in Lung Cancer. Biomolecules (2020) 10(4):524. doi: 10.3390/biom10040524
30. Kobayashi K, Bolatkan A, Shiina S, Hamamoto R. Fully-Connected Neural Networks With Reduced Parameterization for Predicting Histological Types of Lung Cancer From Somatic Mutations. Biomolecules (2020) 10(9):1249. doi: 10.3390/biom10091249
31. Takahashi S, Asada K, Takasawa K, Shimoyama R, Sakai A, Bolatkan A, et al. Predicting Deep Learning Based Multi-Omics Parallel Integration Survival Subtypes in Lung Cancer Using Reverse Phase Protein Array Data. Biomolecules (2020) 10(10):1460.
32. Srivastava N, Salakhutdinov R. Multimodal Learning With Deep Boltzmann Machines. J Mach Learn Res (2014) 15:2949–80.
33. Zhu B, Song N, Shen R, Arora A, Machiela MJ, Song L, et al. Integrating Clinical and Multiple Omics Data for Prognostic Assessment Across Human Cancers. Sci Rep (2017) 7(1):16954. doi: 10.1038/s41598-017-17031-8
34. Chaudhary K, Poirion OB, Lu L, Garmire LX. Deep Learning-Based Multi-Omics Integration Robustly Predicts Survival in Liver Cancer. Clin Cancer Res (2018) 24(6):1248–59. doi: 10.1158/1078-0432.CCR-17-0853
35. Lee SI, Celik S, Logsdon BA, Lundberg SM, Martins TJ, Oehler VG, et al. A Machine Learning Approach to Integrate Big Data for Precision Medicine in Acute Myeloid Leukemia. Nat Commun (2018) 9(1):42. doi: 10.1038/s41467-017-02465-5
36. Gonen M, Margolin AA. Drug Susceptibility Prediction Against a Panel of Drugs Using Kernelized Bayesian Multitask Learning. Bioinformatics (2014) 30(17):i556–63. doi: 10.1093/bioinformatics/btu464
37. Yuan H, Paskov I, Paskov H, Gonzalez AJ, Leslie CS. Multitask Learning Improves Prediction of Cancer Drug Sensitivity. Sci Rep (2016) 6:31619. doi: 10.1038/srep31619
38. Xiao Y, Wu J, Lin Z, Zhao X. a Semi-Supervised Deep Learning Method Based on Stacked Sparse Auto-Encoder for Cancer Prediction Using RNA-Seq Data. Comput Methods Programs BioMed (2018) 166:99–105. doi: 10.1016/j.cmpb.2018.10.004
39. Bengio Y, Courville A, Vincent P. Representation Learning: A Review and New Perspectives. IEEE Trans Pattern Anal Mach Intell (2013) 35(8):1798–828. doi: 10.1109/TPAMI.2013.50
40. Shi M, Zhang B. Semi-Supervised Learning Improves Gene Expression-Based Prediction of Cancer Recurrence. Bioinformatics (2011) 27(21):3017–23. doi: 10.1093/bioinformatics/btr502
41. Chapelle O, Sindhwani V, Keerthi SS. Optimization Techniques for Semi-Supervised Support Vector Machines. J Mach Learn Res (2008) 9:203–33.
42. Bengio Y. Learning Deep Architectures for AI. Foundations Trends® Mach Learn (2009) 2(1):p1–p127.
43. Hamamoto R, Suvarna K, Yamada M, Kobayashi K, Shinkai N, Miyake M, et al. Application of Artificial Intelligence Technology in Oncology: Towards the Establishment of Precision Medicine. Cancers (Basel) (2020) 12(12):3532. doi: 10.3390/cancers12123532
44. Consortium, I.T.P.-C.A.o.W.G. Pan-Cancer Analysis of Whole Genomes. Nature (2020) 578(7793):82–93. doi: 10.1038/s41586-020-1969-6
45. Willems T, Zielinski D, Yuan J, Gordon A, Gymrek M, Erlich Y. Genome-Wide Profiling of Heritable and De Novo STR Variations. Nat Methods (2017) 14(6):590–2. doi: 10.1038/nmeth.4267
46. Maretty L, Jensen JM, Petersen B, Sibbesen JA, Liu S, Villesen P, et al. Sequencing and De Novo Assembly of 150 Genomes From Denmark as a Population Reference. Nature (2017) 548(7665):87–91. doi: 10.1038/nature23264
47. Smedley D, Schubach M, Jacobsen JOB, Kohler S, Zemojtel T, Spielmann M, et al. A Whole-Genome Analysis Framework for Effective Identification of Pathogenic Regulatory Variants in Mendelian Disease. Am J Hum Genet (2016) 99(3):595–606. doi: 10.1016/j.ajhg.2016.07.005
48. Poplin R, Chang PC, Alexander D, Schwartz S, Colthurst T, Ku A, et al. A Universal SNP and Small-Indel Variant Caller Using Deep Neural Networks. Nat Biotechnol (2018) 36(10):983–7. doi: 10.1038/nbt.4235
49. Ruzzo EK, Perez-Cano L, Jung JY, Wang LK, Kashef-Haghighi D, Hartl C, et al. Inherited and De Novo Genetic Risk for Autism Impacts Shared Networks. Cell (2019) 178(4):850–66 e26. doi: 10.1016/j.cell.2019.07.015
50. Zhou J, Park CY, Theesfeld CL, Wong AK, Yuan Y, Scheckel C, et al. Whole-Genome Deep-Learning Analysis Identifies Contribution of Noncoding Mutations to Autism Risk. Nat Genet (2019) 51(6):973–80. doi: 10.1038/s41588-019-0420-0
51. Sahraeian SME, Liu R, Lau B, Podesta K, Mohiyuddin M, Lam HYK. Deep Convolutional Neural Networks for Accurate Somatic Mutation Detection. Nat Commun (2019) 10(1):1041. doi: 10.1038/s41467-019-09027-x
52. Eggertsson HP, Jonsson H, Kristmundsdottir S, Hjartarson E, Kehr B, Masson G, et al. Graphtyper Enables Population-Scale Genotyping Using Pangenome Graphs. Nat Genet (2017) 49(11):1654–60. doi: 10.1038/ng.3964
53. Novak AM, Hickey G, Garrison E, Blum S, Connelly A, Dilthey A, et al. Genome Graphs. bioRxiv (2017). doi: 10.1101/101378
54. Ambler JM, Mulaudzi S, Mulder N. Gengraph: A Python Module for the Simple Generation and Manipulation of Genome Graphs. BMC Bioinf (2019) 20(1):519. doi: 10.1186/s12859-019-3115-8
55. Hadi K, Yao X, Behr JM, Deshpande A, Xanthopoulakis C, Tian H, et al. Distinct Classes of Complex Structural Variation Uncovered Across Thousands of Cancer Genome Graphs. Cell (2020) 183(1):197–210.e32. doi: 10.1016/j.cell.2020.08.006
56. Roth A, Khattra J, Yap D, Wan A, Laks E, Biele J, et al. Pyclone: Statistical Inference of Clonal Population Structure in Cancer. Nat Methods (2014) 11(4):396–8. doi: 10.1038/nmeth.2883
57. Caravagna G, Heide T, Williams MJ, Zapata L, Nichol D, Chkhaidze K, et al. Subclonal Reconstruction of Tumors by Using Machine Learning and Population Genetics. Nat Genet (2020) 52(9):898–907. doi: 10.1038/s41588-020-0675-5
58. KaramiNejadRanjbar M, Sharifzadeh S, Wietek NC, Artibani M, El-Sahhar S, Sauka-Spengler T, et al. A Highly Accurate Platform for Clone-Specific Mutation Discovery Enables the Study of Active Mutational Processes. Elife (2020) 9:e55207. doi: 10.7554/eLife.55207
59. Gulhan DC, Lee JJ, Melloni GEM, Cortes-Ciriano I, Park PJ. Detecting the Mutational Signature of Homologous Recombination Deficiency in Clinical Samples. Nat Genet (2019) 51(5):912–9. doi: 10.1038/s41588-019-0390-2
60. Pei G, Hu R, Dai Y, Zhao Z, Jia P. Decoding Whole-Genome Mutational Signatures in 37 Human Pan-Cancers by Denoising Sparse Autoencoder Neural Network. Oncogene (2020) 39(27):5031–41. doi: 10.1038/s41388-020-1343-z
61. Li S, Crawford FW, Gerstein MB. Using Siglasso to Optimize Cancer Mutation Signatures Jointly With Sampling Likelihood. Nat Commun (2020) 11(1):3575. doi: 10.1038/s41467-020-17388-x
62. Mieth B, Kloft M, Rodriguez JA, Sonnenburg S, Vobruba R, Morcillo-Suarez C, et al. Combining Multiple Hypothesis Testing With Machine Learning Increases the Statistical Power of Genome-Wide Association Studies. Sci Rep (2016) 6:36671. doi: 10.1038/srep36671
63. Arloth J, Eraslan G, Andlauer TFM, Martins J, Iurato S, Kuhnel B, et al. Deepwas: Multivariate Genotype-Phenotype Associations by Directly Integrating Regulatory Information Using Deep Learning. PloS Comput Biol (2020) 16(2):e1007616. doi: 10.1371/journal.pcbi.1007616
64. Yin B, Balvert M, van der Spek RAA, Dutilh BE, Bohte S, Veldink J, et al. Using the Structure of Genome Data in the Design of Deep Neural Networks for Predicting Amyotrophic Lateral Sclerosis From Genotype. Bioinformatics (2019) 35(14):i538–i47. doi: 10.1093/bioinformatics/btz369
65. Nakamura K, Oshima T, Morimoto T, Ikeda S, Yoshikawa H, Shiwa Y, et al. Sequence-Specific Error Profile of Illumina Sequencers. Nucleic Acids Res (2011) 39(13):e90. doi: 10.1093/nar/gkr344
66. Shiraishi Y, Sato Y, Chiba K, Okuno Y, Nagata Y, Yoshida K, et al. An Empirical Bayesian Framework for Somatic Mutation Detection From Cancer Genome Sequencing Data. Nucleic Acids Res (2013) 41(7):e89. doi: 10.1093/nar/gkt126
67. Jiao W, Atwal G, Polak P, Karlic R, Cuppen E, Subtypes PT, et al. A Deep Learning System Accurately Classifies Primary and Metastatic Cancers Using Passenger Mutation Patterns. Nat Commun (2020) 11(1):728. doi: 10.1038/s41467-019-13825-8
68. Paten B, Novak AM, Eizenga JM, Garrison E. Genome Graphs and the Evolution of Genome Inference. Genome Res (2017) 27(5):665–76. doi: 10.1101/gr.214155.116
69. Nik-Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, et al. Mutational Processes Molding the Genomes of 21 Breast Cancers. Cell (2012) 149(5):979–93. doi: 10.1016/j.cell.2012.04.024
70. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of Mutational Processes in Human Cancer. Nature (2013) 500(7463):415–21. doi: 10.1038/nature12477
71. Alexandrov LB, Nik-Zainal S, Wedge DC, Campbell PJ, Stratton MR. Deciphering Signatures of Mutational Processes Operative in Human Cancer. Cell Rep (2013) 3(1):246–59. doi: 10.1016/j.celrep.2012.12.008
72. Maura F, Degasperi A, Nadeu F, Leongamornlert D, Davies H, Moore L, et al. a Practical Guide for Mutational Signature Analysis in Hematological Malignancies. Nat Commun (2019) 10(1):2969. doi: 10.1038/s41467-019-11037-8
73. Ozaki K, Ohnishi Y, Iida A, Sekine A, Yamada R, Tsunoda T, et al. Functional Snps in the Lymphotoxin-Alpha Gene That are Associated With Susceptibility to Myocardial Infarction. Nat Genet (2002) 32(4):650–4. doi: 10.1038/ng1047
74. Baylin SB. DNA Methylation and Gene Silencing in Cancer. Nat Clin Pract Oncol (2005) 2 Suppl:1(S4–11. doi: 10.1038/ncponc0354
75. Merlo A, Herman JG, Mao L, Lee DJ, Gabrielson E, Burger PC, et al. 5’ Cpg Island Methylation is Associated With Transcriptional Silencing of the Tumour Suppressor P16/CDKN2/MTS1 in Human Cancers. Nat Med (1995) 1(7):686–92. doi: 10.1038/nm0795-686
76. Stewart GD, Van Neste L, Delvenne P, Delree P, Delga A, McNeill SA, et al. Clinical Utility of an Epigenetic Assay to Detect Occult Prostate Cancer in Histopathologically Negative Biopsies: Results of the Matloc Study. J Urol (2013) 189(3):1110–6. doi: 10.1016/j.juro.2012.08.219
77. Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, et al. a Randomized Trial of Bevacizumab for Newly Diagnosed Glioblastoma. N Engl J Med (2014) 370(8):699–708. doi: 10.1056/NEJMoa1308573
78. Imperiale TF, Ransohoff DF, Itzkowitz SH, Levin TR, Lavin P, Lidgard GP, et al. Multitarget Stool DNA Testing for Colorectal-Cancer Screening. N Engl J Med (2014) 370(14):1287–97. doi: 10.1056/NEJMoa1311194
79. Lamb YN, Dhillon S. Epi Procolon((R)) 2.0 CE: A Blood-Based Screening Test for Colorectal Cancer. Mol Diagn Ther (2017) 21(2):225–32. doi: 10.1007/s40291-017-0259-y
80. Nuzzo PV, Berchuck JE, Korthauer K, Spisak S, Nassar AH, Abou Alaiwi S, et al. Detection of Renal Cell Carcinoma Using Plasma and Urine Cell-Free Dna Methylomes. Nat Med (2020) 26(7):1041–3. doi: 10.1038/s41591-020-0933-1
81. Nassiri F, Chakravarthy A, Feng S, Shen SY, Nejad R, Zuccato JA, et al. Detection and Discrimination of Intracranial Tumors Using Plasma Cell-Free Dna Methylomes. Nat Med (2020) 26(7):1044–7. doi: 10.1038/s41591-020-0932-2
82. Jurmeister P, Bockmayr M, Seegerer P, Bockmayr T, Treue D, Montavon G, et al. Machine Learning Analysis of Dna Methylation Profiles Distinguishes Primary Lung Squamous Cell Carcinomas From Head and Neck Metastases. Sci Transl Med (2019) 11(509):eaaw8513. doi: 10.1126/scitranslmed.aaw8513
83. Macias-Garcia L, Martinez-Ballesteros M, Luna-Romera JM, Garcia-Heredia JM, Garcia-Gutierrez J, Riquelme-Santos JC. Autoencoded DNA Methylation Data to Predict Breast Cancer Recurrence: Machine Learning Models and Gene-Weight Significance. Artif Intell Med (2020) 110:101976. doi: 10.1016/j.artmed.2020.101976
84. Volik S, Alcaide M, Morin RD, Collins C. Cell-Free DNA (Cfdna): Clinical Significance and Utility in Cancer Shaped by Emerging Technologies. Mol Cancer Res (2016) 14(10):898–908. doi: 10.1158/1541-7786.MCR-16-0044
85. Cancer Genome Atlas Research. N. Comprehensive Molecular Profiling of Lung Adenocarcinoma. Nature (2014) 511(7511):543–50. doi: 10.1038/nature13385
86. Saito M, Shiraishi K, Kunitoh H, Takenoshita S, Yokota J, Kohno T. Gene Aberrations for Precision Medicine Against Lung Adenocarcinoma. Cancer Sci (2016) 107(6):713–20. doi: 10.1111/cas.12941
87. Akhtar-Zaidi B, Cowper-Sal-lari R, Corradin O, Saiakhova A, Bartels CF, Balasubramanian D, et al. Epigenomic Enhancer Profiling Defines a Signature of Colon Cancer. Science (2012) 336(6082):736–9. doi: 10.1126/science.1217277
88. Cohen AJ, Saiakhova A, Corradin O, Luppino JM, Lovrenert K, Bartels CF, et al. Hotspots of Aberrant Enhancer Activity Punctuate the Colorectal Cancer Epigenome. Nat Commun (2017) 8:14400. doi: 10.1038/ncomms14400
89. Skene PJ, Henikoff S. An Efficient Targeted Nuclease Strategy for High-Resolution Mapping of DNA Binding Sites. Elife (2017) 6:e21856. doi: 10.7554/eLife.21856
90. Boyle AP, Davis S, Shulha HP, Meltzer P, Margulies EH, Weng Z, et al. High-resolution mapping and characterization of open chromatin across the genome. Cell (2008) 132(2):311–22. doi: 10.1016/j.cell.2007.12.014
91. Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of Native Chromatin for Fast and Sensitive Epigenomic Profiling of Open Chromatin, DNA-Binding Proteins and Nucleosome Position. Nat Methods (2013) 10(12):1213–8. doi: 10.1038/nmeth.2688
92. Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, et al. Comprehensive Mapping of Long-Range Interactions Reveals Folding Principles of the Human Genome. Science (2009) 326(5950):289–93. doi: 10.1126/science.1181369
93. Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, et al. A 3D Map of the Human Genome At Kilobase Resolution Reveals Principles of Chromatin Looping. Cell (2014) 159(7):1665–80. doi: 10.1016/j.cell.2014.11.021
94. Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, et al. Topological Domains in Mammalian Genomes Identified by Analysis of Chromatin Interactions. Nature (2012) 485(7398):376–80. doi: 10.1038/nature11082
95. Fullwood MJ, Ruan Y. Chip-Based Methods for the Identification of Long-Range Chromatin Interactions. J Cell Biochem (2009) 107(1):30–9. doi: 10.1002/jcb.22116
96. Mumbach MR, Rubin AJ, Flynn RA, Dai C, Khavari PA, Greenleaf WJ, et al. Hichip: Efficient and Sensitive Analysis of Protein-Directed Genome Architecture. Nat Methods (2016) 13(11):919–22. doi: 10.1038/nmeth.3999
97. Fang R, Yu M, Li G, Chee S, Liu T, Schmitt AD, et al. Mapping of Long-Range Chromatin Interactions by Proximity Ligation-Assisted Chip-Seq. Cell Res (2016) 26(12):1345–8. doi: 10.1038/cr.2016.137
98. Zhang J, Liu W, Zou C, Zhao Z, Lai Y, Shi Z, et al. Targeting Super-Enhancer-Associated Oncogenes in Osteosarcoma With Thz2, a Covalent Cdk7 Inhibitor. Clin Cancer Res (2020) 26(11):2681–92. doi: 10.1158/1078-0432.CCR-19-1418
99. Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, et al. Master Transcription Factors and Mediator Establish Super-Enhancers At Key Cell Identity Genes. Cell (2013) 153(2):307–19. doi: 10.1016/j.cell.2013.03.035
100. Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, et al. Super-Enhancers in the Control of Cell Identity and Disease. Cell (2013) 155(4):934–47. doi: 10.1016/j.cell.2013.09.053
101. Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, et al. Selective Inhibition of Tumor Oncogenes by Disruption of Super-Enhancers. Cell (2013) 153(2):320–34. doi: 10.1016/j.cell.2013.03.036
102. Khan A, Mathelier A, Zhang X. Super-Enhancers are Transcriptionally More Active and Cell Type-Specific Than Stretch Enhancers. Epigenetics (2018) 13(9):910–22. doi: 10.1080/15592294.2018.1514231
103. Gong Y, Lazaris C, Sakellaropoulos T, Lozano A, Kambadur P, Ntziachristos P, et al. Stratification of TAD Boundaries Reveals Preferential Insulation of Super-Enhancers by Strong Boundaries. Nat Commun (2018) 9(1):542. doi: 10.1038/s41467-018-03017-1
104. Bu H, Hao J, Gan Y, Zhou S, Guan J. DEEPSEN: A Convolutional Neural Network Based Method for Super-Enhancer Prediction. BMC Bioinf (2019) 20(Suppl 15):598. doi: 10.1186/s12859-019-3180-z
105. Atkins M, Potier D, Romanelli L, Jacobs J, Mach J, Hamaratoglu F, et al. an Ectopic Network of Transcription Factors Regulated by Hippo Signaling Drives Growth and Invasion of a Malignant Tumor Model. Curr Biol (2016) 26(16):2101–13. doi: 10.1016/j.cub.2016.06.035
106. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
107. Cheng PF, Shakhova O, Widmer DS, Eichhoff OM, Zingg D, Frommel SC, et al. Methylation-Dependent Sox9 Expression Mediates Invasion in Human Melanoma Cells and is a Negative Prognostic Factor in Advanced Melanoma. Genome Biol (2015) 16:42. doi: 10.1186/s13059-015-0594-4
108. Vizoso M, Ferreira HJ, Lopez-Serra P, Carmona FJ, Martinez-Cardus A, Girotti MR, et al. Epigenetic Activation of a Cryptic Tbc1d16 Transcript Enhances Melanoma Progression by Targeting Egfr. Nat Med (2015) 21(7):741–50. doi: 10.1038/nm.3863
109. Brown PO, Botstein D. Exploring the New World of the Genome With DNA Microarrays. Nat Genet (1999) 21(1 Suppl):33–7. doi: 10.1038/4462
110. Libbrecht MW, Noble WS. Machine Learning Applications in Genetics and Genomics. Nat Rev Genet (2015) 16(6):321–32. doi: 10.1038/nrg3920
111. Wang Y, Liu T, Xu D, Shi H, Zhang C, Mo YY, et al. Predicting DNA Methylation State of Cpg Dinucleotide Using Genome Topological Features and Deep Networks. Sci Rep (2016) 6:19598. doi: 10.1038/srep19598
112. Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, et al. Mapping and Analysis of Chromatin State Dynamics in Nine Human Cell Types. Nature (2011) 473(7345):43–9. doi: 10.1038/nature09906
113. Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, et al. The Accessible Chromatin Landscape of the Human Genome. Nature (2012) 489(7414):75–82. doi: 10.1038/nature11232
114. Andersson R, Gebhard C, Miguel-Escalada I, Hoof I, Bornholdt J, Boyd M, et al. an Atlas of Active Enhancers Across Human Cell Types and Tissues. Nature (2014) 507(7493):455–61. doi: 10.1038/nature12787
115. Yao L, Shen H, Laird PW, Farnham PJ, Berman BP. Inferring Regulatory Element Landscapes and Transcription Factor Networks From Cancer Methylomes. Genome Biol (2015) 16(1):105. doi: 10.1186/s13059-015-0668-3
116. Cao Q, Anyansi C, Hu X, Xu L, Xiong L, Tang W, et al. Reconstruction of Enhancer-Target Networks in 935 Samples of Human Primary Cells, Tissues and Cell Lines. Nat Genet (2017) 49(10):1428–36. doi: 10.1038/ng.3950
117. Hait TA, Amar D, Shamir R, Elkon R. FOCS: A Novel Method for Analyzing Enhancer and Gene Activity Patterns Infers an Extensive Enhancer-Promoter Map. Genome Biol (2018) 19(1):56. doi: 10.1186/s13059-018-1432-2
118. Singh S, Yang Y, Póczos B, Ma J. Predicting Enhancer-Promoter Interaction From Genomic Sequence With Deep Neural Networks. Quantitative Biol (2019) 7(2):122–37. doi: 10.1007/s40484-019-0154-0
119. Zeng W, Wu M, Jiang R. Prediction of Enhancer-Promoter Interactions Via Natural Language Processing. BMC Genomics (2018) 19(Suppl 2):84. doi: 10.1186/s12864-018-4459-6
120. Rennie S, Dalby M, van Duin L, Andersson R. Transcriptional Decomposition Reveals Active Chromatin Architectures and Cell Specific Regulatory Interactions. Nat Commun (2018) 9(1):487. doi: 10.1038/s41467-017-02798-1
122. Zhang S, Chasman D, Knaack S, Roy S. In Silico Prediction of High-Resolution Hi-C Interaction Matrices. Nat Commun (2019) 10(1):5449. doi: 10.1038/s41467-019-13423-8
123. Degner JF, Pai AA, Pique-Regi R, Veyrieras JB, Gaffney DJ, Pickrell JK, et al. Dnase I Sensitivity Qtls are a Major Determinant of Human Expression Variation. Nature (2012) 482(7385):390–4. doi: 10.1038/nature10808
124. Corces MR, Granja JM, Shams S, Louie BH, Seoane JA, Zhou W, et al. The Chromatin Accessibility Landscape of Primary Human Cancers. Science (2018) 362(6413):eaav1898. doi: 10.1126/science.aav1898
125. Schuster-Bockler B, Lehner B. Chromatin Organization is a Major Influence on Regional Mutation Rates in Human Cancer Cells. Nature (2012) 488(7412):504–7. doi: 10.1038/nature11273
126. Polak P, Karlic R, Koren A, Thurman R, Sandstrom R, Lawrence M, et al. Cell-of-Origin Chromatin Organization Shapes the Mutational Landscape of Cancer. Nature (2015) 518(7539):360–4. doi: 10.1038/nature14221
127. Maqbool MA, Pioger L, El Aabidine AZ, Karasu N, Molitor AM, Dao LTM, et al. Alternative Enhancer Usage and Targeted Polycomb Marking Hallmark Promoter Choice During T Cell Differentiation. Cell Rep (2020) 32(7):108048. doi: 10.1016/j.celrep.2020.108048
128. Demircioglu D, Cukuroglu E, Kindermans M, Nandi T, Calabrese C, Fonseca NA, et al. a Pan-Cancer Transcriptome Analysis Reveals Pervasive Regulation Through Alternative Promoters. Cell (2019) 178(6):1465–77 e17. doi: 10.1016/j.cell.2019.08.018
129. Reimer KA, Mimoso CA, Adelman K, Neugebauer KM. Co-Transcriptional Splicing Regulates 3’ End Cleavage During Mammalian Erythropoiesis. Mol Cell (2021) 81(5):998–1012. doi: 10.1016/j.molcel.2020.12.018
130. Wong M, Mayoh C, Lau LMS, Khuong-Quang DA, Pinese M, Kumar A, et al. Whole Genome, Transcriptome and Methylome Profiling Enhances Actionable Target Discovery in High-Risk Pediatric Cancer. Nat Med (2020) 26(11):1742–53. doi: 10.1038/s41591-020-1072-4
131. International HapMap C. A Haplotype Map of the Human Genome. Nature (2005) 437(7063):1299–320. doi: 10.1038/nature04226
132. International HapMap, C, Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, et al. a Second Generation Human Haplotype Map of Over 3.1 Million Snps. Nature (2007) 449(7164):851–61. doi: 10.1038/nature06258
133. International HapMap, C, Altshuler DM, Gibbs RA, Peltonen L, Altshuler DM, Gibbs RA, et al. Integrating Common and Rare Genetic Variation in Diverse Human Populations. Nature (2010) 467(7311):52–8. doi: 10.1038/nature09298
134. Hnisz D, Weintraub AS, Day DS, Valton AL, Bak RO, Li CH, et al. Activation of Proto-Oncogenes by Disruption of Chromosome Neighborhoods. Science (2016) 351(6280):1454–8. doi: 10.1126/science.aad9024
135. Ando M, Saito Y, Xu G, Bui NQ, Medetgul-Ernar K, Pu M, et al. Chromatin Dysregulation and Dna Methylation At Transcription Start Sites Associated With Transcriptional Repression in Cancers. Nat Commun (2019) 10(1):2188. doi: 10.1038/s41467-019-09937-w
136. Bell RE, Golan T, Sheinboim D, Malcov H, Amar D, Salamon A, et al. Enhancer Methylation Dynamics Contribute to Cancer Plasticity and Patient Mortality. Genome Res (2016) 26(5):601–11. doi: 10.1101/gr.197194.115
137. Dozen A, Komatsu M, Sakai A, Komatsu R, Shozu K, Machino H, et al. Image Segmentation of the Ventricular Septum in Fetal Cardiac Ultrasound Videos Based on Deep Learning Using Time-Series Information. Biomolecules (2020) 10(11):1526. doi: 10.3390/biom10111526
138. Jinnai S, Yamazaki N, Hirano Y, Sugawara Y, Ohe Y, Hamamoto R. the Development of a Skin Cancer Classification System for Pigmented Skin Lesions Using Deep Learning. Biomolecules (2020) 10(8):1123. doi: 10.3390/biom10081123
139. Komatsu M, Sakai A, Komatsu R, Matsuoka R, Yasutomi S, Shozu K, et al. Detection of Cardiac Structural Abnormalities in Fetal Ultrasound Videos Using Deep Learning. Appl Sci (2021) 11(1):371.
140. Shozu K, Komatsu M, Sakai A, Komatsu R, Dozen A, Machino H, et al. Model-Agnostic Method for Thoracic Wall Segmentation in Fetal Ultrasound Videos. Biomolecules (2020) 10(12):1691. doi: 10.3390/biom10121691
141. Yamada M, Saito Y, Imaoka H, Saiko M, Yamada S, Kondo H, et al. Development of a Real-Time Endoscopic Image Diagnosis Support System Using Deep Learning Technology in Colonoscopy. Sci Rep (2019) 9(1):14465. doi: 10.1038/s41598-019-50567-5
142. Yasutomi S, Arakaki T, Matsuoka R, Sakai A, Komatsu R, Shozu K, et al. Shadow Estimation for Ultrasound Images Using Auto-Encoding Structures and Synthetic Shadows. Appl Sci (2021) 11(3):1127. doi: 10.3390/app11031127
143. Hamamoto R. Application of Artificial Intelligence for Medical Research. Biomolecules (2021) 11(1):90. doi: 10.3390/biom11010090
144. Takahashi S, Takahashi M, Kinoshita M, Miyake M, Kawaguchi R, Shinojima N, et al. Fine-Tuning Approach for Segmentation of Gliomas in Brain Magnetic Resonance Images With a Machine Learning Method to Normalize Image Differences Among Facilities. Cancers (Basel) (2021) 13(6):1415. doi: 10.3390/cancers13061415
Keywords: artificial intelligence, whole genome analysis, epigenome analysis, machine learning, biomarker discovery, cancer diagnosis and treatment, precision oncology
Citation: Asada K, Kaneko S, Takasawa K, Machino H, Takahashi S, Shinkai N, Shimoyama R, Komatsu M and Hamamoto R (2021) Integrated Analysis of Whole Genome and Epigenome Data Using Machine Learning Technology: Toward the Establishment of Precision Oncology. Front. Oncol. 11:666937. doi: 10.3389/fonc.2021.666937
Received: 11 February 2021; Accepted: 26 April 2021;
Published: 12 May 2021.
Edited by:
Mónica Hebe Vazquez-Levin, Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), ArgentinaReviewed by:
Vandhana Krishnan, Stanford University, United StatesFrancesco Schettini, Institut de Recerca Biomèdica August Pi i Sunyer (IDIBAPS), Spain
Copyright © 2021 Asada, Kaneko, Takasawa, Machino, Takahashi, Shinkai, Shimoyama, Komatsu and Hamamoto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ken Asada, a2VuLmFzYWRhQHJpa2VuLmpw; Ryuji Hamamoto, cmhhbWFtb3RAbmNjLmdvLmpw
†These authors share first authorship