 Sona Ciernikova
Sona Ciernikova Aneta Sevcikova
Aneta Sevcikova Viola Stevurkova1
Viola Stevurkova1 Michal Mego
Michal Mego- 1Department of Genetics, Cancer Research Institute, Biomedical Research Center of Slovak Academy of Sciences, Bratislava, Slovakia
- 22nd Department of Oncology, Faculty of Medicine, Comenius University, Bratislava and National Cancer Institute, Bratislava, Slovakia
The tumor microenvironment (TME) plays a significant role in tumor progression and cancer cell survival. Besides malignant cells and non-malignant components, including immune cells, elements of the extracellular matrix, stromal cells, and endothelial cells, the tumor microbiome is considered to be an integral part of the TME. Mounting evidence from preclinical and clinical studies evaluated the presence of tumor type-specific intratumoral bacteria. Differences in microbiome composition between cancerous tissues and benign controls suggest the importance of the microbiome-based approach. Complex host-microbiota crosstalk within the TME affects tumor cell biology via the regulation of oncogenic pathways, immune response modulation, and interaction with microbiota-derived metabolites. Significantly, the involvement of tumor-associated microbiota in cancer drug metabolism highlights the therapeutic implications. This review aims to summarize current knowledge about the emerging role of tumor microbiome in various types of solid malignancies. The clinical utility of tumor microbiome in cancer progression and treatment is also discussed. Moreover, we provide an overview of clinical trials evaluating the role of tumor microbiome in cancer patients. The research focusing on the communication between the gut and tumor microbiomes may bring new opportunities for targeting the microbiome to increase the efficacy of cancer treatment and improve patient outcomes.
1. Introduction
A variability of microorganism resident in the human body, collectively termed the microbiota, showed playing a critical role in human health and disease. The enormous development of next-generation sequencing technologies and bioinformatic tools enabled accessing the profound impact of microbiomes - the ecosystems created by resident microbes, their genomes, and functional interactions, on cancer development and treatment. Cancer researchers and clinicians aim to uncover the underlying mechanisms in host-microbiota crosstalk, leading to the clinical utility of the microbiome-based approach. A significant impact of the gut microbiome on the host physiology has been described mainly via modulation of the host immune system, specific function in host metabolism, production of microbiota-derived metabolites, and protection against pathogens (1, 2). Changes in the gut microbiome composition related to dysbiosis contribute to severe pathological conditions, including cancer (3, 4). However, the clinical relevance of the organ-specific microbiomes remains unexplored. The studies showed that infectious microorganisms play a role in approximately 20% of human cancers (5, 6). The existence of specific blood and tissue microbial signatures in different malignancies has been uncovered by reexamination of whole genome and transcriptome sequencing studies (7). Recently, the microbial analysis of tumor samples from seven different malignancies (breast, lung, ovary, pancreas, melanoma, bone, and brain cancer) described the tumor type-specific bacteria localized in cancer and immune cells (8).
Since mounting evidence from animal models and clinical studies, several prospective new hallmarks and enabling characteristics have been added to a very well-known comprehensive concept on the hallmarks of cancer. Besides phenotypic plasticity, non-mutational epigenetic reprogramming, senescent cells, also polymorphic microbiomes became incorporated as core components of the hallmarks of cancer conceptualization (9). Research using fecal transfer from cancer patients to mouse models suggested the existence of both cancer-protective and tumor-promoting microbiomes containing particular bacterial taxa that are capable to modulate the tumorigenic and cancer progression pathways, and have an impact on treatment efficacy (10–12).
Recent advances in cancer research highlight that not only genetic and epigenetic alterations in cancer cells, but also dynamic interactions between all components within the tumor microenvironment (TME) have strong associations with tumor development. Apart from complex signaling communication between cellular and non-cellular TME elements, mutual interactions with microbial components showed a considerable effect on tumorigenesis and tumor progression in a local manner. Mounting evidence suggests the direct involvement of the tumor-associated microbiota in oncogenesis and tumor physiology by the activation of oncogenic pathways, promotion of mucosal inflammation, or metabolic and immune dysregulation. Furthermore, the relationship between the gut/tumor microbiome and response to cancer treatment and drug metabolisms has been comprehensively reviewed (13).
In contrast to gastrointestinal cancers, limited evidence exists about the link between the composition of intratumoral microbiota and other malignancies. Herein, we review current data focusing on the role of the tumor microbiome, as an integral part of the TME. The studies of microbial communities within different solid malignancies are discussed. We provide an overview of the clinical trials, evaluating the presence of microbial communities within tumor tissue samples. Moreover, the clinical relevance and possible therapeutic implications of tumor microbiome analysis in disease progression and cancer treatment efficacy will be outlined.
2. The composition of the tumor microenvironment
The composition of the TME differs between tumor types. However, in general, TME consists of proliferating malignant cells and non-malignant components including immune cells such as microglia, macrophages, and lymphocytes, elements of the extracellular matrix (ECM), stromal cells, and endothelial cells (14). The TME contributes to the promotion of angiogenesis, aiming to overcome hypoxic conditions and restore supplementation with oxygen and nutrients (15). Tumors interact with the surrounding microenvironment and other host organs through the blood or lymphatic circulatory system (16).
Immune cells, important TME constituents affecting cancer growth and progression (17), can be divided into two specific categories; innate immune cell types (dendritic cells, innate lymphoid cells, macrophages, myeloid-derived suppressor cells, natural killer cells, neutrophils) and adaptive immune cell types (B and T cells). The presence of infiltrated innate and adaptive cells within tumors can support either the anti or pro-tumorigenic process (18). The ECM is a highly dynamic non-cellular component of the TME, predominantly composed of collagen, fibronectin, elastin, and laminin (15). A key role of ECM is to direct both cell migration and proliferation (19) and can affect the track and speed of cell migration via its topography and physical properties. According to the findings, tumor cells, as well as cancer-associated fibroblasts (CAFs) are the main source of ECM molecules (20, 21). In many solid tumors, ECM forms almost 60% of the tumor mass and changes during cancer progression and metastasis (22). Stromal cells, including CAFs, create the majority of tumor stroma and promote not only tumor initiation but also angiogenesis and cancer progression. Studies confirmed that CAFs contribute to therapeutic resistance in breast cancer (23). Lakins et al. showed that CAFs directly suppress anti-tumor T-cells within the TME (24). The tumor vasculature is irregular and chaotic in contrast to the normal vasculature of arranged and differentiated arteries, arterioles, capillaries, venules, and veins. Blood vessels are more represented at the tumor interface with decreased vascularity towards the central region, leading to zones of ischemia and necrotic area (25). The vascular density decreased with the tumor growth, which consequently leads to tumor necrosis. The unbalanced secretion of proangiogenic vascular endothelial growth factor-A (VEGF-A) results in the formation of blood vessels (26, 27).
Cancer cell proliferation and metastatic spreading are associated with reprogramming the TME, aiming to support the supplementation of tumor cells with nutrients (28). Complex interactions are determined by the structural and biochemical properties, and also by mutual communication (29). Cross-talk within the TME is mediated by the mixture of cytokines, chemokines, growth factors, and inflammatory mediators together with matrix remodeling enzymes. Moreover, novel ways of communication via extracellular vesicles (EV) including exosomes and apoptotic bodies as well as exosome-derived miRNAs are being explored (30, 31).
Recent research documented a significant impact of the host gut microbiome on shaping the TME. The microbiome regulates the immune and hormonal signaling, having an impact on components of the TME due to the modulation of the processes included in tumor-promoting. In addition, microbiota-derived metabolites can enter the TME via circulation and become a part of the microenvironment (32). Numerous studies confirmed the presence of tumor-type specific bacteria within the TME, suggesting their involvement in several pathways related to tumorigenesis and cancer progression (Figure 1).
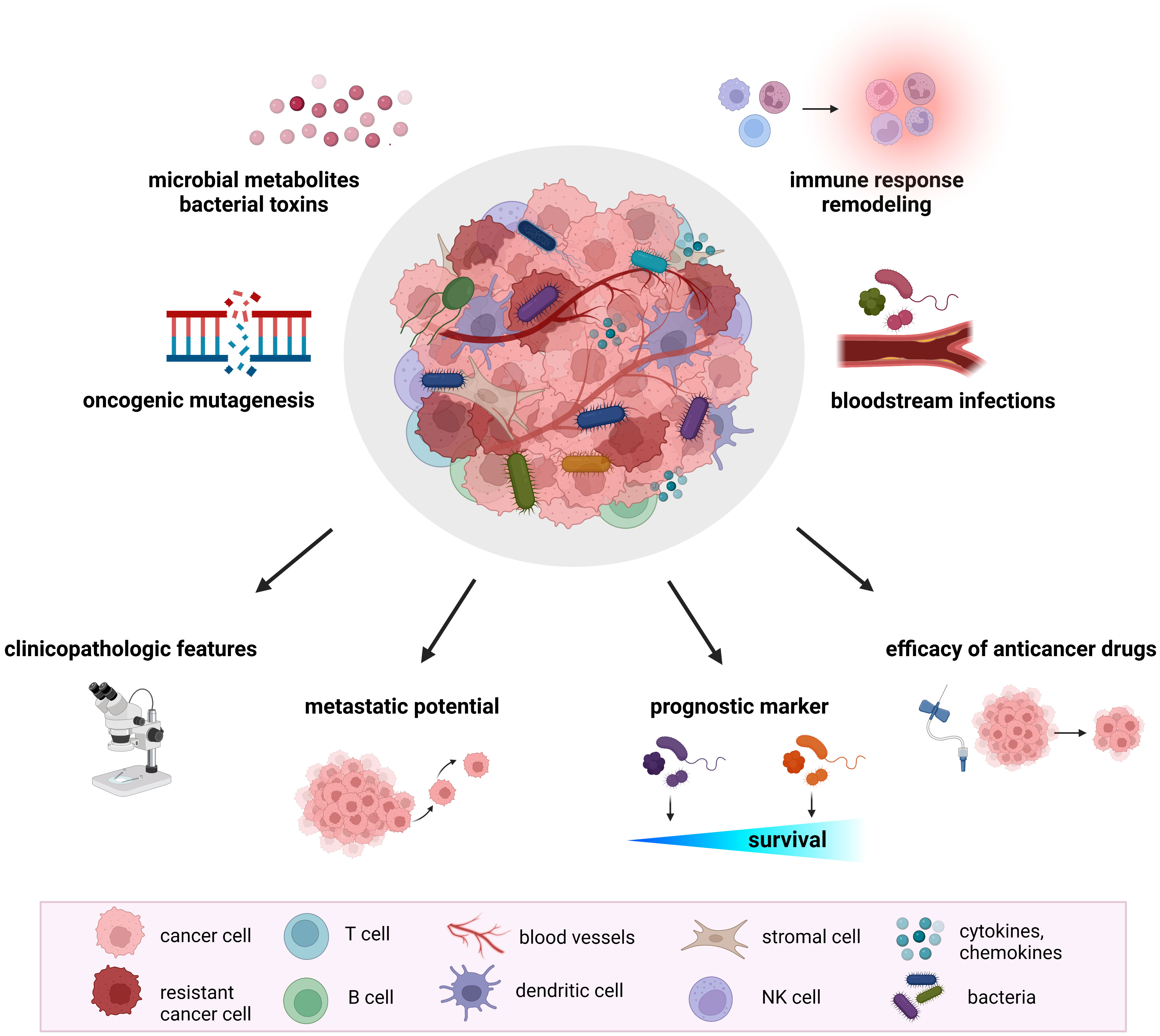
Figure 1 The role of tumor microbiome in cancer development and treatment. Proposed mechanisms by which the tumor microbiome affects tumorigenesis, cancer progression, and response to therapeutic agents include increased mutagenesis, regulation of oncogenes and oncogenic pathways, modulation of host immune response pathways, cancer drug metabolism, production of bacterial toxins and microbiota-derived metabolites. Mounting evidence from animal models and clinical studies revealed the association of tumor microbiome with clinicopathologic features, cancer treatment efficacy, metastatic potential, and cancer survival.
3. The study of tumor microbiome in various solid malignancies
The occurrence of bacteria in tumors was reported more than 100 years ago but it is still unclear if their presence is useful for tumors or bacteria themselves. The study on mucosa-associated bacteria documented the presence of intracellular Escherichia coli in the colonic mucosa of patients with colorectal adenoma and carcinoma compared to controls (33). More recently, Nejman et al. focused on the validation of bacteria in a large group of 1010 tumor samples and 516 normal adjacent tissues. The tumor microbiome was analyzed in seven solid tumor types (glioblastoma multiforme, breast, ovary, bone, pancreas, melanoma, and lung cancer). The study revealed bacterial lipopolysaccharide (LPS) and 16S rRNA in all tumor types. The microbiome of breast tumors was more diverse than the microbiome of other tumor types. In every breast tumor sample was detected 16.4 bacterial species while less than 9 bacterial species were presented in other tumor samples. Proteobacteria and Firmicutes were the most presented in all tumor types while the Actinobacteria phylum including Corynebacteriaceae and Micrococcaceae families dominated in non-gastrointestinal tumors (8).
The overview of ongoing clinical trials concerning the role of tumor microbiome in different cancer types is shown in Table 1.
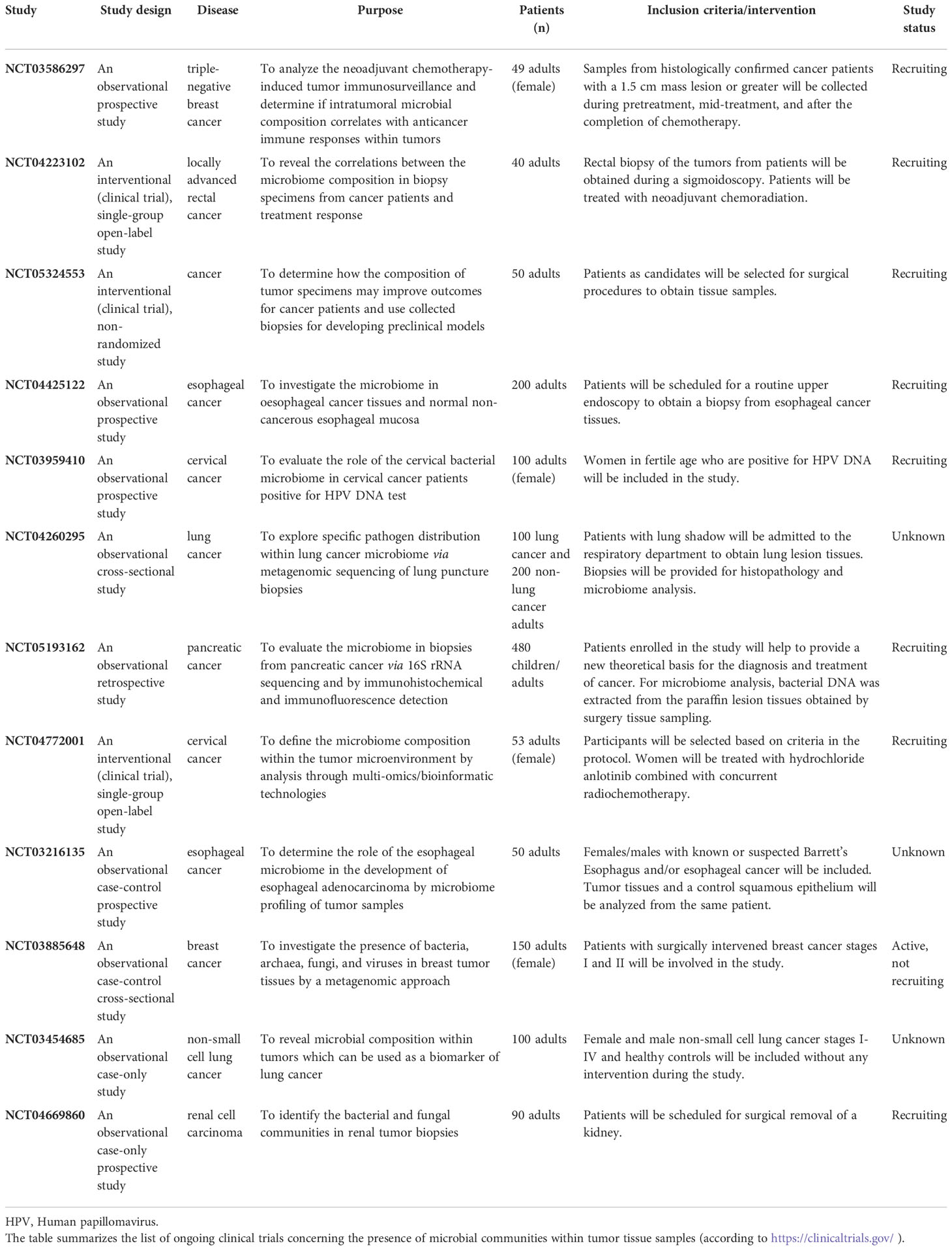
Table 1 Evaluating the role of tumor microbiome in different cancer types.
3.1. Colorectal tumor microbiome
Colorectal cancer (CRC) represents the malignancy with the most evident connection between the microbiome and cancer development, based on more than 50 years of investigation (34). Thus, microbiota modifications consider useful as a potential preventive and therapeutic tool in CRC tumorigenesis (35).
A higher abundance of Fusobacterium nucleatum was reported in colorectal adenomas and carcinomas compared to control tissues (36, 37). Additionally, Fusobacterium positively correlated with metastatic spreading since 74% (29/39) of patients with enrichment of this bacterium developed lymph node metastases (37). Mima et al. also found an increased amount of F. nucleatum in colorectal carcinomas, stage II-IV (38). The microbial differences between samples obtained from colon tumor tissues and adjacent non-malignant mucosa found overexpression of Slackia and Collinsella with decreased levels of Citrobacter, Shigella, Cronobacter, Kluyvera, Serratia, and Salmonella spp. in carcinomas (39). The findings from a study performed by Burns et al. revealed an increased amount of pathogenic bacteria Providencia and Fusobacterium in colon tumor tissues. It is known that Fusobacterium is implicated in colorectal carcinogenesis while Providencia showed to play a role in infections (40, 41). Both bacterial taxa share similar phenotypic characteristics, including the ability to affect and damage colorectal tissues. Patient-matched normal and tumor colon tissue samples showed differences in the abundance of Firmicutes, Bacteroidetes, and Proteobacteria. In particular, Proteobacteria dominated, whereas Firmicutes and Bacteroidetes were depleted in colon tumors. Moreover, the tumor microbiome characterizes decreased amounts of Lachnospiraceae, Ruminococcaceae, Faecalibacterium prausnitzii, Bacteroides, Rikenellaceae, and Bacteroides uniformis (41).
Geng et al. detected a lower abundance of Microbacterium and Anoxybacillus together with Roseburia enrichment in tumor tissues of Chinese colorectal cancer patients (42). The composition of the tumor microbiome was significantly different from adjacent non-neoplastic tissues, with an increase in Firmicutes and Fusobacteria. Overall, a higher number of bacterial genera were observed in cancerous samples. Lactococcus, Bacteroides, Fusobacterium, Prevotella, and Streptococcus dominated in tumors while Pseudomonas in adjacent non-cancerous tissues. The results also showed that the microbiota of proximal colon tumors was similar to that of distal colon cancer. These similarities are partially caused by stool passing through the intestinal tract (43). The analysis of intracellular bacteria reported an increased abundance of intracellular Escherichia coli in adenoma and carcinoma samples compared to control tissues obtained during routine colonoscopy (44). In addition, Klebsiella pneumoniae, Pseudomonas aeruginosa, Enterococcus faecalis, and Bacillus cereus were presented in biopsies from patients with colorectal adenomas (44). More recently, lower amounts of Lanchnospiraceae, Ruminococcaceae/Faecalibacterium were identified in colorectal carcinomas compared to polyps, adenomas, or control samples. A significantly higher level of Bacteroides/Prevotella and Bacteroides/Porphyromonas, especially Porphyromonas asaccharolytica, was detected in cancerous tissue (45). Fusobacterium DNA was detected in 181 colorectal cancer liver metastases. A lower density of CD8+ cytotoxic T cells and a higher density of myeloid-derived suppressor cells were observed in Fusobacterium-positive colorectal cancer liver metastases (46).
Animal models and clinical studies reported the role of microbiota-derived metabolites in colorectal carcinogenesis. A decreased butyrate and increased amounts of lactate, glutamate, alanine, and succinate were found in fecal samples and tumor tissues in CRC patients. Cancer-associated changes affected the metabolic pathways, including glucose and glycolytic metabolism, glutaminolysis, the tricarboxylic acid (TCA) cycle, and SCFA metabolism. A lower amount of fecal acetate correlated with alterations in glucose and myo-inositol levels. Based on the results, acetate showed the diagnostic potential as a single fecal biomarker for colorectal cancer detection (47). An earlier study from Lin and colleagues documented a decreased acetate, propionate, butyrate, glucose, and glutamine, while elevated proline, succinate, isoleucine, leucine, valine, alanine, glutamate, dimethylglycine, and lactate in stool samples of stage I/II CRC patients compared to healthy participants. Metabolomics significantly differentiated between I/II stage CRC patients and later stages of the disease. Changes in metabolite abundance participate in the disruption of bacterial ecology, malabsorption of nutrients, and increased glycolysis and glutaminolysis (48). Yang et al. confirmed that microbial metabolites differed between high-fat diet-fed mice and control diet-fed mice with an intraperitoneal injection of azoxymethane to mimic CRC. Glycerophospholipids, including lysophosphatidylcholine and lysophosphatidic acid, were elevated in mice with a high-fat diet. According to the findings, Alistipes spp. were enriched in the gut microbiome, while probiotic Parabacteroides distasonis was absent in high-fat-diet-fed mice. Antibiotic depletion of the gut microbiome retarded tumor formation in the high-fat diet-fed mice with decreased lysophosphatidylcholine and lysophosphatidic acid (49). The level of hydrogen sulfide was higher, coupled with weakened detoxification activity for this metabolite in CRC patients compared to healthy individuals (50). Hydrogen sulfide is known for its role in the formation of colon cancer due to induced DNA damage, inflammation of colonic mucosa, mucus synthesis, and DNA methylation (51). Increased bioactive lipids, including polyunsaturated fatty acids, secondary bile acids, and sphingolipids, were detected in patients with advanced colorectal adenomas compared to matched controls (52).
3.2. Pancreatic tumor microbiome
Mounting studies documented an emerging role of the pancreatic tumor microbiome in cancer progression and modulating the treatment response (53). Helicobacter DNA was detected in pancreatic tumors or surrounding tissue samples but not in controls suggesting the implication of Helicobacter spp. in the development of chronic pancreatitis and pancreatic cancer (54). Co-cultivation of pancreatic cells with Helicobacter pylori enhanced their malignant potential, leading to a higher activity of NF-κB and AP-1 with consequently dysregulated cellular processes (55). Although Fusobacterium nucleatum was described mainly in colorectal tumors, this bacterium was also enriched in breast and pancreatic tumors (8, 56). Mitsuhashi et al. documented the presence of Fusobacterium species in 8.8% of pancreatic ductal adenocarcinoma (PDAC) tissues, showing a correlation with shorter survival rates in cancer patients (57). The examination of 113 human PDAC samples detected the presence of bacterial DNA in 76% of tumor samples. According to the findings, the majority of bacteria belong to the Gammaproteobacteria, responsible for gemcitabine resistance (58). However, the study by Thomas et al. did not confirm statistically significant differences in the pancreatic microbiome between normal pancreatic tissues compared to tumor samples (59).
In 2018, Pushalkar et al. reported the involvement of the gut and tumor microbiome in PDAC tumor-promoting potential. Bacterial ablation by orally administered antibiotics caused reprogramming of PDAC in mice with a protective effect against tumor progression. The gut microbiome of PDAC patients showed a higher abundance of Proteobacteria, Actinobacteria, Fusobacteria, and Verrucomicrobia compared to healthy controls (60). Interestingly, the analysis of tumor microbiome in PDAC patients showed a potential translocation of Proteobacteria into tumors since the abundance of this bacterial phylum represented 50% in the cancerous tissue (60).
Riquelme et al. reported that prolonged survival of PDAC patients was associated with higher bacterial diversity in the tumor microbiome. The abundance of Saccharopolyspora, Pseudoxanthomonas, and Streptomyces in tumors served as a predictive marker of long-term survivorship with better outcomes. Intratumoral bacteria can modulate the immune tumor microenvironment, whereas the bacterial diversity of the tumor microbiome can contribute to the anti-tumor immune responses with the activation of specific immune cells. Higher levels of Granzyme B+, CD3+, and CD8+ T cells were detected in long-term survivors compared to short-term survivors (61). Guo et al. documented that the basal-like subtype of PDAC was enriched in Acinetobacter, Pseudomonas, and Sphingopyxis. The presence of three bacterial genera positively correlated with DNA replication and the K-ras signaling pathway but negatively correlated with the metabolism of bile acids (62).
Panebianco et al. described that butyrate treatment affected lipidome and metabolome in pancreatic cancer nude BALB/c mice. In addition, butyrate supplementation led to an increase in saturated palmitic acid and stearic acid in gemcitabine-treated mice (63). High expression of aryl hydrocarbon receptor (AhR) correlated with cancer progression. Lactobacillus produced tryptophan metabolite active AhR in tumor macrophages. Although, the elimination of dietary tryptophane decreased the function of AhR, leading to the accumulation of TNFα+IFNγ+CD8+ T cells within tumors (64). The animal study showed that high-fat diet-supplemented obese pancreatic tumor-bearing mice did not respond to gemcitabine and paclitaxel in contrast to control diet-supplemented mice. Queuosine-producing bacteria dominated the gut microbiome of obese mice, while S-adenosyl methionine (SAM)-producing bacteria were elevated in control diet-fed mice. Fecal transfer from the normal into obese mice restored the chemotherapy effect to induce oxidative stress and cause cancer cell death (65). Mirji et al. noted that gut metabolite trimethylamine N-oxide (TMAO) might improve the efficacy of immunotherapy in pancreatic cancer patients. Intraperitoneal administration of TMAO reduced tumor growth in pancreatic tumor-bearing mice and led to increased levels of effector immune T cells within the tumor microenvironment. The combined therapy of TMAO and anti-PD-1 improved survival in pancreatic tumor-bearing mice (66).
3.3. Gastric tumor microbiome
Infection with Helicobacter pylori is a known risk factor for the development of gastric cancer. The presence of this bacterium is associated with changes in stomach acidity leading to differences in bacterial taxa composition (67, 68). Moreover, other biological factors might participate in gastric cancer development by playing the role in the maintenance of the cancerous lesion microenvironment. The data by Chen et al. showed that microbial alpha diversity was higher in tumor samples of patients over 60 years compared to younger participants (69).
Evaluating the gastric microbiome in normal, peritumoral, and tumoral tissues revealed an increased Proteobacteria in peritumoral samples while elevated Firmicutes and Fusobacteria in tumoral specimens. Specifically, an abundance of Halomonas, Shewanella, Enterococcus, Brevundimonas, and decreased Legionella was detected in peritumoral samples. Streptococcus, Peptostreptococcus, Lactobacillus, Bifidobacterium, Neisseria, Veillonella, and Shewanella dominated in tumoral samples compared to normal gastric mucosa tissues. According to the findings, immune system downregulation correlated with increased levels of immunosuppressive cells BDCA2+pDCs and Foxp3+Tregs within the tumor microenvironment (70). Li et al. sequenced biopsies from 33 participants with Helicobacter pylori-associated chronic gastritis, gastric intestinal metaplasia, gastric adenocarcinoma, and controls. The presence of Flavobacterium, Klebsiella, Serratia marcescens, Stenotrophomnonas, Achromobacter, and Pseudomonas characterized the tumor samples. In addition, H. pylori dominated the samples from H. pylori-positive subjects, while Haemophilus, Serratia, Neisseria, and Stenotrophomonas were more abundant in H. pylori-negative samples. The microbial composition of tumor and adjacent non-tumor samples were similar (71).
The analysis of alpha and beta diversity among participants with chronic gastritis and gastric carcinoma showed reduced diversity with dysbiotic potential in gastric carcinomas. Proteobacteria, including Phyllobacterium, Achromobacter, Xanthomonadaceae, and Enterobacteriaceae dominated the gastric cancer microbiome. Based on the results, Helicobacter and Neisseria were also present in tumor tissues but at a lower level. Data revealed that a specific nitrosating bacterial community within gastric carcinoma had the genotoxic potential (72). Proteobacteria dominated almost 90% of cancerous samples obtained by subtotal gastrectomy in 62 gastric cancer patients. Peptostreptococcus, Streptococcus, and Fusobacterium were found in tumor samples. On the other hand, adjacent non-tumor tissue samples contained lactic acid-producing bacteria (69). Specific metabolic pathways, including nucleotide-, energy-, and carbohydrate metabolism were described in cancerous samples [52]. 454 pyrosequencing of the 16S rRNA gene revealed higher nitrate-reducing bacteria in biopsies from cancer patients compared to controls, but the differences were not significant (73).
Wang et al. noted that Actinobacteria, Bacteroides, Firmicutes, Fusobacteria, SR1, and TM7 dominated gastric mucosal biopsies from intraepithelial neoplasia and tumor tissue compared to healthy controls. At the genus level, Granulicatella, Porphyromonas, unclassified Gemellaceae, Rothia, and Fusobacterium were abundant in intraepithelial neoplasia. Further, Helicobacter and Lactobacillus were increased in tumor tissues. Specific differences in gastric microbial composition might serve as a potential predictor of disease stages (74). In addition to obtained data, an extensive investigation needs to elucidate the emerging role of gastric microbiome and the gastric tumor microenvironment in diagnosis, prevention, and cancer treatment (75).
Dai et al. performed metabolome profiling of gastric tumor samples and matched non-tumor tissue samples. Higher bacterial diversity, together with increased levels of amino acids, carbohydrates, carbohydrate conjugates, glycerophospholipids, and nucleosides, were detected in tumor tissue samples. Several metabolites might be considered as biomarkers for the difference between tumor and non-tumor tissues, including 1-methylnicotinamide and N-acetyl-D-glucosamine-6-phosphate. Helicobacter was decreased, while Lactobacillus, Streptococcus, Acinetobacter, Prevotella, Sphingomonas, Bacteroides, Fusobacterium, Comamonas, Empedobacter, and Faecalibacterium were enriched in tumor samples. All detected bacteria were associated with metabolome profiling in cancer tissue samples due to different metabolites in the pathway of amino sugar and nucleotide sugar metabolism (76). Metabolites involved in the lipid metabolism and peroxisome proliferator-activated receptor signaling pathway differed between control mice and the model group of N-Methyl-N0-nitro-N-nitrosoguanidine (MNNG)-induced murine gastric precancerous lesions. Lactobacillus and Bifidobacterium dominated the model group. Turicibacter, Romboutsia, Ruminococcaceae_UCG-014, Ruminococcaceae_UCG-005, and Ruminococcus_1 decreased in the model group. Metabolites, including N-arachidonoyl tyrosine, cucurbitacin D, phosphatidyl inositols, tryptophylhydroxyproline, and 10-epoxy-12-octadecenoic acid positively correlated with the presence of Lactobacillus and Bifidobacterium. On the other hand, a negative correlation was observed between ursodeoxycholic acid, β-amyrin, and phenylalanylvaline and those two bacterial genera (77). Yang et al. did not observe significant differences in bacterial diversity between proximal and distal gastric tumor tissues. Methylobacterium_ and Methylorubrum were elevated in distal tumors. Rikenellaceae_RC9_gut_group, Porphyromonas, Catonella, Proteus, Oribacterium, and Moraxella were more prevalent in proximal tumors. Metabolomics showed 30 discriminative metabolites between proximal vs. distal tumors. Purine metabolism, D-glutamine, and D-glutamate metabolism, sphingolipid signaling pathway, taurine, and hypotaurine metabolism, arginine biosynthesis, alanine, aspartate, glutamate metabolism, β-alanine metabolism, butanoate metabolism, ascorbate, and aldarate metabolism, and nicotinate and nicotinamide metabolism were enriched pathways in distal tumors. Lactobacillus, Muribaculaceae, Rikenellaceae_RC9_gut_group, and Morganella were positively associated with amino acid metabolic pathway and glucose metabolism in proximal tumors (78).
3.4. Breast tumor microbiome
Mounting evidence highlights the role of the breast microbiome in women’s health and disease (79). The microbiome analysis of mammary tissues from lumpectomies, mastectomies, or breast reduction revealed that Bacillus, Acinetobacter, Enterobacteriaceae, Pseudomonas, Staphylococcus, Propionibacterium, Comamonadaceae, Gammaproteobacteria, and Prevotella dominated in the Canadian mammary samples, whereas Enterobacteriaceae, Staphylococcus, Listeria welshimeri, Propionibacterium, and Pseudomonas were the most abundant in Irish cohort (80).
Microbiota-induced changes in metabolic pathways are associated with the heterogeneity of breast cancer. Modified composition of the gut and breast microbiome promotes the progression of breast cancer (81, 82). In this context, different breast microbiome was observed in women with benign or malignant disease. Breast tissues from women with invasive breast cancer showed an increased abundance of Fusobacterium, Atopobium, Hydrogenophaga, Gluconacetobacter, and Lactobacillus. Increased cysteine and methionine metabolism, glycosyltransferases, and fatty acid synthesis were observed in benign tissues while cancerous tissues showed reduced inositol phosphate metabolism (79). Xuan and colleagues detected Methylobacterium radiotolerans in 100% of analyzed breast tumor tissues whereas Sphingomonas yanoikuyae was enriched in paired normal tissues from the same estrogen receptor (ER)-positive breast cancer patient (83). Glycosphingolipid ligands expressed by Sphingomonas yanoikuyae activate invariant Natural killer T (iNKT) cells. iNKT cells have a protective role and promote antitumor immunity in regulating breast cancer metastasis (84, 85).
A comparison of healthy vs. cancer-associated breast microbiomes showed that Bacillus, Staphylococcus, Enterobacteriaceae Comamondaceae, and Bacteroidetes were more abundant in breast tumors from cancer patients while Prevotella, Lactococcus, Streptococcus, Corynebacterium, and Micrococcus dominated in normal tissues of healthy participants. Escherichia coli belonging to the Enterobacteriaceae family was more prevalent in cancer patients but without significant differences in microbial composition between breast cancer and adjacent tissues. Further in vitro experiments reported a higher level of DNA double-stranded breaks in HeLa cells after exposure to Escherichia coli isolates from normal adjacent tissues of breast tumors (86). Hadzega et al. performed RNA-seq analysis of breast tumors and normal tissues from 23 Slovak individuals. In addition, 91 samples obtained from the SRA database and originating in China were included in the study. As results showed, Proteobacteria, Firmicutes, and Actinobacteria were dominant constituents in both cohorts. However, Bacteroides in Slovak samples and Cyanobacteria in Chinese samples were differently abundant (87).
Chiba et al. noted that chemotherapy altered the breast tumor microbiome with a shift in specific microbes. Based on the results, the presence of tumor-specific bacteria might correlate with cancer recurrence (88). Neoadjuvant chemotherapy regimens as a combination of anthracycline, alkylating agents, and taxanes are used to shrink the breast tumor before the surgical procedure (89). Data indicated that neoadjuvant chemotherapy led to an elevated level of Pseudomonas and decreased Prevotella in breast tumors. The abundance of Brevundimonas and Staphylococcus was detected in primary tumors from cancer patients who developed the metastatic disease (88). Pseudomonas aeruginosa was found in 56% of primary breast tumors and 20% of the normal surrounding mammary tissues. Further analysis showed that Pseudomonas aeruginosa modulated cancer cell proliferation and doxorubicin-mediated cell death (88).
More data concerning the role of breast microbiota-derived metabolites in carcinogenesis are warranted. Cadaverine, succinate, and p-cresol might be used as diagnostic markers of breast cancer (90). Wang et al. confirmed that Clostridiales and related metabolite TMAO were prevalent in triple-negative breast tumors. Detected levels of plasma TMAO correlated with better response to PD-1 blockade. In vivo experiments showed that TMAO can activate endoplasmic reticulum stress kinase, which leads to pyroptosis in cancer cells and elevated anti-cancer immunity via CD8+ T cells (91). The gut microbiome is a producer of bile acids. Breast cancer cells are not in direct contact with gut bacteria. However, bacterial metabolites produced by the gut might enter human circulation and then transport to the breast tissue. Production of lithocholic acid was detected as reduced in the early stages of breast cancer (92). Juan et al. showed that probiotic supplementation decreased the occurrence of chemotherapy-related cognitive impairment (CRCI) with modulated plasma metabolites, including p-mentha-1,8-dien-7-ol, linoelaidyl carnitine and 1-aminocyclopropane-1-carboxylic acid in breast cancer patients (Stage I-III) (93).
3.5. Lung tumor microbiome
Although the lungs of healthy individuals were considered sterile, new culture-independent methods and advances in molecular analysis helped to reveal the presence of the lung microbiome (94). In healthy lungs, the studies identified a rich and diverse microbiome with a high abundance of Bacteroidetes, Firmicutes, and Proteobacteria, and the prominent genera Prevotella, Veillomella, Streptococcus, Neisseria, Haemophilus, and Fusobacterium (95–98).
Peters et al. noted that the microbiome of paired normal lung tissues might serve as a biomarker of lung cancer prognosis, presenting a lower diversity in tumor samples compared to paired normal tissues. The results showed that increased levels of Koribacteraceae and Sphingomonadaceae in paired normal lung tissues correlated with increased survival while the abundance of Bacteroidaceae, Lachnospiraceae, and Ruminococcaceae indicated reduced survival. However, the composition of the lung cancer microbiome was not associated with recurrence-free or disease-free survival (99). A study concerning the differences in specific squamous cell lung tumor microbiome between smokers and non-smokers revealed a higher abundance of Acidovorax, Ruminococcus, Oscillospira, Duganella, Ensifer, and Rhizobium in smokers´ samples. As noted, Acidovorax, Klebsiella, Rhodoferax, Comamonas, and Polarmonas were more prevalent in squamous cell lung tumors with TP53 mutations, showing an association between the enrichment in selected microbial taxa and TP53 mutation status (100).
16S rRNA sequencing of lung tumor samples from 89 patients with non-small cell lung cancer (NSCLC) showed a dominance of Actinobacteria, Proteobacteria, Firmicutes, and Bacteroidetes in tumors and adjacent tissue samples. However, no significant differences were observed at the phylum level. In late-stage cancer, the amount of Pseudomonas, Burkholderia, and Aquabacterium was lower while Corynebacterium, Sphingomonas, Streptococcus, Neisseria, Halomonas, Kocuria, Parvimonas, and Rothia were more presented compared to early-stage tumors (101). Yu et al. revealed that alpha diversity was higher in non-malignant lung samples in contrast to malignant samples. Proteobacteria, Firmicutes, Bacteroidetes, and Actinobacteria were presented in 80% of non-cancerous lung tissues. At the genus level, the results showed the presence of Acinetobacter, Pseudomonas, Ralstonia, and two unknown genus-level groups in non-malignant lung tissue samples. Thermus was increased, and Ralstonia was reduced in adenocarcinomas compared to squamous cell carcinoma samples. According to the findings, Thermus levels were higher in lung cancer stages IIIB and IV (102).
Recently, a study on lung cancer patients showed reduced levels of Corynebacterium, Lachnoanaerobaculum, and Halomonas in lung tumor samples (103). Conversely, a study performed by Apopa and colleagues identified that Actinobacteria, Firmicutes, Cyanobacteria, Acidobacteria, and Chloroflexi were more abundant in lung tumors. Moreover, phyla Bacteriodetes and Proteobacteria dominated in lung cancer samples, with a relative abundance of 57% and 24%, respectively (104). Huang et al. documented the high prevalence of Veillonella and Rothia and the significant reduction of Streptococcus in squamous cell lung carcinoma with distal metastasis compared to samples without metastatic spreading (105). Liu observed that Streptococcus was more abundant in lung cancer cases and Staphylococcus was highly-presented in lung samples from controls who consented to bronchoscopy examination (106).
Analysis of the salivary microbiome in NSCLC patients revealed a higher abundance of Veillonella and Streptococcus while decreasing Bacteroides, Fusobacterium, Prevotella, and Faecalibacterium. Pathways associated with xenobiotics biodegradation and metabolism were overexpressed in cancer patients. On the contrary, folate metabolism-related pathways were downregulated (107). Disrupted gut microbiome in lung cancer is associated with under-represented pathways involved in energy metabolism and ABC-type transport system signaling pathways (108).
Ge et al. described 23 murine microbial metabolites with antiproliferative activity in human lung cancer cells, including Pyrrospirone F, chrysophanol, physcion, and purpuride G. Purpuride G is a newly discovered sesquiterpene lactone with the function to block the cell cycle and induce cell death in lung cancer cells (109). Analysis of gut microbial composition and serum metabolite profiles in 30 lung cancer patients confirmed that L-valine reduced with disease progression. L-valine correlated with the presence of Lachnospiraceae_UCG-006 (110). Lee et al. noted that Enterococcus and Lactobacillus were high in feces from NSCLC patients. The presence of Bifidobacterium bifidum was associated with responders to therapy, while Akkermansia muciniphila and Blautia obeum correlated with non-responders. According to the findings, serum metabolic profile of murine model with anti-PD-1 and Bifidobactderium bifidum KCTC3357 was enriched by L-tryptophan, uric acid, and N-acetyl zonisamide (111).
3.6. Glioblastoma tumor microbiome
Since the existence of the blood-brain barrier prevents the diffusion of toxic biological materials and bacteria into the brain environment, the brain has been historically considered a sterile organ. However, several studies detected microbial sequences in pathological and non-pathological human brain samples. Massive parallel sequencing of cerebral white matter-derived RNA from HIV/AIDS patients, other disease controls, and surgical resections for epilepsy reported that Proteobacteria was the most abundant phylum in all brain samples. On the other hand, representatives from Firmicutes were undetectable in most brain-derived RNA samples (112). Previous in vitro studies documented the internalization properties of gut bacteria in HIV/AIDS patients (113). In the human and mouse brain, the presence of members belonging to Firmicutes, Proteobacteria, and Bacteroidetes were identified under noninfectious or nontraumatic conditions (114).
As shown, specific bacteria play a role in the development of numerous pathologies, including cancer, but the occurrence of bacteria in central nervous system (CNS) tumors has not been widely investigated. A combination of immunohistochemistry, fluorescence in situ hybridization (FISH), electron microscopy, 16S rRNA sequencing, and culturomics brought the results describing the tumor-associated microbiota in solid tumors, including glioblastoma (GBM) (8). The microbiome analysis of 40 GBM samples, coupled with the set of negative controls to avoid possible contaminations, detected bacterial DNA in over 40% of the samples. A total of 22 bacterial taxa were identified in GBM tumors by 16S rRNA sequencing. The localization of bacteria mainly within tumor cells was confirmed by a combination of immunohistochemical staining with antibodies against bacterial LPS, lipoteichoic acid (LTA), and 16S rRNA in situ hybridization assay (115). Zhao et al. developed a direct 3D quantitative in situ imaging of bacterial LPS within gliomas to minimize the risk of contamination by sample collection and processing. A contamination-free manner of the study was achieved by a combination of tissue clearing, immunofluorescent labeling, optical sectioning microscopy, and image processing. Immunohistochemical LPS and LTA staining and 16S rRNA FISH analysis were performed to complement 3D information for tissues from the same samples. According to the findings, bacterial components were predominantly localized in the intercellular space or close to nuclear membranes (116).
Tumor-associated bacteria in GBM raises the question of whether the tumor microbiome is associated with the development of gliomas or if it is the result of damaged blood-brain barrier permeability and glioma-associated immunosuppressive microenvironment. As described, gut microbiota-derived metabolites contribute to the pathogenesis of neurodegenerative diseases via disruption of blood-brain barrier integrity, affecting brain functions (117). The emerging evidence of the role of the gut-brain axis in neuro-oncology has been recently reviewed (118). Patrizz et al. documented that glioma development led to a changed gut microbiome in the tumor-bearing murine model. Glioma growth was responsible for an increased presence of the Akkermansia genus and Verrucomicrobia phylum in stool samples (119).
The changes in the gut microbiome composition and subsequently altered gut metabolites affect the immune system and CNS immunity (120). Pommepuy et al. evaluated the effect of the fungal metabolite Brefeldin A to induce apoptosis and cell growth inhibition in glioblastoma cell lines. The metabolite was responsible for 60% of cell growth inhibition, and cell cycle arrest in the early G0/G1 phase with a decrease in the S cell population. Based on the data, Brefeldin A was a potential inducer of apoptosis, a modulator of the cell cycle, and might be considered a promising candidate for the treatment of glioblastomas (121). Aerobic abscesses can be differentiated from glioblastomas according to the metabolite ratio. The choline was increased in the enhancing rim of glioblastomas, while creatine and N-acetylaspartate were decreased compared to normal brains. Acetate was not detected in aerobic bacteria abscesses. Lactate and lipid were observed in 8 patients with aerobic abscesses and 5 patients with a tumor (122). Himmelreich et colleagues showed that metabolite profiling was similar between gliomas and Staphylococcus aureus abscesses. However, N-acetylaspartate was under-represented, while lipid and lactate, glutamine, and/or glutamate levels were increased in Staphylococcus aureus abscesses (123).
3.7. Tumor microbiome in gynecological cancers
The role of microbiome in gynecological cancers is documented mainly in studies of vaginal microbiome composition (124, 125). Microbiota influences gynecological carcinogenesis by regulating estrogen levels, immune response modulation, interference with carbohydrate metabolism, and production of microbiota-derived metabolites and bacterial toxins (126).
3.7.1. Ovarian tumor microbiome
Different bacterial composition was identified within cancerous and normal non-cancerous ovarian tissues. The microbiome analysis of ovarian tumor tissues by 16S rRNA sequencing reported a decreased diversity in tumor samples compared to healthy distal fallopian tube samples. A higher level of Proteobacteria was observed in ovarian cancer tissues, whereas Firmicutes and Acidobacteria were down-regulated. At the genus level, Acinetobacter, Sphingomonas, and Methylobacterium were found in high levels among tumor samples, while Lactococcus was significantly reduced (127). Wang et al. detected a decreased amount of Crenarchaeota and elevated levels of Aquificae and Planctomycetes in cancerous samples. At the species level, normal tissues were enriched in Halobacteroides halobius (14%), Gemmata obscuriglobus (11%), and Methyloprofundus sedimenti (10%). The results showed that Gemmata obscuriglobus (13%), Halobacteroides halobius (11%), and Methyloprofundus sedimenti (11%) dominated tumor samples (128). In a study of 99 ovarian cancer samples, 20 matched control samples from non-cancerous adjacent tissue, and 20 unmatched controls found that Proteobacteria and Firmicutes predominated in cancer ovarian samples. In addition, a lower amount of Bacteroidetes, Actinobacteria, Chlamydiae, Fusobacteria, and Tenericutes was detected in cancer samples. As shown, no common bacterium was detected between cancer, pathological noncancerous tumor-adjacent tissue, and non-matched control samples (129).
Nene et al. reported that lactobacilli did not dominate in cervicovaginal microbiome of ovarian cancer patients and BRCA1/2 mutation carriers younger than 50 years. According to the findings, specific vaginal microbial community with less than 50% of Lactobacillus spp. might be causal for ovarian cancer development (130). More recently, a correlation was found between the microbiome composition of the upper reproductive tract and ovarian cancer status. Acidovorax, Acinetobacter, Aeromonas, Cloacibacterium, Conexibacter, Mariomonas, Methylobacterium, Propionibacterium, Pseudoalteromonas, Vibrio, and Xanthomonas spp. were less presented in upper reproductive tract tissues of ovarian cancer patients, while Bosea, Mesorhizobium, Mycobacterium, Ralstonia, and Variovorax were more prevalent in cancer tissues compared to control tissues (131). The study concerning the oncobiotic peritoneal microbiome demonstrated that ovarian cancer patients showed a unique peritoneal microbial profile compared to patients with a benign mass. Moreover, 18 highly-specific microbial features were identified by a machine learning algorithm (132).
Antibiotic supplementation affected circulating gut microbes-derived metabolites in mice with injected epithelial ovarian cancer (133). Terrein purified from the fermentation metabolites of Aspergillus terreus strain PF26 inhibited the proliferation of ovarian cancer stem-like cells with cell cycle arrest. This studied metabolite might be used as a potential candidate for ovarian cancer treatment (134). Many urinary metabolites were under-represented in ovarian cancer patients. Methanol, with a 65% decrease, was observed in ovarian cancer patients compared to healthy participants. Metabolites, including propylene glycol and mannitol were not changed in healthy and cancer participants (135). In ovarian cancer, diamine oxidase increased in plasma and tumor tissues. Diamine oxidase caused a higher accumulation of produced gamma-aminobutyric acid (136). D’Amico et al. suggested that selected gut microbiome members might affect the level of lactate metabolite. Lactate producers such as Coriobacteriaceae and Bifidobacterium dominated the gut microbiome of platinum-resistant ovarian cancer patients (137).
3.7.2. Endometrial tumor microbiome
Still, a few studies have focused on endometrial microbiome composition (138). Lactobacillus was the most presented genus within the healthy endometrium milieu in most studies. However, some findings reported that other bacterial taxa formed a large part of the microbiome, including Pseudomonas, Acinetobacter, Vagococcus, Sphingobium (139), and Klebsiella pneumoniae, Clostridium botulinum, Pasteurella multocida, and Hydrogenophaga spp. NH-16 (140).
The association of endometrial Pelomonas and Prevotella with tumor burden was described among endometrial cancer patients. Additionally, reduced alpha diversity was observed in endometrial samples from cancer patients (141). Lu et al. noted that Micrococcus was increased in endometrial microbiome of women with endometrial cancer undergoing hysterectomy while Pseudoramibacter_Eubacterium, Rhodobacter, Vogesella, Bilophila, Rheinheimera, and Megamonas were more abundant in endometrial microbiome of women with benign uterine lesions (142). Differences in uterine microbiome between patients with endometrial cancer and patients with the benign disease showed that Porphyromonas somerae was presented in all patients with Type II endometrial malignancy compared to 57% of patients with endometrial hyperplasia (143). Results from the study on 17 patients with endometrial cancer, 4 patients with endometrial hyperplasia, and 10 patients with benign condition also revealed the link between Porphyromonas spp. and endometrial cancer development (144).
Currently, Hawkins et al. presented that selected bacteria might contribute to the pathogenesis of endometrial cancer. Acidorovax, Bradyrhizobium, Flavobacterium, Hyphomicrobium, Pelomonas, and Pseudomonas differentiated samples from endometrial cancer and benign uterus. A comparison of endometrial tumor microbiome in obese women detected a higher abundance of Firmicutes and Cyanobacteria, and a decrease in genera Dietzia and Geobacillus in tumors from black women compared to the samples from white patients. Firmicutes were higher in tumors of non-obese white women than in obese white women (145). According to the previous results, Firmicutes and Actinobacteria were more prevalent in endometrioid carcinoma of African-American women than in Caucasian women. Microbial profiles containing a high prevalence of Actinobacteria, Bacteroidetes, Firmicutes, and Proteobacteria in tumor tissues from obese women correlated with the results from a murine model (146). Caselli et al. evaluated the impact of Atopobium vaginae and Porphyromons somerae on human HEC-1A endometrial adenocarcinoma cells. The results confirmed that co-cultivation with selected bacteria caused the release of inflammatory cytokines and chemokines by endometrial cells (147).
Studies showed the potential involvement of microbiome-produced bacterial toxins and cancer-promoting metabolites in endometrial cancer (148). Metabolite profiling of serum confirmed the lower levels of threonine but increased long-chain fatty acids (LCFA) C16:1, C18:1, C20:1, C20:2, C22:6, C24, and C24:1 in endometrial cancer patients. Levels of C16:1 and C20:2 correlated with Ruminococcus spp., Prevotella spp., and Anaerostipes caccae in cancer patients. The role of C16:1 might be to stimulate endometrial cancer cell proliferation and metastasis via mTOR pathways (149). Alauddin et al. analyzed the effect of treatment with Urolithin A (gut-produced microbial metabolite) in the Ishikawa cell line established from an endometrial adenocarcinoma. Treatment with Urolithin A caused changes in cellular properties in tumor cells. Specifically, Urolithin A treatment significantly decreased Rac1 and PAK1 activity and influenced actin polymerization dynamics (150). The presence of Erysipelatoclostridium and its related metabolite ptilosteroid A was associated with the development of grade 2 radiation-induced intestinal injury (RIII) in endometrial cancer patients. The results showed that both might be used as diagnostic biomarkers for grade 2 RIII (151).
3.8. The urinary tumor microbiome
In the long-term, the bladder and urine were considered sterile in healthy individuals (152). A novel comprehensive approach found that Corynebacterium, Staphylococcus, and Streptococcus dominated in the urine of men with or without a biopsy-proven diagnosis of prostate cancer (153). Pearce et al. noted differences in the urinary microbiomes of women with or without urinary incontinence. Lactobacillus and Gardnerella were the most frequently identified bacterial taxa in both groups (154).
3.8.1. Prostate tumor microbiome
Besides urinary tract microbes from the external environment, the prostate microbiome might be affected by intestinal microbiome. Additionally, surgical procedures could be a source of microbes from the external environment (155). Mycoplasma and its bacterial proteins consider to play a role in prostate cancer due to its presence in the tumor microenvironment, but limited data is still available (155, 156).
Most men do not die from prostate cancer due to improved treatment. Death from prostate cancer can be related mainly to metastatic spreading into the pelvic area, spinal cord, bladder, rectum, bone, and brain (157). An early study concerning the presence or absence of bacteria in prostate samples revealed no bacterial DNA in prostate samples from 18 organ donor controls. Bacteria were detected in prostate samples from radical and simple prostatectomy for prostate cancer and benign prostatic hyperplasia, respectively (158). Recently, mounting studies confirmed that the prostate is not a sterile environment (159). Massive ultradeep pyrosequencing identified the differences in the microbial composition between tumor, peritumor, and non-tumor prostate tissue samples. Staphylococcus spp. dominated in tumor and peritumor samples. Whereas, Streptococcus spp. was presented in non-tumor prostate samples, which supported the idea that bacteria are part of healthy prostate microbiome (160). Another study confirmed that Escherichia, Propionibacterium, Acinetobacter, and Pseudomonas were abundant in prostate tumors and matched benign tissue samples from 65 Chinese patients who underwent prostatectomy (159). Shannon et al. proposed that Propionibacterium acnes as a carcinogen is involved in prostate cancer. Propionibacterium acnes has been linked with the development of prostate cancer because of its high presentation in the urinary male tract after puberty where this bacterium can cause infection (161). Alanee et al. also described modifications in the urinary microbiome within patients with prostate cancer compared to controls. Prostate cancer patients exhibited a higher level of Veillonella, Streptococcus, and Bacteroides with a lower abundance of Faecalibacterium, lactobacilli, and Acinetobacter (162).
The growth and progression of prostate cancer might be influenced by the gut microbiome and produced bacterial metabolites that indicate the existence of an axis “gut-prostate” (163). Liss et al. reported alternations of folate and arginine pathways. According to the bacterial composition, Bacteroides and Streptococcus were more abundant in the microbiome of prostate cancer patients (164). In 2021, Matsushita et al. revealed that SCFA-producing bacteria, including Alistipes and Lachnospira, were more abundant in prostate cancer men compared to non-cancer participants. Metabolic pathways involved in starch and sucrose metabolism, phenylpropanoid biosynthesis, phenylalanine, tyrosine, and tryptophan biosynthesis, cyanoamino acid metabolism, and histidine metabolism were more prevalent in cancer patients (165). A study comparing metabolic profiles in prostate cancer patients vs. healthy subjects found 28 prostate cancer-specific metabolites, including two nucleosides – pseudouridine and uridine (166). Slackia sp. strain NATTS, an equol-producing bacterium, was detected in 34% of prostate cancer patients and 24% of healthy participants. Serum level of equol produced from a bacterial metabolite of daidzein correlated with the level of Slackia in feces from healthy and prostate cancer patients (167).
3.8.2. Bladder cancer microbiome
The risk of bladder cancer is lower in women compared to men. Different bladder-associated microbial communities might be the reason why the disease affects fewer women (168, 169). The analysis of 32 bladder tumor samples collected during resection/radical cystectomy identified lower levels of Actinobacteria in tumor tissues. Following the findings that higher bacterial diversity indicates healthy microbiome, increased diversity was observed in non-tumor tissues. Tumor-associated mucosa of 13 patients was enriched in Barnesiella, Parabacteroides, Prevotella, Alistipes, and Lachnospiracea_incertae_sedis. On the other hand, Staphylococcus dominated the tumor microbiome of 6 patients. Interestingly, a higher abundance of Enterococcus showed in low-grade tumors. Based on these findings, detected microorganisms might play a role in the initial development of bladder cancer (170). Mansour et al. analyzed bacterial community in the urine and bladder carcinoma samples to improve the accuracy of diagnostic in bladder tumor research. According to the findings, Akkermansia, Bacteroides, Clostridium sensu stricto, Enterobacter, and Klebsiella were more present in tissues compared to the urine samples. Description of changes in bacterial composition during bladder cancer progression might be used as a monitoring tool for the disease (171).
Yumba-Mpanga et colleagues identified 24 urine metabolites specific for bladder cancer compared to healthy individuals. The detected level of gluconic acid was statistically different between healthy participants and bladder cancer patients. This metabolite is associated with the pentose phosphate pathway (166). Uridine was observed in lower abundance in urine samples from bladder cancer patients (172). Mager et al. studied inosine levels in the duodenum, jejunum, and cecum of Bifidobacterium pseudolongum-colonized mice. According to the findings, the highest level of inosine was in the duodenum, with a decrease along the intestinal tract. Accordingly, products of inosine, such as xanthine and hypoxanthine, were increased in serum. Orally administered inosine in combination with anti-CTLA-4 and CpG treatment in the bladder tumor-bearing mice led to reduced tumor size and correlated with higher levels of IFN-γ+CD4+ and IFN-γ+CD8+ T cells in the spleen. Gavage of live Bifidobacterium pseudolongum led to increased efficacy of anti-CTLA-4 in SPF mice. However, heat-killed bacterium was not able to enhance the immunotherapy due to its inability to produce inosine metabolite (173). In the past, Abdel-Tawab assessed the association between the excretion of tryptophan metabolites and the amount of N-nitrosamine in the urine samples from bilharzial bladder cancer patients. The presence of N-nitrosamines was detected in 45% of control participants and 93% of patients. 64% of bladder cancer patients metabolized the tryptophan abnormally (174). 40 day-saccharin consumption led to an increased level of indican - tryptophan metabolite formed by bacterial action and p-cresol in rats. The development of bladder tumors correlated with greater excretion of indican. As shown, saccharine caused biochemical and physiological disruptions associated with altered electrolytes and microbial amino-acid metabolites, contributing to bladder tumor formation (175).
3.9. Melanoma microbiome
Due to existing microbial similarities between humans and pigs, porcine models have been used for the study of microbiome composition in melanoma samples, suggesting a potential therapeutic approach. The level of Fusobacterium and Trueperella was higher in melanoma samples of the Melanoma-bearing Libechov Minipig (MeLiM) model. In addition, the amount of Fusobacterium nucleatum significantly increased in the samples with progressive melanoma compared to animals with a regressive form of the disease (176). Mekadim et al. documented that Fusobacterium, Trueperella, Staphylococcus, Streptococcus, and Bacteroides dominated in melanoma tissue samples of MeLiM pigs while Lactobacillus, Clostridium sensu stricto 1, and Corynebacterium 1 showed higher abundance in healthy skin samples. Different bacterial diversity was detected in samples collected from specific areas, including melanoma tissue, surface, and healthy skin tissue. A decrease in diversity was observed in melanoma tissues from the MeLiM progressive melanoma group compared to MeLiM regressive animals (177).
Salava et al. noted that Propionibacterium, Staphylococcus, and Corynebacterium dominated in melanoma samples obtained during surgery using a sterile flocked swab (178). The analysis of bacterial cultures using swab sampling identified the dominance of Corynebacterium in 76.9% melanoma of patients with stage III/IV and 28.6% melanoma of patients with stage I/II, respectively (179). Analysis of 17 melanoma metastases revealed HLA-I and HLA-II peptides derived from 41 bacterial species which can induce immune reactivity. 11 recurrent HLA-I-associated peptides were derived from Fusobacterium nucleatum, Staphylococcus aureus, and Staphylococcus capitis. Microbiota richness was higher in tumors compared to blood samples (180). The findings concerning the relationship between melanoma microbiome and cancer treatment revealed that tumors from metastatic melanoma patients who responded to immune checkpoint inhibitors were enriched in Clostridium (8). Accordingly, Zhu et al. detected an enrichment of Lachnoclostridium within cutaneous melanoma of patients with improved overall survival. Higher amounts of Lachnoclostridium positively correlated with the abundance of infiltrating CD8+ T cells, which brought benefits or improved outcomes for patients (181).
Frankel et al. reported that melanoma patient response to immunotherapy correlated with gut microbial composition, metabolic profile, previous antibiotic supplementation, and patient diet and lifestyle. A higher level of anacardic acid was observed in feces from responding metastatic melanoma patients to immunotherapy. Bacterial enzymes, which played a role in fatty acid synthesis and inositol phosphate metabolism, dominated treatment responders. In total, changes in 83 metabolites were detected between responders vs. patients with progressive disease (182). According to Coutzac et al., lower amounts of baseline butyrate and propionate correlated with longer progression-free survival (PFS) in patients with metastatic melanoma treated with ipilimumab (183). The role of indole-3-carboxaldehyde (3-IAld) is to maintain normal gut epithelial integrity. Renga et al. confirmed the effect of 3-IAld to protect the gut barrier in melanoma tumor-bearing mice with immunotherapy-induced colitis. Administered 3-IAld helped to improve survival rate, maintain murine weight and protect against developed colitis due to reduced therapy-induced gut damage and toxicity (184). Anthraquinone derivatives termstrin A, B, C, and D were isolated from the Streptomyces sp. BYF63. Termstrin B showed a cytotoxic function against melanoma cell line A375 (185). 3,5-dihydroxy Q1 -4-ethyl-trans-stilbene (DETS) is a known bioactive bacterial secondary metabolite isolated from Bacillus cereus. DETS helped to downregulate pathways involved in melanoma progression in cells treated with DETS. However, in vivo studies that will confirm the antioxidant and anti-cancer properties of DETS are needed (186).
As discussed, elucidating the emerging role of microbes within tumors is still an issue of intensive research. Bacterial communities were found in numerous solid malignancies, mainly in ovarian, endometrial, prostate, bladder, breast, pancreatic, colorectal, gastric, melanoma, and lung cancer (Figure 2).
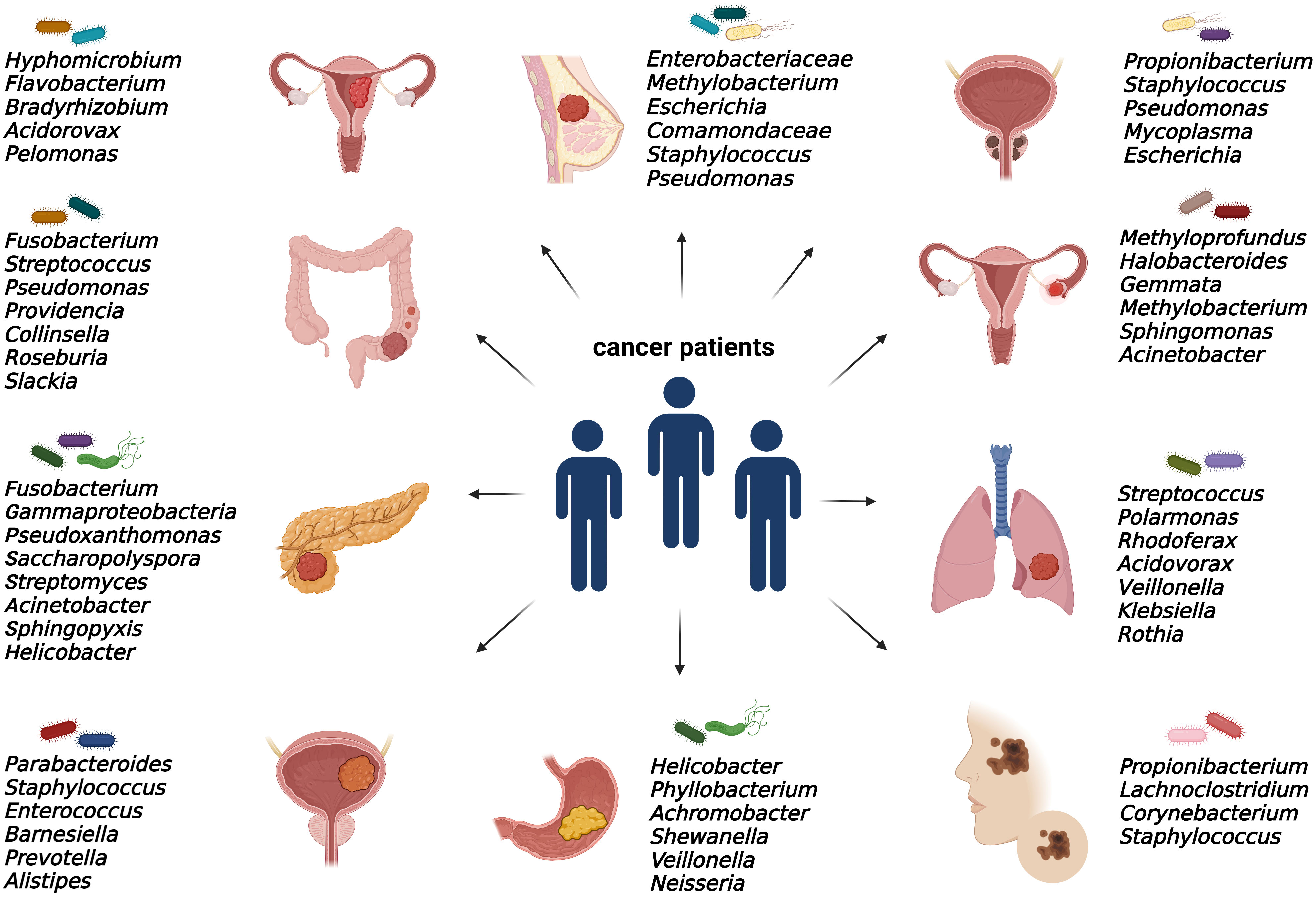
Figure 2 Specific intratumoral bacteria within different types of tumors. The presence of cancer type-specific bacteria within the tumor microenvironment was revealed mainly by 16S rRNA or metagenomic sequencing and via immunohistochemical and immunofluorescence detection methods.
4. The clinical utility of tumor microbiome-based approach
The results from preclinical models and clinical studies reported significant differences in microbiome composition between tumor samples and healthy tissues. Tumor-associated microbiota contributes to carcinogenesis by activation of oncogenic signaling pathways, such as Wnt/β-catenin signaling. The studies demonstrated that Helicobacter pylori, Fusobacterium nucleatum, and enterotoxigenic Bacteroides fragilis secrete cytotoxin-associated gene A (CagA) protein (187), adhesin A (FadA) (188) and toxin Bft (189), respectively. In addition, intratumoral microbiota inhibits innate and adaptive immune responses (190). A comprehensive approach identified unique microbial cancer-related signatures in tissues and blood (7) by analyzing genome and whole transcriptome sequencing studies from 33 cancer types within The Cancer Genome Atlas (191). This raises the possibility of using tumor-associated microbial biomarkers in cancer screening and diagnostics.
Specific intratumoral bacteria correlated with prognostic clinicopathologic features and metastatic potential. One of the largest studies to date evaluated the role of breast microbiome in fresh-frozen surgical specimens from 221 patients with breast cancer, 18 individuals predisposed to breast cancer, and 69 controls. Microbiome analysis using 16S rRNA sequencing, identified significant differences in the relative abundance of specific bacterial taxa regarding tissue type, cancer stage, grade, histologic subtype, receptor status, lymphovascular invasion, and node-positive status (192). For instance, Pseudomonas, Proteus, Porphyromonas, and Azomonas were enriched in tumor samples, while a higher abundance of Propionibacterium and Staphylococcus was observed in healthy control, high-risk, and tumor-adjacent normal tissues. Regarding the association between bacterial genera and prognostic breast tumor features, Porphyromonas, Lacibacter, Ezakiella, and Fusobacterium were related to higher stage tumors and lower levels of Alkanindiges, Micrococcus, Caulobacter, Proteus, Brevibacillus, Kocuria, and Parasediminibacterium were linked to estrogen receptor (ER)-positive status (192). Primary breast tumors from patients who developed distant metastases displayed an increased tumoral abundance of Brevundimonas and Staphylococcus (88).
According to the findings, the Fusobacterium nucleatum gDNA and abundant levels Gal-GalNAc were found not only in colorectal but also in breast cancer tissue samples. Parhi and colleagues noted that fusobacterial colonization is associated with accelerated tumor growth and metastatic progression of breast cancer (193). In a very recent study on murine breast-tumor model MMTV-PyMT, a depletion of intratumoral bacteria was significantly related to the reduction of lung metastasis. As shown, intracellular microbiota reorganized the actin cytoskeleton network within tumor cells, leading to increased survival during breast cancer metastatic spreading (194). Moreover, the metastatic potential of breast carcinomas was increased in tumor models after intratumoral administration of specific genera, isolated from tumor-associated bacteria (194).
Noticeably, several studies described the crucial impact of tumor microbiome on the response to cancer treatment modalities. As noted by Nejman and colleagues, Gardnerella vaginalis was more abundant in tumor samples from non-responders (8). In CRC patients, modulation of the innate immune system by Fusobacterium nucleatum is considered to play a role in chemoresistance. A preclinical study found a link between Fusobacterium-mediated resistance to chemotherapeutics (oxaliplatin and 5-fluorouracil) and autophagy modulation via the TLR4 and MYD88 signaling pathway (195). Pancreatic cancer tissues from PDAC samples showed an abundance of intratumoral Enterobacteriaceae and Pseudomonadaceae families (58). As shown, Gammaproteobacteria were able to enzymatically inactivate gemcitabine by expressing the bacterial cytidine deaminase, and antibiotic treatment with ciprofloxacin overcame the gemcitabine resistance (58). The presence of Mycoplasma hyorhinis in the TME suggested decreasing the cytostatic activity of gemcitabine Mycoplasma-infected tumor cell lines (196).
5. Conclusions, limitations and future directions
In conclusion, targeting the gut and tumor microbiome represents an emerging trend in cancer development and treatment. While the associations between intestinal microbiome, carcinogenesis and the response to anti-cancer therapies have been intensively studied, the impact of tumor microbiome requires further investigation. Distinct microbial communities reside within TME, and tumor-associated microbiota showed having tumor-specific character. Mounting evidence from solid cancers described differences in microbiome composition between cancerous and non-cancerous tissues. Recently, studies reported the correlation between the tumor microbiome and the clinical features of corresponding tumors. Further clinical research focusing on the impact of intratumoral bacteria and microbiota-derived metabolites on therapeutic outcomes is highly warranted. A deep understanding of complex interactions between tumor microbiome, cancer cells and TME components might bring clinically relevant information. In addition, comprehensive studies on large cohorts of tumor samples may identify microbial biomarkers predicting metastatic spreading. Precise characterization of tumor microbiome signatures may be used to stratify cancer patients leading to the development of more effective, individualized, tumor-specific therapies. Importantly, the progress in machine learning algorithms can help to uncover the underlying mechanisms and signaling networks. This could lead to identification of potentially novel targets for predicting the treatment response. However, the considerable variation in microbiome composition between patients, the existence of confounding factors, and heterogeneity in host genetic susceptibility, should be carefully considered.
Although an increasing number of microbiome studies, limits in taxonomic resolutions represent an important issue for proper understanding of functional impact of the microbiome. In this context, more comprehensive approach, including metagenomics, metatrascriptomics, metabolomics, and metaproteomics, should more precisely evaluate the role of the microbiome in cancer. One of the most serious concerns in terms of tumor microbiome studies is the risk of microbial contamination during sample collection, storage, and processing. In the future, procedures controlling the development of 3D imaging methods may allow the analysis of direct interactions between the microbial community and other components of host TME. Recent studies focus mainly on relative abundances of bacterial taxa in each sample. This represents a kind of limitation since changes in total abundance might bring more accurate information about the real impact of microbiota and microbiota-derived metabolites on host physiology. In addition, present data describes the occurrence of intratumoral bacteria but does not distinguish whether their presence is causal in terms of cancer development or whether they co-exist in TME due to leaky vasculature and immunosuppressed microenvironment.
Importantly, the co-cultivation of specific bacterial taxa with organoids prepared from pluripotent stem cells or tissues may represent an effective tool to study host–bacteria interactions and evaluate the potential mechanisms behind them. The existence of crosstalk between intratumor and gut microbiomes brings the possibility of modulation-based therapies, including prebiotic and probiotic administration, fecal microbiota transplantation, and others. Thus, personalized determination of patient gut and tumor microbiome composition might represent a potential diagnostic and prognostic tool, and restoration of balance in microbial community may improve treatment efficacy and patient outcomes.
Author contributions
SC designed the review and prepared the original draft. AS and SC analyzed the literature and wrote the manuscript. SC reviewed and edited the manuscript. AS, SC, and VS prepared the Figures and Table. MM reviewed the manuscript before submission. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the Scientific Grant Agency of the Ministry of Education, Science, Research and Sport of the Slovak Republic and Slovak Academy of Sciences (VEGA), contract No. 2/0069/22. The funding source had no influence on the writing of the manuscript.
Acknowledgments
We would greatly thank Joe Gracik for reading the manuscript carefully and helping with language editing. Figures 1 and 2 were created with BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Lynch SV, Pedersen O. The human intestinal microbiome in health and disease. N Engl J Med (2016) 375(24):2369–79. doi: 10.1056/NEJMra1600266
2. Haase S, Haghikia A, Wilck N, Muller DN, Linker RA. Impacts of microbiome metabolites on immune regulation and autoimmunity. Immunology (2018) 154(2):230–8. doi: 10.1111/imm.12933
3. Mima K, Ogino S, Nakagawa S, Sawayama H, Kinoshita K, Krashima R, et al. The role of intestinal bacteria in the development and progression of gastrointestinal tract neoplasms. Surg Oncol (2017) 26(4):368–76. doi: 10.1016/j.suronc.2017.07.011
4. Picardo SL, Coburn B, Hansen AR. The microbiome and cancer for clinicians. Crit Rev Oncol Hematol (2019) 141:1–12. doi: 10.1016/j.critrevonc.2019.06.004
5. de Martel C, Ferlay J, Franceschi S, Vignat J, Bray F, Forman D, et al. Global burden of cancers attributable to infections in 2008: A review and synthetic analysis. Lancet Oncol (2012) 13(6):607–15. doi: 10.1016/S1470-2045(12)70137-7
6. Dalmasso G, Cougnoux A, Delmas J, Darfeuille-Michaud A, Bonnet R. The bacterial genotoxin colibactin promotes colon tumor growth by modifying the tumor microenvironment. Gut Microbes (2014) 5(5):675–80. doi: 10.4161/19490976.2014.969989
7. Poore GD, Kopylova E, Zhu Q, Carpenter C, Fraraccio S, Wandro S, et al. Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature (2020) 579(7800):567–74. doi: 10.1038/s41586-020-2095-1
8. Nejman D, Livyatan I, Fuks G, Gavert N, Zwang Y, Geller LT, et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science (2020) 368(6494):973–80. doi: 10.1126/science.aay9189
9. Hanahan D. Hallmarks of cancer: New dimensions. Cancer Discov (2022) 12(1):31–46. doi: 10.1158/2159-8290.CD-21-1059
10. Sears CL, Garrett WS. Microbes, microbiota, and colon cancer. Cell Host Microbe (2014) 15(3):317–28. doi: 10.1016/j.chom.2014.02.007
11. Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti-Pd-1 immunotherapy in melanoma patients. Science (2018) 359(6371):97–103. doi: 10.1126/science.aan4236
12. Davar D, Dzutsev AK, McCulloch JA, Rodrigues RR, Chauvin JM, Morrison RM, et al. Fecal microbiota transplant overcomes resistance to anti-Pd-1 therapy in melanoma patients. Science (2021) 371(6529):595–602. doi: 10.1126/science.abf3363
13. Sevcikova A, Izoldova N, Stevurkova V, Kasperova B, Chovanec M, Ciernikova S, et al. The impact of the microbiome on resistance to cancer treatment with chemotherapeutic agents and immunotherapy. Int J Mol Sci (2022) 23(1):488. doi: 10.3390/ijms23010488
14. Baghban R, Roshangar L, Jahanban-Esfahlan R, Seidi K, Ebrahimi-Kalan A, Jaymand M, et al. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun Signal (2020) 18(1):59. doi: 10.1186/s12964-020-0530-4
15. Anderson NM, Simon MC. The tumor microenvironment. Curr Biol (2020) 30(16):R921–R5. doi: 10.1016/j.cub.2020.06.081
16. Korneev KV, Atretkhany KN, Drutskaya MS, Grivennikov SI, Kuprash DV, Nedospasov SA. Tlr-signaling and proinflammatory cytokines as drivers of tumorigenesis. Cytokine (2017) 89:127–35. doi: 10.1016/j.cyto.2016.01.021
17. LeBleu VS. Imaging the tumor microenvironment. Cancer J (2015) 21(3):174–8. doi: 10.1097/PPO.0000000000000118
18. Hinshaw DC, Shevde LA. The tumor microenvironment innately modulates cancer progression. Cancer Res (2019) 79(18):4557–66. doi: 10.1158/0008-5472.CAN-18-3962
19. Walker C, Mojares E, Del Rio Hernandez A. Role of extracellular matrix in development and cancer progression. Int J Mol Sci (2018) 19(10):3028. doi: 10.3390/ijms19103028
20. Casey T, Bond J, Tighe S, Hunter T, Lintault L, Patel O, et al. Molecular signatures suggest a major role for stromal cells in development of invasive breast cancer. Breast Cancer Res Treat (2009) 114(1):47–62. doi: 10.1007/s10549-008-9982-8
21. Naba A, Clauser KR, Hoersch S, Liu H, Carr SA, Hynes RO. The matrisome: In silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol Cell Proteomics (2012) 11(4):M111 014647. doi: 10.1074/mcp.M111.014647
22. Henke E, Nandigama R, Ergun S. Extracellular matrix in the tumor microenvironment and its impact on cancer therapy. Front Mol Biosci (2019) 6:160. doi: 10.3389/fmolb.2019.00160
23. Mao Y, Keller ET, Garfield DH, Shen K, Wang J. Stromal cells in tumor microenvironment and breast cancer. Cancer Metastasis Rev (2013) 32(1-2):303–15. doi: 10.1007/s10555-012-9415-3
24. Lakins MA, Ghorani E, Munir H, Martins CP, Shields JD. Cancer-associated fibroblasts induce antigen-specific deletion of Cd8 (+) T cells to protect tumour cells. Nat Commun (2018) 9(1):948. doi: 10.1038/s41467-018-03347-0
25. Peterson HI. The microcirculation of tumors. Microcirculation Cancer Metastasis United States: CRC Press (1991), 277–98.
26. Dvorak HF. Rous-Whipple award lecture. How tumors make bad blood vessels and stroma. Am J Pathol (2003) 162(6):1747–57. doi: 10.1016/s0002-9440(10)64309-x
27. Nagy JA, Dvorak AM, Dvorak HF. Vegf-a and the induction of pathological angiogenesis. Annu Rev Pathol (2007) 2:251–75. doi: 10.1146/annurev.pathol.2.010506.134925
28. Hanahan D, Coussens LM. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell (2012) 21(3):309–22. doi: 10.1016/j.ccr.2012.02.022
29. Ungefroren H, Sebens S, Seidl D, Lehnert H, Hass R. Interaction of tumor cells with the microenvironment. Cell Commun Signal (2011) 9:18. doi: 10.1186/1478-811X-9-18
30. Pascut D, Pratama MY, Vo NVT, Masadah R, Tiribelli C. The crosstalk between tumor cells and the microenvironment in hepatocellular carcinoma: The role of exosomal micrornas and their clinical implications. Cancers (Basel) (2020) 12(4):823. doi: 10.3390/cancers12040823
31. Chen Q, Li Y, Gao W, Chen L, Xu W, Zhu X. Exosome-mediated crosstalk between tumor and tumor-associated macrophages. Front Mol Biosci (2021) 8:764222. doi: 10.3389/fmolb.2021.764222
32. Rossi T, Vergara D, Fanini F, Maffia M, Bravaccini S, Pirini F. Microbiota-derived metabolites in tumor progression and metastasis. Int J Mol Sci (2020) 21(16):5786. doi: 10.3390/ijms21165786
33. Swidsinski A, Khilkin M, Kerjaschki D, Schreiber S, Ortner M, Weber J, et al. Association between intraepithelial escherichia coli and colorectal cancer. Gastroenterology (1998) 115(2):281–6. doi: 10.1016/s0016-5085(98)70194-5
34. Johns MS, Petrelli NJ. Microbiome and colorectal cancer: A review of the past, present, and future. Surg Oncol (2021) 37:101560. doi: 10.1016/j.suronc.2021.101560
35. Ciernikova S, Mego M, Hainova K, Adamcikova Z, Stevurkova V, Zajac V. Modification of microflora imbalance: Future directions for prevention and treatment of colorectal cancer? Neoplasma (2015) 62(3):345–52. doi: 10.4149/neo_2015_042
36. Kostic AD, Chun E, Robertson L, Glickman JN, Gallini CA, Michaud M, et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe (2013) 14(2):207–15. doi: 10.1016/j.chom.2013.07.007
37. Castellarin M, Warren RL, Freeman JD, Dreolini L, Krzywinski M, Strauss J, et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res (2012) 22(2):299–306. doi: 10.1101/gr.126516.111
38. Mima K, Sukawa Y, Nishihara R, Qian ZR, Yamauchi M, Inamura K, et al. Fusobacterium nucleatum and T cells in colorectal carcinoma. JAMA Oncol (2015) 1(5):653–61. doi: 10.1001/jamaoncol.2015.1377
39. Marchesi JR, Dutilh BE, Hall N, Peters WH, Roelofs R, Boleij A, et al. Towards the human colorectal cancer microbiome. PloS One (2011) 6(5):e20447. doi: 10.1371/journal.pone.0020447
40. Shah MM, Odoyo E, Ichinose Y. Epidemiology and pathogenesis of providencia alcalifaciens infections. Am J Trop Med Hyg (2019) 101(2):290–3. doi: 10.4269/ajtmh.18-0376
41. Burns MB, Lynch J, Starr TK, Knights D, Blekhman R. Virulence genes are a signature of the microbiome in the colorectal tumor microenvironment. Genome Med (2015) 7(1):55. doi: 10.1186/s13073-015-0177-8
42. Geng J, Fan H, Tang X, Zhai H, Zhang Z. Diversified pattern of the human colorectal cancer microbiome. Gut Pathog (2013) 5(1):2. doi: 10.1186/1757-4749-5-2
43. Gao Z, Guo B, Gao R, Zhu Q, Qin H. Microbiota disbiosis is associated with colorectal cancer. Front Microbiol (2015) 6:20. doi: 10.3389/fmicb.2015.00020
44. Wachsmannova L, Majek J, Zajac V, Stevurkova V, Ciernikova S. The study of bacteria in biopsies from Slovak colorectal adenoma and carcinoma patients. Neoplasma (2018) 65(4):644–8. doi: 10.4149/neo_2018_1801174N34
45. Wachsmannova L, Soltys K, Stevurkova V, Ciernikova S. Gut microbiome analysis in biopsies from Slovak colorectal adenomas and carcinomas patients. J Clin Gastroenterol (2020) 54(Supplement 1):S16–6. doi: 10.1097/MCG.0000000000001292
46. Sakamoto Y, Mima K, Ishimoto T, Ogata Y, Imai K, Miyamoto Y, et al. Relationship between fusobacterium nucleatum and antitumor immunity in colorectal cancer liver metastasis. Cancer Sci (2021) 112(11):4470–7. doi: 10.1111/cas.15126
47. Lin Y, Ma C, Bezabeh T, Wang Z, Liang J, Huang Y, et al. (1) h nmr-based metabolomics reveal overlapping discriminatory metabolites and metabolic pathway disturbances between colorectal tumor tissues and fecal samples. Int J Cancer (2019) 145(6):1679–89. doi: 10.1002/ijc.32190
48. Lin Y, Ma C, Liu C, Wang Z, Yang J, Liu X, et al. Nmr-based fecal metabolomics fingerprinting as predictors of earlier diagnosis in patients with colorectal cancer. Oncotarget (2016) 7(20):29454–64. doi: 10.18632/oncotarget.8762
49. Yang J, Wei H, Zhou Y, Szeto CH, Li C, Lin Y, et al. High-fat diet promotes colorectal tumorigenesis through modulating gut microbiota and metabolites. Gastroenterology (2022) 162(1):135–49.e2. doi: 10.1053/j.gastro.2021.08.041
50. Nguyen LH, Ma W, Wang DD, Cao Y, Mallick H, Gerbaba TK, et al. Association between sulfur-metabolizing bacterial communities in stool and risk of distal colorectal cancer in men. Gastroenterology (2020) 158(5):1313–25. doi: 10.1053/j.gastro.2019.12.029
51. Tjalsma H, Scholler-Guinard M, Lasonder E, Ruers TJ, Willems HL, Swinkels DW. Profiling the humoral immune response in colon cancer patients: Diagnostic antigens from streptococcus bovis. Int J Cancer (2006) 119(9):2127–35. doi: 10.1002/ijc.22116
52. Kim M, Vogtmann E, Ahlquist DA, Devens ME, Kisiel JB, Taylor WR, et al. Fecal metabolomic signatures in colorectal adenoma patients are associated with gut microbiota and early events of colorectal cancer pathogenesis. mBio (2020) 11(1):e03186–19. doi: 10.1128/mBio.03186-19
53. Ciernikova S, Novisedlakova M, Cholujova D, Stevurkova V, Mego M. The emerging role of microbiota and microbiome in pancreatic ductal adenocarcinoma. Biomedicines (2020) 8(12):565. doi: 10.3390/biomedicines8120565
54. Nilsson HO, Stenram U, Ihse I, Wadstrom T. Helicobacter species ribosomal DNA in the pancreas, stomach and duodenum of pancreatic cancer patients. World J Gastroenterol (2006) 12(19):3038–43. doi: 10.3748/wjg.v12.i19.3038
55. Takayama S, Takahashi H, Matsuo Y, Okada Y, Manabe T. Effects of helicobacter pylori infection on human pancreatic cancer cell line. Hepatogastroenterology (2007) 54(80):2387–91.
56. Brennan CA, Garrett WS. Fusobacterium nucleatum - symbiont, opportunist and oncobacterium. Nat Rev Microbiol (2019) 17(3):156–66. doi: 10.1038/s41579-018-0129-6
57. Mitsuhashi K, Nosho K, Sukawa Y, Matsunaga Y, Ito M, Kurihara H, et al. Association of fusobacterium species in pancreatic cancer tissues with molecular features and prognosis. Oncotarget (2015) 6(9):7209–20. doi: 10.18632/oncotarget.3109
58. Geller LT, Barzily-Rokni M, Danino T, Jonas OH, Shental N, Nejman D, et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science (2017) 357(6356):1156–60. doi: 10.1126/science.aah5043
59. Thomas RM, Gharaibeh RZ, Gauthier J, Beveridge M, Pope JL, Guijarro MV, et al. Intestinal microbiota enhances pancreatic carcinogenesis in preclinical models. Carcinogenesis (2018) 39(8):1068–78. doi: 10.1093/carcin/bgy073
60. Pushalkar S, Hundeyin M, Daley D, Zambirinis CP, Kurz E, Mishra A, et al. The pancreatic cancer microbiome promotes oncogenesis by induction of innate and adaptive immune suppression. Cancer Discov (2018) 8(4):403–16. doi: 10.1158/2159-8290.CD-17-1134
61. Riquelme E, Zhang Y, Zhang L, Montiel M, Zoltan M, Dong W, et al. Tumor microbiome diversity and composition influence pancreatic cancer outcomes. Cell (2019) 178(4):795–806.e12. doi: 10.1016/j.cell.2019.07.008
62. Guo W, Zhang Y, Guo S, Mei Z, Liao H, Dong H, et al. Tumor microbiome contributes to an aggressive phenotype in the basal-like subtype of pancreatic cancer. Commun Biol (2021) 4(1):1019. doi: 10.1038/s42003-021-02557-5
63. Panebianco C, Villani A, Pisati F, Orsenigo F, Ulaszewska M, Latiano TP, et al. Butyrate, a postbiotic of intestinal bacteria, affects pancreatic cancer and gemcitabine response in in vitro and in vivo models. BioMed Pharmacother (2022) 151:113163. doi: 10.1016/j.biopha.2022.113163
64. Hezaveh K, Shinde RS, Klotgen A, Halaby MJ, Lamorte S, Ciudad MT, et al. Tryptophan-derived microbial metabolites activate the aryl hydrocarbon receptor in tumor-associated macrophages to suppress anti-tumor immunity. Immunity (2022) 55(2):324–40.e8. doi: 10.1016/j.immuni.2022.01.006
65. Kesh K, Mendez R, Mateo-Victoriano B, Garrido VT, Durden B, Gupta VK, et al. Obesity enriches for tumor protective microbial metabolites and treatment refractory cells to confer therapy resistance in pdac. Gut Microbes (2022) 14(1):2096328. doi: 10.1080/19490976.2022.2096328
66. Mirji G, Worth A, Bhat SA, El Sayed M, Kannan T, Goldman AR, et al. The microbiome-derived metabolite tmao drives immune activation and boosts responses to immune checkpoint blockade in pancreatic cancer. Sci Immunol (2022) 7(75):eabn0704. doi: 10.1126/sciimmunol.abn0704
67. Yang J, Zhou X, Liu X, Ling Z, Ji F. Role of the gastric microbiome in gastric cancer: From carcinogenesis to treatment. Front Microbiol (2021) 12:641322. doi: 10.3389/fmicb.2021.641322
68. Kusters JG, van Vliet AH, Kuipers EJ. Pathogenesis of helicobacter pylori infection. Clin Microbiol Rev (2006) 19(3):449–90. doi: 10.1128/CMR.00054-05
69. Chen XH, Wang A, Chu AN, Gong YH, Yuan Y. Mucosa-associated microbiota in gastric cancer tissues compared with non-cancer tissues. Front Microbiol (2019) 10:1261. doi: 10.3389/fmicb.2019.01261
70. Ling Z, Shao L, Liu X, Cheng Y, Yan C, Mei Y, et al. Regulatory T cells and plasmacytoid dendritic cells within the tumor microenvironment in gastric cancer are correlated with gastric microbiota dysbiosis: A preliminary study. Front Immunol (2019) 10:533. doi: 10.3389/fimmu.2019.00533
71. Li TH, Qin Y, Sham PC, Lau KS, Chu KM, Leung WK. Alterations in gastric microbiota after h. pylori eradication and in different histological stages of gastric carcinogenesis. Sci Rep (2017) 7:44935. doi: 10.1038/srep44935
72. Ferreira RM, Pereira-Marques J, Pinto-Ribeiro I, Costa JL, Carneiro F, Machado JC, et al. Gastric microbial community profiling reveals a dysbiotic cancer-associated microbiota. Gut (2018) 67(2):226–36. doi: 10.1136/gutjnl-2017-314205
73. Jo HJ, Kim J, Kim N, Park JH, Nam RH, Seok YJ, et al. Analysis of gastric microbiota by pyrosequencing: Minor role of bacteria other than helicobacter pylori in the gastric carcinogenesis. Helicobacter (2016) 21(5):364–74. doi: 10.1111/hel.12293
74. Wang Z, Gao X, Zeng R, Wu Q, Sun H, Wu W, et al. Changes of the gastric mucosal microbiome associated with histological stages of gastric carcinogenesis. Front Microbiol (2020) 11:997. doi: 10.3389/fmicb.2020.00997
75. Wen J, Lau HC, Peppelenbosch M, Yu J. Gastric microbiota beyond h. pylori: An emerging critical character in gastric carcinogenesis. Biomedicines (2021) 9(11):1680. doi: 10.3390/biomedicines9111680
76. Dai D, Yang Y, Yu J, Dang T, Qin W, Teng L, et al. Interactions between gastric microbiota and metabolites in gastric cancer. Cell Death Dis (2021) 12(12):1104. doi: 10.1038/s41419-021-04396-y
77. Chu F, Li Y, Meng X, Li Y, Li T, Zhai M, et al. Gut microbial dysbiosis and changes in fecal metabolic phenotype in precancerous lesions of gastric cancer induced with n-Methyl-N'-Nitro-N-Nitrosoguanidine, sodium salicylate, ranitidine, and irregular diet. Front Physiol (2021) 12:733979. doi: 10.3389/fphys.2021.733979
78. Yang Y, Dai D, Jin W, Huang Y, Zhang Y, Chen Y, et al. Microbiota and metabolites alterations in proximal and distal gastric cancer patients. J Transl Med (2022) 20(1):439. doi: 10.1186/s12967-022-03650-x
79. Hieken TJ, Chen J, Hoskin TL, Walther-Antonio M, Johnson S, Ramaker S, et al. The microbiome of aseptically collected human breast tissue in benign and malignant disease. Sci Rep (2016) 6:30751. doi: 10.1038/srep30751
80. Urbaniak C, Cummins J, Brackstone M, Macklaim JM, Gloor GB, Baban CK, et al. Microbiota of human breast tissue. Appl Environ Microbiol (2014) 80(10):3007–14. doi: 10.1128/AEM.00242-14
81. Laborda-Illanes A, Sanchez-Alcoholado L, Dominguez-Recio ME, Jimenez-Rodriguez B, Lavado R, Comino-Mendez I, et al. Breast and gut microbiota action mechanisms in breast cancer pathogenesis and treatment. Cancers (Basel) (2020) 12(9):2465. doi: 10.3390/cancers12092465
82. Rao Malla R, Marni R, Kumari S, Chakraborty A, Lalitha P. Microbiome assisted tumor microenvironment: Emerging target of breast cancer. Clin Breast Cancer (2022) 22(3):200–11. doi: 10.1016/j.clbc.2021.09.002
83. Xuan C, Shamonki JM, Chung A, Dinome ML, Chung M, Sieling PA, et al. Microbial dysbiosis is associated with human breast cancer. PloS One (2014) 9(1):e83744. doi: 10.1371/journal.pone.0083744
84. Kinjo Y, Wu D, Kim G, Xing GW, Poles MA, Ho DD, et al. Recognition of bacterial glycosphingolipids by natural killer T cells. Nature (2005) 434(7032):520–5. doi: 10.1038/nature03407
85. Hix LM, Shi YH, Brutkiewicz RR, Stein PL, Wang CR, Zhang M. Cd1d-expressing breast cancer cells modulate nkt cell-mediated antitumor immunity in a murine model of breast cancer metastasis. PloS One (2011) 6(6):e20702. doi: 10.1371/journal.pone.0020702
86. Urbaniak C, Gloor GB, Brackstone M, Scott L, Tangney M, Reid G. The microbiota of breast tissue and its association with breast cancer. Appl Environ Microbiol (2016) 82(16):5039–48. doi: 10.1128/AEM.01235-16
87. Hadzega D, Minarik G, Karaba M, Kalavska K, Benca J, Ciernikova S, et al. Uncovering microbial composition in human breast cancer primary tumour tissue using transcriptomic rna-seq. Int J Mol Sci (2021) 22(16):9058. doi: 10.3390/ijms22169058
88. Chiba A, Bawaneh A, Velazquez C, Clear KYJ, Wilson AS, Howard-McNatt M, et al. Neoadjuvant chemotherapy shifts breast tumor microbiota populations to regulate drug responsiveness and the development of metastasis. Mol Cancer Res (2020) 18(1):130–9. doi: 10.1158/1541-7786.MCR-19-0451
89. Rubovszky G, Horvath Z. Recent advances in the neoadjuvant treatment of breast cancer. J Breast Cancer (2017) 20(2):119–31. doi: 10.4048/jbc.2017.20.2.119
90. Ravnik Z, Muthiah I, Dhanaraj P. Computational studies on bacterial secondary metabolites against breast cancer. J Biomol Struct Dyn (2021) 39(18):7056–64. doi: 10.1080/07391102.2020.1805361
91. Wang H, Rong X, Zhao G, Zhou Y, Xiao Y, Ma D, et al. The microbial metabolite trimethylamine n-oxide promotes antitumor immunity in triple-negative breast cancer. Cell Metab (2022) 34(4):581–94.e8. doi: 10.1016/j.cmet.2022.02.010
92. Miko E, Vida A, Kovacs T, Ujlaki G, Trencsenyi G, Marton J, et al. Lithocholic acid, a bacterial metabolite reduces breast cancer cell proliferation and aggressiveness. Biochim Biophys Acta Bioenerg (2018) 1859(9):958–74. doi: 10.1016/j.bbabio.2018.04.002
93. Juan Z, Chen J, Ding B, Yongping L, Liu K, Wang L, et al. Probiotic supplement attenuates chemotherapy-related cognitive impairment in patients with breast cancer: A randomised, double-blind, and placebo-controlled trial. Eur J Cancer (2022) 161:10–22. doi: 10.1016/j.ejca.2021.11.006
94. Whiteside SA, McGinniss JE, Collman RG. The lung microbiome: Progress and promise. J Clin Invest (2021) 131(15):e150473. doi: 10.1172/JCI150473
95. Hilty M, Burke C, Pedro H, Cardenas P, Bush A, Bossley C, et al. Disordered microbial communities in asthmatic airways. PloS One (2010) 5(1):e8578. doi: 10.1371/journal.pone.0008578
96. Segal LN, Alekseyenko AV, Clemente JC, Kulkarni R, Wu B, Gao Z, et al. Enrichment of lung microbiome with supraglottic taxa is associated with increased pulmonary inflammation. Microbiome (2013) 1(1):19. doi: 10.1186/2049-2618-1-19
97. Dickson RP, Erb-Downward JR, Freeman CM, McCloskey L, Beck JM, Huffnagle GB, et al. Spatial variation in the healthy human lung microbiome and the adapted island model of lung biogeography. Ann Am Thorac Soc (2015) 12(6):821–30. doi: 10.1513/AnnalsATS.201501-029OC
98. Mathieu E, Escribano-Vazquez U, Descamps D, Cherbuy C, Langella P, Riffault S, et al. Paradigms of lung microbiota functions in health and disease, particularly, in asthma. Front Physiol (2018) 9:1168. doi: 10.3389/fphys.2018.01168
99. Peters BA, Hayes RB, Goparaju C, Reid C, Pass HI, Ahn J. The microbiome in lung cancer tissue and recurrence-free survival. Cancer Epidemiol Biomarkers Prev (2019) 28(4):731–40. doi: 10.1158/1055-9965.EPI-18-0966
100. Greathouse KL, White JR, Vargas AJ, Bliskovsky VV, Beck JA, von Muhlinen N, et al. Author correction: Interaction between the microbiome and Tp53 in human lung cancer. Genome Biol (2020) 21(1):41. doi: 10.1186/s13059-020-01961-0
101. Kovaleva O, Podlesnaya P, Rashidova M, Samoilova D, Petrenko A, Zborovskaya I, et al. Lung microbiome differentially impacts survival of patients with non-small cell lung cancer depending on tumor stroma phenotype. Biomedicines (2020) 8(9)349. doi: 10.3390/biomedicines8090349
102. Yu G, Gail MH, Consonni D, Carugno M, Humphrys M, Pesatori AC, et al. Characterizing human lung tissue microbiota and its relationship to epidemiological and clinical features. Genome Biol (2016) 17(1):163. doi: 10.1186/s13059-016-1021-1
103. Najafi S, Abedini F, Azimzadeh Jamalkandi S, Shariati P, Ahmadi A, Gholami Fesharaki M. The composition of lung microbiome in lung cancer: A systematic review and meta-analysis. BMC Microbiol (2021) 21(1):315. doi: 10.1186/s12866-021-02375-z
104. Apopa PL, Alley L, Penney RB, Arnaoutakis K, Steliga MA, Jeffus S, et al. Parp1 is up-regulated in non-small cell lung cancer tissues in the presence of the cyanobacterial toxin microcystin. Front Microbiol (2018) 9:1757. doi: 10.3389/fmicb.2018.01757
105. Huang D, Su X, Yuan M, Zhang S, He J, Deng Q, et al. The characterization of lung microbiome in lung cancer patients with different clinicopathology. Am J Cancer Res (2019) 9(9):2047–63.
106. Liu HX, Tao LL, Zhang J, Zhu YG, Zheng Y, Liu D, et al. Difference of lower airway microbiome in bilateral protected specimen brush between lung cancer patients with unilateral lobar masses and control subjects. Int J Cancer (2018) 142(4):769–78. doi: 10.1002/ijc.31098
107. Zhang W, Luo J, Dong X, Zhao S, Hao Y, Peng C, et al. Salivary microbial dysbiosis is associated with systemic inflammatory markers and predicted oral metabolites in non-small cell lung cancer patients. J Cancer (2019) 10(7):1651–62. doi: 10.7150/jca.28077
108. Liu F, Li J, Guan Y, Lou Y, Chen H, Xu M, et al. Dysbiosis of the gut microbiome is associated with tumor biomarkers in lung cancer. Int J Biol Sci (2019) 15(11):2381–92. doi: 10.7150/ijbs.35980
109. Ge H, Shi M, Ma M, Lian XY, Zhang Z. Evaluation of the antiproliferative activity of 106 marine microbial metabolites against human lung cancer cells and potential antiproliferative mechanism of purpuride G. Bioorg Med Chem Lett (2021) 39:127915. doi: 10.1016/j.bmcl.2021.127915
110. Chen S, Gui R, Zhou XH, Zhang JH, Jiang HY, Liu HT, et al. Combined microbiome and metabolome analysis reveals a novel interplay between intestinal flora and serum metabolites in lung cancer. Front Cell Infect Microbiol (2022) 12:885093. doi: 10.3389/fcimb.2022.885093
111. Lee SH, Cho SY, Yoon Y, Park C, Sohn J, Jeong JJ, et al. Bifidobacterium bifidum strains synergize with immune checkpoint inhibitors to reduce tumour burden in mice. Nat Microbiol (2021) 6(3):277–88. doi: 10.1038/s41564-020-00831-6
112. Branton WG, Ellestad KK, Maingat F, Wheatley BM, Rud E, Warren RL, et al. Brain microbial populations in Hiv/Aids: Alpha-proteobacteria predominate independent of host immune status. PloS One (2013) 8(1):e54673. doi: 10.1371/journal.pone.0054673
113. Zajac V, Mego M, Martinicky D, Stevurkova V, Ciernikova S, Ujhazy E, et al. Testing of bacteria isolated from Hiv/Aids patients in experimental models. Neuro Endocrinol Lett (2006) 27 Suppl 2:61–4.
114. Roberts RC, Farmer CB, Walker CK. The human brain microbiome; there are bacteria in our brains. Conf Rep at Soc Neurosci Meeting (2018).
115. Yust-Katz S, Gigi E, Rosenberg D, Kanner AA, Laviv Y, Benouaich-Amiel A, et al. TAMI-40. tumor microbiome and glioblastoma (GBM). Neuro-Oncology (2020) 22(Supplement 2):ii221. doi: 10.1093/neuonc/noaa215.928
116. Zhao J, He D, Lai HM, Xu Y, Luo Y, Li T, et al. Comprehensive histological imaging of native microbiota in human glioma. J Biophotonics (2022) 15(4):e202100351. doi: 10.1002/jbio.202100351
117. Parker A, Fonseca S, Carding SR. Gut microbes and metabolites as modulators of blood-brain barrier integrity and brain health. Gut Microbes (2020) 11(2):135–57. doi: 10.1080/19490976.2019.1638722
118. Dono A, Nickles J, Rodriguez-Armendariz AG, McFarland BC, Ajami NJ, Ballester LY, et al. Glioma and the gut-brain axis: Opportunities and future perspectives. Neurooncol Adv (2022) 4(1):vdac054. doi: 10.1093/noajnl/vdac054
119. Patrizz A, Dono A, Zorofchian S, Hines G, Takayasu T, Husein N, et al. Glioma and temozolomide induced alterations in gut microbiome. Sci Rep (2020) 10(1):21002. doi: 10.1038/s41598-020-77919-w
120. Lyu Y, Yang H, Chen L. Metabolic regulation on the immune environment of glioma through gut microbiota. Semin Cancer Biol (2021) 86(Pt 2):990–7. doi: 10.1016/j.semcancer.2021.05.005
121. Pommepuy I, Terro F, Petit B, Trimoreau F, Bellet V, Robert S, et al. Brefeldin a induces apoptosis and cell cycle blockade in glioblastoma cell lines. Oncology (2003) 64(4):459–67. doi: 10.1159/000070307
122. Lai PH, Weng HH, Chen CY, Hsu SS, Ding S, Ko CW, et al. In vivo differentiation of aerobic brain abscesses and necrotic glioblastomas multiforme using proton Mr spectroscopic imaging. AJNR Am J Neuroradiol (2008) 29(8):1511–8. doi: 10.3174/ajnr.A1130
123. Himmelreich U, Accurso R, Malik R, Dolenko B, Somorjai RL, Gupta RK, et al. Identification of staphylococcus aureus brain abscesses: Rat and human studies with 1h Mr spectroscopy. Radiology (2005) 236(1):261–70. doi: 10.1148/radiol.2361040869
124. Chase D, Goulder A, Zenhausern F, Monk B, Herbst-Kralovetz M. The vaginal and gastrointestinal microbiomes in gynecologic cancers: A review of applications in etiology, symptoms and treatment. Gynecol Oncol (2015) 138(1):190–200. doi: 10.1016/j.ygyno.2015.04.036
125. Cheng H, Wang Z, Cui L, Wen Y, Chen X, Gong F, et al. Opportunities and challenges of the human microbiome in ovarian cancer. Front Oncol (2020) 10:163. doi: 10.3389/fonc.2020.00163
126. Borella F, Carosso AR, Cosma S, Preti M, Collemi G, Cassoni P, et al. Gut microbiota and gynecological cancers: A summary of pathogenetic mechanisms and future directions. ACS Infect Dis (2021) 7(5):987–1009. doi: 10.1021/acsinfecdis.0c00839
127. Zhou B, Sun C, Huang J, Xia M, Guo E, Li N, et al. The biodiversity composition of microbiome in ovarian carcinoma patients. Sci Rep (2019) 9(1):1691. doi: 10.1038/s41598-018-38031-2
128. Wang Q, Zhao L, Han L, Fu G, Tuo X, Ma S, et al. The differential distribution of bacteria between cancerous and noncancerous ovarian tissues in situ. J Ovarian Res (2020) 13(1):8. doi: 10.1186/s13048-019-0603-4
129. Banerjee S, Tian T, Wei Z, Shih N, Feldman MD, Alwine JC, et al. The ovarian cancer oncobiome. Oncotarget (2017) 8(22):36225–45. doi: 10.18632/oncotarget.16717
130. Nene NR, Reisel D, Leimbach A, Franchi D, Jones A, Evans I, et al. Association between the cervicovaginal microbiome, Brca1 mutation status, and risk of ovarian cancer: A case-control study. Lancet Oncol (2019) 20(8):1171–82. doi: 10.1016/S1470-2045(19)30340-7
131. Brewster WR, Burkett WC, Ko EM, Bae-Jump V, Nicole McCoy A, Keku TO. An evaluation of the microbiota of the upper reproductive tract of women with and without epithelial ovarian cancer. Gynecol Oncol Rep (2022) 42:101017. doi: 10.1016/j.gore.2022.101017
132. Miao R, Badger TC, Groesch K, Diaz-Sylvester PL, Wilson T, Ghareeb A, et al. Assessment of peritoneal microbial features and tumor marker levels as potential diagnostic tools for ovarian cancer. PloS One (2020) 15(1):e0227707. doi: 10.1371/journal.pone.0227707
133. Chambers LM, Esakov Rhoades EL, Bharti R, Braley C, Tewari S, Trestan L, et al. Disruption of the gut microbiota confers cisplatin resistance in epithelial ovarian cancer. Cancer Res (2022). doi: 10.1158/0008-5472.CAN-22-0455
134. Chen YF, Wang SY, Shen H, Yao XF, Zhang FL, Lai D. The marine-derived fungal metabolite, terrein, inhibits cell proliferation and induces cell cycle arrest in human ovarian cancer cells. Int J Mol Med (2014) 34(6):1591–8. doi: 10.3892/ijmm.2014.1964
135. Slupsky CM, Steed H, Wells TH, Dabbs K, Schepansky A, Capstick V, et al. Urine metabolite analysis offers potential early diagnosis of ovarian and breast cancers. Clin Cancer Res (2010) 16(23):5835–41. doi: 10.1158/1078-0432.CCR-10-1434
136. Nicholson-Guthrie CS, Guthrie GD, Sutton GP, Baenziger JC. Urine gaba levels in ovarian cancer patients: Elevated gaba in malignancy. Cancer Lett (2001) 162(1):27–30. doi: 10.1016/s0304-3835(00)00620-0
137. D'Amico F, Perrone AM, Rampelli S, Coluccelli S, Barone M, Ravegnini G, et al. Gut microbiota dynamics during chemotherapy in epithelial ovarian cancer patients are related to therapeutic outcome. Cancers (Basel) (2021) 13(16):3999. doi: 10.3390/cancers13163999
138. Boutriq S, Gonzalez-Gonzalez A, Plaza-Andrades I, Laborda-Illanes A, Sanchez-Alcoholado L, Peralta-Linero J, et al. Gut and endometrial microbiome dysbiosis: A new emergent risk factor for endometrial cancer. J Pers Med (2021) 11(7):659. doi: 10.3390/jpm11070659
139. Chen C, Song X, Wei W, Zhong H, Dai J, Lan Z, et al. The microbiota continuum along the female reproductive tract and its relation to uterine-related diseases. Nat Commun (2017) 8(1):875. doi: 10.1038/s41467-017-00901-0
140. Sola-Leyva A, Andres-Leon E, Molina NM, Terron-Camero LC, Plaza-Diaz J, Saez-Lara MJ, et al. Mapping the entire functionally active endometrial microbiota. Hum Reprod (2021) 36(4):1021–31. doi: 10.1093/humrep/deaa372
141. Li C, Gu Y, He Q, Huang J, Song Y, Wan X, et al. Integrated analysis of microbiome and transcriptome data reveals the interplay between commensal bacteria and fibrin degradation in endometrial cancer. Front Cell Infect Microbiol (2021) 11:748558. doi: 10.3389/fcimb.2021.748558
142. Lu W, He F, Lin Z, Liu S, Tang L, Huang Y, et al. Dysbiosis of the endometrial microbiota and its association with inflammatory cytokines in endometrial cancer. Int J Cancer (2021) 148(7):1708–16. doi: 10.1002/ijc.33428
143. Walsh DM, Hokenstad AN, Chen J, Sung J, Jenkins GD, Chia N, et al. Postmenopause as a key factor in the composition of the endometrial cancer microbiome (Ecbiome). Sci Rep (2019) 9(1):19213. doi: 10.1038/s41598-019-55720-8
144. Walther-Antonio MR, Chen J, Multinu F, Hokenstad A, Distad TJ, Cheek EH, et al. Potential contribution of the uterine microbiome in the development of endometrial cancer. Genome Med (2016) 8(1):122. doi: 10.1186/s13073-016-0368-y
145. Hawkins GM, Burkett WC, McCoy AN, Nichols HB, Olshan AF, Broaddus R, et al. Differences in the microbial profiles of early stage endometrial cancers between black and white women. Gynecol Oncol (2022) 165(2):248–56. doi: 10.1016/j.ygyno.2022.02.021
146. Hawkins GM, Clark LH, Sullivan SA, Keku TO, Brewster WR, Bae-Jump VL. Impact of ethnicity and obesity on the uterine microbiome in women and mice with endometrial cancer. Gynecol Oncol (2019) 154(1):77. doi: 10.1016/j.ygyno.2019.04.181
147. Caselli E, Soffritti I, D'Accolti M, Piva I, Greco P, Bonaccorsi G. Atopobium vaginae and porphyromonas somerae induce proinflammatory cytokines expression in endometrial cells: A possible implication for endometrial cancer? Cancer Manag Res (2019) 11:8571–5. doi: 10.2147/CMAR.S217362
148. Schwabe RF, Jobin C. The microbiome and cancer. Nat Rev Cancer (2013) 13(11):800–12. doi: 10.1038/nrc3610
149. Zhao SS, Chen L, Yang J, Wu ZH, Wang XY, Zhang Q, et al. Altered gut microbial profile accompanied by abnormal fatty acid metabolism activity exacerbates endometrial cancer progression. Microbiol Spectr: ASM Journals (2022):e0261222. doi: 10.1128/spectrum.02612-22
150. Alauddin M, Okumura T, Rajaxavier J, Khozooei S, Poschel S, Takeda S, et al. Gut bacterial metabolite urolithin a decreases actin polymerization and migration in cancer cells. Mol Nutr Food Res (2020) 64(7):e1900390. doi: 10.1002/mnfr.201900390
151. Cai S, Yang Y, Kong Y, Guo Q, Xu Y, Xing P, et al. Gut bacteria erysipelatoclostridium and its related metabolite ptilosteroid a could predict radiation-induced intestinal injury. Front Public Health (2022) 10:862598. doi: 10.3389/fpubh.2022.862598
152. Aragon IM, Herrera-Imbroda B, Queipo-Ortuno MI, Castillo E, Del Moral JS, Gomez-Millan J, et al. The urinary tract microbiome in health and disease. Eur Urol Focus (2018) 4(1):128–38. doi: 10.1016/j.euf.2016.11.001
153. Shrestha E, White JR, Yu SH, Kulac I, Ertunc O, De Marzo AM, et al. Profiling the urinary microbiome in men with positive versus negative biopsies for prostate cancer. J Urol (2018) 199(1):161–71. doi: 10.1016/j.juro.2017.08.001
154. Pearce MM, Hilt EE, Rosenfeld AB, Zilliox MJ, Thomas-White K, Fok C, et al. The female urinary microbiome: A comparison of women with and without urgency urinary incontinence. mBio (2014) 5(4):e01283–14. doi: 10.1128/mBio.01283-14
155. Che B, Zhang W, Xu S, Yin J, He J, Huang T, et al. Prostate microbiota and prostate cancer: A new trend in treatment. Front Oncol (2021) 11:805459. doi: 10.3389/fonc.2021.805459
156. Tantengco OAG, Aquino IMC, de Castro Silva M, Rojo RD, Abad CLR. Association of mycoplasma with prostate cancer: A systematic review and meta-analysis. Cancer Epidemiol (2021) 75:102021. doi: 10.1016/j.canep.2021.102021
157. Schatten H. Brief overview of prostate cancer statistics, grading, diagnosis and treatment strategies. Adv Exp Med Biol (2018) 1095:1–14. doi: 10.1007/978-3-319-95693-0_1
158. Hochreiter WW, Duncan JL, Schaeffer AJ. Evaluation of the bacterial flora of the prostate using a 16s rrna gene based polymerase chain reaction. J Urol (2000) 163(1):127–30. doi: 10.1016/S0022-5347(05)67987-6
159. Feng Y, Ramnarine VR, Bell R, Volik S, Davicioni E, Hayes VM, et al. Metagenomic and metatranscriptomic analysis of human prostate microbiota from patients with prostate cancer. BMC Genomics (2019) 20(1):146. doi: 10.1186/s12864-019-5457-z
160. Cavarretta I, Ferrarese R, Cazzaniga W, Saita D, Luciano R, Ceresola ER, et al. The microbiome of the prostate tumor microenvironment. Eur Urol (2017) 72(4):625–31. doi: 10.1016/j.eururo.2017.03.029
161. Shannon BA, Garrett KL, Cohen RJ. Links between propionibacterium acnes and prostate cancer. Future Oncol (2006) 2(2):225–32. doi: 10.2217/14796694.2.2.225
162. Alanee S, El-Zawahry A, Dynda D, Dabaja A, McVary K, Karr M, et al. A prospective study to examine the association of the urinary and fecal microbiota with prostate cancer diagnosis after transrectal biopsy of the prostate using 16srna gene analysis. Prostate (2019) 79(1):81–7. doi: 10.1002/pros.23713
163. Matsushita M, Fujita K, Hatano K, De Velasco MA, Uemura H, Nonomura N. Connecting the dots between the gut-Igf-1-Prostate axis: A role of igf-1 in prostate carcinogenesis. Front Endocrinol (Lausanne) (2022) 13:852382. doi: 10.3389/fendo.2022.852382
164. Liss MA, White JR, Goros M, Gelfond J, Leach R, Johnson-Pais T, et al. Metabolic biosynthesis pathways identified from fecal microbiome associated with prostate cancer. Eur Urol (2018) 74(5):575–82. doi: 10.1016/j.eururo.2018.06.033
165. Matsushita M, Fujita K, Motooka D, Hatano K, Fukae S, Kawamura N, et al. The gut microbiota associated with high-Gleason prostate cancer. Cancer Sci (2021) 112(8):3125–35. doi: 10.1111/cas.14998
166. Yumba-Mpanga A, Struck-Lewicka W, Wawrzyniak R, Markuszewski M, Roslan M, Kaliszan R, et al. Metabolomic heterogeneity of urogenital tract cancers analyzed by complementary chromatographic techniques coupled with mass spectrometry. Curr Med Chem (2019) 26(1):216–31. doi: 10.2174/0929867324666171006150326
167. Sugiyama Y, Nagata Y, Fukuta F, Takayanagi A, Masumori N, Tsukamoto T, et al. Counts of slackia sp. strain natts in intestinal flora are correlated to serum concentrations of equol both in prostate cancer cases and controls in Japanese men. Asian Pac J Cancer Prev (2014) 15(6):2693–7. doi: 10.7314/apjcp.2014.15.6.2693
168. Bajic P, Wolfe AJ, Gupta GN. The urinary microbiome: Implications in bladder cancer pathogenesis and therapeutics. Urology (2019) 126:10–5. doi: 10.1016/j.urology.2018.12.034
169. Raoult D. Is there a link between urinary microbiota and bladder cancer? Eur J Epidemiol (2017) 32(3):255. doi: 10.1007/s10654-016-0213-z
170. Parra-Grande M, Ore-Arce M, Martinez-Priego L, D'Auria G, Rossello-Mora R, Lillo M, et al. Profiling the bladder microbiota in patients with bladder cancer. Front Microbiol (2021) 12:718776. doi: 10.3389/fmicb.2021.718776
171. Mansour B, Monyok A, Makra N, Gajdacs M, Vadnay I, Ligeti B, et al. Bladder cancer-related microbiota: Examining differences in urine and tissue samples. Sci Rep (2020) 10(1):11042. doi: 10.1038/s41598-020-67443-2
172. Peng J, Chen YT, Chen CL, Li L. Development of a universal metabolome-standard method for long-term lc-Ms metabolome profiling and its application for bladder cancer urine-Metabolite-Biomarker discovery. Anal Chem (2014) 86(13):6540–7. doi: 10.1021/ac5011684
173. Mager LF, Burkhard R, Pett N, Cooke NCA, Brown K, Ramay H, et al. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science (2020) 369(6510):1481–9. doi: 10.1126/science.abc3421
174. Abdel-Tawab GA, Aboul-Azm T, Ebied SA, el-Toukhy MA, Abdel-Hamied HA, el-Kholy ZA, et al. The correlation between certain tryptophan metabolites and the n-nitrosamine content in the urine of bilharzial bladder cancer patients. J Urol (1986) 135(4):826–30. doi: 10.1016/s0022-5347(17)45869-1
175. Lawrie CA, Renwick AG, Sims J. The urinary excretion of bacterial amino-acid metabolites by rats fed saccharin in the diet. Food Chem Toxicol (1985) 23(4-5):445–50. doi: 10.1016/0278-6915(85)90138-3
176. Mrazek J, Mekadim C, Kucerova P, Svejstil R, Salmonova H, Vlasakova J, et al. Melanoma-related changes in skin microbiome. Folia Microbiol (Praha) (2019) 64(3):435–42. doi: 10.1007/s12223-018-00670-3
177. Mekadim C, Skalnikova HK, Cizkova J, Cizkova V, Palanova A, Horak V, et al. Dysbiosis of skin microbiome and gut microbiome in melanoma progression. BMC Microbiol (2022) 22(1):63. doi: 10.1186/s12866-022-02458-5
178. Salava A, Aho V, Pereira P, Koskinen K, Paulin L, Auvinen P, et al. Skin microbiome in melanomas and melanocytic nevi. Eur J Dermatol (2016) 26(1):49–55. doi: 10.1684/ejd.2015.2696
179. Mizuhashi S, Kajihara I, Sawamura S, Kanemaru H, Makino K, Aoi J, et al. Skin microbiome in acral melanoma: Corynebacterium is associated with advanced melanoma. J Dermatol (2021) 48(1):e15–e6. doi: 10.1111/1346-8138.15633
180. Kalaora S, Nagler A, Nejman D, Alon M, Barbolin C, Barnea E, et al. Identification of bacteria-derived hla-bound peptides in melanoma. Nature (2021) 592(7852):138–43. doi: 10.1038/s41586-021-03368-8
181. Zhu G, Su H, Johnson CH, Khan SA, Kluger H, Lu L. Intratumour microbiome associated with the infiltration of cytotoxic Cd8+ T cells and patient survival in cutaneous melanoma. Eur J Cancer (2021) 151:25–34. doi: 10.1016/j.ejca.2021.03.053
182. Frankel AE, Coughlin LA, Kim J, Froehlich TW, Xie Y, Frenkel EP, et al. Metagenomic shotgun sequencing and unbiased metabolomic profiling identify specific human gut microbiota and metabolites associated with immune checkpoint therapy efficacy in melanoma patients. Neoplasia (2017) 19(10):848–55. doi: 10.1016/j.neo.2017.08.004
183. Coutzac C, Jouniaux JM, Paci A, Schmidt J, Mallardo D, Seck A, et al. Systemic short chain fatty acids limit antitumor effect of ctla-4 blockade in hosts with cancer. Nat Commun (2020) 11(1):2168. doi: 10.1038/s41467-020-16079-x
184. Renga G, Nunzi E, Pariano M, Puccetti M, Bellet MM, Pieraccini G, et al. Optimizing therapeutic outcomes of immune checkpoint blockade by a microbial tryptophan metabolite. J Immunother Cancer (2022) 10(3):e003725. doi: 10.1136/jitc-2021-003725
185. Zhang L, Song T, Wu J, Zhang S, Yin C, Huang F, et al. Antibacterial and cytotoxic metabolites of termite-associated streptomyces sp. Byf63. J Antibiot (Tokyo) (2020) 73(11):766–71. doi: 10.1038/s41429-020-0334-1
186. Nath LR, Kumar SN, Das AA, Nambisan B, Shabna A, Mohandas C, et al. In vitro evaluation of the antioxidant, 3,5-Dihydroxy-4-Ethyl-Trans-Stilbene (Dets) isolated from bacillus cereus as a potent candidate against malignant melanoma. Front Microbiol (2016) 7:452. doi: 10.3389/fmicb.2016.00452
187. Buti L, Spooner E, van der Veen AG, Rappuoli R, Covacci A, Ploegh HL. Helicobacter pylori cytotoxin-associated gene a (Caga) subverts the apoptosis-stimulating protein of P53 (Aspp2) tumor suppressor pathway of the host. Proc Natl Acad Sci U (2011) 108(22):9238–43. doi: 10.1073/pnas.1106200108
188. Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, Han YW. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating e-Cadherin/Beta-Catenin signaling via its fada adhesin. Cell Host Microbe (2013) 14(2):195–206. doi: 10.1016/j.chom.2013.07.012
189. Sears CL. The toxins of bacteroides fragilis. Toxicon (2001) 39(11):1737–46. doi: 10.1016/s0041-0101(01)00160-x
190. Jin C, Lagoudas GK, Zhao C, Bullman S, Bhutkar A, Hu B, et al. Commensal microbiota promote lung cancer development via gammadelta T cells. Cell (2019) 176(5):998–1013.e16. doi: 10.1016/j.cell.2018.12.040
191. Cancer Genome Atlas Research N, Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, et al. The cancer genome atlas pan-cancer analysis project. Nat Genet (2013) 45(10):1113–20. doi: 10.1038/ng.2764
192. Tzeng A, Sangwan N, Jia M, Liu CC, Keslar KS, Downs-Kelly E, et al. Human breast microbiome correlates with prognostic features and immunological signatures in breast cancer. Genome Med (2021) 13(1):60. doi: 10.1186/s13073-021-00874-2
193. Parhi L, Alon-Maimon T, Sol A, Nejman D, Shhadeh A, Fainsod-Levi T, et al. Breast cancer colonization by fusobacterium nucleatum accelerates tumor growth and metastatic progression. Nat Commun (2020) 11(1):3259. doi: 10.1038/s41467-020-16967-2
194. Fu A, Yao B, Dong T, Chen Y, Yao J, Liu Y, et al. Tumor-resident intracellular microbiota promotes metastatic colonization in breast cancer. Cell (2022) 185(8):1356–72.e26. doi: 10.1016/j.cell.2022.02.027
195. Yu T, Guo F, Yu Y, Sun T, Ma D, Han J, et al. Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy. Cell (2017) 170(3):548–63.e16. doi: 10.1016/j.cell.2017.07.008
Keywords: gut microbiota, cancer, tumor microenvironment, tumor microbiome, bacterial diversity, treatment response
Citation: Ciernikova S, Sevcikova A, Stevurkova V and Mego M (2022) Tumor microbiome – an integral part of the tumor microenvironment. Front. Oncol. 12:1063100. doi: 10.3389/fonc.2022.1063100
Received: 06 October 2022; Accepted: 08 November 2022;
Published: 24 November 2022.
Edited by:
Ejaz Ahmad, Michigan Medicine, University of Michigan, United StatesReviewed by:
Nithyananda Thorenoor, Department of Pediatrics, The Pennsylvania State University, United StatesGislane Lelis Vilela de Oliveira, São Paulo State University, Brazil
Copyright © 2022 Ciernikova, Sevcikova, Stevurkova and Mego. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sona Ciernikova, c29uYS5jaWVybmlrb3ZhbEBzYXZiYS5zaw==
†These authors have contributed equally to this work and share first authorship