 Olivia L. Walker
Olivia L. Walker Margaret L. Dahn
Margaret L. Dahn Melanie R. Power Coombs
Melanie R. Power Coombs Paola Marcato
Paola Marcato- 1Pathology, Dalhousie University, Halifax, NS, Canada
- 2Biology, Acadia University, Wolfville, NS, Canada
- 3Microbiology and Immunology, Dalhousie University, Halifax, NS, Canada
Culprits of cancer development, metastasis, and drug resistance, cancer stem cells (CSCs) are characterized by specific markers, active developmental signaling pathways, metabolic plasticity, increased motility, invasiveness, and epithelial-mesenchymal transition. In breast cancer, these cells are often more prominent in aggressive disease, are amplified in drug-resistant tumors, and contribute to recurrence. For breast cancer, two distinct CSC populations exist and are typically defined by CD44+/CD24- cell surface marker expression or increased aldehyde dehydrogenase (ALDH) activity. These CSC populations share many of the same properties but also exhibit signaling pathways that are more active in CD44+/CD24- or ALDH+ populations. Understanding these CSC populations and their shared or specific signaling pathways may lead to the development of novel therapeutic strategies that will improve breast cancer patient outcomes. Herein, we review the current evidence and assess published patient tumor datasets of sorted breast CSC populations for evidence of heightened prostaglandin E2 (PGE2) signaling and activity in these breast CSC populations. PGE2 is a biologically active lipid mediator and in cancer PGE2 promotes tumor progression and poor patient prognosis. Overall, the data suggests that PGE2 signaling is important in propagating breast CSCs by enhancing inherent tumor-initiating capacities. Development of anti-PGE2 signaling therapeutics may be beneficial in inhibiting tumor growth and limiting breast CSC populations.
Introduction
Breast cancer is the most commonly diagnosed cancer among women, with nearly a quarter of all patients eventually succumbing to the illness (1–3). There is a need to increase understanding of this disease with the intent that it will lead to development of improved therapeutic strategies and outcomes. In terms of impact on cancer initiation, development, progression and drug resistance, cancer stem cells (CSCs) have an important role. Possessing stem-like characteristics with increased tumorigenicity, these cells have the capacity to self-renew and differentiate into bulk tumor cells (4–6).
CSC-associated enzymes and signaling pathways may provide novel avenues for therapeutic intervention, since these pathways and enzymes are also mediators of tumorigenicity, metastasis, and therapy resistance. Among the most well-studied breast CSC-associated signaling pathways are the developmental Notch, Wnt and Hedgehog pathways (7–9). There are clinical trials underway evaluating therapeutics which target elements of these pathways and they have been reviewed extensively elsewhere (7, 10–13). In this review, we consider the emerging evidence of the role of the prostaglandin synthesis pathway in breast CSC maintenance, assess published transcriptome data for evidence of increased prostaglandin synthesis pathway activation in breast CSC populations, and discuss how this pathway could be targeted in the treatment of breast cancer and limitation of CSC populations.
The Prostaglandin Synthesis Pathway
Prostanoid prostaglandins and thromboxanes are a class of biologically active lipids that belong to the eicosanoid family (14). They are released in response to tissue trauma and are mediators of pain, inflammation, fever, and uterine contractions. The prostanoids are critical in the maintenance of gastric function and renal blood flow (14) and they initiate cell signaling events that result in changes to cell proliferation, apoptosis, differentiation, and adhesion (15–18).
The prostanoids are produced in multi-step processes involving multiple enzymes (Figure 1A). First, arachidonic acid is liberated from phospholipid bilayers by phospholipase A2 (PLA2) and is oxidized by prostaglandin-endoperoxide synthase 1 and 2 (PTGS1 and PTGS2), which are commonly referred to as the cyclooxygenases: COX-1 and COX-2. The COX enzymes generate precursor prostaglandin G2 (PGG2) and then prostaglandin H2 (PGH2) (19, 20), and are differentially expressed in different tissue types (14). COX-1 is constitutively expressed at low levels in most tissues and by maintaining prostanoid production at basal levels, it contributes to gastric function and renal blood flow homeostasis (14, 21, 22). In contrast, COX-2 is not constitutively expressed and is induced during inflammation and in response to mitogens (21–24).
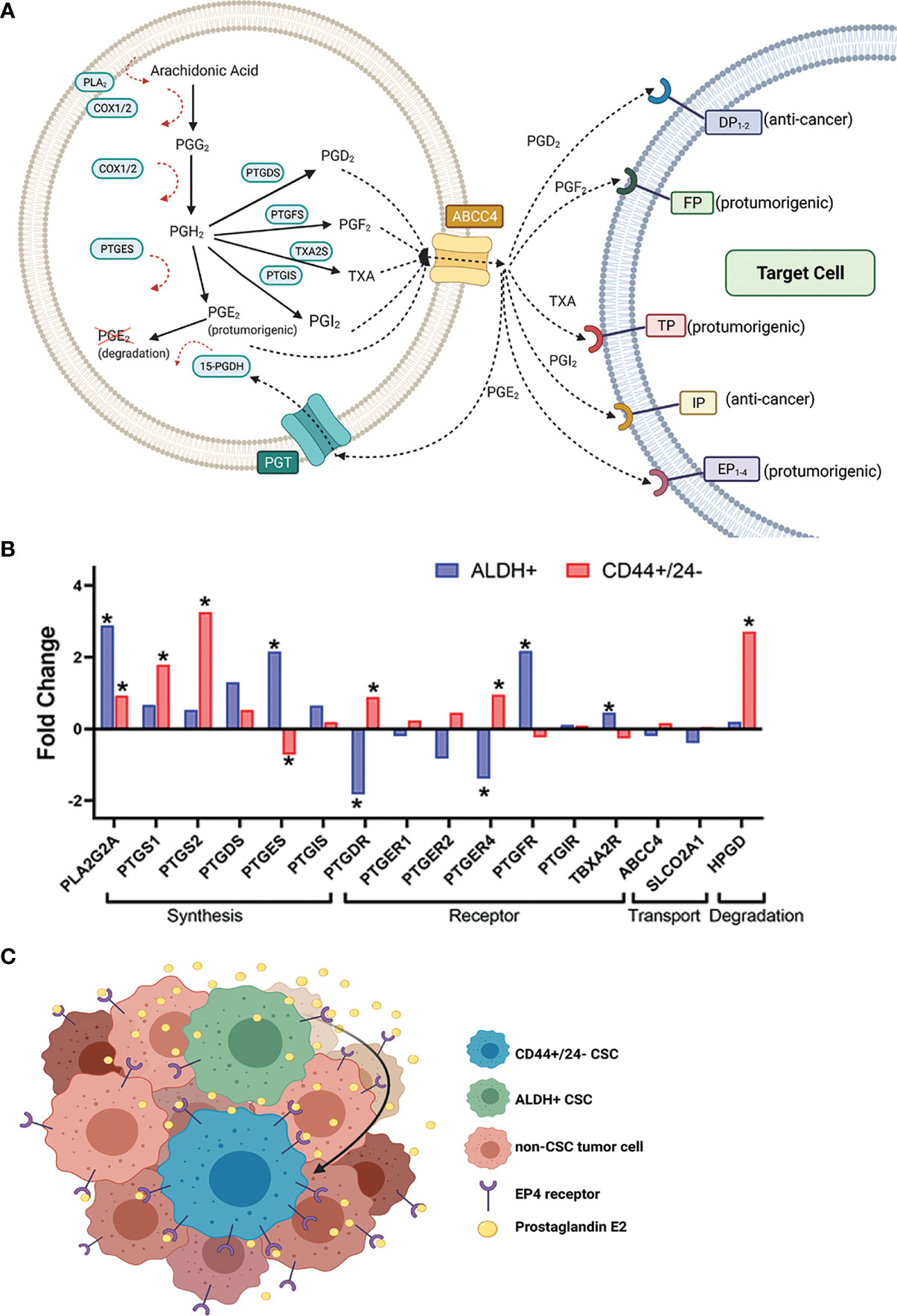
Figure 1 Expression of certain prostaglandin pathway genes in sorted CSC populations from breast cancer patient tumors suggests a hypothetical model for heightened CSC-PGE2 signaling in breast tumors. (A) Overview of the key players in the synthesis of the prostanoids and subsequent prostaglandin signaling (B) Fold change in expression of the prostaglandin synthesis and signaling genes in breast cancer patient tumor cells sorted for ALDH+ versus ALDH- (GSE52327); or CD44+/CD24- versus non-CD44+/CD24- (GSE5713) cells. Significance is noted by *, which represents a p-value <0.05. If more than probe was identified for a gene, the data for the probe with the most significant fold change is shown. Analyzed genes include phospholipase A (PLA2G2A), COX-1 (PTGS1), COX-2 (PTGS2), prostaglandin D synthase (PTGDS), prostaglandin E synthase (PTGES), prostacyclin synthase (PTGIS), DP receptor (PTGDR), EP receptors 1, 2,4 (PTGER1, 2,4), FP receptor (PTGFR), IP receptor (PTGIR), TP receptor (TBXA2R), MRP4 (ABCC4), PGT (SLCO2A1), 15-PGDH (HPGD). Data for the probes against PTGFS and PTGER3 were missing and so they not included in the analysis. Probe IDs for each of the genes included in the analysis are 243928_s_at ABCC4; 203913_s_at HPGD; 203649_s_at PLA2G2A; 215894_at PTGDR; 212187_x_at PTGDS; 214391_x_at PTGER1; 206631_at PTGER2; 204897_at PTGER4; 207388_s_at PTGES; 1555097_a_at PTGFR; 206187_at PTGIR; 211892_s_at PTGIS; 205128_x_at PTGS1; 204748_at PTGS2; 204368_at SLCO2A1; 207555_s_at TBXA2R. (C) Hypothetical model based on the data from (B), where increased PGE2 signaling in the breast tumor microenvironment results from the interplay between ALDH+ and CD44+/CD24- CSC populations.
After the rate-limiting steps of the COX enzymes, the multiple prostaglandin synthase isoforms PTGIS, TXA2S, PTGES, PTGDS, and PTGFS generate the different prostanoids: prostaglandin I2 (PGI2, also called prostacyclin), thromboxane A2 (TXA2), prostaglandin E2 (PGE2), prostaglandin D2 (PGD2), and prostaglandin F2 alpha (PGF2α), respectively from PGH2 (Figure 1A). The prostanoids are secreted by the multidrug resistance protein 4 (MRP4/ABCC4) and exert effects by binding to and activating specific G-protein coupled receptors expressed on target cells (Figure 1A) (25). Secreted PGE2 acts in paracrine or autocrine fashion by binding prostaglandin E2 receptors 1, 2, 3 and 4 (EP1-4), which are respectively encoded by the PTGER1, PTGER2, PTGER3, and PTGER4 genes (Figure 1A) (26, 27). PGE2 signaling termination is mediated by the prostaglandin transporter (PGT) and 15-hydroxyprostaglandin dehydrogenase (15-PGDH) (28). PGT allows the uptake of extracellular PGE2, which is then degraded by 15-PGDH.
As summarized in Table 1, the prostanoids have both overlapping and at times unique and counteracting activities. PGE2 is by far the most implicated of the prostanoids in terms of importance in cancer and will be the major focus of the following sections.
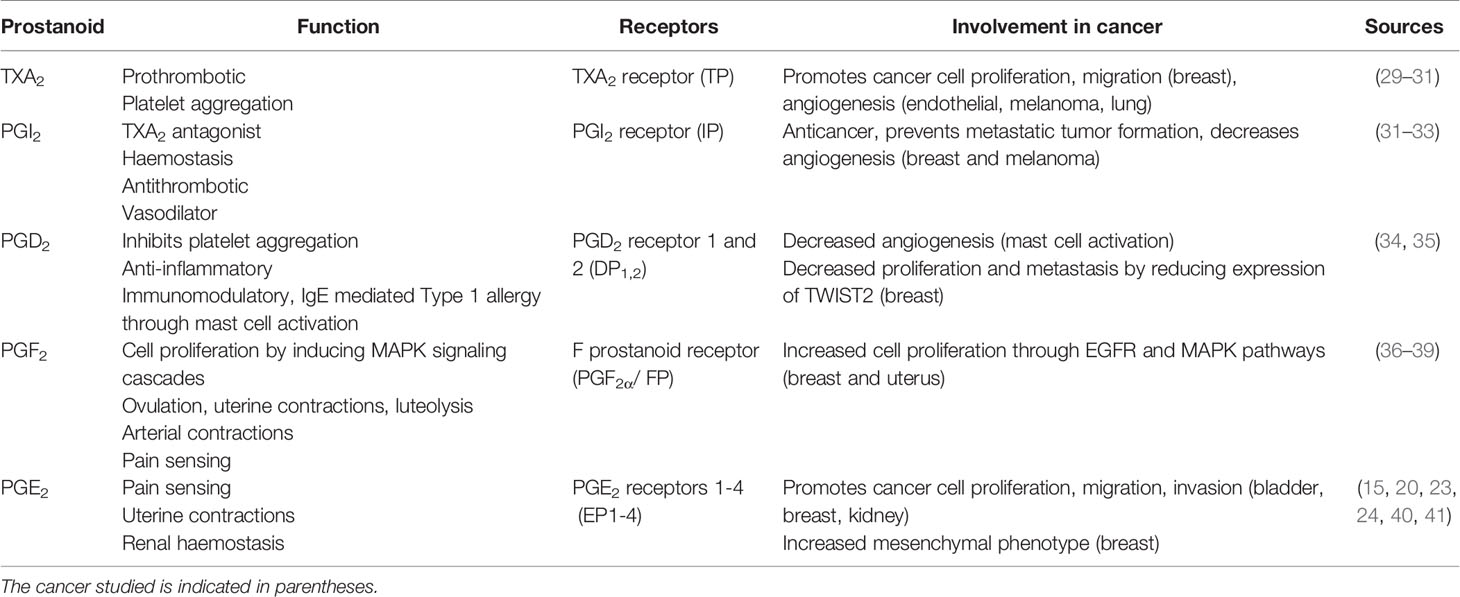
Table 1 Summary of the function of the prostanoid products, their receptors, and role in cancer.
Prostaglandin E2 in Cancer
As the primary mediator of the oncogenic effects of COX-2, PGE2-induced receptor signaling contributes to almost all of the major cancer hallmarks, including angiogenesis, proliferation, epithelial–mesenchymal transition (EMT), and the maintenance of CSC characteristics (42, 43). PGE2 is the most common prostanoid in the tumor microenvironment (44, 45). In breast cancer there is a well-established paradigm of elevated COX-2, low 15-PGDH, increased PGE2, and associations with more aggressive disease and negative outcomes (43, 44, 46–49). COX-2 overexpression induces tumorigenesis via PGE2 production, increased angiogenesis (42), and suppressed tumor immunity (40, 43–45, 50, 51). In patient tumors, COX-2 overexpression is associated with poor prognoses and lower distant disease-free survival time (52). In contrast, the PGE2 degrading enzyme 15-PGDH is tumor suppressive, where its downregulation results in enhanced breast cancer cell proliferation, cell cycle entry in vitro, and enhanced tumorigenicity in vivo (28, 46, 53, 54). Non-transformed healthy cells have high cellular levels of 15-PGDH and corresponding low levels of PGE2 (19); comparatively, cells with low levels of 15-PGDH and corresponding high levels of PGE2 have enhanced tumorigenicity (55). The deregulation of genes like PTGS2 (COX-2) and HPGD (15-PGDH) could be the drivers behind the prostaglandin-induced cell changes associated with cancer.
Among the PGE2 receptors, EP2 and EP4 have the most important roles in breast cancer (56, 57). PGE2 signaling through EP2 can increase tumor angiogenesis and proliferation (20, 26); and can modulate metabolism and promote tumorigenesis through EP4 (26, 58). Stimulation of EP2 increases vascular endothelial growth factor (VEGF) signaling in COX-2 transgenic murine mammary tumors, leading to increased angiogenesis and lymphangiogenesis (59). Additionally, EP2 silencing reduces proliferation and invasion by decreasing the expression of matrix metallopeptidases 2 and 9 (MMP2 and MMP9), and cyclin D; conversely, EP2 overexpression increases tumor volume and metastasis by activating MMPs (60). In in vivo models, metastatic sites had significantly increased levels of EP2 (60).
Binding of PGE2 to EP4 leads to dramatic changes in cell biology, especially in the context of cancer. PGE2 binding to EP4 stimulates the reorganization of actin stress fibres and focal adhesion complexes responsible for epithelial cell attachment to the basement membrane of the extracellular matrix (61). In breast cancer, EP4 stimulation increases proliferation and invasiveness (62), promotes lymphangiogenesis and metastasis (63), and is associated with aggressive phenotypes (64). Breast cancer cell treatment with an EP4 agonist or synthetic PGE2 both stimulated epidermal growth factor receptor (EGFR) and increased tumor spheroid invadopodia, invasion, and extracellular matrix degradation by breast cancer cells (65).
In terms of the downstream molecular signaling events that lead to these EP2 and EP4-mediated phenotypes, EP2 and EP4 both signal through a protein-kinase A (PKA)-dependent manner to increase intracellular levels of cAMP (26, 66); EP4 also signals through the phosphatidylinositol 3-kinase (PI3K)/Akt pathway (58, 66, 67). The stimulation of both receptors can have similar outcomes; however, EP4 stimulation seems to have a more important role in promoting stemness in cancer cells because it can signal through both cAMP/PKA and PI3k/Akt/NOTCH/WNT pathways in COX-2 positive cells. PGE2 stimulation of EP4 through both PKA and PI3k/Akt pathways inhibit glycogen synthase kinase-3 (GSK3), which acts as a negative regulator of Wnt and Notch pathways thereby activating these pathways and inducing the expression of genes associated with stemness, cell cycle, angiogenesis, and lymphangiogenesis (58, 63, 68). Furthermore, induction of hypoxia-inducible factor (HIF)-1 in tumors results in expression of genes involved in angiogenesis, glucose metabolism, and cell survival in breast cancer cells. The synthesis of HIF-1 is regulated by the PI3K and MAPK pathways (55) through which EP4 signals (26, 58). Therefore, PGE2 activates multiple pathways through EP4 that are involved in cancer cell migration and proliferation.
The PGE2 signaling pathway appears to have greater importance in the triple-negative breast cancer (TNBC) subset of breast cancers (64, 69). In addition to staging, breast cancers have distinct prognoses and treatment strategies based on the expression of the hormone receptors estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2). This sub-classifies breast cancers as ER+/PR+, HER2+, or TNBC (those lacking expression of the three receptors). TNBCs represent 10-15% of breast cancers that do not respond to endocrine therapies, have poorer outcomes, and are in most need of novel targeted therapies (70). In a comprehensive study evaluating the players of the PGE2 pathway in breast cancer patient tumors, MRP4, PGT, and 15-PGDH were noted as differentially expressed among distinct breast cancer subtypes (64). High PGE2 in the TNBC tumor microenvironment could be promoted by a combination of high COX-2, high MRP4, low PGT, and low 15-PGDH which was observed in TNBCs (64, 71).
The breast patient tumor data is reflected in cell lines. Secreted PGE2 levels are high in cultured TNBC cells (69, 72). Key for the high secretion of PGE2 by TNBCs is the combination of high MRP4, low PGT, and low 15-PGDH typically found in cell lines of this subtype (64, 69). This is also reflected in breast cancer cell lines; TNBC MDA-MB-231 cell have high EP4, MRP4 and COX-2, while ER+ MCF7 cells have comparably low levels of these PGE2 pathway players (71). It is also noteworthy that TNBCs are enriched for both CD44+/CD24- and aldehyde dehydrogenase positive (ALDH+) breast CSCs compared to other molecular subtypes (73–80), hence the greater impact the PGE2 pathway has in TNBC may also be in part connected to effect on CSC populations.
Evidence for the Role of Prostaglandin Signaling in Breast Cancer Stem Cells
In a 1980 study of breast tumors, Rolland et al. found that in breast cancer patients, high levels of prostaglandins were found in tumor cells present in the lymphatics and nodal tissues (81). They suggested that the tumor cells with high prostaglandin synthesis/response were involved in progressing disease by enhancing migration from the established primary tumor to secondary sites (81). They proposed the existence of a subpopulation of breast tumor cells with high prostaglandin levels that are responsible for driving disease progression; in hindsight they may have been referring to CSCs, which were discovered a couple decades later in leukemia (4, 6).
In 2003, the existence of a highly tumorigenic sub-population of breast cancer cells displaying stem-like characteristics and identified based on CD44+/CD24- cell surface expression was first reported (82). A few years later, a second method for identifying breast CSCs was proposed; increased ALDH activity detected by the Aldefluor assay (83), typically imparted by increased levels ALDH1A3 and/or ALDH1A1 isoforms (84). It is noteworthy that CD44+/CD24- or Aldefluor+/ALDH+ breast cell populations only partly overlap and in fact the two CSC populations have some distinct properties. CD44+/CD24- breast CSCs are more mesenchymal and are associated with tumorigenesis and proliferation; whereas ALDH+ CSCs are more epithelial and are associated with increased metastatic capacity (78, 85–87). The enhanced plasticity of CSCs permits the distinct CD44+/CD24- or ALDH+ CSC populations to transition between the two CSC states which best fits the conditions of the tumor microenvironment. COX-2 derived PGE2 may be a contributing factor to the development of breast CSCs that are associated with tumor initiation (82) and chemotherapy resistance (88, 89). As there exists two distinct populations of CSCs in breast cancer, when reviewing the literature for the potential role of prostaglandin signaling in breast CSC phenotypes it is important to define how the CSCs were identified since there may be a CD44+/CD24- or ALDH+ specific effect.
More studies evaluating PGs in breast CSCs have thus far focused on the CD44+/CD24- phenotype. For example, fibroblasts secreting high levels of PGE2 had enhanced tumor growth and increased proportion of CD44+/CD24- cells (90), and the ability to secrete PGE2 was associated with the ability to expand CD44+/CD24- breast cancer cells in vivo (90). EP4 inhibition of CD44+/CD24- high TNBC MDA-MB-231 cells depleted their drug efflux transporters and CSC-associated proteins like CD44, β-catenin, and fibronectin by inducing their secretion in extracellular vesicles (71). Treatment of ER+ MCF7 cells with the extracellular vesicles from MDA-MB-231 cells transferred the mesenchymal CSC/mesenchymal attributes to MCF7 cells (71). Taken together these studies indicate that increased PGE2/EP4 signaling promote the tumor and mesenchymal activities of CD44+/CD24- breast CSCs; however, any corresponding effect on the more epithelial-like ALDH+ CSCs was not specifically assessed in the studies.
There is evidence of ALDH+ breast CSC associations with increased PGE2 signaling (56, 58). An in situ analysis of breast tumor tissue revealed increased EP4 and COX-2 expression in patients was correlated with increased ALDH1A expression and reduced patient survival (58). COX-2 overexpression in ER+ MCF7 and HER2+ SKBR3 cells increased their ALDH activity, the number of ALDH+ cells, and phenotypes characteristic of CSCs (EMT, spheroid formation, expression of stemness markers, tumorigenicity) (58). These changes were reversed upon treatment with COX-2 inhibitors or EP4 antagonists (58). They also found high expression of COX-2 and EP4 in ALDH+ breast cancer cells. Together the data suggest that ALDH+ CSCs produce more COX-2 derived PGE2 and upregulate the receptors that respond to PGE2 in a way that promotes cell growth and survival (58). EP4 may be a valuable target for ablation of ALDH+ breast CSCs.
The data discussed so far is mostly generated from analysis of breast cancer cell lines; analysis of sorted breast CSCs from patient tumors in the context of prostaglandin synthesis and signaling in breast CSCs would provide additional valuable information. Furthermore, there is no direct comparison yet published assessing how the manipulation of the prostaglandin synthesis pathway may affect CD44+/CD24- versus ALDH+ breast CSC populations. In advance of such evidence, we note that that there are published breast cancer patient tumor transcriptome data available which provides an opportunity for hypothesis generation. We accessed GSE7513 and GSE52327, which consist of gene array data from breast cancer patient tumor cells sorted for CD44+/CD24- or ALDH+ from cells lacking these marker profiles. We specifically assessed the expression of the prostaglandin synthesis and signaling genes in these datasets to test if elements of this pathway are of greater importance in one CSC population versus the other (Figure 1B). We filtered the dataset to focus specifically on the genes in the prostaglandin synthesis pathway (Figure 1B).
This analysis revealed that some of the genes in the prostaglandin pathway are differentially expressed in CSC populations versus non-CSC populations from patient tumors. In both the ALDH+ and CD44+/CD24- sorted cells, PLA2G2A (encodes PLA2) is upregulated (Figure 1B). Given that PLA2 is involved the first step of the synthesis pathway, the upregulation of this gene provides the possibility for greater availability of precursor for potential prostaglandin generation.
More striking is the distinct prostaglandin pathway genes that are differentially expressed in the ALDH+ population compared to CD44+/CD24- sorted patient breast cancer tumor cells. Of note, the genes that encode the rate-limiting COX enzymes PTGS1 and PTGS2, are significantly upregulated in CD44+/CD24- populations. The generator of key pro-tumorigenic PGE2, PTGES is significantly upregulated in ALDH+ cells and conversely downregulated in CD44+/CD24- sorted tumor cells. In contrast, PTGER4 (encodes EP4, the critical receptor for the pro-tumorigenic autocrine and paracrine PGE2 signaling) is downregulated in ALDH+ cells and conversely upregulated in the CD44+/CD24- cells (Figure 1B). Finally, HPGD which encodes the PGE2 signal terminator 15-PGDH is significantly upregulated in the CD44+/CD24- tumor cells; this could negate any increased production of PGE2 cells due to increased PLA2/COX-2, since increased 15-PGDH would degrade produced PGE2. In contrast, there is not significant fold change difference in HPGD levels in ALDH+ over ALDH- cells (Figure 1B).
Together this patient tumor data of CSC sorted populations leads us to hypothesize that if breast CSCs are playing an important role in (and promoted by) heightened PGE2 signaling, there may be a complex interplay in the tumor microenvironment between these two CSC populations (Figure 1C). Potentially ALDH+ CSCs are producing/secreting more PGE2 and the CD44+/CD24- CSCs may be benefiting from this via increased expression of receptor EP4 and paracrine signaling. We hypothesize that in a heterogeneous tumor, certain populations of CSCs may be secreting inflammatory, pro-tumorigenic factors into the tumor microenvironment which can be utilized by all cells in the tumor environment, including other CSC populations to support tumor growth and invasiveness (Figure 1C).
Prostaglandins in Breast CSCs: A Druggable Target?
Taken together, the published studies in the role of prostaglandins in breast cancer and CSCs suggest that targeting this prostaglandin pathway could limit CSC numbers while inhibiting the cancer overall. Treatment with a COX-2 inhibitor can sensitize chemoresistant breast cancer cells to chemotherapy drugs like paclitaxel or doxorubicin (91). This is consistent with prostaglandins playing a key role in chemoresistance and suggests that disrupting the synthesis or signaling of pro-tumorigenic prostaglandins could be a valuable treatment option for reducing drug resistance and CSC development. However, there is a lack of published research investigating the role of PGE2 in breast CSC-mediated drug resistance. One study reported that celecoxib treatment decreased CSC markers and reduced EMT gene signatures in TNBC cell lines (92). This could be important in the context of treatment as the lethality and progression of most patients’ cancers may be reduced through the prevention of an invasive mesenchymal phenotype.
Eicosanoid products are key signaling molecules and modulators of inflammation and pain; they have been the target for pain management drugs like non-steroidal anti-inflammatory drugs (NSAIDs). Inhibition of COX enzymes by NSAIDs are commonly used to control pain and inflammation, but they also prevent the downstream formation of all prostanoid products which can be harmful to the patient. The inhibition of COX-2 activity with long-term use of NSAIDs can have an ulcerative effect on intestinal epithelia (22), is associated with renal toxicity (93), and puts patients at risk of developing cardiotoxicity like blood clots, hypertension, or stroke (19). Therefore, it would be beneficial to find a target further downstream in the prostaglandin synthesis pathway that allows for the blockade of pro-tumorigenic PGE2 without disrupting the cardioprotective prostanoids.
Enzymes involved in PGE2 synthesis and signaling or EP4 receptors have been suggested as drug targets (28) but none are currently approved for clinical use. Inhibition of EP4 by pharmacological agents inhibits the growth of mammospheres, as determined by decreased cellularity in vitro, and in vivo treatment with EP4 antagonists reduced the tumor-forming capacity of breast cancer cells with reduced frequency of CSCs in the tumor and lung metastases (94). EP4 antagonists were as effective as celecoxib at reducing the rate of tumor growth, stem cell markers, spontaneous lung and lymph node metastases, angiogenesis, and lymphangiogenesis (56). This suggests that downstream targets for PGE2 could be as effective as COX-2 inhibitors without the potential side effects.
Targeting the Prostaglandin Synthesis and PGE2 Pathway in Breast Cancer: Clinical Trial Data
There have been several clinical trials using the COX-2 inhibitor celecoxib completed in the treatment of breast cancer (95, 96). One study showed promising results with decreased metastasis, angiogenesis, and inflammation when given perioperatively (96). In a phase II trial, pre-treatment and post-treatment breast biopsies revealed that celecoxib induces transcription of genes associated with anti-tumour and decreased cell proliferation (97). However, in another study patients treated long-term with celecoxib had no significant difference in disease-free survival compared to placebo group (95). Hence, further studies are needed to clarify the potential clinical impact of targeting this pathway. Additionally, inhibiting COX-2 lacks the specificity of targeting the PGE2 pathway specifically, thereby possibly limiting potential therapeutic efficacy with increased potential for the adverse side-effects as described above. It is noteworthy that in these celecoxib clinical trials, the COX-2 inhibitor was well-tolerated, with low rates of reported toxic effects.
Looking ahead, it appears that the clinical data targeting the PGE2 pathway may be forthcoming as several early phase clinical trials are underway investigating EP4 antagonists in cancer. Specific to breast cancer, is a new Phase II trial investigating EP4 antagonist grapiprant in the treatment of metastatic inflammatory breast cancer (NCT05041101). The trial is recruiting patients with a proposed completion date of December 31, 2025. An increase in overall and progression-free survival in patients that receive grapiprant would provide impetus that further clinical investigation is warranted.
Challenges, Advantages, and Limitations
The pre-clinical data supports investigating targeting PGE2 in the treatment of breast cancer, with effects on CSCs. Realistically, obtaining the clinical data that demonstrate therapeutic efficacy of this approach has challenges to overcome and is many years away. Like most clinical trials, we are limited by the potential for demonstrated efficacy based on the eligibility of the patient population enrolled in the trial. For example, having advanced disease which has been refractive to treatment with standard of care is typically an eligibility requisite and this may also limit evidence of therapeutic efficacy. Additionally, a lack of consideration of the tumor composition of the enrolled patients may limit efficacy. Consideration for levels of PGE2, COX-2, EP4, and CSCs present in pre-treatment tumors when enrolling patients may increase the number of patients that achieve objective responses.
Most of the completed pre-clinical breast cancer studies investigating the inhibition of PGE2 downstream of COX-2 have focused on EP4 signaling, yet PGE2 signals through four EP receptors, and while the data thus far suggests that EP4 is the most important in terms of cancer, signaling through EP2 activates some stemness pathways. By focusing solely on EP4, the full potential of specific PGE2 inhibition may be missed.
Specific to our understanding of the role of the prostaglandin signaling pathway in breast CSCs, we are limited by the lack of data utilizing heterogeneous patient tumors and studies that compare the effects of the prostaglandin pathway in both the CD44+/CD24- ALDH+ breast CSC populations in the same study. Given that these CSC populations have distinct properties, this will be necessary to gain a full understanding of the impact of the pathway in the context of breast CSC signaling.
Conclusions
Overall, the evidence strongly suggests that the prostaglandin pathway (specifically PGE2 signaling) in breast cancer is tumor-promoting and enhances the CSC phenotype. The effects of enhanced PGE2 signaling appears to be of greater magnitude in CSC-rich TNBCs. A lack of published research directly comparing the ALDH+ and CD44+/CD24- breast CSC populations in the context of patient tumor samples indicates an opportunity for further study to elucidate the mechanism of prostaglandin signaling in breast CSCs and the tumor microenvironment. Other than one study of fixed patient breast tumors (58), most work on the role of prostaglandins in breast CSCs has been conducted using cell lines. Experiments investigating the role of anti-PGE2 treatment using patient-derived xenografts, followed by a limiting dilution assay to assess CSC numbers would clarify the potential importance of the prostanoid in breast tumors and CSCs.
Other treatment targets, such as specific PGE2 receptor antagonists or synthesis inhibitors (for example, against PTGES), may be valuable in the prevention of tumor proliferation, invasion, and metastasis of breast cancer. PGE2 appears to be important in the progression of many cancer types, so, studying the effect of anti-PGE2 treatment in breast cancer may be advantageous to inform potential broad-spectrum anti-cancer options. The prostaglandin pathway (beyond PGE2-EP4 signaling specifically) could be an important focus in the treatment of breast cancer, especially for TNBCs or tumors enriched in CSCs.
Author Contributions
OW and PM drafted the manuscript and conceptualized the key points. MD and MP provided expert feedback and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
OW is supported by a Genomics in Medicine scholarship from the Dalhousie Medical Research Foundation (DMRF) and a Nova Scotia Graduate Scholarship. Costs of publishing this review are covered by a Canadian Institutes of Health Research grant (CIHR, PJT 162313) to PM.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
Figures 1A, C were created with Biorender.com.
References
1. Ferlay J, Colombet M, Soerjomataram I, Dyba T, Randi G, Bettio M, et al. Cancer Incidence and Mortality Patterns in Europe: Estimates for 40 Countries and 25 Major Cancers in 2018. Eur J Cancer (2018) 103:356–87. doi: 10.1016/j.ejca.2018.07.005
2. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA: A Cancer J Clin (2021) 71(1):7–33. doi: 10.3322/caac.21654
3. DeSantis CE, Ma J, Gaudet MM, Newman LA, Miller KD, Sauer AG, et al. Breast Cancer Statistics, 2019. CA: A Cancer J Clin (2019) 69(6):438–51. doi: 10.3322/caac.21583
4. Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A Cell Initiating Human Acute Myeloid Leukaemia After Transplantation Into SCID Mice. Nature (1994) 367(6464):645–8. doi: 10.1038/367645a0
5. Larochelle A, Vormoor J, Hanenberg H, Wang JC, Bhatia M, Lapidot T, et al. Identification of Primitive Human Hematopoietic Cells Capable of Repopulating NOD/SCID Mouse Bone Marrow: Implications for Gene Therapy. Nat Med (1996) 2(12):1329–37. doi: 10.1038/nm1296-1329
6. Bonnet D, Dick JE. Human Acute Myeloid Leukemia Is Organized as a Hierarchy That Originates From a Primitive Hematopoietic Cell. Nat Med (1997) 3(7):730–7. doi: 10.1038/nm0797-730
7. Bhateja P, Cherian M, Majumder S, Ramaswamy B. The Hedgehog Signaling Pathway: A Viable Target in Breast Cancer? Cancers (Basel) (2019) 11(8):1126. doi: 10.3390/cancers11081126
8. Yan Y, Liu F, Han L, Zhao L, Chen J, Olopade OI, et al. HIF-2α Promotes Conversion to a Stem Cell Phenotype and Induces Chemoresistance in Breast Cancer Cells by Activating Wnt and Notch Pathways. J Exp Clin Cancer Res (2018) 37:256. doi: 10.1186/s13046-018-0925-x
9. Koike Y, Ohta Y, Saitoh W, Yamashita T, Kanomata N, Moriya T, et al. Anti-Cell Growth and Anti-Cancer Stem Cell Activities of the Non-Canonical Hedgehog Inhibitor GANT61 in Triple-Negative Breast Cancer Cells. Breast Cancer (2017) 24(5):683–93. doi: 10.1007/s12282-017-0757-0
10. Rangel MC, Bertolette D, Castro NP, Klauzinska M, Cuttitta F, Salomon DS. Developmental Signaling Pathways Regulating Mammary Stem Cells and Contributing to the Etiology of Triple-Negative Breast Cancer. Breast Cancer Res Treat (2016) 156:211–26. doi: 10.1007/s10549-016-3746-7
11. Carter JL, Hege K, Yang J, Kalpage HA, Su Y, Edwards H, et al. Targeting Multiple Signaling Pathways: The New Approach to Acute Myeloid Leukemia Therapy. Sig Transduct Target Ther (2020) 5(1):1–29. doi: 10.1038/s41392-020-00361-x
12. Janovská P, Normant E, Miskin H, Bryja V. Targeting Casein Kinase 1 (CK1) in Hematological Cancers. Int J Mol Sci (2020) 21(23):9026. doi: 10.3390/ijms21239026
13. Moore G, Annett S, McClements L, Robson T. Top Notch Targeting Strategies in Cancer: A Detailed Overview of Recent Insights and Current Perspectives. Cells (2020) 9(6):1503. doi: 10.3390/cells9061503
14. O’Neill GP, Ford-Hutchinson AW. Expression of mRNA for Cyclooxygenase-1 and Cyclooxygenase-2 in Human Tissues. FEBS Lett (1993) 330(2):157–60. doi: 10.1016/0014-5793(93)80263-T
15. Tsujii M, DuBois RN. Alterations in Cellular Adhesion and Apoptosis in Epithelial Cells Overexpressing Prostaglandin Endoperoxide Synthase 2. Cell (1995) 83(3):493–501. doi: 10.1016/0092-8674(95)90127-2
16. Serrero G, Lepak NM. Prostaglandin F2alpha Receptor (FP Receptor) Agonists are Potent Adipose Differentiation Inhibitors for Primary Culture of Adipocyte Precursors in Defined Medium. Biochem Biophys Res Commun (1997) 233(1):200–2. doi: 10.1006/bbrc.1997.6433
17. Kimura M, Osumi S, Ogihara M. Stimulation of DNA Synthesis and Proliferation by Prostaglandins in Primary Cultures of Adult Rat Hepatocytes. Eur J Pharmacol (2000) 404(3):259–71. doi: 10.1016/S0014-2999(00)00594-X
18. Tsubouchi Y, Sano H, Kawahito Y, Mukai S, Yamada R, Kohno M, et al. Inhibition of Human Lung Cancer Cell Growth by the Peroxisome Proliferator-Activated Receptor-γ Agonists Through Induction of Apoptosis. Biochem Biophys Res Commun (2000) 270(2):400–5. doi: 10.1006/bbrc.2000.2436
19. Howe LR. Inflammation and Breast Cancer. Cyclooxygenase/prostaglandin Signaling and Breast Cancer. Breast Cancer Res (2007) 9(4):210. doi: 10.1186/bcr1678
20. Kalinski P. Regulation of Immune Responses by Prostaglandin E2. J Immunol (2012) 188(1):21–8. doi: 10.4049/jimmunol.1101029
21. Wölfle D. Enhancement of Carcinogen-Induced Malignant Cell Transformation by Prostaglandin F2α. Toxicology (2003) 188(2):139–47. doi: 10.1016/S0300-483X(03)00077-5
22. Williams CS, DuBois RN. Prostaglandin Endoperoxide Synthase: Why Two Isoforms? Am J Physiology-Gastrointestinal Liver Physiol (1996) 270(3):G393–400. doi: 10.1152/ajpgi.1996.270.3.G393
23. Kurtova AV, Xiao J, Mo Q, Pazhanisamy S, Krasnow R, Lerner SP, et al. Blocking PGE 2 -Induced Tumour Repopulation Abrogates Bladder Cancer Chemoresistance. Nature (2015) 517(7533):209–13. doi: 10.1038/nature14034
24. Schrey MP, Patel KV. Prostaglandin E2 Production and Metabolism in Human Breast Cancer Cells and Breast Fibroblasts. Regulation by Inflammatory Mediators. Br J Cancer (1995) 72(6):1412–9. doi: 10.1038/bjc.1995.523
25. Donepudi AC, Lee Y, Lee J-Y, Schuetz JD, Manautou JE. Multidrug Resistance-Associated Protein 4 (Mrp4) is a Novel Genetic Factor in the Pathogenesis of Obesity and Diabetes. FASEB J (2021) 35(2):e21304. doi: 10.1096/fj.202001299RR
26. Narumiya S. Physiology and Pathophysiology of Prostanoid Receptors. Proc Jpn Acad Ser B Phys Biol Sci (2007) 83(9–10):296–319. doi: 10.2183/pjab.83.296
27. Richards JA, Petrel TA, Brueggemeier RW. Signaling Pathways Regulating Aromatase and Cyclooxygenases in Normal and Malignant Breast Cells. J Steroid Biochem Mol Biol (2002) 80(2):203–12. doi: 10.1016/S0960-0760(01)00187-X
28. Wolf I, O’Kelly J, Rubinek T, Tong M, Nguyen A, Lin BT, et al. 15-Hydroxyprostaglandin Dehydrogenase Is a Tumor Suppressor of Human Breast Cancer. Cancer Res (2006) 66(15):7818–23. doi: 10.1158/0008-5472.CAN-05-4368
29. Li H, Lee M-H, Liu K, Wang T, Song M, Han Y, et al. Inhibiting Breast Cancer by Targeting the Thromboxane A2 Pathway. NPJ Precis Oncol (2017) 1:8. doi: 10.1038/s41698-017-0011-4
30. Nie D, Lamberti M, Zacharek A, Li L, Szekeres K, Tang K, et al. Thromboxane A(2) Regulation of Endothelial Cell Migration, Angiogenesis, and Tumor Metastasis. Biochem Biophys Res Commun (2000) 267(1):245–51. doi: 10.1006/bbrc.1999.1840
31. Kij A, Kus K, Smeda M, Zakrzewska A, Proniewski B, Matyjaszczyk K, et al. Differential Effects of Nitric Oxide Deficiency on Primary Tumour Growth, Pulmonary Metastasis and Prostacyclin/Thromboxane A2 Balance in Orthotopic and Intravenous Murine Models of 4T1 Breast Cancer. J Physiol Pharmacol (2018) 69(6):911–9. doi: 10.26402/jpp.2018.6.05
32. Klein T, Benders J, Roth F, Baudler M, Siegle I, Kömhoff M. Expression of Prostacyclin-Synthase in Human Breast Cancer: Negative Prognostic Factor and Protection Against Cell Death In Vitro. Mediators Inflamm (2015) 2015:864136. doi: 10.1155/2015/864136
33. Honn KV, Cicone B, Skoff A. Prostacyclin: A Potent Antimetastatic Agent. Science (1981) 212(4500):1270–2. doi: 10.1126/science.7015512
34. Lewis RA, Soter NA, Diamond PT, Austen KF, Oates JA, Roberts LJ. Prostaglandin D2 Generation After Activation of Rat and Human Mast Cells With Anti-IgE. J Immunol (1982) 129(4):1627–31.
35. Pan J, Zhang L, Huang J. Prostaglandin D2 Synthase/Prostaglandin D2/TWIST2 Signaling Inhibits Breast Cancer Proliferation. Anticancer Drugs (2021) 32(10):1029–37. doi: 10.1097/CAD.0000000000001111
36. Byrns MC, Penning TM. Type 5 17β-Hydroxysteroid Dehydrogenase/Prostaglandin F Synthase (AKR1C3): Role in Breast Cancer and Inhibition by non-Steroidal Anti-Inflammatory Drug Analogs. Chemico-Biological Interactions (2009) 178(1):221–7. doi: 10.1016/j.cbi.2008.10.024
37. Saito O, Guan Y, Qi Z, Davis LS, Kömhoff M, Sugimoto Y, et al. Expression of the Prostaglandin F Receptor (FP) Gene Along the Mouse Genitourinary Tract. Am J Physiol-Renal Physiol (2003) 284(6):F1164–70. doi: 10.1152/ajprenal.00441.2002
38. Kunori S, Matsumura S, Mabuchi T, Tatsumi S, Sugimoto Y, Minami T, et al. Involvement of Prostaglandin F 2 Alpha Receptor in ATP-Induced Mechanical Allodynia. Neuroscience (2009) 163(1):362–71. doi: 10.1016/j.neuroscience.2009.05.069
39. Sales KJ, Milne SA, Williams ARW, Anderson RA, Jabbour HN. Expression, Localization, and Signaling of Prostaglandin F2α Receptor in Human Endometrial Adenocarcinoma: Regulation of Proliferation by Activation of the Epidermal Growth Factor Receptor and Mitogen-Activated Protein Kinase Signaling Pathways. J Clin Endocrinol Metab (2004) 89(2):986–93. doi: 10.1210/jc.2003-031434
40. Gomez PF, Pillinger MH, Attur M, Marjanovic N, Dave M, Park J, et al. Resolution of Inflammation: Prostaglandin E 2 Dissociates Nuclear Trafficking of Individual NF-κb Subunits (P65, P50) in Stimulated Rheumatoid Synovial Fibroblasts. J Immunol (2005) 175(10):6924–30. doi: 10.4049/jimmunol.175.10.6924
41. Nørregaard R, Kwon T-H, Frøkiær J. Physiology and Pathophysiology of Cyclooxygenase-2 and Prostaglandin E2 in the Kidney. Kidney Res Clin Pract (2015) 34(4):194–200. doi: 10.1016/j.krcp.2015.10.004
42. Chang S-H, Liu CH, Conway R, Han DK, Nithipatikom K, Trifan OC, et al. Role of Prostaglandin E2-Dependent Angiogenic Switch in Cyclooxygenase 2-Induced Breast Cancer Progression. PNAS (2004) 101(2):591–6. doi: 10.1073/pnas.2535911100
43. Greenhough A, Smartt HJM, Moore AE, Roberts HR, Williams AC, Paraskeva C, et al. The COX-2/PGE2 Pathway: Key Roles in the Hallmarks of Cancer and Adaptation to the Tumour Microenvironment. Carcinogenesis (2009) 30(3):377–86. doi: 10.1093/carcin/bgp014
44. Miao J, Lu X, Hu Y, Piao C, Wu X, Liu X, et al. Prostaglandin E2 and PD-1 Mediated Inhibition of Antitumor CTL Responses in the Human Tumor Microenvironment. Oncotarget (2017) 8(52):89802–10. doi: 10.18632/oncotarget.21155
45. Gómez-Valenzuela F, Escobar E, Pérez-Tomás R, Montecinos VP. The Inflammatory Profile of the Tumor Microenvironment, Orchestrated by Cyclooxygenase-2, Promotes Epithelial-Mesenchymal Transition. Front Oncol (2021) 11:686792. doi: 10.3389/fonc.2021.686792
46. Zhang Y, Desai A, Yang SY, Bae KB, Antczak MI, Fink SP, et al. Inhibition of the Prostaglandin-Degrading Enzyme 15-PGDH Potentiates Tissue Regeneration(2015). Available at: https://science.sciencemag.org/content/348/6240/aaa2340 (Accessed cited 2020 Nov 11).
47. Boland GP, Butt IS, Prasad R, Knox WF, Bundred NJ. COX-2 Expression is Associated With an Aggressive Phenotype in Ductal Carcinoma in Situ. Br J Cancer (2004) 90(2):423–9. doi: 10.1038/sj.bjc.6601534
48. Half E, Tang XM, Gwyn K, Sahin A, Wathen K, Sinicrope FA. Cyclooxygenase-2 Expression in Human Breast Cancers and Adjacent Ductal Carcinoma in Situ. Cancer Res (2002) 62(6):1676–81.
49. Mizuno R, Kawada K, Sakai Y. Prostaglandin E2/EP Signaling in the Tumor Microenvironment of Colorectal Cancer. Int J Mol Sci (2019) 20(24):6254. doi: 10.3390/ijms20246254
50. Ricciotti E, FitzGerald GA. Prostaglandins and Inflammation. Arterioscler Thromb Vasc Biol (2011) 31(5):986–1000. doi: 10.1161/ATVBAHA.110.207449
51. Robertson FM, Parrett ML, Joarder FS, Ross M, Abou-Issa HM, Alshafie G, et al. Ibuprofen-Induced Inhibition of Cyclooxygenase Isoform Gene Expression and Regression of Rat Mammary Carcinomas. Cancer Lett (1998) 122(1):165–75. doi: 10.1016/S0304-3835(97)00387-X
52. Ristimäki A, Sivula A, Lundin J, Lundin M, Salminen T, Haglund C, et al. Prognostic Significance of Elevated Cyclooxygenase-2 Expression in Breast Cancer. Cancer Res (2002) 62(3):632–5.
53. Volpato M, Cummings M, Shaaban AM, Abderrahman B, Hull MA, Maximov PY, et al. Downregulation of 15-Hydroxyprostaglandin Dehydrogenase During Acquired Tamoxifen Resistance and Association With Poor Prognosis in Erα-Positive Breast Cancer. Explor Target Antitumor Ther (2020) 1:355–71. doi: 10.37349/etat.2020.00021
54. Wu R, Liu T, Yang P, Liu X, Liu F, Wang Y, et al. Association of 15-Hydroxyprostaglandin Dehydrogenate and Poor Prognosis of Obese Breast Cancer Patients. Oncotarget (2017) 8(14):22842–53. doi: 10.18632/oncotarget.15280
55. Bocca C, Ievolella M, Autelli R, Motta M, Mosso L, Torchio B, et al. Expression of Cox-2 in Human Breast Cancer Cells as a Critical Determinant of Epithelial-to-Mesenchymal Transition and Invasiveness. Expert Opin Ther Targets (2014) 18(2):121–35. doi: 10.1517/14728222.2014.860447
56. Majumder M, Xin X, Liu L, Girish GV, Lala PK. Prostaglandin E2 Receptor EP4 as the Common Target on Cancer Cells and Macrophages to Abolish Angiogenesis, Lymphangiogenesis, Metastasis, and Stem-Like Cell Functions. Cancer Sci (2014) 105(9):1142–51. doi: 10.1111/cas.12475
57. Reader J, Holt D, Fulton A. Prostaglandin E2 EP Receptors as Therapeutic Targets in Breast Cancer. Cancer Metastasis Rev (2011) 30(0):449–63. doi: 10.1007/s10555-011-9303-2
58. Majumder M, Xin X, Liu L, Tutunea-Fatan E, Rodriguez-Torres M, Vincent K, et al. COX-2 Induces Breast Cancer Stem Cells via EP4/PI3K/AKT/NOTCH/WNT Axis. Stem Cells (2016) 34(9):2290–305. doi: 10.1002/stem.2426
59. Chang S-H, Liu CH, Wu M-T, Hla T. Regulation of Vascular Endothelial Cell Growth Factor Expression in Mouse Mammary Tumor Cells by the EP2 Subtype of the Prostaglandin E2 Receptor. Prostaglandins Other Lipid Mediat (2005) 76(1–4):48–58. doi: 10.1016/j.prostaglandins.2004.12.001
60. Cheuk IW, Shin VY, Siu MT, Tsang JY, Ho JC, Chen J, et al. Association of EP2 Receptor and SLC19A3 in Regulating Breast Cancer Metastasis. Am J Cancer Res (2015) 5(11):3389–99.
61. Buchanan FG, Wang D, Bargiacchi F, DuBois RN. Prostaglandin E2 Regulates Cell Migration via the Intracellular Activation of the Epidermal Growth Factor Receptor. J Biol Chem (2003) 278(37):35451–7. doi: 10.1074/jbc.M302474200
62. Robertson FM, Simeone A-M, Mazumdar A, Shah AH, McMurray JS, Ghosh S, et al. Molecular and Pharmacological Blockade of the EP4 Receptor Selectively Inhibits Both Proliferation and Invasion of Human Inflammatory Breast Cancer Cells. J Exp Ther Oncol (2008) 7(4):299–312.
63. Nandi P, Girish GV, Majumder M, Xin X, Tutunea-Fatan E, Lala PK. PGE2 Promotes Breast Cancer-Associated Lymphangiogenesis by Activation of EP4 Receptor on Lymphatic Endothelial Cells. BMC Cancer (2017) 17(1):11. doi: 10.1186/s12885-016-3018-2
64. Kochel TJ, Goloubeva OG, Fulton AM. Upregulation of Cyclooxygenase-2/Prostaglandin E2 (COX-2/PGE2) Pathway Member Multiple Drug Resistance-Associated Protein 4 (MRP4) and Downregulation of Prostaglandin Transporter (PGT) and 15-Prostaglandin Dehydrogenase (15-PGDH) in Triple-Negative Breast Cancer. Breast Cancer (Auckl) (2016) 10:61–70. doi: 10.4137/BCBCR.S38529
65. Tönisen F, Perrin L, Bayarmagnai B, van den Dries K, Cambi A, Gligorijevic B. EP4 Receptor Promotes Invadopodia and Invasion in Human Breast Cancer. Eur J Cell Biol (2017) 96(2):218–26. doi: 10.1016/j.ejcb.2016.12.005
66. Sugimoto Y, Narumiya S. Prostaglandin E Receptors*. J Biol Chem (2007) 282(16):11613–7. doi: 10.1074/jbc.R600038200
67. Fujino H, Xu W, Regan JW. Prostaglandin E2 Induced Functional Expression of Early Growth Response Factor-1 by EP4, But Not EP2, Prostanoid Receptors via the Phosphatidylinositol 3-Kinase and Extracellular Signal-Regulated Kinases. J Biol Chem (2003) 278(14):12151–6. doi: 10.1074/jbc.M212665200
68. Hunter S, Nault B, Ugwuagbo KC, Maiti S, Majumder M. Mir526b and Mir655 Promote Tumour Associated Angiogenesis and Lymphangiogenesis in Breast Cancer. Cancers (Basel) (2019) 11(7):E938. doi: 10.3390/cancers11070938
69. Kochel TJ, Reader JC, Ma X, Kundu N, Fulton AM. Multiple Drug Resistance-Associated Protein (MRP4) Exports Prostaglandin E2 (PGE2) and Contributes to Metastasis in Basal/Triple Negative Breast Cancer. Oncotarget (2017) 8(4):6540–54. doi: 10.18632/oncotarget.14145
70. Lee K-L, Kuo Y-C, Ho Y-S, Huang Y-H. Triple-Negative Breast Cancer: Current Understanding and Future Therapeutic Breakthrough Targeting Cancer Stemness. Cancers (2019) 11(9):1334. doi: 10.3390/cancers11091334
71. Lin M-C, Chen S-Y, He P-L, Herschman H, Li H-J. PGE2/EP4 Antagonism Enhances Tumor Chemosensitivity by Inducing Extracellular Vesicle-Mediated Clearance of Cancer Stem Cells. Int J Cancer (2018) 143(6):1440–55. doi: 10.1002/ijc.31523
72. Basudhar D, Glynn SA, Greer M, Somasundaram V, No JH, Scheiblin DA, et al. Coexpression of NOS2 and COX2 Accelerates Tumor Growth and Reduces Survival in Estrogen Receptor-Negative Breast Cancer. Proc Natl Acad Sci USA (2017) 114(49):13030–5. doi: 10.1073/pnas.1709119114
73. Peddi PF, Ellis MJ, Ma C. Molecular Basis of Triple Negative Breast Cancer and Implications for Therapy. Int J Breast Cancer (2012) 2012:217185. doi: 10.1155/2012/217185
74. Li H, Ma F, Wang H, Lin C, Fan Y, Zhang X, et al. Stem Cell Marker Aldehyde Dehydrogenase 1 (ALDH1)-Expressing Cells are Enriched in Triple-Negative Breast Cancer. Int J Biol Markers (2013) 28(4):e357–64. doi: 10.5301/JBM.5000048
75. Idowu MO, Kmieciak M, Dumur C, Burton RS, Grimes MM, Powers CN, et al. CD44(+)/CD24(-/Low) Cancer Stem/Progenitor Cells are More Abundant in Triple-Negative Invasive Breast Carcinoma Phenotype and are Associated With Poor Outcome. Hum Pathol (2012) 43(3):364–73. doi: 10.1016/j.humpath.2011.05.005
76. Giatromanolaki A, Sivridis E, Fiska A, Koukourakis MI. The CD44+/CD24- Phenotype Relates to “Triple-Negative” State and Unfavorable Prognosis in Breast Cancer Patients. Med Oncol (2011) 28(3):745–52. doi: 10.1007/s12032-010-9530-3
77. Wu Y, Sarkissyan M, Elshimali Y, Vadgama JV. Triple Negative Breast Tumors in African-American and Hispanic/Latina Women are High in CD44+, Low in CD24+, and Have Loss of PTEN. PloS One (2013) 8(10):e78259. doi: 10.1371/journal.pone.0078259
78. Ricardo S, Vieira AF, Gerhard R, Leitão D, Pinto R, Cameselle-Teijeiro JF, et al. Breast Cancer Stem Cell Markers CD44, CD24 and ALDH1: Expression Distribution Within Intrinsic Molecular Subtype. J Clin Pathol (2011) 64(11):937–46. doi: 10.1136/jcp.2011.090456
79. Tsang JYS, Huang Y-H, Luo M-H, Ni Y-B, Chan S-K, Lui PCW, et al. Cancer Stem Cell Markers are Associated With Adverse Biomarker Profiles and Molecular Subtypes of Breast Cancer. Breast Cancer Res Treat (2012) 136(2):407–17. doi: 10.1007/s10549-012-2271-6
80. Dalerba P, Cho RW, Clarke MF. Cancer Stem Cells: Models and Concepts. Annu Rev Med (2007) 58:267–84. doi: 10.1146/annurev.med.58.062105.204854
81. Rolland PH, Martin PM, Jacquemier J, Rolland AM, Toga M. Prostaglandin in Human Breast Cancer: Evidence Suggesting That an Elevated Prostaglandin Production Is a Marker of High Metastatic Potential for Neoplastic Cells2. JNCI: J Natl Cancer Institute (1980) 64(5):1061–70.
82. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective Identification of Tumorigenic Breast Cancer Cells. Proc Natl Acad Sci USA (2003) 100(7):3983–8. doi: 10.1073/pnas.0530291100
83. Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, et al. ALDH1 is a Marker of Normal and Malignant Human Mammary Stem Cells and a Predictor of Poor Clinical Outcome. Cell Stem Cell (2007) 1(5):555–67. doi: 10.1016/j.stem.2007.08.014
84. Marcato P, Dean CA, Pan D, Araslanova R, Gillis M, Joshi M, et al. Aldehyde Dehydrogenase Activity of Breast Cancer Stem Cells Is Primarily Due To Isoform ALDH1A3 and Its Expression Is Predictive of Metastasis. Stem Cells (2011) 29(1):32–45. doi: 10.1002/stem.563
85. Park SY, Lee HE, Li H, Shipitsin M, Gelman R, Polyak K. Heterogeneity for Stem Cell-Related Markers According to Tumor Subtype and Histologic Stage in Breast Cancer. Clin Cancer Res (2010) 16(3):876. doi: 10.1158/1078-0432.CCR-09-1532
86. Li W, Ma H, Zhang J, Zhu L, Wang C, Yang Y. Unraveling the Roles of CD44/CD24 and ALDH1 as Cancer Stem Cell Markers in Tumorigenesis and Metastasis. Sci Rep (2017) 7:13856. doi: 10.1038/s41598-017-14364-2
87. Sheridan C, Kishimoto H, Fuchs RK, Mehrotra S, Bhat-Nakshatri P, Turner CH, et al. CD44+/CD24- Breast Cancer Cells Exhibit Enhanced Invasive Properties: An Early Step Necessary for Metastasis. Breast Cancer Res (2006) 8(5):R59. doi: 10.1186/bcr1610
88. Yoshida A, Davé V, Han H, Scanlon KJ. Enhanced Transcription of the Cytosolic ALDH Gene in Cyclophosphamide Resistant Human Carcinoma Cells. In: Weiner H, Crabb DW, Flynn TG, Editors. Enzymol Mol Biol Carbonyl Metab (1993) 4:63–72. doi: 10.1007/978-1-4615-2904-0_8
89. Tomita H, Tanaka K, Tanaka T, Hara A. Aldehyde Dehydrogenase 1A1 in Stem Cells and Cancer. Oncotarget (2016) 7(10):11018–32. doi: 10.18632/oncotarget.6920
90. Rudnick JA, Arendt LM, Klebba I, Hinds JW, Iyer V, Gupta PB, et al. Functional Heterogeneity of Breast Fibroblasts Is Defined by a Prostaglandin Secretory Phenotype That Promotes Expansion of Cancer-Stem Like Cells. PloS One (2011) 6(9):e24605. doi: 10.1371/journal.pone.0024605
91. Zatelli MC, Luchin A, Tagliati F, Leoni S, Piccin D, Bondanelli M, et al. Cyclooxygenase-2 Inhibitors Prevent the Development of Chemoresistance Phenotype in a Breast Cancer Cell Line by Inhibiting Glycoprotein P-170 Expression. Endocrine-Related Cancer (2007) 14(4):1029–38. doi: 10.1677/ERC-07-0114
92. Huang C, Chen Y, Liu H, Yang J, Song X, Zhao J, et al. Celecoxib Targets Breast Cancer Stem Cells by Inhibiting the Synthesis of Prostaglandin E2 and Down-Regulating the Wnt Pathway Activity. Oncotarget (2017) 8(70):115254–69. doi: 10.18632/oncotarget.23250
93. Dinchuk JE, Car BD, Focht RJ, Johnston JJ, Jaffee BD, Covington MB, et al. Renal Abnormalities and an Altered Inflammatory Response in Mice Lacking Cyclooxygenase II. Nature (1995) 378(6555):406–9. doi: 10.1038/378406a0
94. Kundu N, Ma X, Kochel T, Goloubeva O, Staats P, Thompson K, et al. Prostaglandin E Receptor EP4 is a Therapeutic Target in Breast Cancer Cells With Stem-Like Properties. Breast Cancer Res Treat (2014) 143(1):19–31. doi: 10.1007/s10549-013-2779-4
95. Coombes RC, Tovey H, Kilburn L, Mansi J, Palmieri C, Bartlett J, et al. Effect of Celecoxib vs Placebo as Adjuvant Therapy on Disease-Free Survival Among Patients With Breast Cancer: The REACT Randomized Clinical Trial. JAMA Oncol (2021) 7(9):1291–301. doi: 10.1001/jamaoncol.2021.2193
96. Shaashua L, Shabat-Simon M, Haldar R, Matzner P, Zmora O, Shabtai M, et al. Perioperative COX-2 and β-Adrenergic Blockade Improves Metastatic Biomarkers in Breast Cancer Patients in a Phase-II Randomized Trial. Clin Cancer Res (2017) 23(16):4651–61. doi: 10.1158/1078-0432.CCR-17-0152
Keywords: prostaglandin E2, breast cancer, cancer stem cells, aldehyde dehydrogenase, CD44+/CD24-, EP receptors
Citation: Walker OL, Dahn ML, Power Coombs MR and Marcato P (2022) The Prostaglandin E2 Pathway and Breast Cancer Stem Cells: Evidence of Increased Signaling and Potential Targeting. Front. Oncol. 11:791696. doi: 10.3389/fonc.2021.791696
Received: 08 October 2021; Accepted: 27 December 2021;
Published: 19 January 2022.
Edited by:
George Yip, National University of Singapore, SingaporeReviewed by:
Martin Chi Hang Cheung, The University of Hong Kong, Hong Kong SAR, ChinaLeilei Chen, National University of Singapore, Singapore
Copyright © 2022 Walker, Dahn, Power Coombs and Marcato. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paola Marcato, cGFvbGEubWFyY2F0b0BkYWwuY2E=