 Cheng Qiu1,2†
Cheng Qiu1,2† Tianyi Liu
Tianyi Liu Dongyang Luan
Dongyang Luan Lin Cheng
Lin Cheng- 1Department of Orthopaedic Surgery, Qilu Hospital, Cheeloo College of Medicine, Shandong University, Jinan, China
- 2Cheeloo College of Medicine, Shandong University, Jinan, China
- 3Department of Medical Oncology, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China
- 4College of Stomatology, Qingdao University, Qingdao, China
- 5Department of Emergency Medicine, Qilu Hospital of Shandong University, Jinan, China
Ferroptosis has recently been discovered as an iron-dependent and non-apoptotic regulated mechanism of cell death. The induction of ferroptosis in tumor cells improves tumor treatment, making it a current research hotspot. Mechanistically, it starts by lipid peroxidation, iron accumulation, reactive oxygen species (ROS) production, and glutathione deprivation, highlighting novel treatment opportunities for many tumors and neurodegenerative disorders. Several tumor cell lines are resistant to ferroptosis inducers, even when the ferroptosis key enzyme glutathione peroxidase 4 (GPX4) is blocked, indicating that other important elements are also involved in this process. Ferroptosis-suppressor-protein 1 (FSP1) was discovered to be one of these elements in addition to a few others such as ferroptotic gatekeepers like GTP cyclohydrolase 1 (GCH1) and dihydroorotate dehydrogenase (DHODH). Osteosarcoma is the most common primary malignant bone tumor observed most frequently in children and adolescents. Several studies demonstrated that ferroptosis plays a critical role in the treatment of osteosarcoma, in particular drug-resistant osteosarcoma cells. We outlined four primary regulators involved in ferroptosis in this article, reviewed previously published studies of ferroptosis in osteosarcoma to provide covert insights about osteosarcoma treatment, and highlighted several critical issues to point out future research possibilities.
Introduction
Osteosarcoma is the most common primary malignant osseous tumor accounting for the largest proportion (60%) of orthopedic malignant tumors that commonly affect children and those younger than 20 years (1, 2). Distal femur and proximal tibia are the most common sites of osteosarcoma burst. However, the pathogenesis of osteosarcoma remains unclear, and it is considered to be related to the combination of genetic susceptibility, virus infection, ionizing radiation, and chemical toxins (3). Clinically, the main manifestations are swelling, pain, and dysfunction of adjacent joints, which can aggravate pain and affect patients’ sleep at night (4). Several studies demonstrate that 80% of patients with osteosarcoma have a local invasion or distant metastasis when diagnosed (5). The lung is the most common organ for tumor metastasis, accounting for 85% of cases, and 90% of patients with tumors die because of metastasis (6). Current therapeutic strategies for osteosarcoma include surgical resection, radiotherapy, chemotherapy, and immunotherapy with a five-year survival rate of 70% (7). The prognosis of osteosarcoma is still unoptimistic. Tumor cells are resistant to chemotherapeutic drugs. Drug resistance is a critical factor contributing to therapeutic failure and tumor recurrence. Therefore, extensive research on elucidating the mechanisms involved in osteosarcoma and identifying relative molecular targets as well as treatment methods is warranted.
Cell death is a fundamental biological process that pervasively takes place in all living organisms (8). Cancer cells evade quintessential immune surveillance-mediated cell death and then, due to overwhelming proliferation, eventually cause dysregulation in the body (9). The five widely accepted forms of cell death are necrosis, apoptosis, necroptosis, pyroptosis, and ferroptosis (8). Pressing engagement of unknown stimulation or toxic factors concerning the unit of life could trigger uncontrolled necrosis. Additional aforementioned types are accordingly ascribed to regulated cell death (RCD) (10). As a newly identified type of cell death, ferroptosis is a research hotspot, and it mechanistically occurs via lipid peroxidation, iron accumulation, reactive oxygen species (ROS) production, and glutathione deprivation, highlighting the novel treatment opportunities for drug-resistant tumors and neurodegenerative disorders (11). The gatekeepers of ferroptosis include glutathione peroxidase 4 (GPX4) (12), ferroptosis suppressor protein 1 (FSP1) (13, 14), GTP cyclohydrolase 1 (GCH1) (15, 16), and dihydroorotate dehydrogenase (DHODH) (17). Except for these four, other pathways such as acyl-CoA synthetase long-chain family member 4 (ACSL4), adenosine monophosphate activated protein kinase (AMPK)-ACC2, and NF2-YAP are also important in regulating ferroptosis; these regulate polyunsaturated fatty acid (PUFA) metabolism and cellular phospholipid composition (18–20). Lipoxygenases (ALOXs) and cytochrome P450 oxidoreductase (POR) have been known to affect ferroptosis by driving lipid peroxidation, which acts in ways opposite to GSH-GPX4, FSP1-CoQ10, GCH1-BH4, and DHODH (21). Looking at the current treatment options for osteosarcoma, we highlighted advances in ferroptosis and proposed several approaches to contribute to the basic study of osteosarcoma.
Ferroptosis: A Novel Type of Regulated Necrosis
This newly discovered type of regulated cell death, characterized by iron accumulation, glutathione (GSH) deprivation, and lethal lipid peroxidation, was first coined by Dixon et al. in 2012 (11). It was termed ferroptosis suggesting that this form of cell death was induced in an iron-dependent way, which made it morphologically, biochemically, and genetically distinct from the two well-known forms of cell death, namely, apoptosis and necroptosis (22). Hitherto, numerous efforts have been made to unravel this biological process and to establish a correlation between ferroptosis and diseases, including acute kidney injury (23), stroke (24), tuberculosis (25), ischemia/reperfusion injury (26), cardiomyopathy (27, 28), and spinal cord injury (29). Moreover, induction of ferroptosis could be considered as an effective strategy for treating tumor entities (30–35). Ferroptosis has become a research hotspot due to its extraordinary involvement in several untreatable diseases, and three major pathways have been identified (Figure 1).
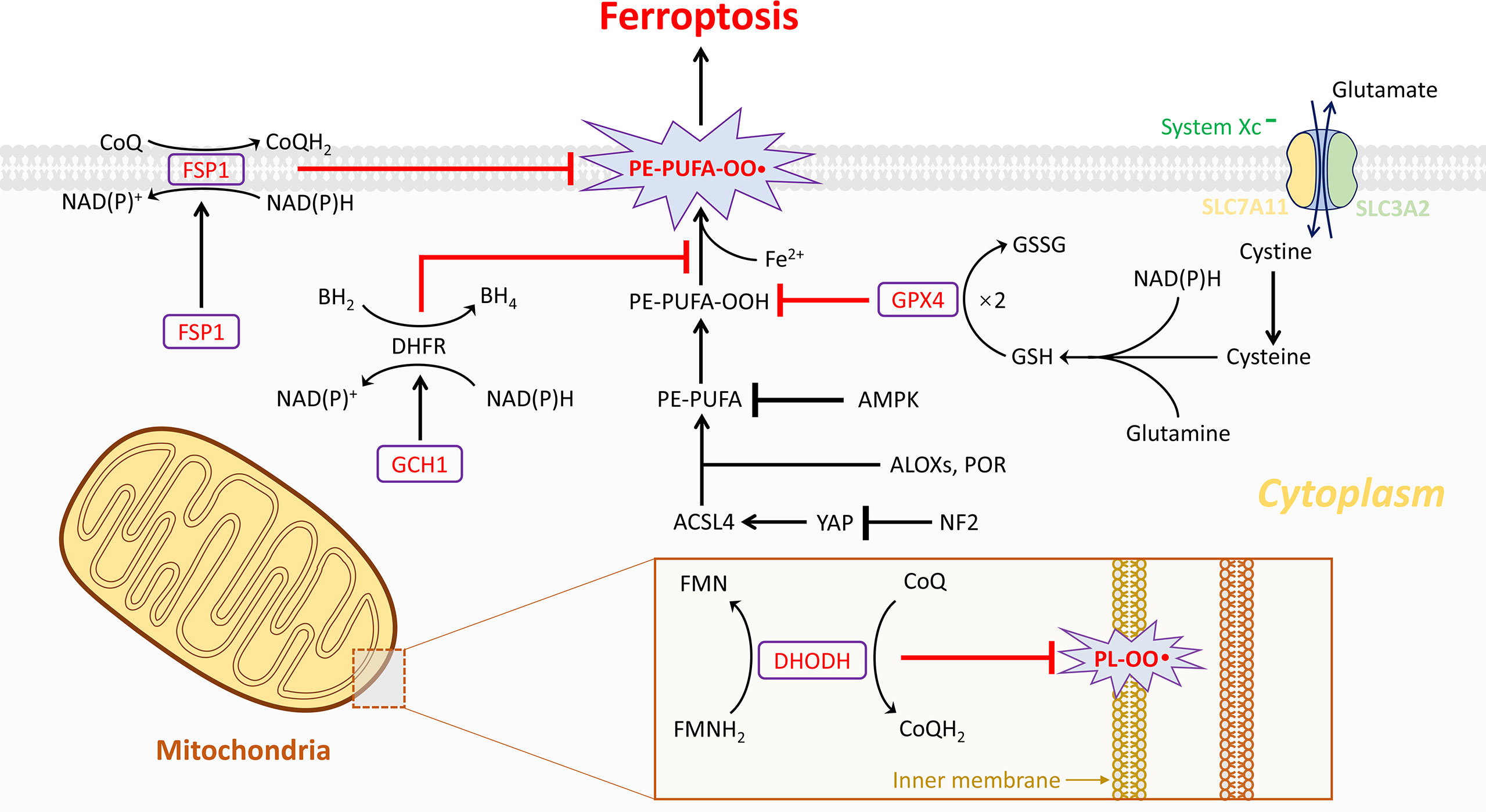
Figure 1 The four current known regulation systems in ferroptosis. Overall, lipid peroxidation in ferroptosis is under control of GPX4-, FSP1-, GCH1-, and DHODH-dependent systems. GPX4 is the most important gatekeeper for ferroptosis and bolstered through the sustainment of GSH and cystine transportation of system Xc- activation. System Xc- is composed of two essential subunits, SLC7A11 and SLC3A2. Generally, ferroptosis could be triggered by GPX4 inhibition directly or indirectly. Nonetheless, several cancer cell lines are resistant to GPX4 inhibition through activating additional regulation systems like FSP1/CoQ10 and GCH1/BH4 systems in the cytoplasm. These two independent manners play a critical role in mitigating cellular ferroptosis especially during loss of GPX4. However, the dysfunction of the three abovementioned systems is observed in organelles such as mitochondria. Notably, then the fourth antioxidant system DHODH-mediated ferroptosis protection in mitochondria is revealed. In the inner membrane of mitochondria, DHODH suppresses ferroptosis via the conversion of ubiquinone to ubiquinol that fights against oxidative damage on the phospholipid membrane. A total of four gatekeepers presumably serve as potential targets for the treatment of osteosarcomas. Except the four pathways, other pathways are also important in regulating ferroptosis, such as ACSL4, AMPK-ACC2, and NF2-YAP pathways, which have been known to affect ferroptosis by regulating PUFA metabolism and cellular phospholipid composition. Lipoxygenases (ALOXs) and POR have been known to affect ferroptosis by driving lipid peroxidation, which play opposite roles to GSH-GPX4, FSP1-CoQ10, GCH1-BH4, and DHODH.
Among them, the GPX4-GSH pathway is the most studied. With system Xc- transferring glutamate and cystine, the synthesis of GSH depends on NAD(P)H thus reducing GPX4. The second pathway FSP1-CoQ10 (FSP1) could catalyze the reduction of CoQ10 through the oxidation of NAD(P)H to generate ubiquinol. In the GCH1-BH4 pathway, GCH1 could upregulate the level of BH4. All three abovementioned products can suppress ferroptosis and therefore have been discussed in more detail in this article (Figure 1).
Ferroptosis Gatekeepers
GPX4
Glutathione peroxidase (GPX), a classical catalysis family bolstered by GSH oxidation to reduce hydrogen peroxide (H2O2) to water, is composed of eight paramount members containing GPX1 to GPX8 (36). Selenocysteine is considered a unique component serving as the central active site for the first four enzymes and is also indispensable for the function of GPX4 (37). In coordination with GSH, GPX4 is important for regulating ferroptosis, as it can neutralize ROS and counter oxidation (Figure 1) (12, 37). In the GSH/GPX4 antioxidant system, upstream system Xc- comprises two subunits, solute carrier family 7 member 11 (SLC7A11) and SLC3A2, required for GSH/GPX4 biosynthesis, the antiporter transporting glutamate and cystine (38). The light chain SLC7A11 is demonstrated to be an effective therapeutic target for tumor treatment due to its unusual overexpression in many cancers. Generally, cellular GPX4 is prone to be inactivated by conventional class I ferroptosis inducers such as erastin, which specifically targets system Xc- to cause GSH depletion, indirectly resulting in iron-dependent lipid ROS peroxidation and subsequent ferroptosis (39, 40). However, class II ferroptosis inducers such as RSL3 could directly react with GPX4 to cause anti-oxidation disorders and elicit ferroptosis (12). Mechanistically, GPX4 mainly plays a specific role in counteracting ROS generated from the Fenton reaction, a classical chemical reaction involving ferrous iron and H2O2, to prevent ferroptosis (41).
FSP1
Similar to GPX4, another promising protein named ferroptosis suppressor protein 1 (FSP1), earlier called apoptosis-inducing factor mitochondrial 2 (AIFM2), was discovered to mediate the ferroptosis protection pathway (13, 14). AIFM2 has been previously demonstrated to induce cell death in a caspase-independent manner, emerging as a potential therapeutic target for tumor treatment (42–44). Few reports have validated the key role of AIFM2 in inhibiting ferroptosis parallel to but independent of GPX4, whose name was then changed to FSP1. FSP1 is located on the human chromosome 10q22.1 and mediates p53-independent apoptosis (42). A majority of FSP1 protein adheres to the outer membrane of mitochondria, whereas the other part of FSP1 protein resides in the cytoplasm. Thus, the cytosolic FSP1 moves to the membrane when myristoylated to inhibit lipid peroxidation and arrest ferroptosis (13, 14). The N-myristoylation signal and flavoprotein oxidoreductase domain of FSP1 are essential to compete against ferroptosis. Moreover, FSP1 functions in collaboration with ubiquinone, also known as coenzyme Q10 (CoQ10). With a substantial utilization of NAD(P)H, FSP1 acting as an NADH-dependent CoQ10 oxidoreductase reduces ubiquinone to ubiquinol, which deals with oxidation through radical scavenging, thereby limiting lipid peroxidation and preventing the occurrence of ferroptosis (Figure 1). Overall, the deficiency of FSP1 and the restrained FSP1/CoQ10 signaling may play a critical role in causing cell death due to oxidative impingement.
GCH1
Recently, the GTP cyclohydrolase 1/tetrahydrobiopterin (GCH1/BH4) pathway was verified to inhibit ferroptosis in a GPX4-independent manner (15, 16). At the cellular level, BH4 plays a critical role in the activities of various enzymes (45). Mechanically, BH4 counteracts ferroptosis by scavenging lipid peroxidation, providing strong protection during the anti-oxidation process, whereas GCH1 is a rate-limiting enzyme for the de novo biosynthesis of BH4 from guanosine triphosphate (GTP) (46). Likewise, ferroptosis is sensitized by simultaneously inhibiting GPX4 function and a recycling system governed by dihydrofolate reductase (DHFR), whereby BH4 regenerates from BH2 (Figure 1) (15, 16). Theoretically, BH4 confers robust defense against ferroptosis relying on the recycling shuttle loop of DHFR upon continuous replenishment by cellular hydrogen carriers. However, the underlying mechanisms of the GCH1/BH4 pathway are poorly understood in ferroptosis burst.
DHODH
The FSP1-dependent CoQ10 reducing system, with antioxidant property, is termed as the endogenous antiferroptosis rudder (13, 14). The functions of FSP1 are strictly limited to cellular membranes according to current reports, and it was unclear whether mitochondrial membranes are subject to similar mechanisms. This question has now been satisfactorily answered by a recently published observation (17). Initially, in different types of cancer cells, lipid peroxidation drives the synthesis of pyrimidine bases in the presence of a specific enzyme dihydroorotate dehydrogenase (DHODH) located on mitochondria. DHODH catalyzes the conversion of dihydroorotate to orotate through an oxidative reaction by consuming ubiquinone and thereby generates ubiquinol to block ferroptosis, while this occurs independently in pyrimidine synthesis (Figure 1). Mitochondrial GPX4 possibly plays a redundant role in cell survival compared to DHODH; however, it repairs the lipid membrane damage (47). Potent DHODH inhibitors are sensitive to cancer cell lines with low GPX4 expression levels.
Ferroptosis in Osteosarcoma
Recent studies have shown that ferroptosis is involved in tumor growth and malignancy (Table 1). Isani et al. first described ferroptosis-like cell death with iron-dependent and non-apoptotic characteristics in osteosarcoma cell line D-27 (55). After treatment with Artemisinin annua extract, D-27 cells showed cell death presented with ballooning phenotype instead of fragmented nuclei distinct from previous apoptosis as well as necrosis patterns and possessed aberrant iron levels. The study identified that ferroptosis sensitization could be induced by dampening STAT3/Nrf2/GPx4 signaling to enhance the sensitivity of osteosarcoma cells to cisplatin (54). This was the first study focused on drug-resistant osteosarcoma and revealed a novel approach to make osteosarcomas more sensitive to the drug by utilizing ferroptosis inducers or STAT3 inhibitors. The vital gatekeeper of ferroptosis, GPX4, was first reported to be involved in osteosarcoma. Moreover, phenethyl isothiocyanate (PEITC), an isothiocyanate that is effective against various cancers, was reported to stimulate osteosarcoma ferroptosis by impairing iron metabolism, redox balance, and GSH-iron-ROS regulation (52, 53). This form of osteosarcoma ferroptosis was observed to be controlled by the MAPK signaling pathway. Ultrasound-activatable doxorubicin (DOX)-Fe(VI)@HMS-HE-PEG (DFHHP) nanoparticles synergistically boosted the growth suppression of hypoxic osteosarcoma by inducing ferroptosis with notable GPX4 downregulation (51). Thereafter, three independent studies respectively demonstrated that the regulators EF24, KDM4A, and tirapazamine could mediate ferroptosis through GPX4 directly or indirectly (48–50). The overall picture of ferroptosis in osteosarcoma is incomplete, and further studies are warranted.
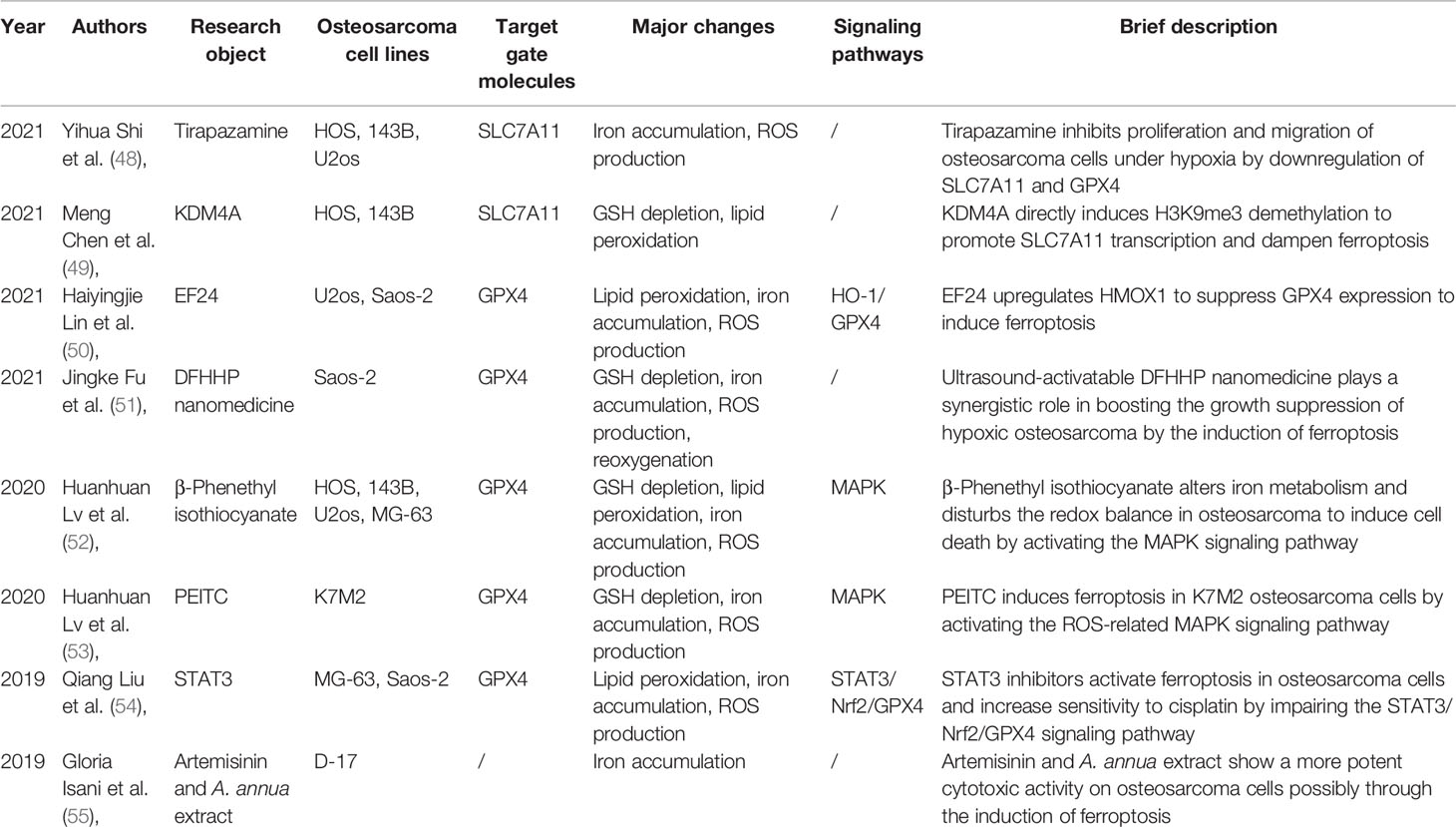
Table 1 Previous published studies regarding ferroptosis in osteosarcoma.
Hopeful Investment in Osteosarcoma
Ferroptosis has been extensively studied in various disorders and exhibits great potential in antitumor therapy (12, 56). Tumor diseases marked by angiogenesis, which accounts for metastasis and poor prognosis, are in most cases treated with surgical resection, radiotherapy, or combined chemotherapy (57). Herein, the evocation of ferroptosis could be regarded as a new approach to halt osteosarcoma growth and could provide support for the treatment of difficult tumors (20, 58). Extensive research has identified several ferroptosis inducers for treating tumors, including erastin modulating system Xc-, sulfasalazine, sorafinib manipulating system Xc-, and RSL3 acting on GPX4 (40, 59). Previous studies on the role of ferroptosis in osteosarcoma mainly targeted GPX4 and reported encouraging results. Some tumor cell lines (possibly containing drug-resistant osteosarcomas) are also found to be resistant to ferroptosis burst when GPX4 is inhibited along with the acyl-CoA synthetase long-chain family member 4 (ACSL4) expression (18, 60). Therefore, induction of ferroptosis by blocking the FSP1/CoQ10 axis with concomitant GPX4 blockade may have positive effects on osteosarcoma treatment (13, 14). In addition, the level of FSP1 was found to be associated with tumor ferroptosis resistance, revealing that inhibitors of FSP1, earlier described as iFSP1, could be utilized to kill osteosarcoma cells, and the treatment could be further potentiated by the loss of GPX4 activity (14). Similarly, DHODH inhibitors are suggested to trigger ferroptosis through mitochondrial lipid peroxidation on those osteosarcoma cells with low GPX4 expression levels (17). However, abolishing the GCH1 network alone is not adequate to promote ferroptosis owing to CoQ10 interaction and the de novo biosynthesis of BH4 from GTP by GCH1 (46). In conclusion, targeting GPX4, FSP1, GCH1, and DHODH has a great potential for combating osteosarcoma.
Discussion
Ferroptosis is a newly discovered form of regulated cell death with already identified four key regulators (61, 62). As a type of programmed cell death, ferroptosis induction offers an emerging strategy to treat tumors and other types of disorders. Although ferroptosis and its modulators, especially GPX4, have been extensively studied, much remains to be understood (12, 37). The mechanism of FSP1- and DHODH-mediated ferroptosis needs to be fully elucidated, and its regulators, inducers, or inhibitors are yet to be investigated. Apart from the aforementioned considerations, different cells may show diverse sensitivity toward ferroptosis or the possible FSP1 and DHODH inhibitors. Consequently, the exact dosage and method of delivering the possible FSP1 and DHODH inhibitors precisely are two major concerns that could limit the clinical application.
FSP1-mediated ferroptosis is also involved in inflammatory processes, hinting that ferroptosis is capable of recruiting immune cells in the tumor microenvironment (22, 63). The interaction between osteosarcoma cells and antitumor drugs is complicated. These drugs could induce cell death in various ways. The inflammatory process is an important component involved in osteosarcoma growth and development as tumor immunity is accompanied by inflammatory reactions (64). Although cell death types are multifarious, several forms could be induced under the same situation. During the process of tumor cell death, inflammation could be induced and related inflammatory cytokines such as TNF-α, IL-1β, and IL-6 further cause immune cell infiltration, which further results in the elimination of dead cells. FSP1 is a bidirectional protein associated with apoptosis or ferroptosis (13, 14, 42, 43, 65). It was previously identified to induce apoptosis in a caspase-independent and p53-independent manner, while it also protects cells from ferroptosis. Therefore, FSP1 may be considered as a decision-maker for the mechanism of cell death. Overall, a high FSP1 expression level confers protection against ferroptosis while inducing cell death through apoptosis. This could explain why almost all normal cells have low FSP1 expression levels, and low FSP1 expression can avoid cell apoptosis. Ferroptosis is an inflammation-regulated necrosis contrary to apoptosis. It can trigger inflammation, which helps purge tumor cells. Targeting FSP1 to promote ferroptosis is an interesting and theoretical strategy in osteosarcoma therapy. A future direction can be set to study the synergistic effect of FSP1 inhibitors and immunotherapy in the treatment of tumors.
In conclusion, the concept of FSP1 and its fast growth have shed new light on antitumor therapies, drastically changing the landscape of tumor treatment. However, further research on the correlation of FSP1 with osteosarcoma growth and progression and the underlying mechanism of FSP1 in osteosarcoma pathogenesis is required. Further research will be crucial for developing ferroptosis-based therapeutic regimens (64).
Author Contributions
SW and LC raised the idea for the article and critically revised the manuscript. CQ, TL, DL, and DYL performed the literature search and data analysis. CQ, TL, LC, and SW were the major contributors in the drafting of the work. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank all the reviewers who participated in the review, as well as MJEditor (www.mjeditor.com) for providing English editing services during the preparation of this manuscript.
References
1. Yang J, Zhang W. New Molecular Insights Into Osteosarcoma Targeted Therapy. Curr Opin Oncol (2013) 25(4):398–406. doi: 10.1097/CCO.0b013e3283622c1b
2. Ottaviani G, Jaffe N. The Epidemiology of Osteosarcoma. Cancer Treat Res (2009) 152:3–13. doi: 10.1007/978-1-4419-0284-9_1
3. Gill J, Gorlick R. Advancing Therapy for Osteosarcoma. Nat Rev Clin Oncol (2021) 18(10):609–24. doi: 10.1038/s41571-021-00519-8
4. Moriarity BS, Otto GM, Rahrmann EP, Rathe SK, Wolf NK, Weg MT, et al. A Sleeping Beauty Forward Genetic Screen Identifies New Genes and Pathways Driving Osteosarcoma Development and Metastasis. Nat Genet (2015) 47(6):615–24. doi: 10.1038/ng.3293
5. Faisham WI, Mat Saad AZ, Alsaigh LN, Nor Azman MZ, Kamarul Imran M, Biswal BM, et al. Prognostic Factors and Survival Rate of Osteosarcoma: A Single-Institution Study. Asian J Clin Oncol (2017) 13(2):e104–e10. doi: 10.1111/ajco.12346
6. Meyers PA, Schwartz CL, Krailo M, Kleinerman ES, Betcher D, Bernstein ML, et al. Osteosarcoma: A Randomized, Prospective Trial of the Addition of Ifosfamide and/or Muramyl Tripeptide to Cisplatin, Doxorubicin, and High-Dose Methotrexate. J Clin Oncol Off J Am Soc Clin Oncol (2005) 23(9):2004–11. doi: 10.1200/jco.2005.06.031
7. Isakoff MS, Bielack SS, Meltzer P, Gorlick R. Osteosarcoma: Current Treatment and a Collaborative Pathway to Success. J Clin Oncol Off J Am Soc Clin Oncol (2015) 33(27):3029–35. doi: 10.1200/jco.2014.59.4895
8. Green DR. The Coming Decade of Cell Death Research: Five Riddles. Cell (2019) 177(5):1094–107. doi: 10.1016/j.cell.2019.04.024
9. Green DR, Evan Gi. A Matter of Life and Death. Cancer Cell (2002) 1(1):19–30. doi: 10.1016/s1535-6108(02)00024-7
10. Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated Necrosis: The Expanding Network of non-Apoptotic Cell Death Pathways. Nat Rev Mol Cell Biol (2014) 15(2):135–47. doi: 10.1038/nrm3737
11. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell (2012) 149(5):1060–72. doi: 10.1016/j.cell.2012.03.042
12. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell (2014) 156(1-2):317–31. doi: 10.1016/j.cell.2013.12.010
13. Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a Glutathione-Independent Ferroptosis Suppressor. Nature (2019) 575(7784):693–8. doi: 10.1038/s41586-019-1707-0
14. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ Oxidoreductase FSP1 Acts Parallel to GPX4 to Inhibit Ferroptosis. Nature (2019) 575(7784):688–92. doi: 10.1038/s41586-019-1705-2
15. Kraft VAN, Bezjian CT, Pfeiffer S, Ringelstetter L, Müller C, Zandkarimi F, et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis Through Lipid Remodeling. ACS Cent Sci (2020) 6(1):41–53. doi: 10.1021/acscentsci.9b01063
16. Soula M, Weber RA, Zilka O, Alwaseem H, La K, Yen F, et al. Metabolic Determinants of Cancer Cell Sensitivity to Canonical Ferroptosis Inducers. Nat Chem Biol (2020) 16(12):1351–60. doi: 10.1038/s41589-020-0613-y
17. Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, et al. DHODH-Mediated Ferroptosis Defence is a Targetable Vulnerability in Cancer. Nature (2021) 593(7860):586–90. doi: 10.1038/s41586-021-03539-7
18. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 Dictates Ferroptosis Sensitivity by Shaping Cellular Lipid Composition. Nat Chem Biol (2017) 13(1):91–8. doi: 10.1038/nchembio.2239
19. Lee H, Zandkarimi F, Zhang Y, Meena JK, Kim J, Zhuang L, et al. Energy-Stress-Mediated AMPK Activation Inhibits Ferroptosis. Nat Cell Biol (2020) 22(2):225–34. doi: 10.1038/s41556-020-0461-8
20. Wu J, Minikes AM, Gao M, Bian H, Li Y, Stockwell BR, et al. Intercellular Interaction Dictates Cancer Cell Ferroptosis via NF2-YAP Signalling. Nature (2019) 572(7769):402–6. doi: 10.1038/s41586-019-1426-6
21. Yan B, Ai Y, Sun Q, Ma Y, Cao Y, Wang J, et al. Membrane Damage During Ferroptosis Is Caused by Oxidation of Phospholipids Catalyzed by the Oxidoreductases POR and CYB5R1. Mol Cell (2021) 81(2):355–69.e10. doi: 10.1016/j.molcel.2020.11.024
22. Zheng J, Conrad M. The Metabolic Underpinnings of Ferroptosis. Cell Metab (2020) 32(6):920–37. doi: 10.1016/j.cmet.2020.10.011
23. Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, et al. Inactivation of the Ferroptosis Regulator Gpx4 Triggers Acute Renal Failure in Mice. Nat Cell Biol (2014) 16(12):1180–91. doi: 10.1038/ncb3064
24. Alim I, Caulfield JT, Chen Y, Swarup V, Geschwind DH, Ivanova E, et al. Selenium Drives a Transcriptional Adaptive Program to Block Ferroptosis and Treat Stroke. Cell (2019) 177(5):1262–79.e25. doi: 10.1016/j.cell.2019.03.032
25. Amaral EP, Costa DL, Mittereder L, Mayer-Barber KD, Andrade BB, Namasivayam S, et al. A Major Role for Ferroptosis in Mycobacterium Tuberculosis-Induced Cell Death and Tissue Necrosis. J Exp Med (2019) 216(3):556–70. doi: 10.1084/jem.20181776
26. Li Y, Cao Y, Xiao J, Shang J, Tan Q, Ping F, et al. Inhibitor of Apoptosis-Stimulating Protein of P53 Inhibits Ferroptosis and Alleviates Intestinal Ischemia/Reperfusion-Induced Acute Lung Injury. Cell Death Differ (2020) 27(9):2635–50. doi: 10.1038/s41418-020-0528-x
27. Fang X, Wang H, Han D, Xie E, Yang X, Wei J, et al. Ferroptosis as a Target for Protection Against Cardiomyopathy. Proc Natl Acad Sci USA (2019) 116(7):2672–80. doi: 10.1073/pnas.1821022116
28. Fang X, Cai Z, Wang H, Han D, Cheng Q, Zhang P, et al. Loss of Cardiac Ferritin H Facilitates Cardiomyopathy via Slc7a11-Mediated Ferroptosis. Circ Res (2020) 127(4):486–501. doi: 10.1161/circresaha.120.316509
29. Yao X, Zhang Y, Hao J, Duan HQ, Zhao CX, Sun C, et al. Deferoxamine Promotes Recovery of Traumatic Spinal Cord Injury by Inhibiting Ferroptosis. Neural Regen Res (2019) 14(3):532–41. doi: 10.4103/1673-5374.245480
30. Badgley MA, Kremer DM, Maurer HC, DelGiorno KE, Lee HJ, Purohit V, et al. Cysteine Depletion Induces Pancreatic Tumor Ferroptosis in Mice. Sci (New York NY) (2020) 368(6486):85–9. doi: 10.1126/science.aaw9872
31. Shen Z, Song J, Yung BC, Zhou Z, Wu A, Chen X. Emerging Strategies of Cancer Therapy Based on Ferroptosis. Adv Mater (Deerfield Beach Fla) (2018) 30(12):e1704007. doi: 10.1002/adma.201704007
32. Hassannia B, Vandenabeele P, Vanden Berghe T. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell (2019) 35(6):830–49. doi: 10.1016/j.ccell.2019.04.002
33. Yee PP, Wei Y, Kim SY, Lu T, Chih SY, Lawson C, et al. Neutrophil-Induced Ferroptosis Promotes Tumor Necrosis in Glioblastoma Progression. Nat Commun (2020) 11(1):5424. doi: 10.1038/s41467-020-19193-y
34. Ubellacker JM, Tasdogan A, Ramesh V, Shen B, Mitchell EC, Martin-Sandoval MS, et al. Lymph Protects Metastasizing Melanoma Cells From Ferroptosis. Nature (2020) 585(7823):113–8. doi: 10.1038/s41586-020-2623-z
35. Zou Y, Henry WS, Ricq EL, Graham ET, Phadnis VV, Maretich P, et al. Plasticity of Ether Lipids Promotes Ferroptosis Susceptibility and Evasion. Nature (2020) 585(7826):603–8. doi: 10.1038/s41586-020-2732-8
36. Friedmann Angeli JP, Conrad M. Selenium and GPX4, a Vital Symbiosis. Free Radic Biol Med (2018) 127:153–9. doi: 10.1016/j.freeradbiomed.2018.03.001
37. Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, Buday K, et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell (2018) 172(3):409–22.e21. doi: 10.1016/j.cell.2017.11.048
38. Wang H, An P, Xie E, Wu Q, Fang X, Gao H, et al. Characterization of Ferroptosis in Murine Models of Hemochromatosis. Hepatol (Baltimore Md) (2017) 66(2):449–65. doi: 10.1002/hep.29117
39. Hadian K, Stockwell BR. SnapShot: Ferroptosis. Cell (2020) 181(5):1188–.e1. doi: 10.1016/j.cell.2020.04.039
40. Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, et al. Ferroptosis: Process and Function. Cell Death Differ (2016) 23(3):369–79. doi: 10.1038/cdd.2015.158
41. Shen Z, Liu T, Li Y, Lau J, Yang Z, Fan W, et al. Fenton-Reaction-Acceleratable Magnetic Nanoparticles for Ferroptosis Therapy of Orthotopic Brain Tumors. ACS Nano (2018) 12(11):11355–65. doi: 10.1021/acsnano.8b06201
42. Ohiro Y, Garkavtsev I, Kobayashi S, Sreekumar KR, Nantz R, Higashikubo BT, et al. A Novel P53-Inducible Apoptogenic Gene, PRG3, Encodes a Homologue of the Apoptosis-Inducing Factor (AIF). FEBS Lett (2002) 524(1-3):163–71. doi: 10.1016/s0014-5793(02)03049-1
43. Wu M, Xu LG, Li X, Zhai Z, Shu HB. AMID, an Apoptosis-Inducing Factor-Homologous Mitochondrion-Associated Protein, Induces Caspase-Independent Apoptosis. J Biol Chem (2002) 277(28):25617–23. doi: 10.1074/jbc.M202285200
44. Gong M, Hay S, Marshall KR, Munro AW, Scrutton NS. DNA Binding Suppresses Human AIF-M2 Activity and Provides a Connection Between Redox Chemistry, Reactive Oxygen Species, and Apoptosis. J Biol Chem (2007) 282(41):30331–40. doi: 10.1074/jbc.M703713200
45. Cronin SJF, Seehus C, Weidinger A, Talbot S, Reissig S, Seifert M, et al. The Metabolite BH4 Controls T Cell Proliferation in Autoimmunity and Cancer. Nature (2018) 563(7732):564–8. doi: 10.1038/s41586-018-0701-2
46. Thöny B, Auerbach G, Blau N. Tetrahydrobiopterin Biosynthesis, Regeneration and Functions. Biochem J (2000) 347 Pt 1(Pt 1):1–16. doi: 10.1042/bj3470001
47. Garcia-Bermudez J, Birsoy K. A Mitochondrial Gatekeeper That Helps Cells Escape Death by Ferroptosis. Nature (2021) 593(7860):514–5. doi: 10.1038/d41586-021-01203-8
48. Shi Y, Gong M, Deng Z, Liu H, Chang Y, Yang Z, et al. Tirapazamine Suppress Osteosarcoma Cells in Part Through SLC7A11 Mediated Ferroptosis. Biochem Biophys Res Commun (2021) 567:118–24. doi: 10.1016/j.bbrc.2021.06.036
49. Chen M, Jiang Y, Sun Y. KDM4A-Mediated Histone Demethylation of SLC7A11 Inhibits Cell Ferroptosis in Osteosarcoma. Biochem Biophys Res Commun (2021) 550:77–83. doi: 10.1016/j.bbrc.2021.02.137
50. Lin H, Chen X, Zhang C, Yang T, Deng Z, Song Y, et al. EF24 Induces Ferroptosis in Osteosarcoma Cells Through HMOX1. Biomed Pharmacotherapy = Biomed Pharmacother (2021) 136:111202. doi: 10.1016/j.biopha.2020.111202
51. Fu J, Li T, Yang Y, Jiang L, Wang W, Fu L, et al. Activatable Nanomedicine for Overcoming Hypoxia-Induced Resistance to Chemotherapy and Inhibiting Tumor Growth by Inducing Collaborative Apoptosis and Ferroptosis in Solid Tumors. Biomaterials (2021) 268:120537. doi: 10.1016/j.biomaterials.2020.120537
52. Lv H, Zhen C, Liu J, Shang P. β-Phenethyl Isothiocyanate Induces Cell Death in Human Osteosarcoma Through Altering Iron Metabolism, Disturbing the Redox Balance, and Activating the MAPK Signaling Pathway. Oxid Med Cell Longevity (2020) 2020:5021983. doi: 10.1155/2020/5021983
53. Lv HH, Zhen CX, Liu JY, Shang P. PEITC Triggers Multiple Forms of Cell Death by GSH-Iron-ROS Regulation in K7M2 Murine Osteosarcoma Cells. Acta Pharmacol Sin (2020) 41(8):1119–32. doi: 10.1038/s41401-020-0376-8
54. Liu Q, Wang K. The Induction of Ferroptosis by Impairing STAT3/Nrf2/GPx4 Signaling Enhances the Sensitivity of Osteosarcoma Cells to Cisplatin. Cell Biol Int (2019) 43(11):1245–56. doi: 10.1002/cbin.11121
55. Isani G, Bertocchi M, Andreani G, Farruggia G, Cappadone C, Salaroli R, et al. Cytotoxic Effects of Artemisia Annua L. And Pure Artemisinin on the D-17 Canine Osteosarcoma Cell Line. Oxid Med Cell Longev (2019) 2019:1615758. doi: 10.1155/2019/1615758
56. Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a P53-Mediated Activity During Tumour Suppression. Nature (2015) 520(7545):57–62. doi: 10.1038/nature14344
57. Li L, Qiu C, Hou M, Wang X, Huang C, Zou J, et al. Ferroptosis in Ovarian Cancer: A Novel Therapeutic Strategy. Front Oncol (2021) 11:665945. doi: 10.3389/fonc.2021.665945
58. Wang W, Green M, Choi JE, Gijón M, Kennedy PD, Johnson JK, et al. CD8(+) T Cells Regulate Tumour Ferroptosis During Cancer Immunotherapy. Nature (2019) 569(7755):270–4. doi: 10.1038/s41586-019-1170-y
59. Liang C, Zhang X, Yang M, Dong X. Recent Progress in Ferroptosis Inducers for Cancer Therapy. Adv Mater (Deerfield Beach Fla) (2019) 31(51):e1904197. doi: 10.1002/adma.201904197
60. Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, et al. Drug-Tolerant Persister Cancer Cells Are Vulnerable to GPX4 Inhibition. Nature (2017) 551(7679):247–50. doi: 10.1038/nature24297
61. Jiang X, Stockwell BR, Conrad M. Ferroptosis: Mechanisms, Biology and Role in Disease. Nat Rev Mol Cell Biol (2021) 22(4):266–82. doi: 10.1038/s41580-020-00324-8
62. Chen X, Kang R, Kroemer G, Tang D. Broadening Horizons: The Role of Ferroptosis in Cancer. Nat Rev Clin Oncol (2021) 18(5):280–96. doi: 10.1038/s41571-020-00462-0
63. Xie Z, Hou H, Luo D, An R, Zhao Y, Qiu C. ROS-Dependent Lipid Peroxidation and Reliant Antioxidant Ferroptosis-Suppressor-Protein 1 in Rheumatoid Arthritis: A Covert Clue for Potential Therapy. Inflammation (2021) 44(1):35–47. doi: 10.1007/s10753-020-01338-2
64. Zhong Z, Sanchez-Lopez E, Karin M. Autophagy, Inflammation, and Immunity: A Troika Governing Cancer and Its Treatment. Cell (2016) 166(2):288–98. doi: 10.1016/j.cell.2016.05.051
Keywords: ferroptosis, osteosarcoma, drug resistance, GPX4, FSP1, GCH1, BH4, DHODH
Citation: Qiu C, Liu T, Luo D, Luan D, Cheng L and Wang S (2022) Novel Therapeutic Savior for Osteosarcoma: The Endorsement of Ferroptosis. Front. Oncol. 12:746030. doi: 10.3389/fonc.2022.746030
Received: 23 July 2021; Accepted: 28 February 2022;
Published: 24 March 2022.
Edited by:
Matiullah Khan, AIMST University, MalaysiaReviewed by:
Zhiyuan Zhang, National Institute of Biological Sciences (NIBS), ChinaChunying Li, Georgia State University, United States
Copyright © 2022 Qiu, Liu, Luo, Luan, Cheng and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lin Cheng, Y2hlbmdsaW4yMDA3QGVtYWlsLnNkdS5lZHUuY24=; Songgang Wang, cWlsdXdhbmdzb25nZ2FuZ0AxNjMuY29t
†These authors have contributed equally to this work