 Yanni Ma1,2
Yanni Ma1,2 Ronghui Xia
Ronghui Xia Robert L. Judson-Torres
Robert L. Judson-Torres Hanlin Zeng
Hanlin Zeng- 1Department of Oncology, Ninth People’s Hospital, Shanghai Jiaotong University School of Medicine, Shanghai, China
- 2Shanghai Institute of Precision Medicine, Shanghai, China
- 3Department of Oral Pathology, Ninth People’s Hospital, Shanghai Jiaotong University School of Medicine, Shanghai, China
- 4Department of Oral & Maxillofacial - Head and Neck Oncology, Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China
- 5Department of Dermatology, University of Utah, Salt Lake City, UT, United States
- 6Huntsman Cancer Institute, Salt Lake City, UT, United States
Mucosal melanoma (MM) is a rare melanoma subtype that originates from melanocytes within sun-protected mucous membranes. Compared with cutaneous melanoma (CM), MM has worse prognosis and lacks effective treatment options. Moreover, the endogenous or exogenous risk factors that influence mucosal melanocyte transformation, as well as the identity of MM precursor lesions, are ambiguous. Consequently, there remains a lack of molecular markers that can be used for early diagnosis, and therefore better management, of MM. In this review, we first summarize the main functions of mucosal melanocytes. Then, using oral mucosal melanoma (OMM) as a model, we discuss the distinct pathologic stages from benign mucosal melanocytes to metastatic MM, mapping the possible evolutionary trajectories that correspond to MM initiation and progression. We highlight key areas of ambiguity during the genetic evolution of MM from its benign lesions, and the resolution of which could aid in the discovery of new biomarkers for MM detection and diagnosis. We outline the key pathways that are altered in MM, including the MAPK pathway, the PI3K/AKT pathway, cell cycle regulation, telomere maintenance, and the RNA maturation process, and discuss targeted therapy strategies for MM currently in use or under investigation.
Introduction
Melanoma develops due to the unchecked proliferation of melanocytes, which are responsible for the production of pigment. About 90% of melanoma cases are cutaneous melanoma (CM) mainly induced by exposure to ultraviolet (UV) light (1). Non-cutaneous subtypes include uveal melanoma (UM) and mucosal melanoma (MM). MM is a rare type of melanoma that presents on mucosal surfaces of cavities within the body, including the oral, nasal, anorectal, genitourinary, and vulvovaginal region (2). Although MM makes up approximately 1% of all cases of melanoma, it is one of the most aggressive subtypes, and thus exhibits a worse prognosis compared with the common CM (3, 4). Based on a retrospective study, the 5-year survival rate of MM, considering all stages at the time of diagnosis, is 10-20% when compared to 93% for CM (4–6).
There are several possible reasons for this worse prognosis in MM: 1) Both the biology of mucosal melanocytes as well as the risk factors that are related to MM incidence are poorly understood. Exposure to UV is a well-established risk factor for CM but the mutagens that contribute to the development of MM remain unknown. According to epidemiological studies, smoking, ill-fitting dentures, and ingested/inhaled carcinogens such as tobacco and formaldehyde are regarded as potential causative factors for oral and sinonasal mucosal melanoma (2, 7), while chronic inflammatory disease, viral infections as well as chemical irritants are thought to be implicated in vulvar mucosal melanoma and human immunodeficiency virus (HIV) is associated with anorectal mucosal melanoma (2). However, the contributions and mechanisms of the aforementioned factors to MM initiation or progression are not clearly defined. 2) The evolution of MM from precursor lesions is poorly understood. CM is associated with different types of precursor lesions, including benign melanocytic nevi commonly associated with the BRAF V600E mutation and dysplastic nevi associated NRAS alterations and TERT promoter mutations (8). CM can evolve from these benign lesions following additional mutations that drive tumor invasion and metastasis such as loss of CDKN2A, PTEN, or TP53 (9). Characterization of the morphology and molecular landscape of precursors compared to early melanoma has provided candidate molecular biomarkers for early diagnosis in CM (10–13). However, although several forms of mucosal melanocytic benign lesions are reported, there is still a lack of defined MM precursor lesions, leading to a weak understanding of the evolutionary trajectory of MM despite molecular profiles unveiled by recent whole-genome sequencing data (14–16). 3) MM has more diverse mutation patterns with fewer targetable mutations compared to CM. According to the most frequent and mutually exclusive mutations, CM is mainly classified into 4 genomic subtypes: BRAF(52%), RAS(31%), NF1(14%), and a small portion of triple wild-type (17, 18). Hence, co-targeting BRAF and MEK have been proved to achieve a significant response rate for BRAF V600 mutated CM patients in clinical management (19, 20). In contrast, MM has more diverse mutation patterns, with less than 20% of BRAFV600E mutations (16), followed by the majority of mutations that are scattered and difficult to target, including NRAS, NF1, KIT, SF3B1, and SPRED1 (21).
Our goals in conducting this review were to: 1) Summarize the types of mucosal melanocytic benign lesions, aiming to find possible genetic and pathological evolutional patterns from benign mucosal melanocytes lesions to malignant tumors; 2) Discuss the main driver mutations and pathways in MM; and 3) Outline the options of targeted treatment for MM in clinical use or under clinical trials.
Biological Functions of Melanocytes
Melanocytes are neural crest-derived cells that migrate to specific anatomic locations - including skin, eyes, leptomeninges, and mucous membrane - during development. Cutaneous melanocytes have two final destinations: hair follicles and the basal cell layer of epithelium where they conduct their main biological function of melanin production (22, 23). Melanin is a natural pigment in skin that absorbs UV radiation and scavenges cytotoxic free radicals generated from sunlight exposure (23, 24). Synthesized melanin is secreted to the nearby keratinocytes under solar stimulation and protects the genome of keratinocytes from sun damage (25).
In addition to residing in the skin, melanocytes also dwell in many sun-shielded mucosal tissues like respiratory (oral, nasal, pharynx, larynx, and upper esophagus), intestinal, urogenital, and rectal tracts (2, 24, 26–28). As melanocytes located in mucous membranes are not usually directly exposed to sunlight, it is unlikely that photoprotection is the primary and definitive function of mucosal melanocytes. It was hypothesized that melanocytes localized to mucosal tissues due to errors of migration from the neural crest during embryogenesis (6, 26), but recent evidence suggests that mucosal melanocytes might have biological functions besides pigment production. Specifically, since mucosa plays an important role in the innate immune defense system, it is speculated that mucosal melanocytes are also equipped with immunogenic functions (23, 29).
It is reported that melanin has strong toxin binding properties that can neutralize toxins produced by bacteria (30). Meanwhile, aromatic precursors, including quinone and semiquinone intermediates generated during the melanization cascade, can disrupt the lipid bilayer of cell membranes of microorganisms and mediate an anti-bacterial effect (31, 32). The strong binding capacity of melanin is probably due to its specific graphite-like lamellar structure in which four to eight monomers are covalently bound to form a porphyrin-like system (33). As a result, melanin is able to interact with aromatic metabolites or compositions of microorganisms through hydrogen bonds or π-π interactions (34, 35). Another explanation for the anti-bacterial properties of melanin and its intermediates is that they contain high levels of redox-active catechol groups, which can produce reactive oxygen species under light and water stimulation (36). However, since the mucosal melanocytes are in a dark environment with marginal melanin production, it remains unknown whether the pigment levels in mucosal regions are sufficient for antimicrobial effects.
In addition to the anti-bacterial properties of melanin, melanocytes can also participate in the intrinsic and acquired immune system. On the one hand, melanocytes can participate in innate immunity since they are found to express Toll-like receptors, indicating melanocytes can recognize pathogen-associated molecular patterns present in microbes (37, 38). Once being recognized, bacteria and fungi can be engulfed by melanocytes – a phenomenon that has been observed under the microscope - before undergoing possible degradation pathways by lysosome hydrolytic enzymes contained in melanosomes (39, 40). On the other hand, melanocytes may be a component of acquired immunity. Melanocytes have been reported to express MHC class II loaded with mycobacterial peptides (41), suggesting that melanocytes may function as antigen-presenting cells and subsequently activate CD4+ T cells proliferation (42). Although the phagocytotic functions of melanocytes have been observed, the activation of T cells through antigen presented by melanocytes, for instance, should be further verified by investigating the expression of CD86, CD80, or other markers of antigen-presenting cells on the surface of melanocytes. Since melanocytes have the capacity to produce a variety of cytokines, including interleukins and interferons which may be involved in the regulation of the activity of neighboring immune cells under stimulations of exogenous nucleic acids (43–47), the hypothesis that melanocytes activate T cells through secreting specific cytokines, instead of acting as antigen-presenting cells, must also be tested. These collective observations suggest melanocytes likely participate to some extent in the immune defense of the body, but their precise immunological roles in the innate or adaptive immune system need to be dissected in further studies.
Mucosal Melanocytic Benign Lesions
Studies in the pathologic evolution of CM have shown that invasive melanomas can evolve from a variety of benign and intermediate pathological stages including benign nevus, dysplastic nevus, and malignant tumor in situ (8, 9). Melanocytic nevi are benign lesions requiring no further treatment, while atypical melanocytic hyperplasia or atypical nevi are regarded as either indeterminant or premalignant lesions that warrant careful clinical management and long-term follow-up for patients. In contrast, there is no clear definition and characterization of precursor lesions of MM despite the fact that multiple mucosal melanocytic benign lesions are observed and documented in the clinic (Figures 1A, B). Using OMM as the most well-studied example, Table 1 summarizes several benign pigmented lesions including macule, nevus, and melanocanthoma with their specific pathological characteristics.

Table 1 Comparison of benign lesions and malignant oral mucosal melanoma.
Melanotic Macule of the Oral Mucosa
Melanotic macules are one of the most common melanocytic lesions (48, 49) and lentigo simplex is the term used to describe a group of small and round macules (50). The color of macules varies from gray to brown to black. The diversity of pigmentation is thought to be associated with the ratio of eumelanin and pheomelanin (23, 24). Macules are usually regarded as benign lesions since the causative factor of macules is melanin deposition and no Ki-67 positive melanocytes are observed (51, 52). Hence, the diameter of the pigmented lesions is usually less than 1 cm, and their morphology is flat, solitary, and well-circumscribed (Figure 1A). From histological examinations, the basal cell layer of benign macules is exhibited with uniform melanin accumulation without an increase in the density of melanocytes or the presence of nevus (Figures 1E, F). These lesions are asymptomatic and no malignant transformation is reported at this stage. The most frequently observed site for macules in the oral cavity is the vermillion border of the lip at the rate of 30% followed by the gingiva and alveolar ridge (23%), and the buccal (16%) or labial mucosa (9%) (24). Interestingly, the hard palate, which is one of the most common locations for OMM has less chance for macule occurrence (7%) (24). Although there is no evidence that melanotic macules are directly associated with the eventual diagnosis of MM in oral mucosa, some published case reports have recorded the transformation of benign macules to malignant OMM after years of diagnosis (53–55), suggesting the malignant potential of some macular lesions to be considered as precursor lesions. As Ki-67 staining is not routinely requested in the diagnosis of melanotic macules, it is unclear what percentage of lesions contain proliferating melanocytes and may possess malignancy potentiality.
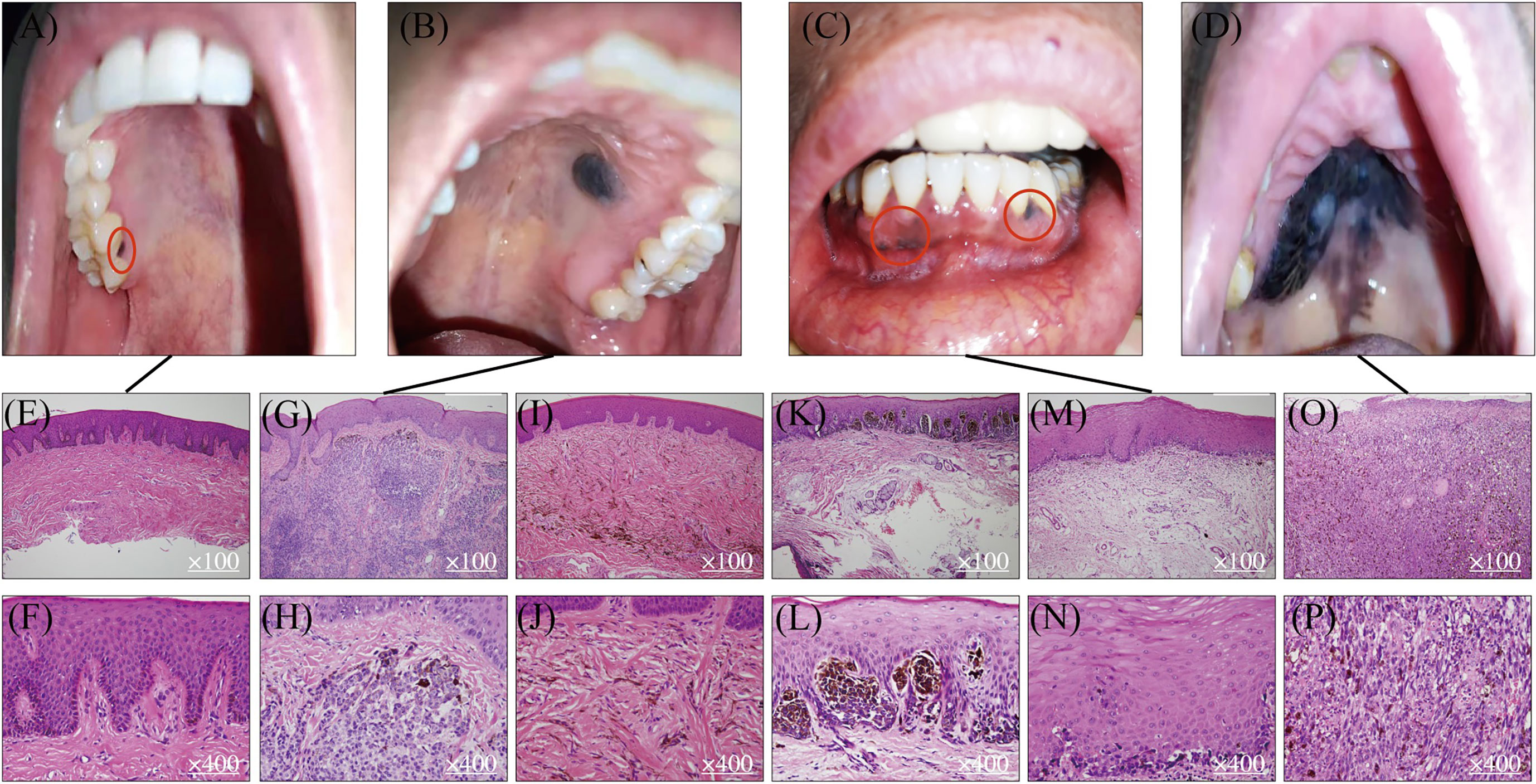
Figure 1 Mucosal melanocytic benign lesions and malignant OMM. Benign hyperpigmented lesions (A, B) and malignant OMM in situ (C, D). Benign macule in gingiva (A) and its HE staining pictures (E, F). Benign intramucosal nevus on the hard palate (B) and its HE staining pictures (G, H). HE staining of blue nevus (I, J). HE staining of junctional nevus (K, L). Lentigo maligma melanoma on mandibular gingiva (C) and its HE staining figures (M, N). Ulcerated malignant MM on the hard palate (D) and its HE staining pictures (O, P).
Oral Mucosal Nevus
Oral nevi are much less common than their counterparts on the skin and their prevalence is about 0.1% in the general population (24). Subepithelial lesions are the most common oral mucosal nevi (55%), followed by blue nevi in submucosal–mucosal junction (Figures 1I, J) (36%), and junctional nevi are the least frequent ones (3%) (7, 56). According to the histologic location of melanocytes, oral nevi can be divided into three categories: junctional nevi (Figures 1K, L) at the tip of the widened and elongated epithelial spikes; compound nevi arranged in nests and belts in the lamina propria; and subepithelial nevi (Figures 1B, G, H) entirely in the subepithelial connective tissue. The formation of nevi in oral mucosa results from the proliferation of melanocytes along with the epithelial basal cell layer, but most are relatively small with a mean diameter of 0.5cm. Similar to macules, nevi harbor clear borders. However, instead of being flat, more than 50% of nevi are elevated pigmented lesions. In addition, about 15% of oral nevi are non-pigmented and the mechanism behind the lack of pigmentation remains unclear (24). From the histopathological point of view, the appearance of nevus cells along epithelial spikes is polygonal and epithelioid. Typical nevus cells have uniformly round or oval nuclei and contain sparse, uniform, and small melanin granules in the cytoplasm. As for nevus cells in the deeper subepithelial tissue, they become smaller with less cytoplasm and dense and deeply stained nucleus-like lymphocytes.
Although there is a lack of case reports unequivocally documenting the transformation of benign nevi to malignant tumors in the oral cavity, the risk of malignancy in some oral nevi cannot be excluded. The deficiency of case reports is partially due to the rare individuals with congenital or acquired nevi and short follow-up periods of objects (57). A clinicopathologic analysis shows that five out of seven OMM patients have junctional nevi, therefore some clinicians recommend a complete excisional biopsy to rule out early OMM for individuals with junctional nevi (58). In addition, nowadays the classification of nevi is mainly based on their histologic positions and lacks criteria based on the degree of malignancy. Only the appearance of dysplastic nevi is considered as an increased risk of melanoma (59–61). Dysplastic nevi are usually larger than normal nevi with macular or popular components and ill-defined borders (62). The current diagnosis of dysplastic nevi mainly depends on their architectural disorder rather than specific biomarkers, which heavily relies on the experience of pathologists and causes a relatively high rate of misdiagnosis. Hence, more refined diagnostic criteria and more sensitive biomarkers are needed clinically to find potential precancerous nevi.
Melanoacanthoma
Melanoacanthoma is a rare form of benign melanotic lesion characterized by benign proliferation of both keratinocytes and melanocytes (24, 63). Microscopic examination can detect the hyperplastic keratinocytes, while positive immunostainings of HMB-45 and S-100 prove the presence of melanocytes abnormal accumulation (64). Compared with macule and nevus, this benign entity is much rarer but may mimic OMM due to its rapid increase in size with diameters of several centimeters being reached in just a few weeks. The lesion is usually flat or slightly raised and most commonly occurs on the buccal mucosa. Histopathologic examination shows many dendritic melanocytes and processes containing melanin in all strata of epithelium. Besides, melanophagocytes, mild lymphocyte infiltration, as well as vasodilation are seen in the lamina proporia. Similar to other benign lesions, once the melanoacanthoma is diagnosed, the site is usually just monitored, as these lesions are highly likely to regress within 2 to 6 months after biopsy (65, 66). However, if a melanoacanthoma enlarges in a very short period of time, it may indicate a sign of malignancy (67). A more comprehensive understanding of what drives mucosal melanocyte proliferation as well as the regression in melanoacanthoma, and how the fast-growing melanoacanthoma transforms into MM, is needed.
Oral macule, nevus, and melanoacanthoma are usually diagnosed as benign lesions, but periodical physical examinations and biopsies of those melanocytic lesions are still recommended because approximately one-third of OMM patients are found to present benign pigmented lesions prior to the emergence of the malignant state (67–69). Additionally, our collaborating clinicians and pathologists at Shanghai Ninth People’s Hospital have observed the development of hyperpigmentations adjacent to OMM in a majority of patients and they suspect that tumors expand through those de novo pigmented lesions (Figures 1D). Based on the above findings, we propose that a subgroup of benign lesions, especially macules, possess the potential to transform to OMM. If true, identification of biomarkers for those cancer-predisposing lesions coupled with more radical surgical excision may improve the outcome of patients. To achieve this goal, a thorough genetic evolution study sequencing not only malignant MMs but also benign lesions and suspected premalignant lesions is needed. Assessment of the genetic evolution of benign and malignant MM subtypes may reveal markers of increased risk of malignant transformation to aid in early diagnosis and clinical management.
Mucosal Melanoma
OMM is one of the most frequent and well-studied MM subtypes. The preferred site for OMM (Figure 1D) is the keratinized mucosa, including the hard palate and maxillary gingiva where the masticatory stress is focused (70). Symptoms include pain, ulceration, bleeding, loose teeth, bone erosion, etc. (71, 72). The MM shows variable color from black to red or white accompanied with asymmetric and irregular morphology (73). Contrasting from CM, which is commonly diagnosed in the radial growth phase, OMM is usually first identified in a vertical growth phase with 30% of lesions at an invasive stage, and 55% of lesions at a combined invasive and in situ stage (7). Also different from CM, MM lacks a clear classification system for subtypes of lesions. Based on the histopathologic patterns and levels of solar damage, there are several different categorization methods for CM (74), whereas the subclassification of MM remains controversial. Currently, it is simply divided into MM in situ, invasive MM, and MM with a mixed pattern. The observing surface architecture of MM ranges from macular to ulcerated and nodular (75). Lentigo maligna melanoma (Figure 1C) is regarded as one form of OMM in situ as it shares similar histopathological traits as typical OMM in situ (Figures 1M, N). From the microscopic perspective, OMM consists of diverse morphological melanocytes including epithelioid, spindle, and plasmacytoid, which typically have a large, vesicular nucleus with prominent nucleoli (Figures 1O, P). They are usually aggregated into sheets or alveolar groups and less commonly neurotropic or desmoplastic configurations are observed. Most of the tumors contain melanin, while only a small proportion is amelanotic (76). As for immunohistochemical features of OMM, there is no single immunohistochemical marker that invariantly identifies all OMM. A variable expression of S-100, Melan-A, MITF, tyrosinase, and HMB-45 has been reported (27, 28, 49, 76). SOX 10 is a new marker, showing high sensitivity (positive in 88-100% of OMM cases) but moderate specificity in MM (77, 78). Hence, identification of biomarkers for OMM with better test characteristics of needed to achieve a consistent accurate diagnosis of MM and its initial lesions.
Mutations and Signaling Pathway Dependency in MM
To date, whole-genome sequencing (WGS) and whole-exome sequencing (WES) has revealed the genomic profile of MM and pinpointed reoccurring aberrant genes that potentially drive the evolution of melanocytes to malignant tumors in the mucosal membrane. In contrast to CM, MM harbors a low single nucleotide mutation burden, but a high number of chromosomal structural variants (16, 79). BRAF and NRAS mutations, which are widely present in CM, are less frequent in MM (16, 79). Instead, activating mutations in SF3B1 and KIT, loss of CDKN2A, PTEN, or SPRED1, as well as amplification of CDK4, TERT, KIT, MDM2, or CCND1, are more common in MM (16). Table 2 compares the genetic profile between CM and MM. The data for altered genes in CM are average from Akbani’s and Hayward’s papers (17, 79), while the figures for MM are obtained from Newell’s paper (16). Figure 2 summarizes the frequency of alterations in possible driver genes based upon WGS data of 67 frozen tumors (16). Those mutated genes correspond to specific cellular pathways that are potentially highly dependent on the initiation and progression of MM, providing potentially effective targets for combined treatment in the clinic.
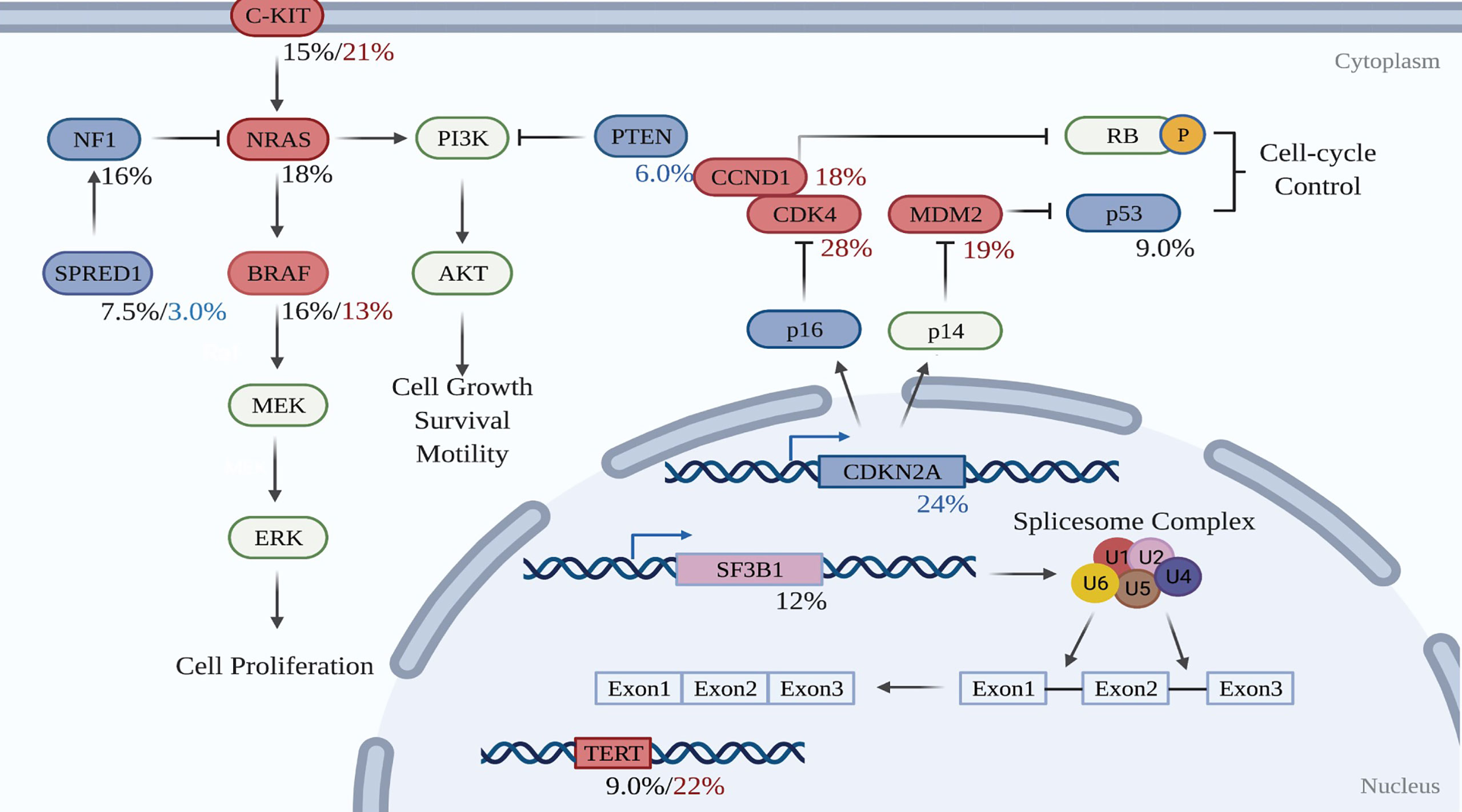
Figure 2 Molecular pathways involved in the development of mucosal melanoma. Red-filled rectangles indicate genes experiencing activating mutation or amplification, while blue-filled rectangles genes undoing suppressing mutation or deletion. Black figures suggest mutation rates, whereas red and blue percentages are respectively amplification and deletion proportions in the test cohort. Created with BioRender.com (2021). Retrieved from https://app.biorender.com/biorender-templates.
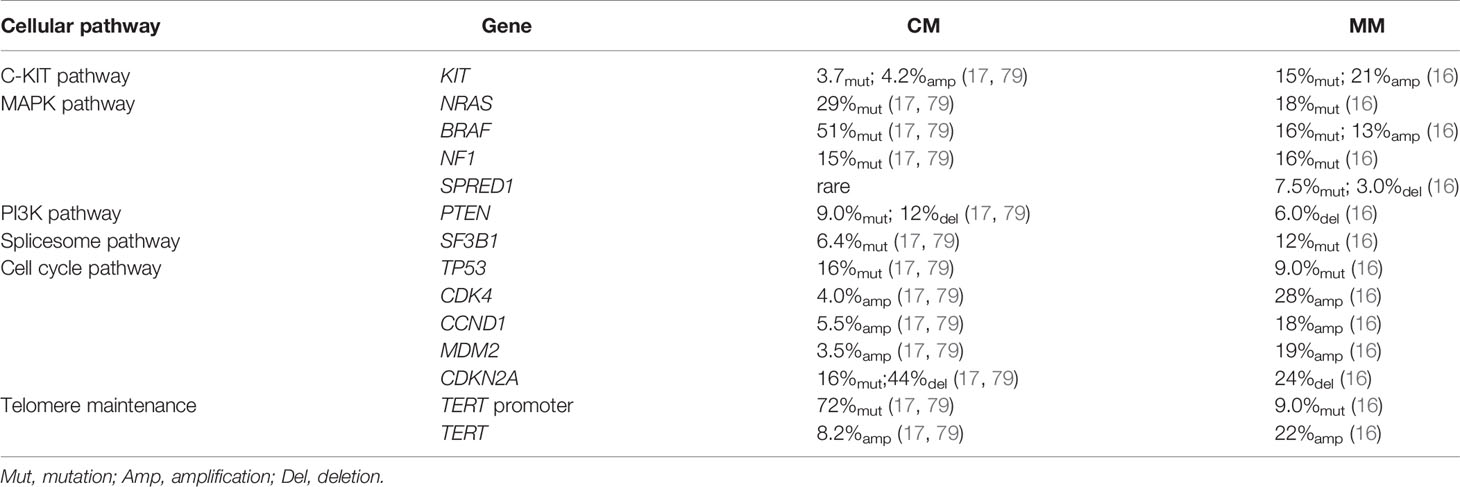
Table 2 Comparison of genetic profiles MM and CM.
KIT Signaling Pathway
C-KIT is a receptor tyrosine kinase located on the membrane of various cell types. The stimulation of the C-KIT receptor by its extracellular ligand leads to downstream activation of the MAPK and PI3K signaling cascades that play an important role in proliferation, survival, and motility of melanoma cells (80). There is a high prevalence of KIT gain-of-function alterations including missense mutation and copy number amplification in patients with MM at rates of 15% and 21% respectively (16), while the corresponding figures in CM are only 3.7% and 4.2% separately (17, 79). MMs with KIT mutations presumably affect the function of juxta-membrane autoinhibitory domain (JMD) (W557R, N566D, V559A, V559D, V560D, V569G, P573L, L576P, K642E) and tyrosine kinase domains (D816H, D820Y, A829P, N822K), causing constitutive activation of C-KIT-regulated pathways (81–84). Among the aforementioned mutations, K642E is the most frequently observed in MM. Although codon 642 is located out of the JMD, amino acid aberrations in this position are thought to destabilize the JMD through amino acid interactions (81). Besides WGS and WES data, immunohistochemistry images show increased protein expression of C-KIT in all in situ MMs and nearly 90% of invasive tissues in a cohort of 18 cases (85), indicating the strengthening of the C-KIT signaling pathway. Compared to CM, the gain-of-function alterations of KIT are more common not only in MM but also in acral melanoma (AM) (86). Although the mechanism explaining why C-KIT is pathogenetically important in sun-protected melanomas remains poorly understood, it does not prevent the protein from being a potentially effective therapeutic target and a series of C-KIT inhibitors are currently under pre-clinical and clinical investigations (82, 87–89).
RAS-RAF-MEK-ERK MAP Kinases Pathway
Stimulation of C-KIT or other receptor tyrosine kinase on the cellular membrane by extracellular growth factors provokes downstream activation of RAS and RAF kinases followed by phosphorylation of MEK and ERK, leading to activation of the MAPK pathway implicated in the regulation of cell proliferation, differentiation, and survival (90). NRAS and BRAF both play a part in the MAPK pathway, which are thought to contribute to melanoma development.
NRAS activating mutations are prevalent in CM at 29% (17, 79), while the mutation frequency in MM is only 18% (16). Furthermore, nearly 90% of NRAS missense mutations occur at codon 61 in CM, compared to 54% for MM. The remaining 46% of mutations are located at codon 12 and codon 13 (91). Compared with NRAS activating mutations at positions 12 and 13, NRAS Q61 mutations exert a stronger activating effect on the MAPK pathway since codon 61 is the catalytic residue for GTP hydrolysis and Q61 mutation impedes the return of RAS to an inactive GDP-bound state (92). For both CM and MM, Q61R and Q61K are the most commonly detected amino acid transitions at codon 61. Similarly, the most common mutations for both types of melanoma at codon 13 are G13D and G13R, although G13R is predominant in CM and G13D in MM (93).
BRAF is a serine-threonine kinase involved in the MAPK signaling pathway. Over 50% of CM cases report activating mutation of the BRAF gene (17, 79), while merely 16% of MMs experience the same alteration (16). In addition to harboring common active mutations, BRAF is distinct from NRAS in MM in that the locus undergoes amplification in 13% of cases (16). Mutations in the BRAF gene are missense mutations and they most frequently occur at codon 600, the activating loop, where amino acids change from valine to glutamic acid (V600E) (93). Besides the activation loop (A-loop), the second most common site for amino acid substitutions is the GSGSFG phosphate-binding loop (P-loop) at residues 464-469 (94). The activity of BRAF kinase is regulated by the interaction formed between A-loop and P-loop, thus mutations in either A-loop or P-loop disrupt the interaction and cause hyperactivation of the kinase (93, 95). In CM, more than 90% of mutations are present on the V600 codon, whereas in MM only 63% are V600 mutations, with the remainder (37%) on the G469, D594, and K601 codons (91, 93). In other words, MM not only has far less frequent BRAF missense mutations but also has more diverse locations for BRAF mutations as compared to CM. However, similar to CM there is nearly no coincidence of NRAS and BRAF missense mutations, suggesting functionally redundant NRAS and BRAF mutations in MM despite more variable mutational locations.
Although the contribution of mutant NRAS and BRAF to MM progression appears to be less common than melanoma of the skin, more components in the MAPK pathway of MM tend to undergo mutations or copy number changes, rendering inhibition of MAPK signaling transduction more challenging. The NF1 gene, for example, encodes neurofibromin 1 protein, a negative regulator of Ras proteins, and can lose its function in both CM and MM. NF1 mutation rates are 16% in MM (16) and 15% in CM (17, 79), suggesting NF1 plays a pivotal role in the biology of both types of melanoma. Loss of NF1 is associated with sustained activation of Ras proteins, leading to hyperactivation of MAPK and PI3K-AKT intracellular signaling pathway that evokes melanogenesis. Similar to CM, NF1 suppression is significantly enriched in tumors lacking either BRAF or NRAS mutations (16, 96). However, in melanomas harboring both BRAF and NF1 mutations, it is more likely that tumors can escape from MAPK inhibiting therapy (97, 98). Meanwhile, NF1 is significantly co-mutated with KIT in 32% of MMs, whereas the co-occurrence level in CM is merely 4% (99), which indicates that the MAPK cascade is upregulated in MM not only by the single protein in the cytoplasm but also by the assistance of C-KIT receptor on the cell membrane.
SPRED1 is another potential driver gene for MM. SPRED1, sprout-related, EVH1 domain containing protein 1, is a tumor suppressor. SPRED1 facilitates the localization of NF1 to the plasma membrane where it suppresses RAS signaling (6). Therefore, the loss of SPRED1 function leads to the activation of MAPK pathway signaling transduction. 7.5% of MM have SPRED1 inactivating mutations and 12% undergo SPRED1 copy number loss (16), whereas SPRED1 alterations are insignificant in CM (17, 79). In MM, SPRED1 loss rarely co-occurs with BRAF mutations, NRAS mutations, or NF1 inactivation mutations (6), indicating those alterations play similar roles in activating MAPK pathway signaling in MM. Analogous to NF1, around 30% of MM cases with SPRED1 inactivation simultaneously exhibit KIT alterations, suggesting that SPRED1 inactivation may be in collaboration with other oncogenic events to stimulate tumor development (100). Based on the pattern of mutually exclusive occurrence of NF1 and SPRED1 and their respective tendency to alter simultaneously with KIT, it is reasonable to speculate that NF1 and SPRED1 loss function similarly in MM. In addition, it has been proposed that the reduced sensitivity and drug resistance to KIT inhibitors partially result from the hyperactivation of MAPK caused by the loss of SPRED1 – a model verified in human melanoma cell lines and in vivo zebrafish model (6, 101), but presently untested in mouse models and patient samples.
PI3K-AKT-mTOR Pathway
The PI3K-AKT-mTOR pathway is another frequently activated oncogenic signaling cascade in MM, which is verified by elevated AKT phosphorylation through immunohistochemical staining (102, 103). The aforementioned abnormal KIT, NRAS, NF1, and SPRED1 genes are able to not only activate the MAPK signaling cascade but also dysregulate the PI3K-AKT-mTOR pathway. Additionally, the PI3K pathway is stimulated by suppression of a negative regulator, phosphatase and tensin homologue (PTEN) (104). Compared with 12% of PTEN loss in CM where deletions and mutations both account for the changes (17, 79), PTEN is deeply deleted in merely 6.0% of MM cases and has hardly any mutations (16) Furthermore, there is rare co-occurrence of deleted PTEN and amplified KIT that possibly implies that loss of PTEN or gain of KIT are redundant for the activation of the PI3K pathway in MM. This is also supported by the fact that besides KIT and PTEN, mutations in PI3K and AKT homologous of PI3K-AKT-mTOR are scarce (4.8% PIK3CA, 3.8% PIK3CG, and 4.8% AKT3) (105). It has been reported that silencing of PTEN cooperates with activated AKT to promote metastasis of melanoma (102, 103, 106, 107), but MM does not show a weaker performance in metastasis than CM in the clinic possibly because there are other gene alterations in the PI3K pathway promoting invasiveness of tumor (108). In a cohort of 91 MM patients, 18% of cases show TSC1 loss-of-function mutations which plays a suppressive role in cellular proliferation initiated by mTOR (109) and a similar result also showed in a recently published meta-analysis review of MM (105). Due to the limited sample size, the alteration levels for TSC need to be further verified. Taking into account the frequency of alterations in known genes involved in PI3K-AKT-mTOR pathway activation, applying PI3K pathway blockers could possibly be an effective target strategy in MM patients.
The Spliceosome Pathway
The spliceosome complex is responsible for the removal of introns from precursor mRNA and the ligation of exons to form mature mRNA. SF3B1 (splicing factor 3b subunit 1) is the largest and core component of the U2 small nuclear ribonucleoprotein (snRNP) and thus SF3B1 mutations directly cause aberrant gene transcripts which eventually lead to mRNA degradation or abnormal protein function or protein decay (110, 111). SF3B1 mutations have been reported in 12% of MM (16), while a very small portion of tested CM patients harbor similar alterations (17, 79, 112). Despite their notable absence in CM, alterations in the SF3B1 gene are not unique to MM - similar mutations have also been detected in uveal melanoma, breast cancer, myelodysplastic syndromes, and chronic lymphocytic leukemia, including mutation hotspots such as codon 700, 622, 625, 662, and 666 (99, 113). To be more specific, SF3B1 mutations at codon 625 are predominately associated with mucosal and uveal melanoma, while alterations at codon 700 are present across myeloid leukemia and chronic lymphocytic leukemia (6), implying disparate SF3B1 mutational preference that is possibly related to distinct etiology. Although SF3B1 mutations are widely present in MM, it is poorly understood which genes alternatively spliced by mutant SF3B1 drive malignant transformation. It has been discovered that BRD9, PPP2R5A, and DVL2, are candidate genes for alternative splicing in SF3B1 K700-mutant chronic lymphocytic leukemia (114–116), while ABCC5, UQCC, and CRNDE are possible targets in three uveal melanoma cases mixed with R625 and K700 mutations (117). Hence, due to the variability of SF3B1 mutations in solid and hematologic cancers, experiments that query the consequence of the SF3B1 R625 mutation in mucosal melanocytes are needed to understand mis-spliced targets and tumorigenic oncogenic mechanisms related to SF3B1 mutations in MM.
Intriguingly, although SF3B1 does not possess a direct role in MAPK pathway signal transduction, there is little overlap between tumors with MAPK pathway mutations and SF3B1-mutated tumors, suggesting that SF3B1 mutations possibly lead to splicing variations of specific genes that can lead to MAPK activation (16). Meanwhile, the MAPK pathway can also regulate splicesome activity. It is reported that activation of the MEK-ERK pathway by Golgi stress enhances the activity of ETS transcriptional factors that have the capacity to regulate the expression of splicesome components, resulting in a switch of MCL1 protein function through different splicing (118). This study indicates that dysregulation of some downstream effectors by the MAPK pathway is able to lead to splicing aberrations. The mechanisms connecting the splicing alternations to signal transduction remain enigmatic. Mutant K700E SF3B1 causes loss of function of phosphatase PP2A, followed by the phosphorylation changes related to signaling cascade (115), thereby providing a potential link between an alternative splicing and signaling pathways. Further investigations are needed to elucidate the specific mis-spliced genes and proteins directly influenced by SF3B1 mutations together with the crosstalk between SF3B1 and MAPK pathway in tumorigenesis of MM, and the findings may provide a new perspective for targeted therapy.
Cell Cycle Pathway
The abrogation of cell cycle checkpoint and apoptosis regulators is widely present in melanoma (119), including CDKN2A loss and CDK4/6 or CCND1 amplification. The CDKN2A locus encodes two distinct tumor suppressors, p16INK4A and p14ARF. The p16INK4A protein suppresses the forward progression of the cell cycle by inhibiting CDK4 or CDK6. The CDK4/6/CCND1 complex phosphorylates and inhibits the retinoblastoma (Rb), which leads to E2F1 transcription activation and G1-S phase cell cycle transmission (120). The other CDKN2A transcript, p14ARF, functions, at least in part, by blocking MDM2 ubiquitylation mediated TP53 degradation, which permits apoptosis escape (121). In MM patients, 24% of tumors exhibit copy number loss of CDKN2A (16). Additionally, CDK4, CCND1, and MDM2 are amplified in 28%, 18%, and 19% of samples respectively, and TP53 mutations occur in 9.0% of MMs (16). Most of these cell cycle components are also commonly disrupted in CM. However, although CM has a much higher CDKN2A loss than MM, MM tends to show a greater frequency of CCND1 and CDK4 amplification (120, 122), implying CDK4 blocking agents may achieve a desired anti-tumor effect on MM. Intriguingly, MM cells without mutations in BRAF or NRAS mutations tend to exhibit CCND1 or CDK4 amplification (122), suggesting that copy number variations of cell cycle regulatory genes act as an alternative driver and can substitute for BRAF or NRAS mediated proliferation signaling pathway activation.
Telomere Maintenance
Telomerase reverse transcriptase, encoded by the gene TERT, is the catalytic subunit of the enzyme telomerase, responsible for lengthening telomeres at the end of chromatin (123, 124). Thus, overexpression of TERT confers the potential of cells to become immortal (125), which is one of the hallmarks of cancer. TERT promoter mutations and TERT amplifications are common genetic events in the early stages of melanoma of the skin (8). For CM, more than two-thirds of tumors exhibit TERT promoter mutation, and only a minority of malignancies present copy number amplification (17, 79). As a comparison, the frequency of TERT activating alteration in MM declines to 30%, and most of them are copy number gain rather than promoter mutations (16). As for why CM and MM present distinct mechanisms of TERT activation, most TERT promoter mutations in CM are C>T mutations or CC>TT di-pyrimidine mutations (126, 127), suggesting that TERT promoter mutations are induced by UV radiation which partially explains why these mutations are rare in sun-shielded MM. Apart from TERT, the gene ATRX is also involved in telomere maintenance. ATRX is associated with alternative lengthening of telomeres as an additional mechanism for telomere maintenance in tumors lacking TERT promoter mutations (128). Despite rare samples associated with ATRX alterations in CM, nonsense mutation and frameshift of ATRX are detected in 11.9% of MMs (16) implying that ATRX is responsible for telomere extension in MM as well. However, the altering level of ATRX needs to be further tested in a larger cohort since the gene are not significantly mutated in other sequencing results except for Newell’s study.
Although CM exhibits a much higher frequency of TERT activation than MM, there is no statistically significant difference in telomere length among CM and MM, and both of them even undergo telomere shortening (8, 79, 129). Those intriguing findings remind us that aberrant TERT might have tumorigenic impacts in melanocytes other than telomere lengthening. It is reported that human TERT (hTERT) is equipped with a telomere protective function independent of its canonical catalytic activity (130). Overexpression of hTERT in melanoma is able to produce a protective complex on DNA damage that leads to the sustained proliferation capacity of cancer cells (130). In addition, phosphorylated TERT at a specific position by CDK1 has an RNA-dependent RNA polymerase (RdRP) activity (131). RdRP generates small interfering RNAs complementary to a tumor suppressor gene FOXO4, degrading mRNAs of FOXO4, reducing protein expression and consequently leading to tumor formation (131, 132). Taken together, these observations suggest potential differences in telomere maintenance mechanisms among different subtypes of melanoma.
Progress in MM Target Therapy
When compared with CM, MM is typically detected at advanced stages, which renders the tumor challenging to treat. Surgical excision is predominately the first choice for MM (133–135). However, due to the lentiginous growth pattern, multifocal nature of MM, and limitations of the specific MM anatomic sites, it is extremely difficult for surgery to achieve wide negative margins, which leads to a high local relapse rate at 50%-90% (2). For unresectable and metastatic MM, targeted therapy and immunotherapy are constrained since MM is deficient in dominant MAPK activating mutations that can be targeted and is less responsive to immunotherapy (136). Therefore, to date, the first-line therapeutic modality for advanced MM remains chemotherapy despite limited efficacy (137).
While targeted therapies for MM are limited, multiple clinical trials targeting aberrant genes in MM are ongoing. Similar to CM, the MAPK cascade is hyperactivated by altered genes in MM including NRAS, BRAF, NF1, and SPRED1, thereby making inhibition of MAPK signal transduction a promising treatment strategy for MM patients. For the minority of MM patients with BRAF mutations, combined inhibition of BRAF and MEK is an attractive strategy because the combination therapy shows an impressive response rate at 76% and has a 5-year survival rate of 33% for BRAFV600E/K positive CM patients (138). Although there is no clinical trial underway specifically evaluating the safety and efficacy of combination therapy of BRAF inhibitor plus MEK inhibitor in MM, a retrospective study in Japan showed that MM/AM and CM exhibited similar response rates to combined BRAF and MEK suppression (64.3% vs 76.5%) (139), suggesting the potential efficiency of dual repression of BRAF and MEK in MM. For patients without BRAF mutations but with NRAS, NF1, or SPRED1 alterations, targeting the downstream protein MEK is another strategy for MM patients. The safety and efficacy of MEK blocking agents in MM have been confirmed in an ongoing phase 2 study where 20% NRAS-mutated melanoma patients showed partial response to MEK inhibitor binimetinib with tolerated and manageable adverse events (140). However, for both monotherapy of MEK inhibitor and combined treatment of BRAF and MEK inhibitors, acquired resistance through reactivation of the MAPK pathway can restrict their therapeutic efficacy (141). To overcome this resistance, it is necessary to inhibit downstream proteins like ERK or develop new molecules targeting aberrant MAPK signaling. Meanwhile, besides independent suppression of MAPK pathway, MEK blockers have also been combined with mTOR1/2, AKT, or CDK4/6 inhibitors in preclinical models or in clinical trials of MM (142–144). Although therapeutic parameters do not significantly improve compared to the single MAPK inhibition, dual signaling pathway blocking still provides a new perspective for MM targeted therapy.
Interestingly, compared to CM, MM patients tend to harbor more activating mutations or amplifications in the receptor tyrosine kinase KIT, providing a rationale for targeting C-KIT. Imatinib, sunitinib, dasatinib, nilotinib, and masitinib are approved C-KIT inhibitors in different cancer types and their anti-cancer effects for MM are currently in the clinical research stage (145–148). Imatinib is the most widely investigated C-KIT inhibitor. In a recent trial of 78 melanoma patients harboring KIT alterations, the median overall survival for imatinib is 13.1 months and the objective response rate is 21.8% (149). Additionally, it has been discovered that C-KIT inhibitor imatinib harbors high efficacy against melanoma with KIT mutations, but not with KIT amplification only (54% vs 0% partial response) (148, 150). To be more specific, MMs with KIT mutations in exon 11 (L576P) and exon 13 (K642E) tend to have a better and longer response to C-KIT inhibitors than other mutations (84, 151). Despite the strong anti-tumor effect of C-KIT inhibitors, MM patients who respond to the inhibiting agents well at the beginning will frequently experience a brief period of disease response before developing resistance to KIT inhibitors that eventually leads to progressive disease (152, 153). The acquired resistance to KIT inhibitors is possibly conferred from pre-existing concomitant mutations in other oncogenes like NRAS or secondary KIT mutations during the use of drugs. For instance, secondary A829P KIT mutation renders melanoma cells resistant to imatinib but has no influence on nilotinib and dasatinib, while the T670I KIT mutation exhibits resistance to imatinib, nilotinib as well as dasatinib, but can still be suppressed by sunitinib (154). Considering the promising performance of C-KIT inhibitors in MM, now more efforts have been focused on the understanding of the acquired resistance mechanism and the development of new blocking agents to overcome resistance, offering hope for patients with advanced MM and limited treatment options.
In the future, targeted therapy could offer an alternative adjuvant therapy option for a group of patients based on their gene sequencing results. If actionable driver mutations are identified in an individual MM, targeted therapies for the driver genes or proteins could be utilized on a patient-by-patient basis. Until now there are only a few available targeted therapies for MM clinical trials: BRAF, MEK, CDK4/6 and, C-KIT inhibitors, with limited clinical use and efficacy. Therefore, it requires more efforts on developing other alternative targeting strategies based on mutated genes in MM such as splicesome complex components, telomerase, and DNA repair pathway. H3B-8800, for instance, is the blocker for splicing modulator of SF3B complex and it at present is in phase I study of myeloid cancers (155). Considering the MM specific SF3B1 hotspot mutation in R625, developing strategies that can specifically target R625 mutant SF3B1 might may achieve benefit MM patients with low side effects.
Discussion
MM is a rare but aggressive malignancy. Due at least in part to delayed diagnosis at the advanced stage and the lack of efficient therapeutic strategies, this subtype of melanoma is associated with a worse prognosis than melanoma arising from the skin. In contrast to CM, the etiology, risk factors, and pathogenesis of MM are poorly understood, partially explaining the deficiency of effective treatment options and extremely poor prognosis. This review takes OMM as a model and attempts to identify commonalities in etiology, pathogenesis, mutation patterns, and corresponding pathway dependency. Besides cigarette smoking, denture irritation, and alcohol, chronic infections caused by microorganisms and mechanical stress generated by routine activities may have an impact on tumorigenesis in the mucosal membrane. However, the oral microflora is in a dynamic process of change and is influenced by many internal and external factors, including the host’s physical conditions, diet, and hygiene habits. A more comprehensive study investigating the relationship between flora and cancer, the selection of patients, sampling locations, and control settings will be needed.
Since there is limited knowledge about pre-MM lesions and a lack of corresponding molecular pathological biomarkers, early diagnosis, as well as early intervention becomes extremely challenging, leading to the short life expectancy in MM patients. Due to the unclear relationships between benign lesions and precursor lesions, histopathological information alone cannot thoroughly define and accurately discriminate them. It is reported that one patient died of OMM after 63 months of misdiagnosed premalignant atypical melanocytic hyperplasia as a benign lentigo simplex (50). Therefore, it is an urgent need to discover biomarkers for lesions with a greater tendency of malignant transformation. To achieve this goal, a thorough genomic and transcriptomic profiling of the evolutionary trajectories of MM starting from benign lesions and potential intermediate lesions is worth pursuit. Another strategy for studying cancer evolution is to establish transgenic mice that capture the evolution process of MM. However, unlike CM, there is a lack of animal models that can recapitulate the oncogenesis process accompanied with the accumulation of genetic alterations in MM. By stepwise introduction of BRAF V600E mutation, CDKN2A loss, PTEN loss and mTOR activation, CM precursor lesions followed by CM formation was observed in mice (156–159). Likewise, decoding the accumulative mutation pattern based on MM patient samples will pave the path to the generation of MM transgenic mice model, which not only contribute to understanding the pathogenesis of MM but also serve as functional tools to evaluate the efficacy of novel therapeutic modalities.
Recent sequencing studies have identified significant alterations in NRAS, BRAF, NF1, KIT, SF3B1, TP53, and SPRED1, informing potential targeted therapeutic strategies for MM (14–16, 160, 161). Firstly, MM patients have shown similar pathway dependency although with divergent mutation patterns. Compared to CM, fewer NRAS, BRAF mutations are seen in MM, but more SF3B1 mutation and KIT alterations are found. Since targetable BRAF mutations are far less frequent in MM, target validation of other alterations in the MAPK pathway is needed. The sequencing results of 67 MMs show that mutations of NRAS, BRAF, KIT, and SF3B1 are mutually exclusive, implying those mutations may converge on activating the MAPK pathways (16). Further studies about how SF3B1 mutations are involved in MAPK pathway activation are needed. Secondly, MM has gained fewer genetic mutations for cell cycle regulators but more copy number changes than CM. While CDKN2A copy number loss is a frequently observed event in both CM and MM, MM presents more CDK4 and CCND1 amplifications, which makes targeting CDK4 promising in MM.
It is worth mentioning that MM has a much higher level of structure variation and chromosomal instability compared to CM. As a result, specific attention should be paid to targeting the chromosomal rearrangements. Targeting genes involved in DNA damage repair response including PARP, DNA-PKcs, ATR, ATM, CHK1, WEE1 might achieve unexpected clinical response in MM patients (162–164). Olaparib, for instance, is an FDA-approved inhibitor of the enzyme poly ADP ribose polymerase (PARP) which can efficiently kill BRCA mutant tumor cells, a successful synthetic lethality based targeting strategy used in breast cancers and ovarian cancers (165). Although MM rarely shows BRCA mutations, the significantly high level of structure variation indicates the deficiency of homologous recombination repair (HRR) capacity, which makes MM potentially responsive to PARP inhibition. Nevertheless, a more comprehensive understanding about the mutation signatures as well as signatures of chromosome structure variation in MM are needed. A more stringent validation of PARP inhibitor response in MM cell lines, PDX, and early clinical trials are supposed to conduct to better understand the pharmacological mechanism of drug response in MM.
Other than targeted therapy, immune checkpoint blocker (ICB) based immunotherapy has shown a strong anti-tumor effect on metastatic CM. Ipilimumab against cytotoxic T-lymphocyte antigen 4 (CTLA4), nivolumab and pembrolizumab against programmed death 1 (PD1) as well as atezolizumab against programmed death-ligand 1 (PD-L1) are approved by the FDA for the treatment for advanced melanoma either as monotherapy or combination therapy (166). The overall response rate (ORR) for CM patients to ipilimumab, nivolumab, pembrolizumab and atezolizumab is 12%, 40%, 33% and 30% respectively (167–169), while the combined treatment of ipilimumab with nivolumab significantly improves the ORR to 61% with median progression-free survival (PFS) of 11.5 months (170, 171). However, those ICBs do not exert a satisfactorily inhibitory impact on MM as they do on CM, showing the ORR to anti-CTLA4 or anti-PD1/PDL1 as the single agent from 7% to 35% (136, 172–174). Even the combination regimen of anti-CTLA4 (ipilimumab) and anti-PD1 (nivolumab) agents merely witness a slight increased ORR to 37% with PFS at 5.9 month (136, 175). The limited response to ICBs in MM is mainly because of low mutation burden and limited immune cell infiltration compared to CM (3, 176, 177). To further overcome unsatisfactory performance of ICBs in MM, combination of ICBs with different targeted therapy strategies has been tested in clinical trials. For example, a phase Ib trial using PD-1 antibody toripalimab and vascular endothelial growth factor receptors (VEGFR) inhibitor axitinib showed a dramatical improvement in ORR and PFS to 61% and 9.1 months separately (178, 179), although the safety and efficacy of this combination strategy needs to be further validated. Meanwhile, the combination of toripalimab and vorolanib which targets and inhibits multi-tyrosine kinase including VEGF and C-KIT are ongoing in MM trial (180).
In summary, both basic research and drug discoveries in CM have achieved enormous progress, whereas little is known about either how MM initiates or how to target MM. As a result, patients of MM are suffering from limited treatment options and undesirable response rates that lead to extremely poor prognoses. Here we summarize the current state of knowledge regarding initiation and progression of MM and the risk factors and treatment options for MM. In doing so, we highlight current gaps in our knowledge regarding MM progression, and propose important future research directions, includes studying the genetic evolution trajectory of MM from benign precursor lesions and evaluating new targeting strategies specifically for MM, such as targeting CDK4, SF3B1 or PARP, either as single agent or in combinations with ICBs. We hope these efforts will give more comprehensive knowledge about how MM initiates and progresses, provide more specific biomarkers for MM early diagnosis, offer more potentially effective treatment options for MM and, in the end, improve the life expectancy and quality for MM patients.
Author Contributions
YM wrote the main body of the paper. HZ and RLJ revised the review. RX and XM provided significant intellectual contribution on clinical observations and pathological description of melanocytic lesions. HZ conceived and directed the idea of the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. D’Orazio J, Jarrett S, Amaro-Ortiz A, Scott T. UV Radiation and the Skin. Int J Mol Sci (2013) 14(6):12222–48. doi: 10.3390/ijms140612222
2. Carvajal RD, Spencer SA, Lydiatt W. Mucosal Melanoma: A Clinically and Biologically Unique Disease Entity. J Natl Compr Canc Netw (2012) 10(3):345–56. doi: 10.6004/jnccn.2012.0034
3. Tyrrell H, Payne M. Combatting Mucosal Melanoma: Recent Advances and Future Perspectives. Melanoma Manag (2018) 5(3):MMT11. doi: 10.2217/mmt-2018-0003
4. Hahn HM, Lee KG, Choi W, Cheong SH, Myung KB, Hahn HJ. An Updated Review of Mucosal Melanoma: Survival Meta-Analysis. Mol Clin Oncol (2019) 11(2):116–26. doi: 10.3892/mco.2019.1870
5. Gershenwald JE, Scolyer RA, Hess KR, Sondak VK, Long GV, Ross MI, et al. Melanoma Staging: Evidence-Based Changes in the American Joint Committee on Cancer Eighth Edition Cancer Staging Manual. CA Cancer J Clin (2017) 67(6):472–92. doi: 10.3322/caac.21409
6. Nassar KW, Tan AC. The Mutational Landscape of Mucosal Melanoma. Semin Cancer Biol (2020) 61:139–48. doi: 10.1016/j.semcancer.2019.09.013
7. Femiano F, Lanza A, Buonaiuto C, Gombos F, Di Spirito F, Cirillo N. Oral Malignant Melanoma: A Review of the Literature. J Oral Pathol Med (2008) 37(7):383–8. doi: 10.1111/j.1600-0714.2008.00660.x
8. Shain AH, Yeh I, Kovalyshyn I, Sriharan A, Talevich E, Gagnon A, et al. The Genetic Evolution of Melanoma From Precursor Lesions. N Engl J Med (2015) 373(20):1926–36. doi: 10.1056/NEJMoa1502583
9. Shain AH, Bastian BC. From Melanocytes to Melanomas. Nat Rev Cancer (2016) 16(6):345–58. doi: 10.1038/nrc.2016.37
10. Bastian BC, Olshen AB, LeBoit PE, Pinkel D. Classifying Melanocytic Tumors Based on DNA Copy Number Changes. Am J Pathol (2003) 163(5):1765–70. doi: 10.1016/S0002-9440(10)63536-5
11. Clarke LE, Flake DD 2nd, Busam K, Cockerell C, Helm K, McNiff J, et al. An Independent Validation of a Gene Expression Signature to Differentiate Malignant Melanoma From Benign Melanocytic Nevi. Cancer (2017) 123(4):617–28. doi: 10.1002/cncr.30385
12. Hilliard NJ, Krahl D, Sellheyer K. P16 Expression Differentiates Between Desmoplastic Spitz Nevus and Desmoplastic Melanoma. J Cutan Pathol (2009) 36(7):753–9. doi: 10.1111/j.1600-0560.2008.01154.x
13. Lezcano C, Jungbluth AA, Nehal KS, Hollmann TJ, Busam KJ. PRAME Expression in Melanocytic Tumors. Am J Surg Pathol (2018) 42(11):1456–65. doi: 10.1097/PAS.0000000000001134
14. Lyu J, Song Z, Chen J, Shepard MJ, Song H, Ren G, et al. Whole-Exome Sequencing of Oral Mucosal Melanoma Reveals Mutational Profile and Therapeutic Targets. J Pathol (2018) 244(3):358–66. doi: 10.1002/path.5017
15. Mikkelsen LH, Maag E, Andersen MK, Kruhoffer M, Larsen AC, Melchior LC, et al. The Molecular Profile of Mucosal Melanoma. Melanoma Res (2020) 30(6):533–42. doi: 10.1097/CMR.0000000000000686
16. Newell F, Kong Y, Wilmott JS, Johansson PA, Ferguson PM, Cui C, et al. Whole-Genome Landscape of Mucosal Melanoma Reveals Diverse Drivers and Therapeutic Targets. Nat Commun (2019) 10(1):3163. doi: 10.1038/s41467-019-11107-x
17. Cancer Genome Atlas N. Genomic Classification of Cutaneous Melanoma. Cell (2015) 161(7):1681–96. doi: 10.1016/j.cell.2015.05.044
18. Conway JR, Dietlein F, Taylor-Weiner A, AlDubayan S, Vokes N, Keenan T, et al. Integrated Molecular Drivers Coordinate Biological and Clinical States in Melanoma. Nat Genet (2020) 52(12):1373–83. doi: 10.1038/s41588-020-00739-1
19. Larkin J, Ascierto PA, Dreno B, Atkinson V, Liszkay G, Maio M, et al. Combined Vemurafenib and Cobimetinib in BRAF-Mutated Melanoma. N Engl J Med (2014) 371(20):1867–76. doi: 10.1056/NEJMoa1408868
20. Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, et al. Clinical Efficacy of a RAF Inhibitor Needs Broad Target Blockade in BRAF-Mutant Melanoma. Nature (2010) 467(7315):596–9. doi: 10.1038/nature09454
21. Rabbie R, Ferguson P, Molina-Aguilar C, Adams DJ, Robles-Espinoza CD. Melanoma Subtypes: Genomic Profiles, Prognostic Molecular Markers and Therapeutic Possibilities. J Pathol (2019) 247(5):539–51. doi: 10.1002/path.5213
22. Cichorek M, Wachulska M, Stasiewicz A, Tyminska A. Skin Melanocytes: Biology and Development. Postepy Dermatol Alergol (2013) 30(1):30–41. doi: 10.5114/pdia.2013.33376
23. Feller L, Masilana A, Khammissa RA, Altini M, Jadwat Y, Lemmer J. Melanin: The Biophysiology of Oral Melanocytes and Physiological Oral Pigmentation. Head Face Med (2014) 10:8. doi: 10.1186/1746-160X-10-8
24. Hicks MJ, Flaitz CM. Oral Mucosal Melanoma: Epidemiology and Pathobiology. Oral Oncol (2000) 36(2):152–69. doi: 10.1016/s1368-8375(99)00085-8
25. Lin JY, Fisher DE. Melanocyte Biology and Skin Pigmentation. Nature (2007) 445(7130):843–50. doi: 10.1038/nature05660
26. Mihajlovic M, Vlajkovic S, Jovanovic P, Stefanovic V. Primary Mucosal Melanomas: A Comprehensive Review. Int J Clin Exp Pathol (2012) 5(8):739–53. doi: 10.3109/15513815.2012.659410
27. Pontes FSC, de Souza LL, de Abreu MC, Fernandes LA, Rodrigues ALM, do Nascimento DM, et al. Sinonasal Melanoma: A Systematic Review of the Prognostic Factors. Int J Oral Maxillofac Surg (2020) 49(5):549–57. doi: 10.1016/j.ijom.2019.11.001
28. Santos RS, Andrade MF, Alves Fde A, Kowalski LP, Perez DE. Metastases of Melanoma to Head and Neck Mucosa: A Report of Short Series. Clin Exp Otorhinolaryngol (2016) 9(1):80–4. doi: 10.21053/ceo.2016.9.1.80
29. Feller L, Chandran R, Kramer B, Khammissa RA, Altini M, Lemmer J. Melanocyte Biology and Function With Reference to Oral Melanin Hyperpigmentation in HIV-Seropositive Subjects. AIDS Res Hum Retroviruses (2014) 30(9):837–43. doi: 10.1089/AID.2014.0062
30. Plonka PM, Picardo M, Slominski AT. Does Melanin Matter in the Dark? Exp Dermatol (2017) 26(7):595–7. doi: 10.1111/exd.13171
31. Urabe K, Aroca P, Tsukamoto K, Mascagna D, Palumbo A, Prota G, et al. The Inherent Cytotoxicity of Melanin Precursors: A Revision. Biochim Biophys Acta (1994) 1221(3):272–8. doi: 10.1016/0167-4889(94)90250-x
32. Man MQ, Lin TK, Santiago JL, Celli A, Zhong L, Huang ZM, et al. Basis for Enhanced Barrier Function of Pigmented Skin. J Invest Dermatol (2014) 134(9):2399–407. doi: 10.1038/jid.2014.187
33. Tolleson WH. Human Melanocyte Biology, Toxicology, and Pathology. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev (2005) 23(2):105–61. doi: 10.1080/10590500500234970
34. Hong L, Simon JD. Current Understanding of the Binding Sites, Capacity, Affinity, and Biological Significance of Metals in Melanin. J Phys Chem B (2007) 111(28):7938–47. doi: 10.1021/jp071439h
35. Jakubiak P, Lack F, Thun J, Urtti A, Alvarez-Sanchez R. Influence of Melanin Characteristics on Drug Binding Properties. Mol Pharm (2019) 16(6):2549–56. doi: 10.1021/acs.molpharmaceut.9b00157
36. Rahmani Eliato T, Smith JT, Tian Z, Kim ES, Hwang W, Andam CP, et al. Melanin Pigments Extracted From Horsehair as Antibacterial Agents. J Mater Chem B (2021) 9(6):1536–45. doi: 10.1039/d0tb02475a
37. Ahn JH, Park TJ, Jin SH, Kang HY. Human Melanocytes Express Functional Toll-Like Receptor 4. Exp Dermatol (2008) 17(5):412–7. doi: 10.1111/j.1600-0625.2008.00701.x
38. Yu N, Zhang S, Zuo F, Kang K, Guan M, Xiang L. Cultured Human Melanocytes Express Functional Toll-Like Receptors 2-4, 7 and 9. J Dermatol Sci (2009) 56(2):113–20. doi: 10.1016/j.jdermsci.2009.08.003
39. Le Poole IC, van den Wijngaard RM, Westerhof W, Verkruisen RP, Dutrieux RP, Dingemans KP, et al. Phagocytosis by Normal Human Melanocytes In Vitro. Exp Cell Res (1993) 205(2):388–95. doi: 10.1006/excr.1993.1102
40. Dell’Angelica EC, Mullins C, Caplan S, Bonifacino JS. Lysosome-Related Organelles. FASEB J (2000) 14(10):1265–78. doi: 10.1096/fj.14.10.1265
41. Le Poole IC, Mutis T, van den Wijngaard RM, Westerhof W, Ottenhoff T, de Vries RR, et al. A Novel, Antigen-Presenting Function of Melanocytes and its Possible Relationship to Hypopigmentary Disorders. J Immunol (1993) 151(12):7284–92.
42. van Tuyn J, Jaber-Hijazi F, MacKenzie D, Cole JJ, Mann E, Pawlikowski JS, et al. Oncogene-Expressing Senescent Melanocytes Up-Regulate MHC Class II, a Candidate Melanoma Suppressor Function. J Invest Dermatol (2017) 137(10):2197–207. doi: 10.1016/j.jid.2017.05.030
43. Swope VB, Sauder DN, McKenzie RC, Sramkoski RM, Krug KA, Babcock GF, et al. Synthesis of Interleukin-1 Alpha and Beta by Normal Human Melanocytes. J Invest Dermatol (1994) 102(5):749–53. doi: 10.1111/1523-1747.ep12376970
44. Mattei S, Colombo MP, Melani C, Silvani A, Parmiani G, Herlyn M. Expression of Cytokine/Growth Factors and Their Receptors in Human Melanoma and Melanocytes. Int J Cancer (1994) 56(6):853–7. doi: 10.1002/ijc.2910560617
45. Wang S, Liu D, Ning W, Xu A. Cytosolic dsDNA Triggers Apoptosis and Pro-Inflammatory Cytokine Production in Normal Human Melanocytes. Exp Dermatol (2015) 24(4):298–300. doi: 10.1111/exd.12621
46. Besch R, Poeck H, Hohenauer T, Senft D, Hacker G, Berking C, et al. Proapoptotic Signaling Induced by RIG-I and MDA-5 Results in Type I Interferon-Independent Apoptosis in Human Melanoma Cells. J Clin Invest (2009) 119(8):2399–411. doi: 10.1172/JCI37155
47. Satomi H, Wang B, Fujisawa H, Otsuka F. Interferon-Beta From Melanoma Cells Suppresses the Proliferations of Melanoma Cells in an Autocrine Manner. Cytokine (2002) 18(2):108–15. doi: 10.1006/cyto.2002.1028
48. Buchner A, Merrell PW, Carpenter WM. Relative Frequency of Solitary Melanocytic Lesions of the Oral Mucosa. J Oral Pathol Med (2004) 33(9):550–7. doi: 10.1111/j.1600-0714.2004.00238.x
49. Tavares TS, Meirelles DP, de Aguiar MCF, Caldeira PC. Pigmented Lesions of the Oral Mucosa: A Cross-Sectional Study of 458 Histopathological Specimens. Oral Dis (2018) 24(8):1484–91. doi: 10.1111/odi.12924
50. Umeda M, Komatsubara H, Shibuya Y, Yokoo S, Komori T. Premalignant Melanocytic Dysplasia and Malignant Melanoma of the Oral Mucosa. Oral Oncol (2002) 38(7):714–22. doi: 10.1016/s1368-8375(02)00008-8
51. Rivera RS, Nagatsuka H, Siar CH, Gunduz M, Tsujigiwa H, Han PP, et al. Heparanase and Vascular Endothelial Growth Factor Expression in the Progression of Oral Mucosal Melanoma. Oncol Rep (2008) 19(3):657–61. doi: 10.3892/or.19.3.657
52. Buery RR, Siar CH, Katase N, Fujii M, Liu H, Kubota M, et al. Clinico-Pathological Evaluation of Oral Melanotic Macule, Oral Pigmented Nevus and Oral Mucosal Melanoma. J Hard Tissue Biol (2010) 19(1):57–64. doi: 10.2485/jhtb.19.57
53. Garzino-Demo P, Fasolis M, Maggiore GM, Pagano M, Berrone S. Oral Mucosal Melanoma: A Series of Case Reports. J Craniomaxillofac Surg (2004) 32(4):251–7. doi: 10.1016/j.jcms.2003.12.007
54. Kahn MA, Weathers DR, Hoffman JG. Transformation of a Benign Oral Pigmentation to Primary Oral Melanoma. Oral Surg Oral Med Oral Pathol Oral Radiol Endod (2005) 100(4):454–9. doi: 10.1016/j.tripleo.2005.01.018
55. Lu S-Y, Lin C-F, Huang S-C. Metastatic Oral Malignant Melanoma Transformed From Pre-Existing Pigmented Lesions in Mandibular Gingiva: Report of an Unusual Case. J Dental Sci (2013) 8(3):328–32. doi: 10.1016/j.jds.2012.11.005
56. Patrick RJ, Fenske NA, Messina JL. Primary Mucosal Melanoma. J Am Acad Dermatol (2007) 56(5):828–34. doi: 10.1016/j.jaad.2006.06.017
57. Meleti M, Mooi WJ, Casparie MK, van der Waal I. Melanocytic Nevi of the Oral Mucosa - No Evidence of Increased Risk for Oral Malignant Melanoma: An Analysis of 119 Cases. Oral Oncol (2007) 43(10):976–81. doi: 10.1016/j.oraloncology.2006.11.013
58. Liu W, Wang Y, Du G, Zhou Z, Yang X, Shi L. Potential Association Between Oral Mucosal Nevus and Melanoma: A Preliminary Clinicopathologic Study. Oral Dis (2020) 26(6):1240–5. doi: 10.1111/odi.13335
59. Ferreira L, Jham B, Assi R, Readinger A, Kessler HP. Oral Melanocytic Nevi: A Clinicopathologic Study of 100 Cases. Oral Surg Oral Med Oral Pathol Oral Radiol (2015) 120(3):358–67. doi: 10.1016/j.oooo.2015.05.008
60. Psaty EL, Scope A, Halpern AC, Marghoob AA. Defining the Patient at High Risk for Melanoma. Int J Dermatol (2010) 49(4):362–76. doi: 10.1111/j.1365-4632.2010.04381.x
61. Kim CC, Berry EG, Marchetti MA, Swetter SM, Lim G, Grossman D, et al. Risk of Subsequent Cutaneous Melanoma in Moderately Dysplastic Nevi Excisionally Biopsied But With Positive Histologic Margins. JAMA Dermatol (2018) 154(12):1401–8. doi: 10.1001/jamadermatol.2018.3359
62. Clark WH Jr., Reimer RR, Greene M, Ainsworth AM, Mastrangelo MJ. Origin of Familial Malignant Melanomas From Heritable Melanocytic Lesions. ‘The B-K Mole Syndrome’. Arch Dermatol (1978) 114(5):732–8. doi: 10.1001/archderm.114.5.732
63. Rosebush MS, Briody AN, Cordell KG. Black and Brown: Non-Neoplastic Pigmentation of the Oral Mucosa. Head Neck Pathol (2019) 13(1):47–55. doi: 10.1007/s12105-018-0980-9
64. Datta A, Lamba AK, Tandon S, Urs A, Lnu M. A Unique Presentation of Gingival Melanoacanthoma: Case Report and Review of Literature. Cureus (2020) 12(3):e7315. doi: 10.7759/cureus.7315
65. Lakshminarayanan V, Ranganathan K. Oral Melanoacanthoma: A Case Report and Review of the Literature. J Med Case Rep (2009) 3:11. doi: 10.1186/1752-1947-3-11
66. Rohilla K, Ramesh V, Balamurali P, Singh N. Oral Melanoacanthoma of a Rare Intraoral Site: Case Report and Review of Literature. Int J Clin Pediatr Dent (2013) 6(1):40–3. doi: 10.5005/jp-journals-10005-1185
67. Olszewska M, Banka A, Gorska R, Warszawik O. Dermoscopy of Pigmented Oral Lesions. J Dermatol Case Rep (2008) 2(3):43–8. doi: 10.3315/jdcr.2008.1015
68. Magremanne M, Vervaet C. [Melanoma of the Oral Mucosa]. Rev Stomatol Chir Maxillofac (2008) 109(3):175–7. doi: 10.1016/j.stomax.2008.02.003
69. Hashemi Pour MS. Malignant Melanoma of the Oral Cavity: A Review of Literature. Indian J Dent Res (2008) 19(1):47–51. doi: 10.4103/0970-9290.38932
70. Rambhia PH, Stojanov IJ, Arbesman J. Predominance of Oral Mucosal Melanoma in Areas of High Mechanical Stress. J Am Acad Dermatol (2019) 80(4):1133–5. doi: 10.1016/j.jaad.2018.07.064
71. Tacastacas JD, Bray J, Cohen YK, Arbesman J, Kim J, Koon HB, et al. Update on Primary Mucosal Melanoma. J Am Acad Dermatol (2014) 71(2):366–75. doi: 10.1016/j.jaad.2014.03.031
72. Lopez F, Rodrigo JP, Cardesa A, Triantafyllou A, Devaney KO, Mendenhall WM, et al. Update on Primary Head and Neck Mucosal Melanoma. Head Neck (2016) 38(1):147–55. doi: 10.1002/hed.23872
73. Warszawik-Hendzel O, Slowinska M, Olszewska M, Rudnicka L. Melanoma of the Oral Cavity: Pathogenesis, Dermoscopy, Clinical Features, Staging and Management. J Dermatol Case Rep (2014) 8(3):60–6. doi: 10.3315/jdcr.2014.1175
74. Elder DE, Bastian BC, Cree IA, Massi D, Scolyer RA. The 2018 World Health Organization Classification of Cutaneous, Mucosal, and Uveal Melanoma: Detailed Analysis of 9 Distinct Subtypes Defined by Their Evolutionary Pathway. Arch Pathol Lab Med (2020) 144(4):500–22. doi: 10.5858/arpa.2019-0561-RA
75. Wu Y, Zhong Y, Li C, Song H, Guo W, Ren G. Neck Dissection for Oral Mucosal Melanoma: Caution of Nodular Lesion. Oral Oncol (2014) 50(4):319–24. doi: 10.1016/j.oraloncology.2014.01.008
76. de-Andrade BA, Toral-Rizo VH, Leon JE, Contreras E, Carlos R, Delgado-Azanero W, et al. Primary Oral Melanoma: A Histopathological and Immunohistochemical Study of 22 Cases of Latin America. Med Oral Patol Oral Cir Bucal (2012) 17(3):e383–8. doi: 10.4317/medoral.17588
77. Williams MD. Update From the 4th Edition of the World Health Organization Classification of Head and Neck Tumours: Mucosal Melanomas. Head Neck Pathol (2017) 11(1):110–7. doi: 10.1007/s12105-017-0789-y
78. Miettinen M, McCue PA, Sarlomo-Rikala M, Biernat W, Czapiewski P, Kopczynski J, et al. Sox10–a Marker for Not Only Schwannian and Melanocytic Neoplasms But Also Myoepithelial Cell Tumors of Soft Tissue: A Systematic Analysis of 5134 Tumors. Am J Surg Pathol (2015) 39(6):826–35. doi: 10.1097/PAS.0000000000000398
79. Hayward NK, Wilmott JS, Waddell N, Johansson PA, Field MA, Nones K, et al. Whole-Genome Landscapes of Major Melanoma Subtypes. Nature (2017) 545(7653):175–80. doi: 10.1038/nature22071
80. Ronnstrand L. Signal Transduction via the Stem Cell Factor Receptor/C-Kit. Cell Mol Life Sci (2004) 61(19-20):2535–48. doi: 10.1007/s00018-004-4189-6
81. Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic Activation of KIT in Distinct Subtypes of Melanoma. J Clin Oncol (2006) 24(26):4340–6. doi: 10.1200/JCO.2006.06.2984
82. Satzger I, Schaefer T, Kuettler U, Broecker V, Voelker B, Ostertag H, et al. Analysis of C-KIT Expression and KIT Gene Mutation in Human Mucosal Melanomas. Br J Cancer (2008) 99(12):2065–9. doi: 10.1038/sj.bjc.6604791
83. Omholt K, Grafstrom E, Kanter-Lewensohn L, Hansson J, Ragnarsson-Olding BK. KIT Pathway Alterations in Mucosal Melanomas of the Vulva and Other Sites. Clin Cancer Res (2011) 17(12):3933–42. doi: 10.1158/1078-0432.CCR-10-2917
84. Carvajal RD, Antonescu CR, Wolchok JD, Chapman PB, Roman RA, Teitcher J, et al. KIT as a Therapeutic Target in Metastatic Melanoma. JAMA (2011) 305(22):2327–34. doi: 10.1001/jama.2011.746
85. Rivera RS, Nagatsuka H, Gunduz M, Cengiz B, Gunduz E, Siar CH, et al. C-Kit Protein Expression Correlated With Activating Mutations in KIT Gene in Oral Mucosal Melanoma. Virchows Arch (2008) 452(1):27–32. doi: 10.1007/s00428-007-0524-2
86. Yun J, Lee J, Jang J, Lee EJ, Jang KT, Kim JH, et al. KIT Amplification and Gene Mutations in Acral/Mucosal Melanoma in Korea. APMIS (2011) 119(6):330–5. doi: 10.1111/j.1600-0463.2011.02737.x
87. Meng D, Carvajal RD. KIT as an Oncogenic Driver in Melanoma: An Update on Clinical Development. Am J Clin Dermatol (2019) 20(3):315–23. doi: 10.1007/s40257-018-0414-1
88. Boichuk S, Galembikova A, Dunaev P, Valeeva E, Shagimardanova E, Gusev O, et al. A Novel Receptor Tyrosine Kinase Switch Promotes Gastrointestinal Stromal Tumor Drug Resistance. Molecules (2017) 22(12):2152. doi: 10.3390/molecules22122152
89. Martorana A, Lauria A. Design of Antitumor Drugs Targeting C-Kit Receptor by a New Mixed Ligand-Structure Based Method. J Mol Graph Model (2020) 100:107666. doi: 10.1016/j.jmgm.2020.107666
90. Reddy BY, Miller DM, Tsao H. Somatic Driver Mutations in Melanoma. Cancer (2017) 123(S11):2104–17. doi: 10.1002/cncr.30593
91. Alicea GM, Rebecca VW. Emerging Strategies to Treat Rare and Intractable Subtypes of Melanoma. Pigment Cell Melanoma Res (2021) 34(1):44–58. doi: 10.1111/pcmr.12880
92. Burd CE, Liu W, Huynh MV, Waqas MA, Gillahan JE, Clark KS, et al. Mutation-Specific RAS Oncogenicity Explains NRAS Codon 61 Selection in Melanoma. Cancer Discov (2014) 4(12):1418–29. doi: 10.1158/2159-8290.CD-14-0729
93. Dumaz N, Jouenne F, Delyon J, Mourah S, Bensussan A, Lebbe C. Atypical BRAF and NRAS Mutations in Mucosal Melanoma. Cancers (Basel) (2019) 11(8):1133. doi: 10.3390/cancers11081133
94. Karoulia Z, Gavathiotis E, Poulikakos PI. New Perspectives for Targeting RAF Kinase in Human Cancer. Nat Rev Cancer (2017) 17(11):676–91. doi: 10.1038/nrc.2017.79
95. Lavoie H, Therrien M. Regulation of RAF Protein Kinases in ERK Signalling. Nat Rev Mol Cell Biol (2015) 16(5):281–98. doi: 10.1038/nrm3979
96. Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, et al. A Landscape of Driver Mutations in Melanoma. Cell (2012) 150(2):251–63. doi: 10.1016/j.cell.2012.06.024
97. Whittaker SR, Theurillat JP, Van Allen E, Wagle N, Hsiao J, Cowley GS, et al. A Genome-Scale RNA Interference Screen Implicates NF1 Loss in Resistance to RAF Inhibition. Cancer Discov (2013) 3(3):350–62. doi: 10.1158/2159-8290.CD-12-0470
98. Gibney GT, Smalley KS. An Unholy Alliance: Cooperation Between BRAF and NF1 in Melanoma Development and BRAF Inhibitor Resistance. Cancer Discov (2013) 3(3):260–3. doi: 10.1158/2159-8290.CD-13-0017
99. Hintzsche JD, Gorden NT, Amato CM, Kim J, Wuensch KE, Robinson SE, et al. Whole-Exome Sequencing Identifies Recurrent SF3B1 R625 Mutation and Comutation of NF1 and KIT in Mucosal Melanoma. Melanoma Res (2017) 27(3):189–99. doi: 10.1097/CMR.0000000000000345
100. Ablain J, Xu M, Rothschild H, Jordan RC, Mito JK, Daniels BH, et al. Human Tumor Genomics and Zebrafish Modeling Identify SPRED1 Loss as a Driver of Mucosal Melanoma. Science (2018) 362(6418):1055–60. doi: 10.1126/science.aau6509
101. Ablain J, Liu S, Moriceau G, Lo RS, Zon LI. SPRED1 Deletion Confers Resistance to MAPK Inhibition in Melanoma. J Exp Med (2021) 218(3):e20201097. doi: 10.1084/jem.20201097
102. Soares CD, Borges CF, Sena-Filho M, Almeida OP, Stelini RF, Cintra ML, et al. Prognostic Significance of Cyclooxygenase 2 and Phosphorylated Akt1 Overexpression in Primary Nonmetastatic and Metastatic Cutaneous Melanomas. Melanoma Res (2017) 27(5):448–56. doi: 10.1097/CMR.0000000000000368
103. Soares C, Melo de Lima Morais T, Carlos R, Mariano FV, Altemani A, Freire de Carvalho MG, et al. Phosphorylated Akt1 Expression is Associated With Poor Prognosis in Cutaneous, Oral and Sinonasal Melanomas. Oncotarget (2018) 9(99):37291–304. doi: 10.18632/oncotarget.26458
104. Wu H, Goel V, Haluska FG. PTEN Signaling Pathways in Melanoma. Oncogene (2003) 22(20):3113–22. doi: 10.1038/sj.onc.1206451
105. Broit N, Johansson PA, Rodgers CB, Walpole ST, Newell F, Hayward NK, et al. Meta-Analysis and Systematic Review of the Genomics of Mucosal Melanoma. Mol Cancer Res (2021) 19(6):991–1004. doi: 10.1158/1541-7786.MCR-20-0839
106. Damsky WE, Curley DP, Santhanakrishnan M, Rosenbaum LE, Platt JT, Gould Rothberg BE, et al. Beta-Catenin Signaling Controls Metastasis in Braf-Activated Pten-Deficient Melanomas. Cancer Cell (2011) 20(6):741–54. doi: 10.1016/j.ccr.2011.10.030
107. Cho JH, Robinson JP, Arave RA, Burnett WJ, Kircher DA, Chen G, et al. AKT1 Activation Promotes Development of Melanoma Metastases. Cell Rep (2015) 13(5):898–905. doi: 10.1016/j.celrep.2015.09.057
108. Tas F, Keskin S, Karadeniz A, Dagoglu N, Sen F, Kilic L, et al. Noncutaneous Melanoma Have Distinct Features From Each Other and Cutaneous Melanoma. Oncology (2011) 81(5-6):353–8. doi: 10.1159/000334863
109. Ma M, Dai J, Xu T, Yu S, Yu H, Tang H, et al. Analysis of TSC1 Mutation Spectrum in Mucosal Melanoma. J Cancer Res Clin Oncol (2018) 144(2):257–67. doi: 10.1007/s00432-017-2550-z
110. Darman RB, Seiler M, Agrawal AA, Lim KH, Peng S, Aird D, et al. Cancer-Associated SF3B1 Hotspot Mutations Induce Cryptic 3’ Splice Site Selection Through Use of a Different Branch Point. Cell Rep (2015) 13(5):1033–45. doi: 10.1016/j.celrep.2015.09.053
111. Zhou Z, Gong Q, Wang Y, Li M, Wang L, Ding H, et al. The Biological Function and Clinical Significance of SF3B1 Mutations in Cancer. Biomark Res (2020) 8:38. doi: 10.1186/s40364-020-00220-5
112. Kong Y, Krauthammer M, Halaban R. Rare SF3B1 R625 Mutations in Cutaneous Melanoma. Melanoma Res (2014) 24(4):332–4. doi: 10.1097/CMR.0000000000000071
113. Alsafadi S, Houy A, Battistella A, Popova T, Wassef M, Henry E, et al. Cancer-Associated SF3B1 Mutations Affect Alternative Splicing by Promoting Alternative Branchpoint Usage. Nat Commun (2016) 7:10615. doi: 10.1038/ncomms10615
114. Inoue D, Chew GL, Liu B, Michel BC, Pangallo J, D’Avino AR, et al. Spliceosomal Disruption of the non-Canonical BAF Complex in Cancer. Nature (2019) 574(7778):432–6. doi: 10.1038/s41586-019-1646-9
115. Liu Z, Yoshimi A, Wang J, Cho H, Chun-Wei Lee S, Ki M, et al. Mutations in the RNA Splicing Factor SF3B1 Promote Tumorigenesis Through MYC Stabilization. Cancer Discov (2020) 10(6):806–21. doi: 10.1158/2159-8290.CD-19-1330
116. Wang L, Brooks AN, Fan J, Wan Y, Gambe R, Li S, et al. Transcriptomic Characterization of SF3B1 Mutation Reveals Its Pleiotropic Effects in Chronic Lymphocytic Leukemia. Cancer Cell (2016) 30(5):750–63. doi: 10.1016/j.ccell.2016.10.005
117. Furney SJ, Pedersen M, Gentien D, Dumont AG, Rapinat A, Desjardins L, et al. SF3B1 Mutations are Associated With Alternative Splicing in Uveal Melanoma. Cancer Discov (2013) 3(10):1122–9. doi: 10.1158/2159-8290.CD-13-0330
118. Baumann J, Ignashkova TI, Chirasani SR, Ramirez-Peinado S, Alborzinia H, Gendarme M, et al. Golgi Stress-Induced Transcriptional Changes Mediated by MAPK Signaling and Three ETS Transcription Factors Regulate MCL1 Splicing. Mol Biol Cell (2018) 29(1):42–52. doi: 10.1091/mbc.E17-06-0418
119. Si L, Wang X, Guo J. Genotyping of Mucosal Melanoma. Chin Clin Oncol (2014) 3(3):34. doi: 10.3978/j.issn.2304-3865.2014.07.03
120. Xu L, Cheng Z, Cui C, Wu X, Yu H, Guo J, et al. Frequent Genetic Aberrations in the Cell Cycle Related Genes in Mucosal Melanoma Indicate the Potential for Targeted Therapy. J Transl Med (2019) 17(1):245. doi: 10.1186/s12967-019-1987-z
121. Williams GH, Stoeber K. The Cell Cycle and Cancer. J Pathol (2012) 226(2):352–64. doi: 10.1002/path.3022
122. Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner H, et al. Distinct Sets of Genetic Alterations in Melanoma. N Engl J Med (2005) 353(20):2135–47. doi: 10.1056/NEJMoa050092
123. Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA. Highly Recurrent TERT Promoter Mutations in Human Melanoma. Science (2013) 339(6122):957–9. doi: 10.1126/science.1229259
124. Vinagre J, Almeida A, Populo H, Batista R, Lyra J, Pinto V, et al. Frequency of TERT Promoter Mutations in Human Cancers. Nat Commun (2013) 4:2185. doi: 10.1038/ncomms3185
125. Bell RJ, Rube HT, Xavier-Magalhaes A, Costa BM, Mancini A, Song JS, et al. Understanding TERT Promoter Mutations: A Common Path to Immortality. Mol Cancer Res (2016) 14(4):315–23. doi: 10.1158/1541-7786.MCR-16-0003
126. Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, Greenman CD, et al. A Comprehensive Catalogue of Somatic Mutations From a Human Cancer Genome. Nature (2010) 463(7278):191–6. doi: 10.1038/nature08658
127. Griewank KG, Murali R, Puig-Butille JA, Schilling B, Livingstone E, Potrony M, et al. TERT Promoter Mutation Status as an Independent Prognostic Factor in Cutaneous Melanoma. J Natl Cancer Inst (2014) 106(9):dju246. doi: 10.1093/jnci/dju246
128. Clynes D, Jelinska C, Xella B, Ayyub H, Scott C, Mitson M, et al. Suppression of the Alternative Lengthening of Telomere Pathway by the Chromatin Remodelling Factor ATRX. Nat Commun (2015) 6:7538. doi: 10.1038/ncomms8538
129. Chiba K, Lorbeer FK, Shain AH, McSwiggen DT, Schruf E, Oh A, et al. Mutations in the Promoter of the Telomerase Gene TERT Contribute to Tumorigenesis by a Two-Step Mechanism. Science (2017) 357(6358):1416–20. doi: 10.1126/science.aao0535
130. Perera ON, Sobinoff AP, Teber ET, Harman A, Maritz MF, Yang SF, et al. Telomerase Promotes Formation of a Telomere Protective Complex in Cancer Cells. Sci Adv (2019) 5(10):eaav4409. doi: 10.1126/sciadv.aav4409
131. Yasukawa M, Ando Y, Yamashita T, Matsuda Y, Shoji S, Morioka MS, et al. CDK1 Dependent Phosphorylation of hTERT Contributes to Cancer Progression. Nat Commun (2020) 11(1):1557. doi: 10.1038/s41467-020-15289-7
132. Maida Y, Yasukawa M, Masutomi K. De Novo RNA Synthesis by RNA-Dependent RNA Polymerase Activity of Telomerase Reverse Transcriptase. Mol Cell Biol (2016) 36(8):1248–59. doi: 10.1128/MCB.01021-15
133. Khan S, Carvajal RD. Mucosal Melanoma: Current Strategies and Future Directions. Expert Opin Orphan Drugs (2019) 7(10):427–34. doi: 10.1080/21678707.2019.1672534
134. Seifried S, Haydu LE, Quinn MJ, Scolyer RA, Stretch JR, Thompson JF. Melanoma of the Vulva and Vagina: Principles of Staging and Their Relevance to Management Based on a Clinicopathologic Analysis of 85 Cases. Ann Surg Oncol (2015) 22(6):1959–66. doi: 10.1245/s10434-014-4215-3
135. Lazarev S, Gupta V, Hu K, Harrison LB, Bakst R. Mucosal Melanoma of the Head and Neck: A Systematic Review of the Literature. Int J Radiat Oncol Biol Phys (2014) 90(5):1108–18. doi: 10.1016/j.ijrobp.2014.03.042
136. D’Angelo SP, Larkin J, Sosman JA, Lebbe C, Brady B, Neyns B, et al. Efficacy and Safety of Nivolumab Alone or in Combination With Ipilimumab in Patients With Mucosal Melanoma: A Pooled Analysis. J Clin Oncol (2017) 35(2):226–35. doi: 10.1200/JCO.2016.67.9258
137. Postow MA, Hamid O, Carvajal RD. Mucosal Melanoma: Pathogenesis, Clinical Behavior, and Management. Curr Oncol Rep (2012) 14(5):441–8. doi: 10.1007/s11912-012-0244-x
138. Long GV, Eroglu Z, Infante J, Patel S, Daud A, Johnson DB, et al. Long-Term Outcomes in Patients With BRAF V600-Mutant Metastatic Melanoma Who Received Dabrafenib Combined With Trametinib. J Clin Oncol (2018) 36(7):667–73. doi: 10.1200/JCO.2017.74.1025
139. Fujisawa Y, Ito T, Kato H, Irie H, Kaji T, Maekawa T, et al. Outcome of Combination Therapy Using BRAF and MEK Inhibitors Among Asian Patients With Advanced Melanoma: An Analysis of 112 Cases. Eur J Cancer (2021) 145:210–20. doi: 10.1016/j.ejca.2020.12.021
140. Ascierto PA, Schadendorf D, Berking C, Agarwala SS, van Herpen CM, Queirolo P, et al. MEK162 for Patients With Advanced Melanoma Harbouring NRAS or Val600 BRAF Mutations: A non-Randomised, Open-Label Phase 2 Study. Lancet Oncol (2013) 14(3):249–56. doi: 10.1016/S1470-2045(13)70024-X
141. Trunzer K, Pavlick AC, Schuchter L, Gonzalez R, McArthur GA, Hutson TE, et al. Pharmacodynamic Effects and Mechanisms of Resistance to Vemurafenib in Patients With Metastatic Melanoma. J Clin Oncol (2013) 31(14):1767–74. doi: 10.1200/JCO.2012.44.7888
142. Algazi AP, Muthukumar AH, O’Brien K, Lencioni A, Tsai KK, Kadafour M, et al. Phase II Trial of Trametinib in Combination With the AKT Inhibitor GSK 2141795 in BRAF Wild-Type Melanoma. Am Soc Clin Oncol (2015) 33(15):suppl 9068–9068. doi: 10.1200/jco.2015.33.15_suppl.9068
143. Sosman JA, Kittaneh M, Lolkema MP, Postow MA, Schwartz G, Franklin C, et al. A Phase 1b/2 Study of LEE011 in Combination With Binimetinib (MEK162) in Patients With NRAS-Mutant Melanoma: Early Encouraging Clinical Activity. Am Soc Clin Oncol (2014) 32(15):suppl 9068–9068. doi: 10.1200/jco.2015.33.15_suppl.9068
144. Wei BR, Hoover SB, Peer CJ, Dwyer JE, Adissu HA, Shankarappa P, et al. Efficacy, Tolerability and Pharmacokinetics of Combined Targeted MEK and Dual Mtorc1/2 Inhibition in a Preclinical Model of Mucosal Melanoma. Mol Cancer Ther (2020) 19(11):2308–18. doi: 10.1158/1535-7163.MCT-19-0858
145. Minor DR, Kashani-Sabet M, Garrido M, O’Day SJ, Hamid O, Bastian BC. Sunitinib Therapy for Melanoma Patients With KIT Mutations. Clin Cancer Res (2012) 18(5):1457–63. doi: 10.1158/1078-0432.CCR-11-1987
146. Kluger HM, Dudek AZ, McCann C, Ritacco J, Southard N, Jilaveanu LB, et al. A Phase 2 Trial of Dasatinib in Advanced Melanoma. Cancer (2011) 117(10):2202–8. doi: 10.1002/cncr.25766
147. Cho JH, Kim KM, Kwon M, Kim JH, Lee J. Nilotinib in Patients With Metastatic Melanoma Harboring KIT Gene Aberration. Invest New Drugs (2012) 30(5):2008–14. doi: 10.1007/s10637-011-9763-9
148. Carvajal RD, Lawrence DP, Weber JS, Gajewski TF, Gonzalez R, Lutzky J, et al. Phase II Study of Nilotinib in Melanoma Harboring KIT Alterations Following Progression to Prior KIT Inhibition. Clin Cancer Res (2015) 21(10):2289–96. doi: 10.1158/1078-0432.CCR-14-1630
149. Wei X, Mao L, Chi Z, Sheng X, Cui C, Kong Y, et al. Efficacy Evaluation of Imatinib for the Treatment of Melanoma: Evidence From a Retrospective Study. Oncol Res (2019) 27(4):495–501. doi: 10.3727/096504018X15331163433914
150. Hodi FS, Corless CL, Giobbie-Hurder A, Fletcher JA, Zhu M, Marino-Enriquez A, et al. Imatinib for Melanomas Harboring Mutationally Activated or Amplified KIT Arising on Mucosal, Acral, and Chronically Sun-Damaged Skin. J Clin Oncol (2013) 31(26):3182–90. doi: 10.1200/JCO.2012.47.7836
151. Guo J, Carvajal RD, Dummer R, Hauschild A, Daud A, Bastian BC, et al. Efficacy and Safety of Nilotinib in Patients With KIT-Mutated Metastatic or Inoperable Melanoma: Final Results From the Global, Single-Arm, Phase II TEAM Trial. Ann Oncol (2017) 28(6):1380–7. doi: 10.1093/annonc/mdx079
152. Woodman SE, Davies MA. Targeting KIT in Melanoma: A Paradigm of Molecular Medicine and Targeted Therapeutics. Biochem Pharmacol (2010) 80(5):568–74. doi: 10.1016/j.bcp.2010.04.032
153. Spencer KR, Mehnert JM. Mucosal Melanoma: Epidemiology, Biology and Treatment. Cancer Treat Res (2016) 167:295–320. doi: 10.1007/978-3-319-22539-5_13
154. Todd JR, Becker TM, Kefford RF, Rizos H. Secondary C-Kit Mutations Confer Acquired Resistance to RTK Inhibitors in C-Kit Mutant Melanoma Cells. Pigment Cell Melanoma Res (2013) 26(4):518–26. doi: 10.1111/pcmr.12107
155. Steensma DP, Wermke M, Klimek VM, Greenberg PL, Font P, Komrokji RS, et al. Results of a Clinical Trial of H3B-8800, a Splicing Modulator, in Patients With Myelodysplastic Syndromes (MDS), Acute Myeloid Leukemia (AML) or Chronic Myelomonocytic Leukemia (CMML). Am Soc Hematol Washington DC (2019) 134(Supplement_1):673. doi: 10.1182/blood-2019-123854
156. Damsky WE, Bosenberg M. Melanocytic Nevi and Melanoma: Unraveling a Complex Relationship. Oncogene (2017) 36(42):5771–92. doi: 10.1038/onc.2017.189
157. Dhomen N, Reis-Filho JS, da Rocha Dias S, Hayward R, Savage K, Delmas V, et al. Oncogenic Braf Induces Melanocyte Senescence and Melanoma in Mice. Cancer Cell (2009) 15(4):294–303. doi: 10.1016/j.ccr.2009.02.022
158. Damsky W, Micevic G, Meeth K, Muthusamy V, Curley DP, Santhanakrishnan M, et al. Mtorc1 Activation Blocks BrafV600E-Induced Growth Arrest But is Insufficient for Melanoma Formation. Cancer Cell (2015) 27(1):41–56. doi: 10.1016/j.ccell.2014.11.014
159. Patton EE, Mueller KL, Adams DJ, Anandasabapathy N, Aplin AE, Bertolotto C, et al. Melanoma Models for the Next Generation of Therapies. Cancer Cell (2021) 39(5):610–31. doi: 10.1016/j.ccell.2021.01.011
160. Furney SJ, Turajlic S, Stamp G, Nohadani M, Carlisle A, Thomas JM, et al. Genome Sequencing of Mucosal Melanomas Reveals That They are Driven by Distinct Mechanisms From Cutaneous Melanoma. J Pathol (2013) 230(3):261–9. doi: 10.1002/path.4204
161. Wroblewska JP, Mull J, Wu CL, Fujimoto M, Ogawa T, Marszalek A, et al. SF3B1, NRAS, KIT, and BRAF Mutation; CD117 and cMYC Expression; and Tumoral Pigmentation in Sinonasal Melanomas: An Analysis With Newly Found Molecular Alterations and Some Population-Based Molecular Differences. Am J Surg Pathol (2019) 43(2):168–77. doi: 10.1097/PAS.0000000000001166
162. Ronco C, Martin AR, Demange L, Benhida R. ATM, ATR, CHK1, CHK2 and WEE1 Inhibitors in Cancer and Cancer Stem Cells. Medchemcomm (2017) 8(2):295–319. doi: 10.1039/c6md00439c
163. Brown JS, O’Carrigan B, Jackson SP, Yap TA. Targeting DNA Repair in Cancer: Beyond PARP Inhibitors. Cancer Discov (2017) 7(1):20–37. doi: 10.1158/2159-8290.CD-16-0860
164. Rundle S, Bradbury A, Drew Y, Curtin NJ. Targeting the ATR-CHK1 Axis in Cancer Therapy. Cancers (Basel) (2017) 9(5):41. doi: 10.3390/cancers9050041
165. Lord CJ, Ashworth A. PARP Inhibitors: Synthetic Lethality in the Clinic. Science (2017) 355(6330):1152–8. doi: 10.1126/science.aam7344
166. Vaddepally RK, Kharel P, Pandey R, Garje R, Chandra AB. Review of Indications of FDA-Approved Immune Checkpoint Inhibitors Per NCCN Guidelines With the Level of Evidence. Cancers (Basel) (2020) 12(3):738. doi: 10.3390/cancers12030738
167. Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab Versus Ipilimumab in Advanced Melanoma. N Engl J Med (2015) 372(26):2521–32. doi: 10.1056/NEJMoa1503093
168. Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in Previously Untreated Melanoma Without BRAF Mutation. N Engl J Med (2015) 372(4):320–30. doi: 10.1056/NEJMoa1412082
169. Hamid O, Molinero L, Bolen CR, Sosman JA, Munoz-Couselo E, Kluger HM, et al. Safety, Clinical Activity, and Biological Correlates of Response in Patients With Metastatic Melanoma: Results From a Phase I Trial of Atezolizumab. Clin Cancer Res (2019) 25(20):6061–72. doi: 10.1158/1078-0432.CCR-18-3488
170. Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, et al. Nivolumab and Ipilimumab Versus Ipilimumab in Untreated Melanoma. N Engl J Med (2015) 372(21):2006–17. doi: 10.1056/NEJMoa1414428
171. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lao CD, et al. Five-Year Survival With Combined Nivolumab and Ipilimumab in Advanced Melanoma. N Engl J Med (2019) 381(16):1535–46. doi: 10.1056/NEJMoa1910836
172. Del Vecchio M, Di Guardo L, Ascierto PA, Grimaldi AM, Sileni VC, Pigozzo J, et al. Efficacy and Safety of Ipilimumab 3mg/Kg in Patients With Pretreated, Metastatic, Mucosal Melanoma. Eur J Cancer (2014) 50(1):121–7. doi: 10.1016/j.ejca.2013.09.007
173. Shoushtari AN, Wagstaff J, Ascierto PA, Butler MO, Lao CD, Marquez-Rodas I, et al. CheckMate 067: Long-Term Outcomes in Patients With Mucosal Melanoma. J Clin Oncol (2020) 38(15_suppl):10019–. doi: 10.1200/JCO.2020.38.15_suppl.10019
174. Moya-Plana A, Herrera Gomez RG, Rossoni C, Dercle L, Ammari S, Girault I, et al. Evaluation of the Efficacy of Immunotherapy for non-Resectable Mucosal Melanoma. Cancer Immunol Immunother (2019) 68(7):1171–8. doi: 10.1007/s00262-019-02351-7
175. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab Plus Ipilimumab in Advanced Melanoma. N Engl J Med (2013) 369(2):122–33. doi: 10.1056/NEJMoa1302369
176. Johnson DB, Carlson JA, Elvin JA, Vergilio J-A, Suh J, Ramkissoon S, et al. Landscape of Genomic Alterations (GA) and Tumor Mutational Burden (TMB) in Different Metastatic Melanoma (MM) Subtypes. J Clin Oncol (2017) 35(15_suppl):9536–. doi: 10.1200/JCO.2017.35.15_suppl.9536
177. Klemen ND, Wang M, Rubinstein JC, Olino K, Clune J, Ariyan S, et al. Survival After Checkpoint Inhibitors for Metastatic Acral, Mucosal and Uveal Melanoma. J Immunother Cancer (2020) 8(1):e000341. doi: 10.1136/jitc-2019-000341
178. Taylor MH, Vogelzang NJ, Cohn AL, Stepan DE, Shumaker RC, Dutcus CE, et al. Phase Ib/II Trial of Lenvatinib Plus Pembrolizumab in Advanced Melanoma. J Clin Oncol (2019) 37(8_suppl):15–. doi: 10.1200/JCO.2019.37.8_suppl.15
179. Guo J, Sheng X, Si L, Kong Y, Chi Z, Cui C, et al. A Phase Ib Study of JS001, a Humanized IgG4 mAb Against Programmed Death-1 (PD-1) Combination With Axitinib in Patients With Metastatic Mucosal Melanoma. J Clin Oncol (2018) 36(15_suppl):9528–. doi: 10.1200/JCO.2018.36.15_suppl.9528
Keywords: mucosal melanoma, mucosal melanocytes, melanocytic lesions, mutations, signaling dependency, targeted therapy
Citation: Ma Y, Xia R, Ma X, Judson-Torres RL and Zeng H (2021) Mucosal Melanoma: Pathological Evolution, Pathway Dependency and Targeted Therapy. Front. Oncol. 11:702287. doi: 10.3389/fonc.2021.702287
Received: 29 April 2021; Accepted: 02 July 2021;
Published: 19 July 2021.
Edited by:
Vladimir Spiegelman, Penn State Milton S. Hershey Medical Center, United StatesReviewed by:
Moran Amit, University of Texas MD Anderson Cancer Center, United StatesCiro Dantas Soares, Laboratório de Citopatologia, Brazil
Copyright © 2021 Ma, Xia, Ma, Judson-Torres and Zeng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hanlin Zeng, aGFubGluLnplbmdAc2hzbXUuZWR1LmNu