 Jialan Niu
Jialan Niu Danyue Peng
Danyue Peng Lingbo Liu
Lingbo Liu- Institute of Hematology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
Acute myeloid leukemia (AML) is a polyclonal and heterogeneous hematological malignancy. Relapse and refractory after induction chemotherapy are still challenges for curing AML. Leukemia stem cells (LSCs), accepted to originate from hematopoietic stem/precursor cells, are the main root of leukemogenesis and drug resistance. LSCs are dynamic derivations and possess various elusive resistance mechanisms. In this review, we summarized different primary resistance and remolding mechanisms of LSCs after chemotherapy, as well as the indispensable role of the bone marrow microenvironment on LSCs resistance. Through a detailed and comprehensive review of the spectacle of LSCs resistance, it can provide better strategies for future researches on eradicating LSCs and clinical treatment of AML.
Introduction
Acute myeloid leukemia (AML) is a hematopoietic disease characterized by malignant proliferation of myeloid stem/progenitor cells and differentiation arrest. Standard intensive chemotherapy-based regimens remain central in inducing complete remission (CR) in AML, with CR rates of 60–85% for adults younger than 60 years of age and 40–60% for older patients aged ≥60 years (1). Furthermore, with an advanced understanding of the pathogenesis and drug-resistant mechanisms, new therapeutic agents in clinical development target abnormal karyotypes, such as FMS-like tyrosine kinase 3 (FLT3) inhibitors (2), isocitrate dehydrogenase 1 or 2 (IDH1/2) inhibitors (3, 4), and target effector molecules of apoptosis, metabolism, epigenetics, stemness, and immune escape, such as B-cell lymphoma 2 (BCL2) inhibitors (5, 6), hypomethylating agents (HMA) (7), smoothened (SMO) inhibitors (8), CD33 monoclonal antibody (9), have improved the overall survival of patients, especially elderly patients with AML who are not candidates for intensive chemotherapy (10). However, there are still induction failures in some patients with AML with high-risk karyotypes, and short- or long-term relapse occurs after complete remission in most patients, based on different relapse/refractory mechanisms (11).
Lapidot and Bonnet et al. successively identified a rare population of leukemia cells capable of initiating leukemia when transplanted into severe combined immune-deficient (SCID) mice. The rare subset that marked the CD34+CD38- population was the same as that of normal hematopoietic stem cells (HSCs), and was defined as leukemia stem cells (LSCs). These LSCs possessed self-renewal and unlimited proliferation potential (12, 13). Several subsequent studies also confirmed the existence of this cell population (14–17). The established views described that current chemotherapy drugs could only eradicate most of the AML blast cells, but not LSCs, and LSCs were the root of AML relapse (18, 19). Furthermore, there were also treatment-resistant cells carrying leukemia clones with different characteristics from the originally diagnosed LSCs (19–21). Chemotherapy-induced leukemia repopulating cells, some of which derive from LSCs present intrinsic mechanisms of chemotherapy resistance, and are termed primary resistances while others derive from leukemia cells that regain stemness under chemotherapy stress, and are termed secondary resistance mechanisms (22). Therefore, identifying both primary and secondary mechanisms of chemotherapy resistance is essential for the targeted elimination of LSCs and ultimately the cure of AML. This review mainly summarizes the primary and secondary resistance mechanisms of different leukemia clones identified in recent years (Figure 1), including (1) the inherent dormancy of LSCs that protects them from cell cycle-specific agents (CCSA) ;(2) the overexpression of multiple ATP-binding cassettes (ABC) transporters in LSCs transports cytotoxic drugs out of the cell ;(3) LSCs with the defects in apoptotic signals lead to chemotherapy resistance ;(4) senescent resistance mechanisms also result in chemotherapeutic failure ;(5) metabolic reprogramming of LSCs allows them to adjust to energy changes ;(6) epigenetic alternations and reprogramming refresh the stemness of LSCs; and (7) bone marrow (BM) niches that provide a sanctuary for LSCs from therapeutic drugs.
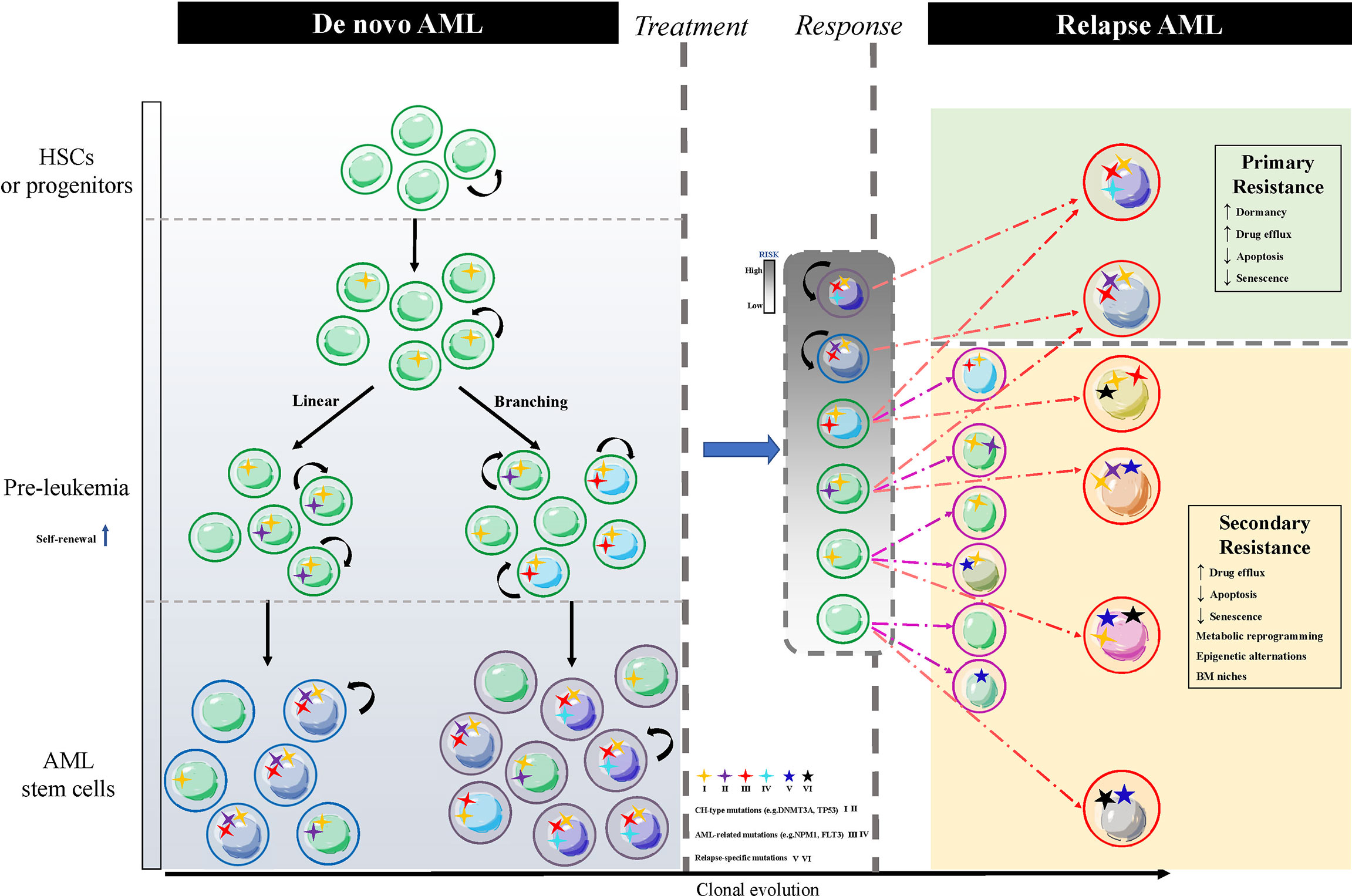
Figure 1 The schematic of LSCs clonal diversification resulting in AML relapse. The normal HSCs or progenitor cells carry CH-type mutations without proliferative potentials, such as ‘DTA’ mutations, denoted by the number I, and convert to pre-leukemia HSCs or progenitor cells with enhanced self-renewal through acquiring additional genetic mutations, which are divided into linear and branching patterns of clonal evolution. The former stepwise acquires CH-type mutations like TP53, IDH1/2 and AML-related mutations like FLT3, NPM1. The latter parallelly acquires CH-type mutations and AML-related mutations in different cell populations. Different colors of nuclei show heterogeneous LSCs clones. Residual leukemia cells (grey box) after treatment include dominant clones LSCs and subclones leukemia cells. Relapsed AML arises from these two types of cells corresponding to the primary resistance (green box) of LSCs and the secondary resistance (yellow box) of reprogramming leukemia cells via acquiring a new relapse-driven clone. AML, acute myeloid leukemia; LSCs, AML stem cells; HSCs, hematopoietic stem cells; CH, clonal hematopoiesis; BM, bone marrow; ‘DTA’ mutations, mutations in DNMT3A, TET2 and ASXL1.
Dormancy of LSCs and Drug Resistance
In short, the dormant cancer cells are defined as being in a state of reversible nonproliferation, which includes both the quiescent cell populations in the primary tumor and residual tumor cells after chemotherapy-induced stress, both are closely associated with tumor recurrence. Thus, dormant cancer cells include not only cancer stem cells (CSCs) but also cancer cells that have acquired stemness (23). In AML patients, CD34+CD38- LSCs are enriched with stemness-related genes and persist after chemotherapy, and are closely correlated with a poor prognosis (14, 18, 24, 25). LSCs, derived from hematopoietic stem cells or progenitor cells, acquire self-renewal capacity and stemness in the process of clone formation and often remain in a quiescent state (26). LSCs are in the G0 phase prior to transplantation into nonobese diabetic(NOD)-SCID mice, and some remain quiescent and self-renewal after several consecutive transplants (27). With different methods such as the use of granulocyte colony-stimulating factor (G-CSF) to induce dormant LSCs into the cell cycle, initially slow-proliferating LSCs become susceptible to chemotherapy (28). Generally, AML recurrences are mainly attributed to quiescent LSCs, because conventional CCSA only act on proliferating cells but not on quiescent LSCs. Herein, we mainly describe the regulation of LSCs quiescence by cellular molecular mechanisms and their surrounding microenvironment.
The activation of the Wnt signaling pathway is one of the most important pathways for the maintenance of LSCs quiescence (Figure 2). Wnt/β-catenin activation is required for the conversion of HSCs as well as relatively mature granulocyte-macrophage progenitor cells (GMPs) into LSCs, regulates LSCs stemness, self-renewal and proliferation (29). R-spondin 3 (RSPO3) activates β-catenin by binding to LGR4 (leucine-rich repeat-containing G protein-coupled receptors) high-expressed on the surface of LSCs, further promotes the expression of stemness and self-renewal genes such as MYC, CCND1 and HOXA clusters. The anti-RSPO3 antibody can reliably debilitate the ability of LSCs to restart leukemia through serial transplantation in mice (30). Recently, it has been discovered that the transcription factor FOXM1 activates the Wnt/β-catenin molecular pathway through stabilizing β-catenin expression to mediate quiescence and self-renewal of MLL-AF9-LSCs (31). In particular, FOXM1 inhibitors do not significantly alter downstream Nurr1 expression [one of the most important molecules regulating HSCs quiescence (32)] and have a lower impact on HSCs, providing a therapeutic window for the targeted elimination of LSCs. Other studies have shown that the long non-coding RNA (lncRNA)-DANCR (differentiation antagonizing non-protein coding RNA) is involved in the regulation of the Wnt signaling pathway. LncRNA DANCR is relatively highly expressed in CD34+ AML primary cells, and knocking down of lncRNA DANCR expression reduces the quiescence and self-renewal of LSCs in vitro and in vivo by down-regulating MYC expression, but not that of normal hematopoietic stem and progenitor cells (HSPCs) (33). The differential regulation of MYC expression by lncRNA DANCR illustrates the important role of the Wnt signaling pathway in LSCs resistance. Although lncRNA DANCR is differentially regulated in HSCs and LSCs, the resistance mechanisms in LSCs require a thorough understanding. Further research is needed on how to clinically inhibit the Wnt signaling pathway to provide a therapeutic window for the targeted clearance of LSCs (34).
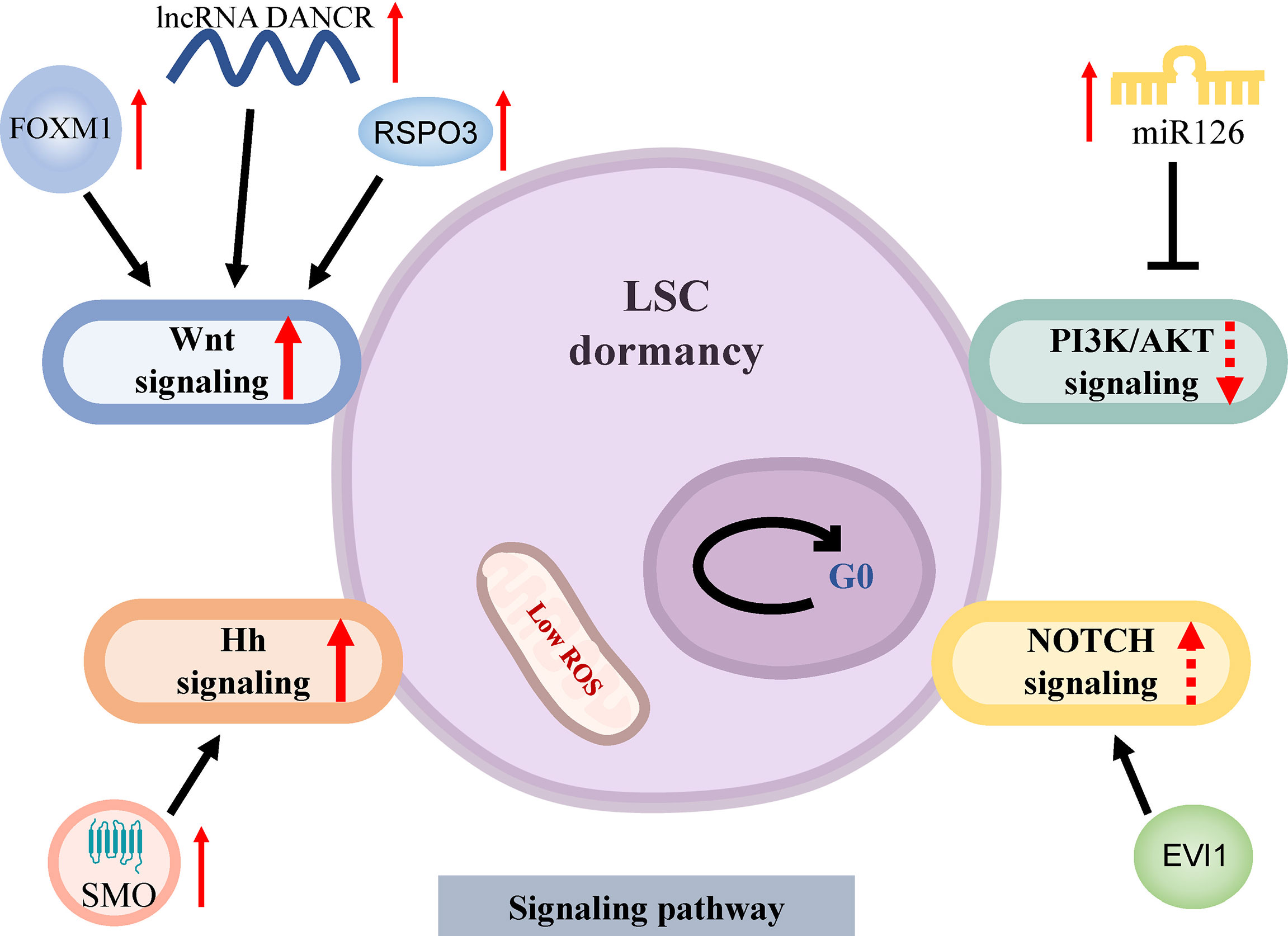
Figure 2 Signaling pathways of dormancy in LSCs. LSCs arrest the cell cycle at G0 phase in a low level of ROS. The Wnt signaling pathway is activated through overexpression of FOXM1 which stabilizes β-catenin, up-regulated RSPO3 by activating β-catenin and increased lncRNA DANCR which upregulates MYC expression to induce LSCs dormancy. The repression of PI3K/AKT signaling pathway by miR126 mediates the stemness of LSCs and drug resistance. The abnormal activation of the Hh and NOTCH signaling pathways plays an important role in maintaining LSCs quiescence. AML, acute myeloid leukemia; LSCs, AML stem cells; ROS, reactive oxygen species; Hh, Hedgehog; SMO, smoothened; EVI1, Ecotropic virus integration site 1.
The other important mechanism of quiescent maintenance of LSCs involves the PI3K/AKT signaling pathway, LSCs rely on the relative low-activity AKT to keep quiescent (Figure 2). PI3K/AKT signaling pathway is usually activated during leukemogenesis to promote cell proliferation and is associated with specific genetic alterations like mutations of FLT3 and NPM1 (35). Thus, the state of LSCs depends on rigorous AKT regulation. Micro(miR)-126 is an important negative regulator of the PI3K/AKT pathway in LSCs. The overexpression of miR-126 in LSCs reduces AKT activity by targeted down-regulation of CDK3, a regulator of G0/G1 conversion, to maintain cell cycle quiescence, improving stemness and resistance to chemotherapy (36). Recent studies have shown that miR-126 is highly expressed in the endothelial cells (ECs) surrounding BM arterioles relative to sinusoids. In the FLT3-ITD+ AML mouse model, treatment with tyrosine kinase inhibitors (TKIs) inhibited the AML-mediated remodeling of arterioles to sinusoids, resulting in the increase of miR-126 in the BM, which further enhanced LSCs quiescence, whereas, the inhibition of miR-126 can effectively eradicate LSCs and prolong survival (37). This may partly explain the greater therapeutic resistance of AML after relapse. Altogether, these findings suggest that miR-126 has a great potential in targeting quiescent LSCs.
Other signal pathways involved in LSCs quiescence are the Hedgehog (Hh) and NOTCH pathways (Figure 2). The Hh pathway plays an important role in maintaining quiescence, self-renewal, survival, and chemoresistance of BCR-ABL+ LSCs (38). The abnormal activation of the Hh signaling pathway has been observed in AML (39). The transmembrane protein smoothened (SMO), an essential regulator of Hh signal transduction, is highly expressed in CD34+ leukemia cells and can activate downstream glioma-associated oncoprotein (GLI) transcription factors to maintain the dormancy and chemoresistance of LSCs (40). Preclinical studies found that PF-913, an inhibitor of SMO, can decrease the proportion of CD34+ leukemia cells, induce dormant LSCs into the cell cycle, reduce leukemia initiation, and exert synergistic effects with cytarabine (AraC) (41). Phase II trial of glasdegib (SMO inhibitor) plus conventional chemotherapy in patients with AML achieved a better complete remission (CR) rate and prolonged overall survival (8) (Table 1). On the other hand, NOTCH signaling pathway can also drive the expression of stemness-related genes and maintain the pool of LSCs. LSCs with high expression of Ecotropic virus integration site 1 (EVI1) were enriched in G0 phase in vivo and in vitro, and all-trans retinoic acid (atRA) further enhanced LSCs quiescence in an EVI1-dependent manner, and EVI1 and atRA synchronously activate NOTCH signal pathway through targeting downstream molecule NOTCH4 (57). Paradoxically, the finding by Lobry showed that activation of the NOTCH signaling pathway induced cell cycle arrest and apoptosis in LSCs with MLL-AF9 fusion, in part due to Tet methylcytosine dioxygenase 2 (TET2)-mediated epigenetic alterations (58). As a result, precise modulation of NOTCH threshold to induce LSCs entry into the cell cycle and jointly promote AML blast clearance may be an interesting direction.
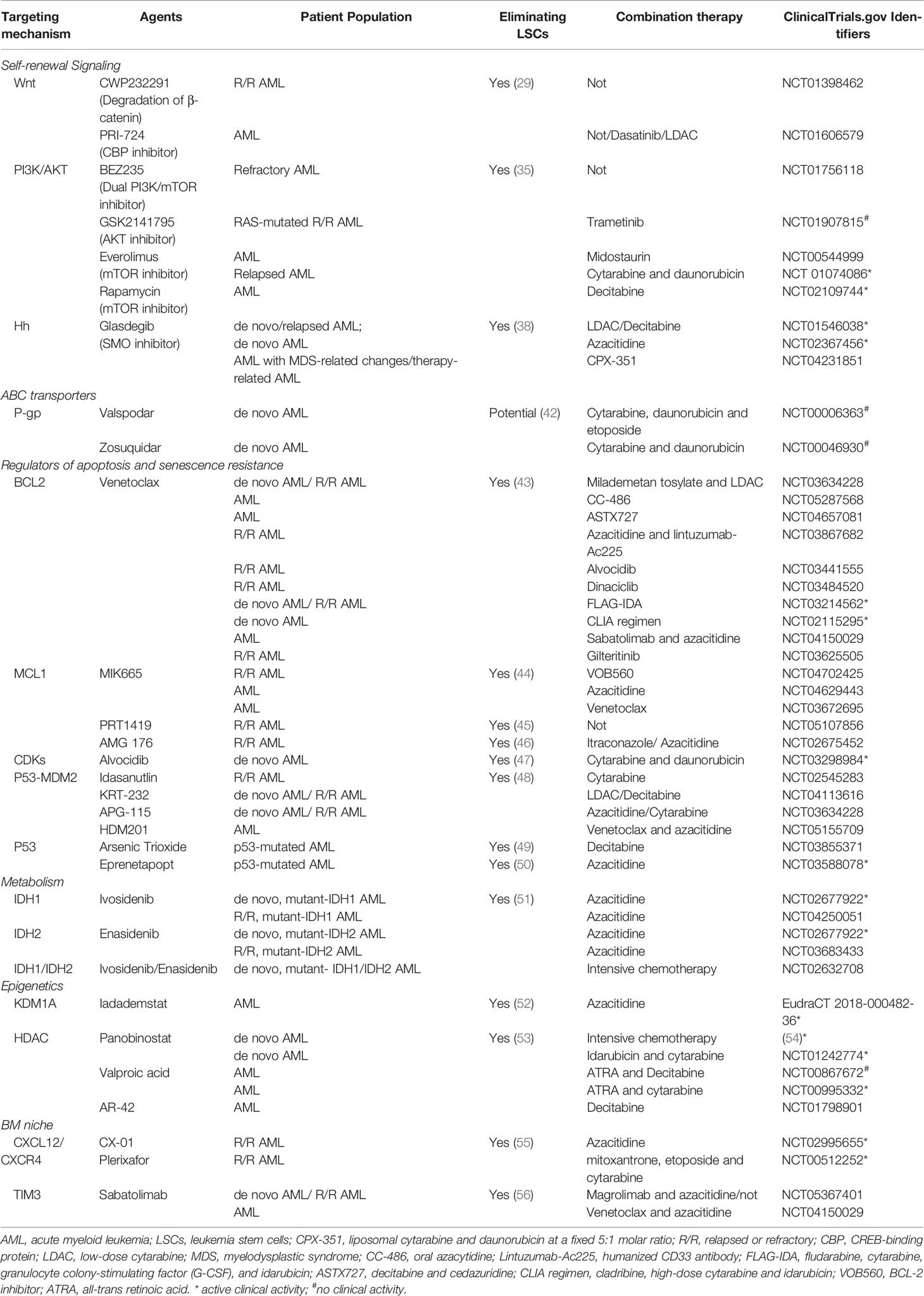
Table 1 Clinical trials of targeted therapies in AML.
In addition, the BM microenvironment also plays a vital role in maintaining LSCs quiescence. The interaction of the CXCR4/CXCL12 axis induced the localization of LSCs in the endosteal region where LSCs could maintain a quiescent state, which was closely related to resistance to chemotherapy and adverse prognosis (55). Hypoxia is essential for HSCs dormancy based on the regulation of hypoxia-inducible factors (HIFs) (59). However, the role of HIFs in LSCs is contradictory (60). The expression of HIF-1α in LSCs was increased. HIF-1α deletion led to the elimination of LSCs (61), in contrast, HIF-1α deletion promoted the conversion of pre-leukemic cells to LSCs, and HIF-1α knockout in MLL-AF9 mice showed LSCs increase and AML progression after chemotherapy (62). Recently, reactive oxygen species (ROS) have been proposed to represent the LSCs population (43). Cell populations with low ROS were rich in quiescent LSCs, which was associated with drug resistance/recurrence, compared to cell populations with high ROS (63). Eimear OR et al. simulated BM-induced dormancy of KG1a cells in vitro by using a hydrogel-based layered co-culture system based on bone mesenchymal stem cells (BMSCs), adding TNFβ-1 and HIFs (64). This model is valuable for targeted drugs screening to treat dormant LSCs in the microenvironment. The regulation mechanisms of BM on the quiescence of LSCs need to be further studied. The humanized mesenchymal niche model system established by Waclawiczek et al. is prospective to study the crosstalk between MSCs, LSCs and normal hematopoiesis (65).
The maintenance of LSCs quiescence and self-renewal depend on a complex intracellular molecular signaling network, which overall maintains low expression of cell cycle-related genes and proliferation-related genes, with elusive crosstalk between various stemness-related pathways. It was found that GLI1 can promote AML cell proliferation directly through PI3K signaling pathway and can be reversed by AKT inhibitors (66). AKT can enhance FOXM1 expression and form a positive feedback to enhance venetoclax resistance in FLT3 wild-type AML (67). Therefore, FOXM1 inhibitors may be a promising therapeutic strategy due to their dual signaling pathway regulations. Similarly, it was found that the combination of inhibitors targeting PI3K/AKT/mTOR signaling pathway and atRA can almost eliminate cellular MYC to drastically kill leukemia cells of different AML subtypes (68). Additionally, the crosstalk between the Wnt and NOTCH pathways has also been reported. Kang YA et al. verified that NOTCH and Wnt signal cooperatively maintained HSCs lineage differentiation. And it also demonstrated that LSCs characteristically maintain high levels of Wnt and low levels of NOTCH, and that restoring those dysregulated pathway activities can inhibit myeloid differentiation and delay leukemia progression (69). These results present the possibility of strong crosstalk in cell-cycle regulation between both pathways. Furthermore, due to the high heterogeneity within and between patients, there is no single indicator to identify LSCs. Data have shown multiple candidate surface markers including CD123 (70), CD33 (71), CD96 (72), TIM-3 (73), CD44 (74), CLL-1 (75), c-MPL (76), MHDA2 (77), CD25 and CD32 (24) that play a significant role in the stemness properties of LSCs. Of these, CD25, CD32, CD123, c-MPL, and MHDA2 are expressed in quiescent human LSCs, and targeting these surface markers may facilitate the removal of dormant LSCs. A recent study tracking the leukemia cells of AML patients before and after chemotherapy found that CD34+CD38- LSCs and AML blasts were equally eliminated, and LSCs no longer remained quiescent after chemotherapy (21, 63). Although chemotherapy may promote the evolution of some clones, quiescence or dormancy is not sufficient to protect LSCs from chemotherapy-induced cell death (78).
ABC Transporter Family and AML Drug Resistance
A vital mechanism of multiple drug resistance in tumor cells is the transport of cytotoxic drugs outside cells to reduce DNA damage. The ABC transporter family, which currently consists of 48 members, pumps out exogenous substances via ATP-dependent membrane transport pathways (79). A variety of ABC transporters are highly expressed in AML cells, and are closely associated with adverse chemotherapy responses in AML patients. Consecutively, follow-up studies revealed that LSCs also have higher expression of different ABC transporters (42). Therefore, targeting ABC transporters has a positive impact on enhancing the chemosensitivity of AML. ABC transporters showing the greatest correlation with AML chemotherapy resistance are permeability glycoprotein (P-gp), breast cancer resistance protein (BCRP), and multidrug resistance-associated protein 1 (MRP1), respectively, where P-gp and BCRP are the most frequently expressed (Figure 3).

Figure 3 Drug efflux and drug resistance. Multiple anti-tumor drugs enter to kill leukemia cells (upper part), but LSCs upregulate ABC transporters to efflux drugs to overcome the cytotoxic effects of multiple chemotherapeutic drugs, even single blocking of MDR1 does not significantly improve chemotherapy outcomes. Additionally, MRP1 mediates the ATP-dependent efflux of GSH to reduce cellular oxidative stress. BRCP transports excessive PPIX out of the cell to maintain porphyrin homeostasis (lower part). MRP1, multidrug resistance-associated protein 1; MDR1, multidrug resistance gene 1; BRCP, breast cancer resistance protein; PPIX, protoporphyrin IX; GSH, glutathione; ATP, adenosine triphosphate; ADP, adenosine diphosphate.
The best-characterized ABC transporter is P-gp, which is encoded by the multidrug resistance gene 1 (MDR1 or ABCB1). P-gp confers resistance to chemotherapeutic drugs such as doxorubicin, daunorubicin, vincristine, mitoxantrone, and methotrexate (80). Numerous studies have confirmed that leukemia cells overexpressed P-gp, which was strongly associated with the poor prognosis in AML (81–83). Subsequent studies have found that the association was age-dependent, which was more pronounced in patients older than 60 years than in young adults with AML (84). Furthermore, de Figueredo et al. reported that compared with the relatively mature CD34+CD38−CD123− cells, the CD34+CD38−CD123+ subset had a higher P-gp, while CD34+CD38−CD123− cells had an increased P-gp expression than the CD34+CD38+ subset (85). However, the study found that the use of P-gp inhibitors, such as zosuquidar or cyclosporine, failed to improve the CR rate and OS of AML patients (86). The possible cause may be that AML stem cells express multiple ABC transporters, hence, single blocking of a certain ABC transporter does not significantly improve chemotherapy outcomes. In addition, adriamycin stress can increase the expression of ABCB1 enhancers, and this adaptive change of ABCB1 mediated drug resistance in AML (87).
Another important ABC transporter, BCRP, encoded by the ABCG2 on chromosome 4, has been linked to drug resistance in the treatment of AML with chemotherapeutic agents such as doxorubicin, daunorubicin, vincristine, mitoxantrone, and methotrexate (80). Many studies have indicated that higher levels of BCRP expression were associated with lower survival rates of AML, and increased expression of BCRP in patients is observed in patients that fail to achieve CR after chemotherapy (82, 88, 89). Researchers also found that the expression of BCRP has correlated with the expression of side population (SP) cells, a stem cell-enriched fraction, with obvious potential for both self-renewal and proliferation (90). Raaijmakers et al. found that expression of BCRP was predominant in LSCs and blocking of BCRP-mediated drug export by the fumitremorgin C analog KO143 resulted in increased intracellular mitoxantrone accumulation in these cells (91). Therefore, targeting ABCG2 may be a promising therapy to eliminate LSCs.
The ABC-C subfamily encodes multidrug resistance-associated proteins (MRP), of which MRP1 was the first MRP molecule to be discovered. MRP1 is overexpressed in many multidrug-resistant cancer cell lines and confers an extensive drug resistance phenotype, which can develop resistance to multiple chemotherapy agents in AML (80). Previous studies have reported a correlation between MRP1 expression and the prognosis of AML, but the results remain controversial. Some studies have shown that high expression of MRP1 is associated with poor prognosis in AML (83, 92), contrarily, some researchers have found no such correlation (93, 94). There are several possible explanations such as the different methods used to study MRP1 expression, different treatment regimens, and patient characteristics which impact on MRP1 expression. The inconsistency of these findings has also suggested that MRP1is not a major determinant in drug resistance in AML.
Overall, LSCs exhibit a much higher expression of ABC transporters than mature cell populations, which results in chemotherapy resistance (95). Noteworthily, ABC transporters also act as efflux transporters of biomolecules relevant to tumor resistance. MRP1 mediates the ATP-dependent efflux of glutathione (GSH) to reduce cellular oxidative stress (96). BRCP transports excessive protoporphyrin IX (PPIX) out of the cell to maintain porphyrin homeostasis (97). However, clinical trials evaluating the efficacy of ABC transporters-inhibitors have been unsatisfactory. The main reason may be that HSCs also have a high expression of various ABC transporters, as demonstrated by Zhou et al. Mice models engineered to knock-out ABCB1 or ABCG2 genes are particularly sensitive to drugs such as mitoxantrone and vinblastine, so the expression of these genes may represent a protective mechanism for HSCs (98). Inhibitors of ABC transporters may reduce the chemoresistance of leukemia primitive populations and LSCs, but may also render HSCs more sensitive to chemotherapeutic drugs, which compromises normal hematopoietic function. Co-expression or activity of ABC transporters is also particularly important in chemotherapy resistance in AML patients. ABCB1 and ABCG2 mRNA co-expression was significantly associated with higher age, increased CD34 expression and poor prognosis (99). Robinson et al. developed that overexpressing both ABCB1 and ABCG2 in vitro not only exerted the function of substrate transport independently but also additively transport new substrate molecules (100). Therefore, it is of great significance to find ABC transporters that are specifically expressed by LSCs.
Apoptosis Resistance of LSCs is Associated With Drug Resistance
The induction of apoptosis of AML cells by chemotherapy is an effective approach to kill malignant cells. However, recent studies have shown that multiple anti-apoptotic mechanisms exist in LSCs. Defects of apoptosis-related signaling pathways represent an integral mechanism for treatment resistance in AML. The principal apoptosis-regulatory molecules are B-cell lymphoma 2 (BCL-2), myeloid cell leukemia sequence 1 (MCL1) and p53, mutations or abnormalities of which can induce apoptotic resistance of LSCs.
BCL-2, the foremost anti-apoptotic protein in hematological malignancies, is significantly up-regulated in lymphoma, multiple myeloma, and AML (101). BCL-2 can stabilize mitochondria and prevent the activation of pro-apoptotic proteins. The expression of BCL-2 in different AML subtypes is irregular, with much higher levels in M1 and M2 subtypes than in M3, M4, and M5 subtypes (102). Furthermore, higher expression of BCL-2 is associated with a worse response to chemotherapy and inferior survival of patients, indicating that BCL-2 is an important factor contributing to the prognosis of AML (102, 103). BCL-2 is markedly up-regulated in LSCs, and its inhibition can restrain oxidative phosphorylation and selectively eradicate quiescent LSCs (43). Venetoclax, which specifically targets BCL-2, achieves a wide range of anti-leukemia capabilities, partly due to the decreasing apoptotic threshold of AML cells (104). MCL1 is another antiapoptotic protein involved in leukemia cell survival. Overexpression of MCL1 has been associated with drug resistance and a poor prognosis of AML (105). The increased expression of MCL1 is also another important cause of venetoclax-based resistance (106, 107). Several MCL1 inhibitors have currently been evaluated in clinical trials, and are one of the promising molecules for targeted AML treatment and detailed examples will be discussed below. Similarly, the inhibitor of cyclin-dependent kinase 9 (CDK9) induced AML apoptosis by down-regulating the expression of MCL1 and demonstrated anti-leukemia and clinical activity in refractory and relapsed (R/R) AML (47, 108) (Table 1).
Mutation of the anti-oncogene p53 also has a great contribution to apoptosis resistance, and p53 plays a central role in the initiation of apoptosis in different physiological conditions (Figure 4) (109). Although the frequency of p53 mutation is low in newly diagnosed AML, the mutation rate is markedly elevated in therapy-related AML patients (110). Absent apoptosis signals by p53 gene mutation result in chemotherapy resistance (111). Furthermore, p53 may selectively activate the MDR-1 promoter, causing leukemia cells to develop multidrug resistance (112). AML patients with mutated p53 exhibit a poor response to chemotherapy and a poor prognosis. The finding suggested that restoring p53 activity through histone deacetylation could enhance the targeted chemotherapeutics in LSCs (113). PRIMA-1, a small molecule, has been reported to restore mutant p53 activity via binding to DNA and induction of apoptosis in p53-deleted AML, which enhances the sensitivity of chemotherapy drugs (50). In wild-type TP53 AML, overexpression of MDM2 (minute 2 homolog, a negative regulator of p53) leads to inactivation of p53 expression, which also exhibits apoptotic resistance. Indeed, several inhibitors targeting the MDM2-p53 axis to restore p53 function have shown broad anti-leukemia activities (48, 114). P53 activation promotes MCL1 degradation through downregulating the RAS/RAF/MEK/ERK signaling pathway, therefore, MDM2 inhibitor in combination with BCL2 inhibitor overcomes MCL1-mediated apoptotic resistance and shows synergistic lethality of relapsed/refractory AML in preclinical and clinical trials (115) (Table 1).
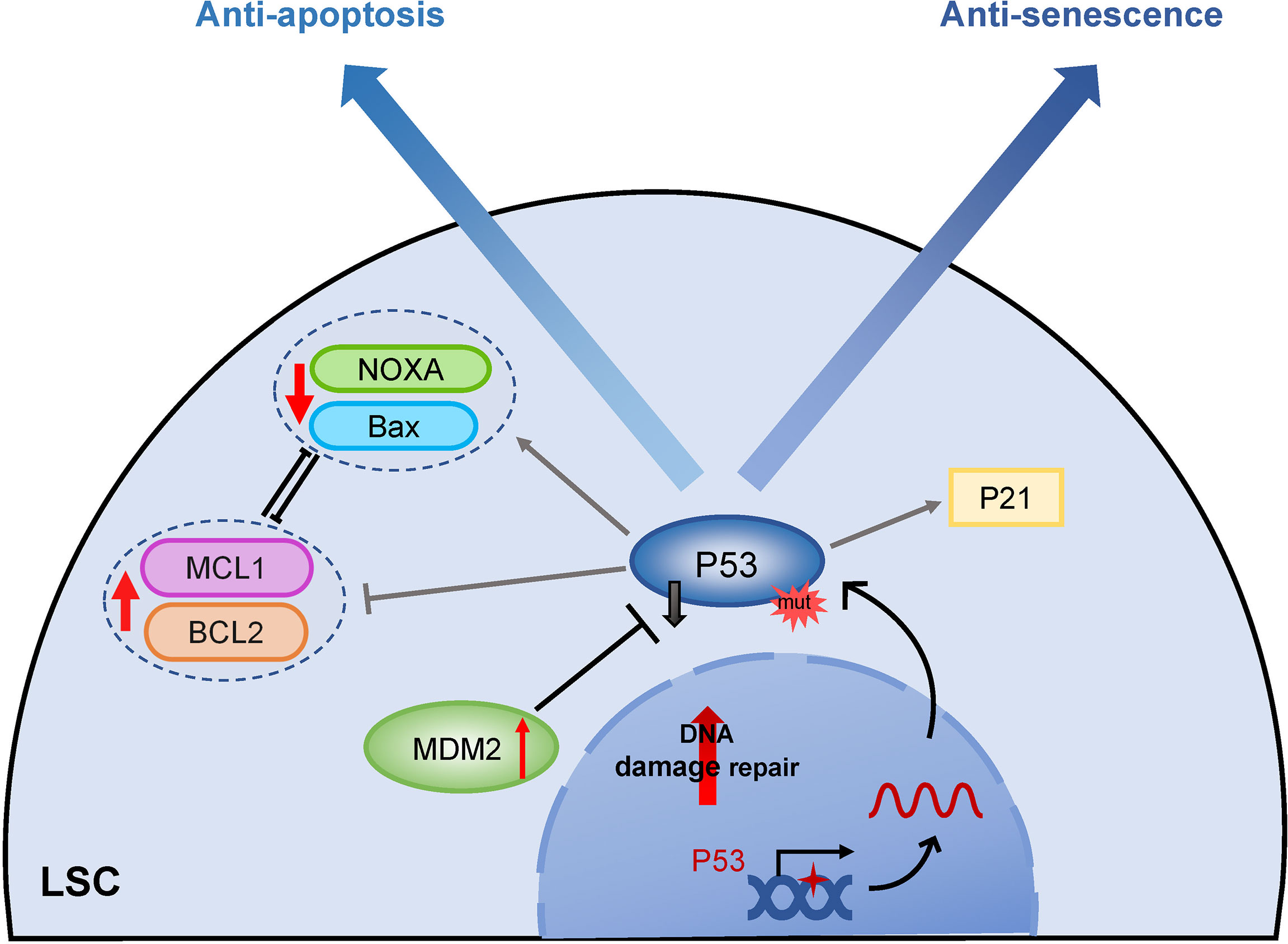
Figure 4 The role of TP53 in apoptosis and senescence. Mutations or inactivation of p53 plays an important role in both apoptosis and senescence resistance of LSCs. While LSCs with mutant P53 are unable to activate downstream P21 to enter the senescent process, they do not target apoptosis pathway proteins including induction of pro-apoptotic proteins like Bax, and apoptosis initiator groups like NOXA and repressing anti-apoptotic members BCL2 and MCL1, eventually fail to initiate apoptosis. MDM2, minute 2 homolog; BCL2, B-cell lymphoma 2; MCL1, myeloid cell leukemia sequence 1; Bax, Bcl-2-associated X protein; NOXA, proapoptotic BH3-only protein.
Senescence Resistance of LSCs Involves Drug Resistance
The observations indicated that stress-induced activation of p38 mitogen-activated protein kinase (p38 MAPK), ROS, and DNA damage response (DDR) could induce HSCs senescence (116). However, a previous study found that LSCs had higher p38 MAPK activity, ROS levels, and increased accumulation of DNA damage but lower cellular senescence compared with HSCs (117). Wajapeyee et al. demonstrated that oncogenes induced senescence of HSCs rather than of LSCs (118). These investigations collectively suggested that senescent resistance mechanisms existed in LSCs. Ablain et al. were the first to identify the mechanisms involving arsenic trioxide in the treatment of acute promyelocytic leukemia (APL). Arsenic trioxide induced senescence through the promyelocytic leukemia gene (PML)-p53 pathway rather than by inducing APL cells apoptosis, which indicated that the induction of senescence was critical for the eradication of LSCs (49). Our recent data firmly demonstrated that lower expression of miR-34c-5p, a central regulator of cell senescence, was probably a significant factor involved in the resistance of LSCs senescence. Up-regulated miR-34c-5p in LSCs induces cellular senescence and promotes the eradication of LSCs in mouse models, which further supported the hypothesis (119).
Most chemotherapy drugs exert their therapeutic effect by inducing senescence of AML cells. However, recent studies have indicated that LSCs present multiple mechanisms of senescence resistance compared with HSCs, and also result in chemotherapy resistance. Until recently, these mechanisms have been poorly understood, for the following potential reasons (1) LSCs present the abnormal expression of both known and unknown molecules related to the senescent signaling pathway; and (2) LSCs activate DNA repair mechanisms leading to chemotherapy-induced DDR that fail to induce cell senescence.
Molecular abnormalities in the p53/p21 or p16/pRb axes, two fundamental signal pathways regulating cell senescence (120), could result in resistance of LSCs senescence. Mutational inactivation of p53 is a significant factor influencing senescence resistance and poor therapeutic outcomes in AML (Figure 4). The mutation rate of p53 in AML patients is less than 10%, and p53 mutation-positive AML patients are mostly treatment-related or are associated with cases of AML with a complex karyotype (110, 121, 122). P53 has been strongly associated with poor chemotherapy efficacy and prognosis of AML, whereas refractory and relapsed (R/R) LSCs are largely dependent on p53 mutation inactivation (112, 123). Currently, few reports have evaluated the expression of senescence regulatory molecules such as p16 and p21 in LSCs, which implicits a research direction to be explored in the future.
DNA damage is considered an important factor inducing cell senescence. Chemotherapy drugs induce DNA damage, and simultaneously DDR occurs in HSCs and LSCs (124). Interestingly, HSCs are more sensitive to chemotherapeutic agents than LSCs. Normal HSCs possess mechanisms able to inhibit DNA damage repair. DNA damage prolongs the S phase of the cell cycle and promotes DNA damage repair, but severe DNA damage can lead to cellular senescence (125). Hence, HSCs may progress to premature senescence induced by DNA damage, which can reduce the accumulation of harmful mutations in HSCs but also disrupts normal hematopoiesis. Conversely, LSCs are able to escape this mechanism, which does not appear to be associated with the phenomenon of premature senescence induced by chemotherapeutic drug-induced DNA damage, because the LSCs population lacks the signals inhibiting DNA damage repair (126). Thus, several DNA damage repair mechanisms are activated in LSCs treated with chemotherapy agents, which protect LSCs from chemotherapy-induced DNA damage and cell senescence (127, 128). Furthermore, sequential chemotherapy cycles also lead to depletion of the normal HSCs pool, and exacerbate the growth advantages of LSCs, resulting in R/R leukemia. Therefore, the restoration of senescence-initiated transmitting signals after chemotherapy-induced DNA damage may represent a novel approach for effectively eradicating AML. Evidence showing that the combined treatment of miR-34c-5p and chemotherapy facilitated LSCs senescence and elimination of LSCs in vivo preliminarily confirmed this hypothesis (119).
A more recent study found that following cytarabine chemotherapy, the enriched-senescent AML cells produced a senescence-associated secretory phenotype (SASP) by increasing NF-κB transcription through the ataxia telangiectasia and Rad3-related (ATR) activities, and regained stemness properties sufficient to restore leukemia (129). These effects may be mediated by changes in the SASP in a specific context. Further, it is important to understand how to eradicate senescent AML cells and overcome the adverse effects of SASP to prevent a recurrence.
Metabolic Reprogramming-Induced Chemoresistance in LSCs
Enhanced glycolysis and an active truncated tricarboxylic acid (TCA) cycle have been described in many malignancies (130). The significant increase in AML blast cells leads to higher glucose uptake and the accumulation of a large amount of lactate, even under unlimited oxygen conditions, noted as the Warburg effect (131). The serum changes of 6 metabolites involved in glucose metabolism, including increased glycerol-3-phosphate, lactate, citrate, and decreased pyruvate, 2-oxoglutarate, 2-hydroxyglutaric acid (2-HG), have independent prognostic values in AML patients, in which poor prognosis was linked to accelerated glycolysis and activation of the TCA cycle (132). Additionally, HSCs reside in the hypoxic BM niche with tightly limited oxidative stress. HSCs specifically utilize glycolysis for energy generation while suppressing the influx of glycolytic metabolites into the mitochondria via pyruvate dehydrogenase kinases (PDKs) activity, which are downstream effectors of HIF-1α (133). Nevertheless, LSCs are particularly dependent on oxidative phosphorylation (OXPHOS) (63). OXPHOS utilizes amino acids, fatty acids, and glucose as energy substrates to generate ATP. It is more efficient to maintain LSCs survival in a hypoxic and nutrient-deprived microenvironment (Figure 5). Increased OXPHOS activity in LSCs has been associated with chemotherapy resistance. In primary human AML, despite the increased mitochondrial mass, LSCs display reduced spare respiratory capacity for OXPHOS compared to mature AML blasts and normal HSCs (134). It appears to be paradoxical that LSCs are maintained in a low ROS state because OXPHOS is prone to produce more ROS. Therefore, the metabolic stringency of LSCs suggests that they are susceptible to OXPHOS disturbances.
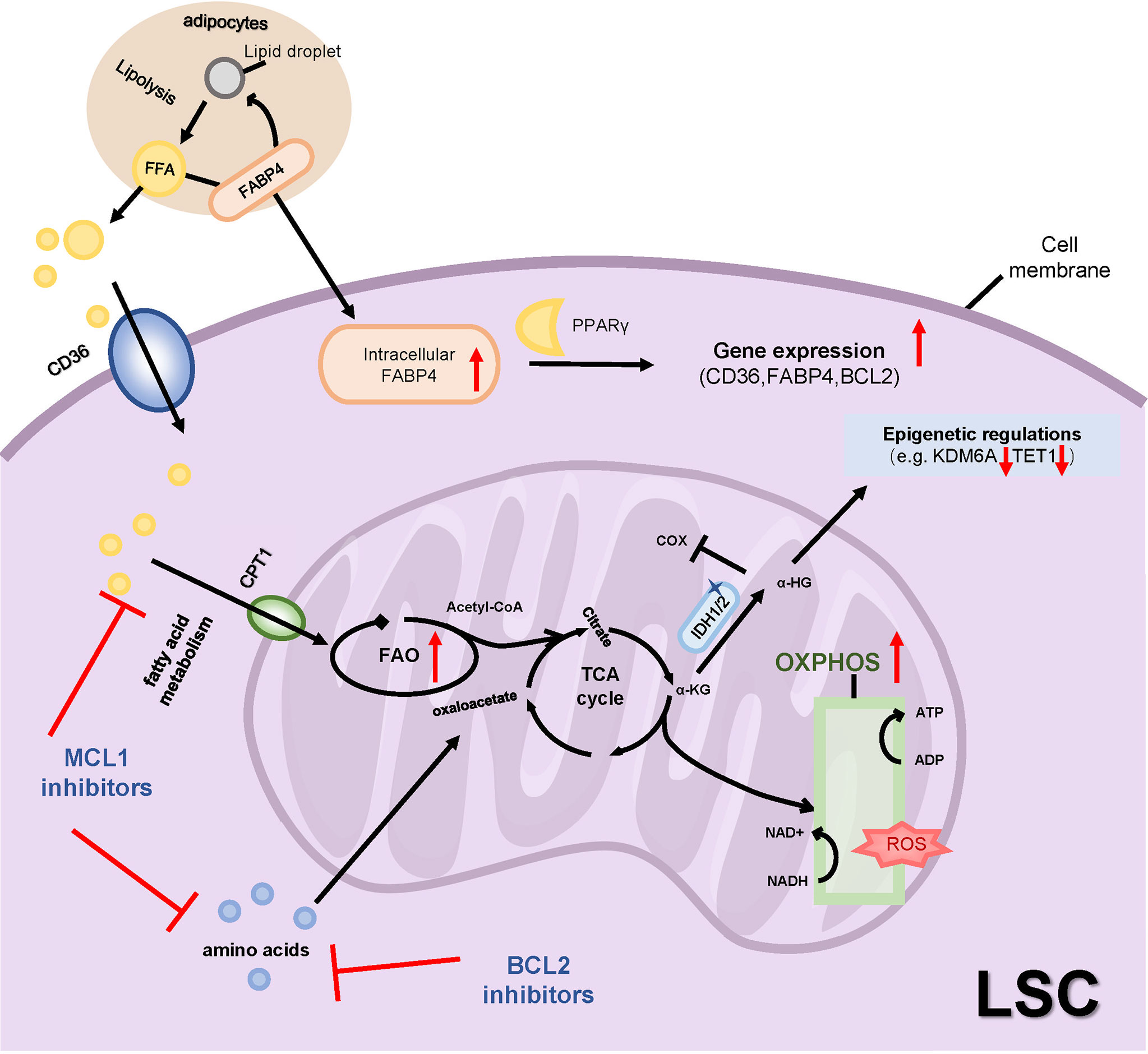
Figure 5 Metabolic alteration and drug resistance. LSCs are particularly dependent on OXPHOS, which utilizes amino acids and fatty acids rather than glucose as energy substrates to generate ATP. Therapy-resistant LSCs exhibit increased FAO through increasing fatty acid transporters such as CD36, CPT1 and FABP4. Inhibitions of MCL1 or BCL2 inhibit amino acid and fatty acid metabolisms to reduce OXPHOS. The mutant IDH1/2 catalyzes the conversion of α-KG to 2-HG, the latter regulates both epigenetic and metabolic changes. OXPHOS, oxidative phosphorylation; FAO, fatty acid oxidation; TCA, tricarboxylic acid cycle; FFA, free fatty acids; FABP4, fatty acid-binding protein-4; PPARγ, peroxisome proliferator-activated receptor γ; CPT1, carnitine O-palmitoyltransferase 1; α-KG, α-ketoglutaric acid; α-HG, 2-hydroxyglutaric acid; IDH1/2, isocitrate dehydrogenase 1 or 2; cox, cytochrome c oxidase; KDM6A, Lysine Demethylase 6A.
BCL2 is highly expressed in LSCs defined by low ROS levels. Venetoclax targets LSCs by inhibiting OXPHOS and impairs energy homeostasis. Jones et al. suggested that LSCs were uniquely dependent on amino acids that catabolize into the intermediates of the TCA to fuel OXPHOS in de novo AML patients. In relapsed AML patients, fatty acid catabolism was alternatively upregulated to maintain OSPHOS levels upon venetoclax plus azacitidine (ven/aza) treatment (135). Another study revealed that R/R AML patients achieved a worse response to ven/aza treatment because of elevated nicotinamide metabolism in LSCs which activated both amino acid and fatty acid metabolism in the TCA cycle to drive OXPHOS. Moreover, inhibition of nicotinamide phosphoribosyltransferase (NAMPT), the rate-limiting enzyme for the biosynthesis of nicotinamide adenine dinucleotide (NAD+), specifically targeted R/R LSCs by decreasing OXPHOS. Further, NAMPT inhibitors decreased glycolysis, but not OXPHOS, in AML blasts with high ROS levels, suggesting a metabolic heterogeneity of AML cells (136). In addition to the metabolic plasticity of LSCs, loss of BCL2 expression is another mechanism of venetoclax drug resistance. In particular, patients with monocytic AML are preferentially dependent on the anti-apoptotic protein MCL1 for OXPHOS activity (106). Inhibition of MCL1 showed a dual restriction on amino acid levels and fatty acid oxidation to reduce OXPHOS. Pre-clinical trials investigating multiple MCL1 inhibitors MIK665 (44), PRT1419 (45), AMG176 (46) and AMG397 (137) have been reported to exert anti-leukemia activity and have reported synergistic effects with venetoclax in murine models of AML (Table 1).
As described previously, ven/aza-resistant LSCs exhibit increased fatty acid oxidation (FAO) compared to ven/aza-sensitive LSCs, including L-carnitine and acyl-carnitines rather than the levels of fatty acids (138). BM adipocytes provide sufficient fatty acids for LSCs survival and proliferation. Inhibition of fatty acid transporters, such as fatty acid transporter CD36, fatty acid mitochondrial transporter CPT1, and fatty acid-binding protein 4 (FABP4), are other mechanisms that reduce FAO to overcome drug resistance. The expression of CD36 was increased in AraC-resistant LSCs and enhanced proliferative activity (63). Studies have shown that CD36+ LSCs were protected from chemotherapy damage by the microenvironment of gonadal adipose tissue (GAT). Drug-resistant LSCs secretes TNF-α, IL-1α, IL-1β, and CSF2 to promote GAT lipolysis and the release of free fatty acids (FFA) in the MLL-AF9 mouse model. CD36+ LSCs increased intracellular FAO levels by uptake of lipolytic free fatty acids (139). And deletion of CD36 in combination with ven/aza showed the potential to eliminate resistant LSCs. The CPT1 inhibitor, etomoxir, similarly decreased LSCs activity only in the presence of ven/aza, indicating that inhibition of both amino acid and fatty acid metabolism is necessary to target LSCs resistant to ven/aza treatment (138). BM adipocytes have higher FABP4 expression and promote lipid transport to support the activities of AML cells, and FABP4 can activate the downstream peroxisome proliferator-activated receptor (PPARγ) to further induce gene transcription of CD36, FABP4, and BCL2 in AML cells (140). These studies explain how adipose tissue and fatty acid transport support AML metabolism and are associated with drug resistance (Figure 5). In addition, the sphingolipid pathway is involved in the regulation of leukemia cell apoptosis, thus targeting sphingolipid metabolism also has considerable clinical significance (141). Xie SZ et al. demonstrated that S1PR3, a receptor for sphingosine-1-phosphate, was specifically overexpressed in the myeloid-like subset of LSCs. S1PR3 regulates myeloid differentiation, activates inflammatory and metabolic-related pathways through TNFα–NF-κB axis which disrupts LSCs stemness (142).
Mutations in isocitrate dehydrogenase 1 or 2 (IDH1/2) occur in 10 to 20% of the patients with AML and play an important role in the occurrence of AML (143). The mutant IDH1/2 catalyzes the conversion of α-ketoglutaric acid (α-KG) to 2-HG. The accumulation of the oncometabolite 2-HG regulates both epigenetic and metabolic changes in AML (Figure 5). 2-HG consistently inhibited the activity of cytochrome c oxidase (COX) and promoted the breakdown of L-glutamine, inducing the dependence of LSCs on BCL2, thus demonstrating the therapeutic sensitivity of venetoclax (51). Importantly, the US Food and Drug Administration (FDA) has approved inhibitors targeting mutant IDH1 or IDH2 to treat AML with relapses or resistance to other drugs. Clinical trials of ivosidenib and enasidenib, respectively mIDH1 and mIDH2 inhibitors, in newly diagnosed and R/R AML patients with IDH1/IDH2 mutations have improved chemotherapy response, and mutation-cleared AML patients had a longer duration of remission and a longer overall survival than patients without mutation clearance (3, 144–146). Other clinical trials reported that IDH1/2 inhibitors plus azacytidine achieved a higher complete response rate and mutation clearance and prolonged survival (147)(Table 1). But in patients with AML with simultaneous FLT3-ITD or RAS mutations, IDH1/2 inhibitors were less effective (148, 149). Mutations in receptor tyrosine kinase (RTK) pathway genes and IDH-related mutations were associated with resistance/relapse of ivosidenib (149). These prompt us to pay attention to the resistance mechanisms of IDH inhibitors and their combined application with other targeted inhibitors.
Epigenetic Alternations Driving Drug Resistance of LSCs
Driver mutations in leukemia often involve epigenetic genes, and subsequent epigenetic abnormalities promote the progression of AML. Specific epigenetic modifications of LSCs are linked to chemotherapy resistance and relapse. Methylation, the most common type of epigenetic modification in tumors, operates at the DNA, RNA, and histone levels as a “reader”, “writer” and “eraser” (Figure 6).
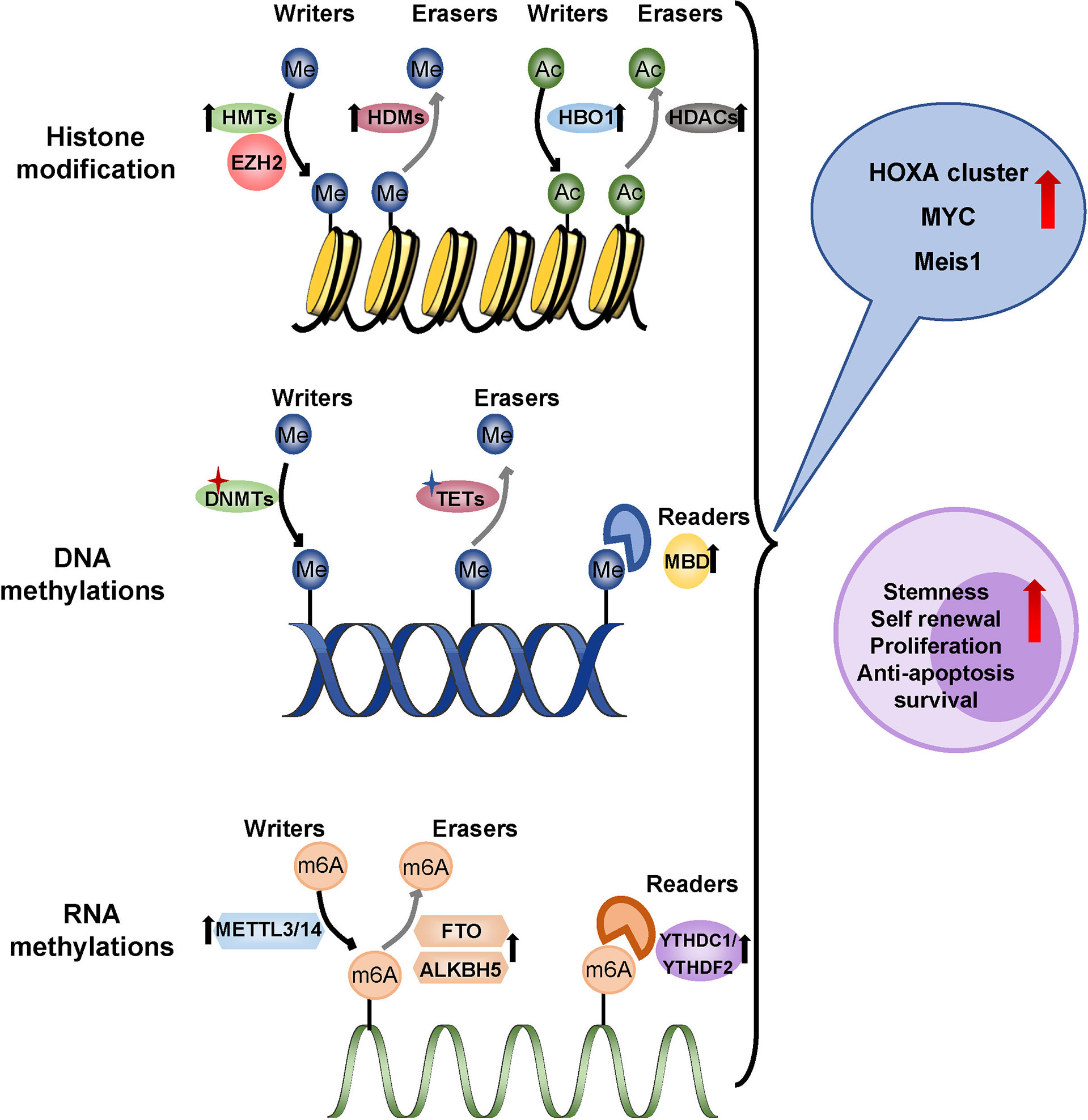
Figure 6 Epigenetic regulation in LSCs. Methylation operates at the DNA, RNA, and histone levels as a ‘reader’, ‘writer’ and ‘eraser’. LSCs showed a hypomethylated state which overexpressed the tumor-promoting gene like HOXA cluster, MYC and MEIS1. Overexpressed HBO1 in LSCs adds acetyl to histone H3K14 up-regulating HOXA gene transcription. Me, methyl; Ac, Acetyl; HMTs, histone methyltransferases; HDMs, histone demethylases; EZH2, zeste homolog 2; HBO1, lysine acetyltransferase 7; HDACs, histone deacetylases; DNMTs, DNA methyltransferases; TETs, ten-eleven translocation; MBD, methyl-CpG binding domain protein 2; m6A, N6-methyladenosine; METTL3/14, methyltransferase-like protein 3/14; FTO, fat mass and obesity-associated protein; ALBH5, ALKB Homolog 5YTHDC1/YTHDF2, YTH family proteins.
Aberrant DNA methylations are common molecular changes, including DNA methylation mutations and hypermethylation of CpG islands, which are associated with the prognosis of patients with AML (150). DNA hypermethylation generally inhibits gene expression, while hypomethylation leads to chromosome instability and abnormal gene activation (151). It was generally believed that the mutation of epigenetic factors was mainly involved in the conversion of HSPCs to pre-LSCs, and involved mainly DNA methyltransferase 3A (DNMT3A) and Tet methyl cytosine dioxygenase 2 (TET2), which made HSCs more prone to leukemogenesis (152). Pre-LSCs mutations that persist after chemotherapy were more likely to relapse of AML. DNMT3A mutations are frequently accompanied by FLT3 and NPM1 mutations in AML. The frequency of LSCs was increased in samples from patients with triple mutations in AML, indicating a poor prognosis (153). The deletion mutations of TET2 coexisted with mutations such as TP53, FLT3, and KRAS, cooperating to drive the occurrence of leukemia, which is related to a poor prognosis (154). Another DNA methylation reader, the methyl-CpG binding domain protein 2 (MBD2), maintained the quiescent state of LSCs by inhibiting the expression of cyclin-dependent kinase inhibitor 2C (CDKN2C) by hypermethylation (155). As a whole, LSCs showed a hypomethylated state rather than mutation dependence, which overexpressed the HOXA cluster, suggesting the possibility of resistance to hypomethylating agents (156).
In recent years, increasing studies have revealed the role of RNA epigenetic modification in the initiation and progression of AML. N6-methyladenosine (m6A) is the most frequent RNA epigenetic modification including METTL3/14 methylase as “writer” proteins, FTO/ALKBH5 demethylase as “eraser” proteins and YTH family proteins as “reader” proteins. Patients with higher m6A scores had a higher survival rate, while those with lower m6A scores had a poor prognosis. Methyltransferase 3(METTL3) increased the translation of downstream leukemia-related genes, such as MYC, BCL2, and PTEN mRNA, in an m6A-dependent manner, and depletion of METTL3 blocked proliferation, induced differentiation, and apoptosis in immunodeficient recipient mice (157). In addition, METTL3 recruitment by the transcription factor CEBPZ to the promoter of SP1, increased its mRNA methylation level, and promoted its protein translation by alleviating ribosomal stasis, which was essential for the maintenance of LSCs (158). STM2457, a METTL3 inhibitor, strongly reduced the frequency of LSCs, inhibited the implantation and progression of AML derived from patient xenografts, and prolonged the life span of recipient mice (159). METTL14 has also been found to play an important role in LSCs. METTL14 can enhance the stability of the oncogenic transcription factor MYB and MYC mRNA and promote translation to progress AML (160). The fat mass and obesity-associated protein (FTO) was the first m6A demethylase identified that removes m6A methylation modifications. FTO was more abundant in CD34+ AML cells. Knockdown of FTO in LSCs achieved a lower frequency of LSCs, impaired their self-renewal ability, and promoted apoptosis and differentiation by decreasing the expression of MYC and CEBPA in an m6A-dependent manner (161). Meanwhile, inhibition of FTO significantly reprogrammed the immune response by inhibiting the expression of the immune checkpoint gene LILRB4, making leukemia cells sensitive to T cell toxicity and overcoming hypomethylation agent-induced immune evasion (162). Additionally, ALKB Homolog 5 (ALKBH5) was significantly higher in LSCs derived from AML patients, which was related to a poorer prognosis. The deletion of ALKBH5 can significantly inhibit proliferation and impair the leukemia initiation potential in the immunodeficient mouse model (163). ALKBH5 acts through the KDM4C-ALKBH5-AXL signal axis in AML. KDM4C increased chromatin accessibility by recruiting MYB and Pol II to the ALKBH5 promoter and reducing H3K9me3. ALKBH5 subsequently promoted the expression of receptor tyrosine kinase (RTK) AXL, further activated downstream PI3K, MAPK, JAK/STAT, and NF-κB pathways, and played an essential role in the development and maintenance of LSCs (164). Furthermore, recent studies have shown that the m6A recognition protein YTHDF2 (YTH N6-Methyladenosine RNA Binding Protein 2) promoted the stemness maintenance of AML LSCs. The deletion of YTHDF2 improved apoptosis to selectively eliminate LSCs (165). Recently, it has been discovered that YTH Domain Containing 1 (YTHDC1) was the top essential m6A reader in AML from a genome-wide CRISPR screen. YTHDC1 enhanced MYC expression and reduced degradation to maintain leukemogenesis. YTHDC1 knockdown also reduced LSCs proliferation, increased differentiation, and delayed the development of leukemia (166).
Histone modification is an essential form of epigenetic regulation. Histone methyltransferases (HMTs) and histone demethylases (HDMs) regulate methylation at different histone locations. The Histone-H3 lysine-79 (H3K79) methyltransferase DOT1L and H3K4 demethylases KDM1A play a key role in the occurrence and development of LSCs harboring the MLL rearrangement by up-regulating stem cell-related genes HOXA and Meis (167, 168). In the primary AML cells, the KDM1A inhibitors inhibit PI3K pathway and activate the MEK pathway, further inhibition of KDM1A sequential the MEK inhibitor significantly promotes leukemia cell apoptosis especially in M5 leukemia, which was associated with protein phosphorylation and RAS mutation (169). The DOT1L inhibitor EPZ5676 (170) and KDM1A inhibitor ORY-1001 (52), respectively, inhibited the proliferation and differentiation of MLL-LSCs and increased the survival of transplanted mice. Nuclear receptor binding SET Domain Protein 1 (NSD1) encodes mono- and di-methylation of H3K36. The NUP98-NSD1 fusion activated HOX gene through H3K36 hypermethylation for leukemogenesis, and was identified as a high-risk factor for AML (171). A recent study reported that targeting inhibitor of NSD1, BT5, reduced the expression levels of HOX cluster and MEIS1 and impaired the colony formation of primary AML cells (172). And inhibition of NUP98-NSD1 reduced leukemia burden and prolonged survival in NUP98-NSD1 patient-derived xenograft model (172, 173).
Other histone methylation modifications at methyl residues include H3K27 methyltransferase component zeste homolog 2 (EZH2) and H3K27 demethylase KDM6B. EZH2 is a member of the Polycomb inhibitory complex 2 (PRC2), which acts as a transcriptional inhibitor (174). Low levels or loss of the EZH2 protein are associated with chemoresistance and a poor prognosis (175). However, with the advanced evolution of AML, EZH2 may play the opposite oncogenic function, which demonstrates the dependence on tumor stage and context (176). Thus, targeted EZH2 therapy should consider leukemia status. 3-Deazaneplanocin A (DZNep), an inhibitor of EZH2 and H3K27me3, targeted LSCs to induce apoptosis (177). Overexpression of KDM6B is correlated with poor OS in AML. GSK-J4, a KDM inhibitor, decreased the level of the cAMP response element-binding protein (CREB) and inhibited the proliferation of LSCs (178). However, KDM6B also maintained self-renewal and quiescence of the HSCs (179). This may limit the clinical translation of GSK-J4.
Histone modification also includes histone acetylation. Generally, increased acetylation is related to up-regulating transcription activity, while decreased acetylation is related to inhibition of gene expression (Figure 6) (151). The MYST acetyltransferase HBO1 and acetylation of histone H3K14, upregulate the HOXA family of genes in MLL-LSCs, which are essential for the maintenance of LSCs. WM-3835 exhibits a remarkable anti-leukemic effect in vitro by the reduction of H3K14ac levels (180). The abnormal recruitment of histone deacetylase (HDAC) proteins into cancer-causing fusion proteins such as AML1-ETO, CBFB-MYTH11, PML-Rα and MLL plays an important role in the development of leukemia (181). However, clinical results have shown that HDAC inhibitors are not effective as a monotherapy for AML, and in vitro experiments have shown that HDAC inhibitors and DNMT inhibitors cooperate to induce the re-expression of silent genes in LSCs (53). Phase I clinical trial showed that panobinostat, a HDAC inhibitor, combined with intensive chemotherapy induced CR/incomplete count recovery (Cri) in 8 elderly AML patients (54). The reversibility of epigenetic modification provides an opportunity to personalize AML with specific epigenetic inhibitors.
The Protection Mechanisms of LSCs Within the BM Microenvironment
Chemotherapy-resistant LSCs home and remodel the BM niche to allow leukemia survival, thus inhibiting normal hematopoiesis. The complex crosstalk between LSCs and their microenvironment contributes to LSCs survival, treatment resistance, and recurrence (Figure 7). Targeting the LSCs microenvironment is a critical avenue for the treatment of AML.
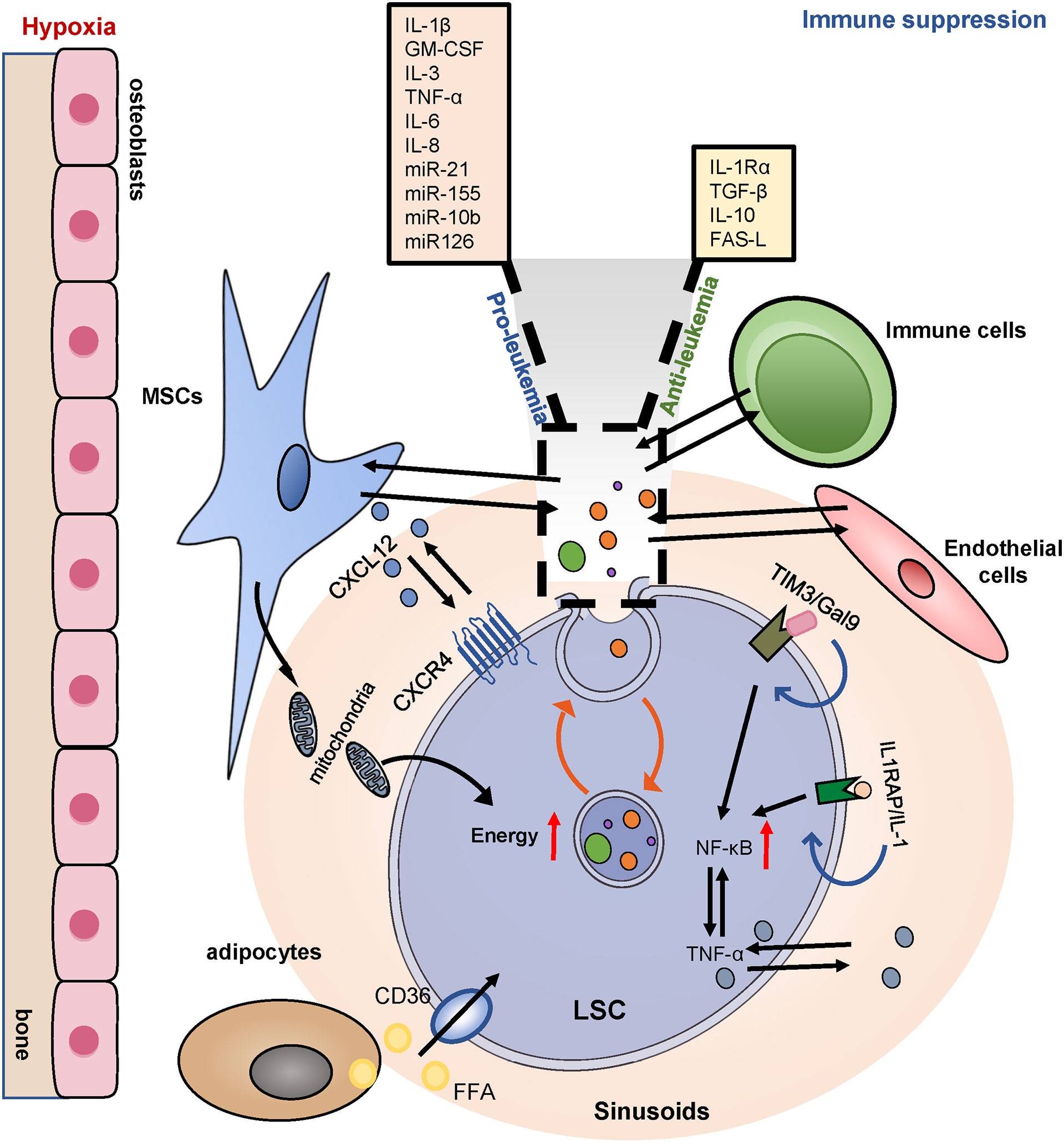
Figure 7 BM niches and drug resistance. LSCs utilize the BM niches for increased survival. LSCs highly express CXCR4, binding to its ligand CXCL12, positioned in a hypoxic area. BMSCs also transfer mitochondria to provide additional energy for the survival of LSCs. Adipocytes provide sufficient fatty acids for LSCs metabolic reprogramming. LSCs globally induce immune tolerance via upregulating TIM3 and inducing multiple chemokines and an inflammatory secretome and thereby promoting leukemia progression. BM, bone marrow; MSCs, mesenchymal stromal cells; CXCR4, CXC chemokine receptor-type 4; CXCL12, CXC motif chemokine ligand 12; IL1RAP, interleukin-1 co-receptor; TIM3/Gal9, T cell immunoglobulin and mucin protein 3/galectin-9; NF-κB, nuclear factor κB. EVs, extracellular vesicles.
BMSCs play a key role in remodeling the leukemia niche. The CXCR4/CXCL12 axis plays a central role in the regulation of LSCs-BMSCs interactions. Blocking the interaction between CXCR4/CXCL12 effectively compromised the homing of LSCs into the BM niche and rendered leukemia cells sensitive to chemotherapy (55). The BMSCs transferred mitochondria to provide additional energy for the survival of both the AML blast cells and the LSCs. And recipient LSCs were resistant to cytarabine-induced apoptosis and had a greater potential for leukemia initiation (182). Furthermore, the accumulation of Nestin+ BMSCs in the bone marrow of AML was critical for the viability and proliferation of LSCs in vitro and in vivo, Nestin+ BMSCs induced chemoresistance by increasing energy production and glutathione-peroxidase (GPX) activity in LSCs (183). BMSCs protect AML cells from chemotherapy by increasing stem-type signaling pathways, such as the Notch and Wnt pathways, and by inhibiting apoptosis (184). In addition, senescent BMSCs increased the survival and proliferation of AML in the form of cytokine secretion (185).
The formation and maintenance of the AML niche depend on the imbalance of the cytokine profile. LSCs existed in a pro-inflammatory environment that was conducive to the survival and proliferation of LSCs. Autocrine tumor necrosis factor α (TNF-α) of LSCs activated NF-κB activity to form the NF-κB/TNF-α positive feedback loop (186). Similarly, LSCs highly expressed the interleukin-1 co-receptor IL1RAP, while deletion of IL1RAP inhibited the stemness of LSCs and increased apoptosis (187). Researchers have found a specific overexpression of T cell immunoglobulin and mucin protein 3 (Tim-3) in LSCs. In addition, the formation of the Tim-3/Gal-9 autocrine loop activated NF-κB and β-catenin pathways to support the self-renewal and survival of AML cells (56). Treatment with anti-Tim-3 antibodies not only reduced LSCs, but also regulated immune balance to prevent AML from immune evasion (188). In BM, several cytokines and soluble factors have been shown to influence the survival and growth of leukemia cells. For example, the pro-inflammatory cytokines IL-1β, GM-CSF, IL-3, TNF-α, and IL-6 appear to promote the proliferation of AML cells, while anti-inflammatory molecules such as IL-1Rα, TGF-β and IL-10 exert inhibitory effects (189). The specific functions of cytokines depend on the crosstalk of multiple complex molecules within the microenvironment.
Extracellular vesicles(EVs), classified into exosomes, microvesicles, and apoptotic bodies, also regulate the interaction between LSCs and the BM niche (190). A growing number of studies have shown that EVs played a key role in drug resistance and regulating immune response regulation. EVs derived from AML bone marrow-derived MSCs promote AML cell proliferation, migration and protect AML cells from treatment with AraC (191). BMSCs secrete exosomes and IL-8 to promote the resistance to etoposide in the AML stem cell line KG1a (192). Chemoresistant AML cells were able to induce increased expression of MRP-1 in chemosensitive AML cells through EVs to enable them to be resistant to chemotherapy drugs (193). EVs contain higher levels of known clinical risk factors, such as TGF-β1, miR-21, miR-155, miR-10b (194, 195). Similarly, exosomes carry Fas ligand (FAS-L), NPM1, FLT3, matrix metallopeptidase 9 (MMP9), insulin-like growth factor type 1 receptor (IGF1-R), CXCR4, and chaperones to alter the BM microenvironment and to improve the survival of leukemia cells, especially LSCs (196, 197). Overall, EVs can directly or indirectly alter the signaling pathways, or increase the mitochondrial content, leading to the proliferation, survival, and drug resistance of AML. Therefore, targeted blocking of EVs in the crosstalk between LSCs and the supportive microenvironment may be considered a promising therapy.
Conclusions
Multiple intracellular and extracellular mechanisms are the root of drug resistance and relapse in AML stem cells, and influence the dormancy, resistance to apoptosis and senescence, epigenetic modification, metabolic reprogramming, and protection of the BM microenvironment. Although corresponding targeted drugs have been developed and are being applied in the clinic, it remains difficult to eradicate LSCs. We are now convinced that AML might alter leukemic clones during treatment, which results in tumor progression. LSCs were located at the top of the clonal heterogeneity, and their clonal diversification is accelerated by the tumor mutation burden (26). AML clone evolution is divided into linear and branching patterns. The former stepwise acquires clonal hematopoiesis (CH)-type mutations and AML-related mutations. The latter parallelly acquires CH-type mutations and AML-related mutations in different cell populations (198). CH is prominent in AML clone evolution, and is assumed to represent a pre-malignant state. DNMT3A, TET2, and ASXL1 (“DTA”) mutations prevalently occur early in pre-leukemia hematopoietic stem and progenitor cells (HSPCs), persist for a long time and are not considered to be associated with poor prognosis. The acquisition and persistence of other CH-type mutations like TP53, IDH1/2 are related to an increased risk of AML relapse (199). AML transformation occurs with acquirement of AML-related mutations like FLT3, NPM1, NRAS/KRAS. The relapse of LSCs clones derives from persistent clonal evolution or new sub-clonal structures post-chemotherapy. Mutations in AML can persist after treatment induction, or new types of AML mutations may appear after chemotherapy, eventually leading to the recurrence of leukemia (20) (Figure 1). LSCs are the root of drug resistance and are prone to the co-existence of multiple mechanisms and crosstalk with the BM microenvironment.
In general, treatment agents must be adjusted to target altered LSCs clones. Future treatment strategies have been proposed to address the dynamics of LSCs clones; thus, real-time monitoring of minimal residual disease (MRD) and timely adjustment of treatment regimens are critical. Finding more LSCs-specific therapeutic targets that can avoid affecting normal hematopoiesis. Furthermore, chemotherapy can induce multiple drug-resistant LSCs clones, and consequently, it is critical to focus on the elimination of naïve LSCs that can minimize the formation of polyclonal LSCs. Regarding relapsed drug-resistant LSCs, combined therapy is necessary due to the different mechanisms of drug resistance. Ultimately. Single-cell combined with multi-omics sequencing will contribute to the specific identification and the discovery of key mechanisms of LSCs. Preliminary clinical trials have shown that the combination of novel agents or with traditional chemotherapeutic agents increased clinical activity, which was beneficial in overcoming primary resistance and second resistance in AML.
Author Contributions
JN and DP reviewed the literature and wrote the manuscript, LL provided concept and guidance and approved the final version to be published. All authors contributed to the article and approved the submitted version.
Funding
The study was supported by grants from the National Natural Science Foundation of China (Nos. 81973999, 81900188, 81770192, 81370660), and the Wuhan Science and Technology Program, China (No. 2017060201010180).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank the Charlesworth Group (www.cwauthors.com.cn) for English language editing.
References
1. Short NJ, Rytting ME, Cortes JE. Acute Myeloid Leukaemia. Lancet (2018) 392(10147):593–606. doi: 10.1016/s0140-6736(18)31041-9
2. Stone RM, Mandrekar SJ, Sanford BL, Laumann K, Geyer S, Bloomfield CD, et al. Midostaurin Plus Chemotherapy for Acute Myeloid Leukemia With a FLT3 Mutation. N Engl J Med (2017) 377(5):454–64. doi: 10.1056/NEJMoa1614359
3. Roboz GJ, DiNardo CD, Stein EM, de Botton S, Mims AS, Prince GT, et al. Ivosidenib Induces Deep Durable Remissions in Patients With Newly Diagnosed IDH1-Mutant Acute Myeloid Leukemia. Blood (2020) 135(7):463–71. doi: 10.1182/blood.2019002140
4. Dinardo CD, Schuh AC, Stein EM, Montesinos P, Wei A, De Botton S, et al. Effect of Enasidenib (ENA) Plus Azacitidine (AZA) on Complete Remission and Overall Response Versus AZA Monotherapy in Mutant-IDH2 (Midh2) Newly Diagnosed Acute Myeloid Leukemia (ND-AML). J Clin Oncol (2020) 38(15):7501–7501. doi: 10.1200/JCO.2020.38.15_suppl.7501
5. Wei AH, Montesinos P, Ivanov V, DiNardo CD, Novak J, Laribi K, et al. Venetoclax Plus LDAC for Newly Diagnosed AML Ineligible for Intensive Chemotherapy: A Phase 3 Randomized Placebo-Controlled Trial. Blood (2020) 135(24):2137–45. doi: 10.1182/blood.2020004856
6. Maiti A, Qiao W, Sasaki K, Ravandi F, Kadia TM, Jabbour EJ, et al. Venetoclax With Decitabine vs Intensive Chemotherapy in Acute Myeloid Leukemia: A Propensity Score Matched Analysis Stratified by Risk of Treatment-Related Mortality. Am J Hematol (2021) 96(3):282–91. doi: 10.1002/ajh.26061
7. Roboz GJ, Montesinos P, Selleslag D, Wei A, Jang JH, Falantes J, et al. Design of the Randomized, Phase III, QUAZAR AML Maintenance Trial of CC-486 (Oral Azacitidine) Maintenance Therapy in Acute Myeloid Leukemia. Future Oncol (2016) 12(3):293–302. doi: 10.2217/fon.15.326
8. Cortes JE, Heidel FH, Hellmann A, Fiedler W, Smith BD, Robak T, et al. Randomized Comparison of Low Dose Cytarabine With or Without Glasdegib in Patients With Newly Diagnosed Acute Myeloid Leukemia or High-Risk Myelodysplastic Syndrome. Leukemia (2019) 33(2):379–89. doi: 10.1038/s41375-018-0312-9
9. Schlenk RF, Paschka P, Krzykalla J, Weber D, Kapp-Schwoerer S, Gaidzik VI, et al. Gemtuzumab Ozogamicin in NPM1-Mutated Acute Myeloid Leukemia: Early Results From the Prospective Randomized AMLSG 09-09 Phase III Study. J Clin Oncol (2020) 38(6):623–32. doi: 10.1200/jco.19.01406
10. Short NJ, Kantarjian H. When Less Is More: Reevaluating the Role of Intensive Chemotherapy for Older Adults With Acute Myeloid Leukemia in the Modern Era. J Clin Oncol (2021) 39(28):3104–8. doi: 10.1200/jco.21.00960
11. Dohner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Buchner T, et al. Diagnosis and Management of AML in Adults: 2017 ELN Recommendations From an International Expert Panel. Blood (2017) 129(4):424–47. doi: 10.1182/blood-2016-08-733196
12. Bonnet D, Dick JE. Human Acute Myeloid Leukemia is Organized as a Hierarchy That Originates From a Primitive Hematopoietic Cell. Nat Med (1997) 3(7):730–7. doi: 10.1038/nm0797-730
13. Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A Cell Initiating Human Acute Myeloid Leukaemia After Transplantation Into SCID Mice. Nature (London) (1994) 367(6464):645–8. doi: 10.1038/367645a0
14. Eppert K, Takenaka K, Lechman ER, Waldron L, Nilsson B, Van Galen P, et al. Stem Cell Gene Expression Programs Influence Clinical Outcome in Human Leukemia. Nat Med (2011) 17(9):1086–93. doi: 10.1038/nm.2415
15. Ng SWK, Mitchell A, Kennedy JA, Chen WC, McLeod J, Ibrahimova N, et al. A 17-Gene Stemness Score for Rapid Determination of Risk in Acute Leukaemia. Nature (2016) 540(7633):433–7. doi: 10.1038/nature20598
16. van Galen P, Hovestadt V, Wadsworth MH, Hughes TK, Griffin GK, Battaglia S, et al. Single-Cell RNA-Seq Reveals AML Hierarchies Relevant to Disease Progression and Immunity. Cell (2019) 176(6):1265–1281.e24. doi: 10.1016/j.cell.2019.01.031
17. Sarry JE, Murphy K, Perry R, Sanchez PV, Secreto A, Keefer C, et al. Human Acute Myelogenous Leukemia Stem Cells are Rare and Heterogeneous When Assayed in NOD/SCID/IL2R Gamma C-Deficient Mice. J Clin Invest (2011) 121(1):384–95. doi: 10.1172/jci41495
18. Hanekamp D, Denys B, Kaspers GJL, Te Marvelde JG, Schuurhuis GJ, De Haas V, et al. Leukaemic Stem Cell Load at Diagnosis Predicts the Development of Relapse in Young Acute Myeloid Leukaemia Patients. Br J Haematol (2018) 183(3):512–6. doi: 10.1111/bjh.14991
19. Ho T-C, Lamere M, Stevens BM, Ashton JM, Myers JR, O’Dwyer KM, et al. Evolution of Acute Myelogenous Leukemia Stem Cell Properties After Treatment and Progression. Blood (2016) 128(13):1671–8. doi: 10.1182/blood-2016-02-695312
20. Shlush LI, Mitchell A, Heisler L, Abelson S, Ng SWK, Trotman-Grant A, et al. Tracing the Origins of Relapse in Acute Myeloid Leukaemia to Stem Cells. Nature (2017) 547(7661):104–8. doi: 10.1038/nature22993
21. Boyd AL, Aslostovar L, Reid J, Ye W, Tanasijevic B, Porras DP, et al. Identification of Chemotherapy-Induced Leukemic-Regenerating Cells Reveals a Transient Vulnerability of Human AML Recurrence. Cancer Cell (2018) 34(3):483–98.e5. doi: 10.1016/j.ccell.2018.08.007
22. Vu LP, Kharas MG. Targeting the Residual Leukemia Cells After Chemotherapy. Cancer Cell (2018) 34(3):353–5. doi: 10.1016/j.ccell.2018.08.012
23. Phan TG, Croucher PI. The Dormant Cancer Cell Life Cycle. Nat Rev Cancer (2020) 20(7):398–411. doi: 10.1038/s41568-020-0263-0
24. Saito Y, Kitamura H, Hijikata A, Tomizawa-Murasawa M, Tanaka S, Takagi S, et al. Identification of Therapeutic Targets for Quiescent, Chemotherapy-Resistant Human Leukemia Stem Cells. Sci Trans Med (2010) 2(17):17ra9. doi: 10.1126/scitranslmed.3000349
25. Hackl H, Steinleitner K, Lind K, Hofer S, Tosic N, Pavlovic S, et al. A Gene Expression Profile Associated With Relapse of Cytogenetically Normal Acute Myeloid Leukemia is Enriched for Leukemia Stem Cell Genes. Leuk Lymphoma (2015) 56(4):1126–8. doi: 10.3109/10428194.2014.944523
26. Vetrie D, Helgason GV, Copland M. The Leukaemia Stem Cell: Similarities, Differences and Clinical Prospects in CML and AML. Nat Rev Cancer (2020) 20(3):158–73. doi: 10.1038/s41568-019-0230-9
27. Hope KJ, Jin LQ, Dick JE. Acute Myeloid Leukemia Originates From a Hierarchy of Leukemic Stem Cell Classes That Differ in Self-Renewal Capacity. Nat Immunol (2004) 5(7):738–43. doi: 10.1038/ni1080
28. Saito Y, Uchida N, Tanaka S, Suzuki N, Tomizawa-Murasawa M, Sone A, et al. Induction of Cell Cycle Entry Eliminates Human Leukemia Stem Cells in a Mouse Model of AML. Nat Biotechnol (2010) 28(3):275–U133. doi: 10.1038/nbt.1607
29. Gruszka AM, Valli D, Alcalay M. Wnt Signalling in Acute Myeloid Leukaemia. Cells (2019) 8(11):1403. doi: 10.3390/cells8111403
30. Salik B, Yi H, Hassan N, Santiappillai N, Vick B, Connerty P, et al. Targeting RSPO3-LGR4 Signaling for Leukemia Stem Cell Eradication in Acute Myeloid Leukemia. Cancer Cell (2020) 38(2):263–78.e6. doi: 10.1016/j.ccell.2020.05.014
31. Sheng Y, Yu C, Liu Y, Hu C, Ma R, Lu X, et al. FOXM1 Regulates Leukemia Stem Cell Quiescence and Survival in MLL-Rearranged AML. Nat Commun (2020) 11(1):928. doi: 10.1038/s41467-020-14590-9
32. Hou Y, Li W, Sheng Y, Li LP, Huang Y, Zhang ZH, et al. The Transcription Factor Foxm1 is Essential for the Quiescence and Maintenance of Hematopoietic Stem Cells. Nat Immunol (2015) 16(8):810–8. doi: 10.1038/ni.3204
33. Bill M, Papaioannou D, Karunasiri M, Kohlschmidt J, Pepe F, Walker CJ, et al. Expression and Functional Relevance of Long non-Coding RNAs in Acute Myeloid Leukemia Stem Cells. Leukemia (2019) 33(9):2169–82. doi: 10.1038/s41375-019-0429-5
34. Jiang X, Mak PY, Mu H, Tao W, Mak DH, Kornblau S, et al. Disruption of Wnt/beta-Catenin Exerts Antileukemia Activity and Synergizes With FLT3 Inhibition in FLT3-Mutant Acute Myeloid Leukemia. Clin Cancer Res (2018) 24(10):2417–29. doi: 10.1158/1078-0432.CCR-17-1556
35. Nepstad I, Hatfield KJ, Gronningsaeter IS, Reikvam H. The PI3K-Akt-mTOR Signaling Pathway in Human Acute Myeloid Leukemia (AML) Cells. Int J Mol Sci (2020) 21(8):2907. doi: 10.3390/ijms21082907
36. Lechman ER, Gentner B, Ng SW, Schoof EM, van Galen P, Kennedy JA, et al. miR-126 Regulates Distinct Self-Renewal Outcomes in Normal and Malignant Hematopoietic Stem Cells. Cancer Cell (2016) 29(2):214–28. doi: 10.1016/j.ccell.2015.12.011
37. Zhang B, Nguyen LXT, Zhao DD, Frankhouser DE, Wang HF, Hoang DH, et al. Treatment-Induced Arteriolar Revascularization and miR-126 Enhancement in Bone Marrow Niche Protect Leukemic Stem Cells in AML. J Hematol Oncol (2021) 14(1):122. doi: 10.1186/s13045-021-01133-y
38. Zhao C, Chen A, Jamieson CH, Fereshteh M, Abrahamsson A, Blum J, et al. Hedgehog Signalling is Essential for Maintenance of Cancer Stem Cells in Myeloid Leukaemia. Nature (2009) 458(7239):776–U117. doi: 10.1038/nature07737
39. Jamieson C, Martinelli G, Papayannidis C, Cortes JE. Hedgehog Pathway Inhibitors: A New Therapeutic Class for the Treatment of Acute Myeloid Leukemia. Blood Cancer Discov (2020) 1(2):134–45. doi: 10.1158/2643-3230.Bcd-20-0007
40. Kobune M, Takimoto R, Murase K, Iyama S, Sato T, Kikuchi S, et al. Drug Resistance is Dramatically Restored by Hedgehog Inhibitors in CD34(+) Leukemic Cells. Cancer Sci (2009) 100(5):948–55. doi: 10.1111/j.1349-7006.2009.01111.x
41. Fukushima N, Minami Y, Kakiuchi S, Kuwatsuka Y, Hayakawa F, Jamieson C, et al. Small-Molecule Hedgehog Inhibitor Attenuates the Leukemia-Initiation Potential of Acute Myeloid Leukemia Cells. Cancer Sci (2016) 107(10):1422–9. doi: 10.1111/cas.13019
42. Vasconcelos FC, de Souza PS, Hancio T, de Faria FCC, Maia RC. Update on Drug Transporter Proteins in Acute Myeloid Leukemia: Pathological Implication and Clinical Setting. Crit Rev Oncol Hematol (2021) 160:103281. doi: 10.1016/j.critrevonc.2021.103281
43. Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, et al. BCL-2 Inhibition Targets Oxidative Phosphorylation and Selectively Eradicates Quiescent Human Leukemia Stem Cells. Cell Stem Cell (2013) 12(3):329–41. doi: 10.1016/j.stem.2012.12.013
44. Halilovic E, Chanrion M, Mistry P, Wartmann M, Qiu SM, Sanghavi S, et al. MIK665/S64315, a Novel Mcl-1 Inhibitor, in Combination With Bcl-2 Inhibitors Exhibits Strong Synergistic Antitumor Activity in a Range of Hematologic Malignancies. Cancer Res (2019) 79(13):4477. doi: 10.1158/1538-7445.Am2019-4477
45. Bhagwat N, Grego A, Gowen-MacDonald W, Wang M, Cowart M, Wu XW, et al. Preclinical Characterization of PRT1419, a Potent, Selective and Orally Available Inhibitor of MCL1. Cancer Res (2021) 81(13): 983. doi: 10.1158/1538-7445.AM2021-983
46. Caenepeel S, Brown SP, Belmontes B, Moody G, Keegan KS, Chui D, et al. AMG 176, a Selective MCL1 Inhibitor, is Effective in Hematological Cancer Models Alone and in Combination With Established Therapies. Cancer Discov (2018) 8(12):1582–1597. doi: 10.1158/2159-8290.cd-18-0387
47. Lee DJ, Zeidner JF. Cyclin-Dependent Kinase (CDK) 9 and 4/6 Inhibitors in Acute Myeloid Leukemia (AML): A Promising Therapeutic Approach. Expert Opin Investig Drugs (2019) 28(11):989–1001. doi: 10.1080/13543784.2019.1678583
48. Kojima K, Konopleva M, Samudio IJ, Shikami M, Cabreira-Hansen M, McQueen T, et al. MDM2 Antagonists Induce P53-Dependent Apoptosis in AML: Implications for Leukemia Therapy. Blood (2005) 106(9):3150–9. doi: 10.1182/blood-2005-02-0553
49. Ablain J, Rice K, Soilihi H, De Reynies A, Minucci S, De Thé H. Activation of a Promyelocytic Leukemia–Tumor Protein 53 Axis Underlies Acute Promyelocytic Leukemia Cure. Nat Med (2014) 20(2):167–74. doi: 10.1038/nm.3441
50. Nahi H, Merup M, Lehmann S, Bengtzen S, Mollgard L, Selivanova G, et al. PRIMA-1 Induces Apoptosis in Acute Myeloid Leukaemia Cells With P53 Gene Deletion. Br J Haematol (2006) 132(2):230–6. doi: 10.1111/j.1365-2141.2005.05851.x
51. Chan SM, Thomas D, Corces-Zimmerman MR, Xavy S, Rastogi S, Hong W-J, et al. Isocitrate Dehydrogenase 1 and 2 Mutations Induce BCL-2 Dependence in Acute Myeloid Leukemia. Nat Med (2015) 21(2):178–84. doi: 10.1038/nm.3788
52. Maes T, Mascaro C, Tirapu I, Estiarte A, Ciceri F, Lunardi S, et al. ORY-1001, a Potent and Selective Covalent KDM1A Inhibitor, for the Treatment of Acute Leukemia. Cancer Cell (2018) 33(3):495–511.e12. doi: 10.1016/j.ccell.2018.02.002
53. Blagitko-Dorfs N, Schlosser P, Greve G, Pfeifer D, Meier R, Baude A, et al. Combination Treatment of Acute Myeloid Leukemia Cells With DNMT and HDAC Inhibitors: Predominant Synergistic Gene Downregulation Associated With Gene Body Demethylation. Leukemia (2019) 33(4):945–56. doi: 10.1038/s41375-018-0293-8
54. Wieduwilt MJ, Pawlowska N, Thomas S, Olin R, Logan AC, Damon LE, et al. Histone Deacetylase Inhibition With Panobinostat Combined With Intensive Induction Chemotherapy in Older Patients With Acute Myeloid Leukemia: Phase I Study Results. Clin Cancer Res (2019) 25(16):4917–23. doi: 10.1158/1078-0432.CCR-19-0171
55. Tavor S, Petit I, Porozov S, Avigdor A, Dar A, Leider-Trejo L, et al. CXCR4 Regulates Migration and Development of Human Acute Myelogenous Leukemia Stem Cells in Transplanted NOD/SCID Mice. Cancer Res (2004) 64(8):2817–24. doi: 10.1158/0008-5472.can-03-3693
56. Kikushige Y, Akashi K. A Tim-3/Gal-9 Autocrine Stimulatory Loop Drives Self-Renewal of Human Myeloid Leukemia Stem Cells and Leukemic Progression. Exp Hematol (2016) 44(9):S36–S. doi: 10.1016/j.exphem.2016.06.033
57. Nguyen CH, Bauer K, Hackl H, Schlerka A, Koller E, Hladik A, et al. All-Trans Retinoic Acid Enhances, and a Pan-RAR Antagonist Counteracts, the Stem Cell Promoting Activity of EVI1 in Acute Myeloid Leukemia. Cell Death Dis (2019) 10(12):944. doi: 10.1038/s41419-019-2172-2
58. Lobry C, Ntziachristos P, Ndiaye-Lobry D, Oh P, Cimmino L, Zhu N, et al. Notch Pathway Activation Targets AML-Initiating Cell Homeostasis and Differentiation. J Exp Med (2013) 210(2):301–19. doi: 10.1084/jem.20121484
59. Takubo K, Goda N, Yamada W, Iriuchishima H, Ikeda E, Kubota Y, et al. Regulation of the HIF-1 Alpha Level Is Essential for Hematopoietic Stem Cells. Cell Stem Cell (2010) 7(3):391–402. doi: 10.1016/j.stem.2010.06.020
60. Vyas P. Targeting HIF Function: The Debate Continues. Blood (2014) 124(24):3510–1. doi: 10.1182/blood-2014-10-605055
61. Wang Y, Liu Y, Malek SN, Zheng P, Liu Y. Targeting HIF1 Alpha Eliminates Cancer Stem Cells in Hematological Malignancies. Cell Stem Cell (2011) 8(4):399–411. doi: 10.1016/j.stem.2011.02.006
62. Velasco-Hernandez T, Soneji S, Hidalgo I, Erlandsson E, Cammenga J, Bryder D. Hif-1 Alpha Deletion May Lead to Adverse Treatment Effect in a Mouse Model of MLL-AF9-Driven AML. Stem Cell Rep (2019) 12(1):112–21. doi: 10.1016/j.stemcr.2018.11.023
63. Farge T, Saland E, de Toni F, Aroua N, Hosseini M, Perry R, et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells But Require Oxidative Metabolism. Cancer Discov (2017) 7(7):716–35. doi: 10.1158/2159-8290.Cd-16-0441
64. O’Reilly E, Zeinabad HA, Nolan C, Sefy J, Williams T, Tarunina M, et al. Recreating the Bone Marrow Microenvironment to Model Leukemic Stem Cell Quiescence. Front Cell Dev Biol (2021) 9:662868. doi: 10.3389/fcell.2021.662868
65. Waclawiczek A, Hamilton A, Rouault-Pierre K, Abarrategi A, Albornoz MG, Miraki-Moud F, et al. Mesenchymal Niche Remodeling Impairs Hematopoiesis via Stanniocalcin 1 in Acute Myeloid Leukemia. J Clin Invest (2020) 130(6):3038–50. doi: 10.1172/jci133187
66. Zhou C, Du J, Zhao L, Liu W, Zhao TM, Liang H, et al. GLI1 Reduces Drug Sensitivity by Regulating Cell Cycle Through PI3K/AKT/GSK3/CDK Pathway in Acute Myeloid Leukemia. Cell Death Dis (2021) 12(3):231. doi: 10.1038/s41419-021-03504-2
67. Chesnokov MS, Borhani S, Halasi M, Arbieva Z, Khan I, Gartel AL. FOXM1-AKT Positive Regulation Loop Provides Venetoclax Resistance in AML. Front Oncol (2021) 11:696532. doi: 10.3389/fonc.2021.696532
68. Stengel S, Petrie KR, Sbirkov Y, Stanko C, Ghazvini Zadegan F, Gil V, et al. Suppression of MYC by PI3K/AKT/mTOR Pathway Inhibition in Combination With All-Trans Retinoic Acid Treatment for Therapeutic Gain in Acute Myeloid Leukaemia. Br J Haematol (2022) 00:1–11. doi: 10.1111/bjh.18187
69. Kang YA, Pietras EM, Passegue E. Deregulated Notch and Wnt Signaling Activates Early-Stage Myeloid Regeneration Pathways in Leukemia. J Exp Med (2020) 217(3):jem.20190787. doi: 10.1084/jem.20190787
70. Jin LQ, Lee EM, Ramshaw HS, Busfield SJ, Peoppl AG, Wilkinson L, et al. Monoclonal Antibody-Mediated Targeting of CD123, IL-3 Receptor Alpha Chain, Eliminates Human Acute Myeloid Leukemic Stem Cells. Cell Stem Cell (2009) 5(1):31–42. doi: 10.1016/j.stem.2009.04.018
71. Taussig DC, Pearce DJ, Simpson C, Rohatiner AZ, Lister TA, Kelly G, et al. Hematopoietic Stem Cells Express Multiple Myeloid Markers: Implications for the Origin and Targeted Therapy of Acute Myeloid Leukemia. Blood (2005) 106(13):4086–92. doi: 10.1182/blood-2005-03-1072
72. Hosen N, Park CY, Tatsumi N, Oji Y, Sugiyama H, Gramatzki M, et al. CD96 is a Leukemic Stem Cell-Specific Marker in Human Acute Myeloid Leukemia. Proc Natl Acad Sci U S A (2007) 104(26):11008–13. doi: 10.1073/pnas.0704271104
73. Kikushige Y, Shima T, Takayanagi S, Urata S, Miyamoto T, Iwasaki H, et al. TIM-3 Is a Promising Target to Selectively Kill Acute Myeloid Leukemia Stem Cells. Cell Stem Cell (2010) 7(6):708–17. doi: 10.1016/j.stem.2010.11.014
74. Jin LQ, Hope KJ, Zhai QL, Smadja-Joffe F, Dick JE. Targeting of CD44 Eradicates Human Acute Myeloid Leukemic Stem Cells. Nat Med (2006) 12(10):1167–74. doi: 10.1038/nm1483
75. van Rhenen A, van Dongen G, Kelder A, Rombouts EJ, Feller N, Moshaver B, et al. The Novel AML Stem Cell-Associated Antigen CLL-1 Aids in Discrimination Between Normal and Leukemic Stem Cells. Blood (2007) 110(7):2659–66. doi: 10.1182/blood-2007-03-083048
76. Li H, Zhao N, Li YH, Xing HY, Chen SY, Xu YX, et al. C-MPL Is a Candidate Surface Marker and Confers Self-Renewal, Quiescence, Chemotherapy Resistance, and Leukemia Initiation Potential in Leukemia Stem Cells. Stem Cells (2018) 36(11):1685–96. doi: 10.1002/stem.2897
77. Wang H, Zhao D, Nguyen LX, Wu H, Li L, Dong D, et al. Targeting Cell Membrane HDM2: A Novel Therapeutic Approach for Acute Myeloid Leukemia. Leukemia (2020) 34(1):75–86. doi: 10.1038/s41375-019-0522-9
78. van Gils N, Denkers F, Smit L. Escape From Treatment; the Different Faces of Leukemic Stem Cells and Therapy Resistance in Acute Myeloid Leukemia. Front Oncol (2021) 11:659253. doi: 10.3389/fonc.2021.659253
79. Robey RW, Pluchino KM, Hall MD, Fojo AT, Bates SE, Gottesman MM. Revisiting the Role of ABC Transporters in Multidrug-Resistant Cancer. Nat Rev Cancer (2018) 18(7):452–64. doi: 10.1038/s41568-018-0005-8
80. De Jonge-Peeters SDPWM, Kuipers F, De Vries EGE, Vellenga E. ABC Transporter Expression in Hematopoietic Stem Cells and the Role in AML Drug Resistance. Crit Rev Oncol Hematol (2007) 62(3):214–26. doi: 10.1016/j.critrevonc.2007.02.003
81. Wuchter C, Karawajew L, Ruppert V, Schrappe M, Buchner T, Schoch C, et al. Clinical Significance of P-Glycoprotein Expression and Function for Response to Induction Chemotherapy, Relapse Rate and Overall Survival in Acute Leukemia. Haematologica (2000) 85(7):711–21.
82. Benderra Z, Faussat AM, Sayada L, Perrot JY, Chaoui D, Marie JP, et al. Breast Cancer Resistance Protein and P-Glycoprotein in 149 Adult Acute Myeloid Leukemias. Clin Cancer Res (2004) 10(23):7896–902. doi: 10.1158/1078-0432.Ccr-04-0795
83. Schaich M, Soucek S, Thiede C, Ehninger G, Illmer T, Grp SAS. MDR1 and MRP1 Gene Expression are Independent Predictors for Treatment Outcome in Adult Acute Myeloid Leukaemia. Br J Haematol (2005) 128(3):324–32. doi: 10.1111/j.1365-2141.2004.05319.x
84. Leith CP, Kopecky KJ, Godwin J, McConnell T, Slovak ML, Chen IM, et al. Acute Myeloid Leukemia in the Elderly: Assessment of Multidrug Resistance (MDR1) and Cytogenetics Distinguishes Biologic Subgroups With Remarkably Distinct Responses to Standard Chemotherapy. A Southwest Oncology Group Study. Blood (1997) 89(9):3323–9. doi: 10.1182/blood.V89.9.3323
85. de Figueiredo-Pontes LL, Pintao MCT, Oliveira LCO, Dalmazzo LFF, Jacomo RH, Garcia AB, et al. Determination of P-Glycoprotein, MDR-Related Protein 1, Breast Cancer Resistance Protein, and Lung-Resistance Protein Expression in Leukemic Stem Cells of Acute Myeloid Leukemia. Cytometry Part B Clin Cytom (2008) 74B(3):163–8. doi: 10.1002/cyto.b.20403
86. Cripe LD, Uno H, Paietta EM, Litzow MR, Ketterling RP, Bennett JM, et al. Zosuquidar, a Novel Modulator of P-Glycoprotein, Does Not Improve the Outcome of Older Patients With Newly Diagnosed Acute Myeloid Leukemia: A Randomized, Placebo-Controlled Trial of the Eastern Cooperative Oncology Group 3999. Blood (2010) 116(20):4077–85. doi: 10.1182/blood-2010-04-277269
87. Williams MS, Amaral FMR, Simeoni F, Somervaille TCP. A Stress-Responsive Enhancer Induces Dynamic Drug Resistance in Acute Myeloid Leukemia. J Clin Invest (2020) 130(3):1217–32. doi: 10.1172/jci130809
88. Uggla B, Stahl E, Wagsater D, Paul C, Karlsson MG, Sirsjo A, et al. BCRP mRNA Expression V. Clinical Outcome in 40 Adult AML Patients. Leuk Res (2005) 29(2):141–6. doi: 10.1016/j.leukres.2004.06.004
89. Damiani D, Tiribelli M, Calistri E, Geromin A, Chiarvesio A, Michelutti A, et al. The Prognostic Value of P-Glycoprotein (ABCB) and Breast Cancer Resistance Protein (ABCG2) in Adults With De Novo Acute Myeloid Leukemia With Normal Karyotype. Haematologica (2006) 91(6):825–8.
90. Huang FF, Wu DS, Zhang L, Yu YH, Yuan XY, Li WJ, et al. Inactivation of PTEN Increases ABCG2 Expression and the Side Population Through the PI3K/Akt Pathway in Adult Acute Leukemia. Cancer Lett (2013) 336(1):96–105. doi: 10.1016/j.canlet.2013.04.006
91. Raaijmakers M, de Grouw E, Heuver LHH, van der Reijden BA, Jansen JH, Scheper RJ, et al. Breast Cancer Resistance Protein in Drug Resistance of Primitive CD34+38-Cells in Acute Myeloid Leukemia. Clin Cancer Res (2005) 11(6):2436–44. doi: 10.1158/1078-0432.Ccr-04-0212
92. Laupeze B, Amiot L, Drenou B, Bernard M, Branger B, Grosset JM, et al. High Multidrug Resistance Protein Activity in Acute Myeloid Leukaemias is Associated With Poor Response to Chemotherapy and Reduced Patient Survival. Br J Haematol (2002) 116(4):834–8. doi: 10.1046/j.0007-1048.2002.03350.x
93. Filipits M, Suchomel RW, Zochbauer S, Brunner R, Lechner K, Pirker R. Multidrug Resistance-Associated Protein in Acute Myeloid Leukemia: No Impact on Treatment Outcome. Clin Cancer Res (1997) 3(8):1419–25.
94. van der Kolk DM, de Vries EGE, van Putten WLJ, Verdonck LF, Ossenkoppele GJ, Verhoef GEG, et al. P-Glycoprotein and Multidrug Resistance Protein Activities in Relation to Treatment Outcome in Acute Myeloid Leukemia. Clin Cancer Res (2000) 6(8):3205–14.
95. de Grouw E, Raaijmakers M, Boezeman JB, van der Reijden BA, van de Locht LTF, de Witte TJM, et al. Preferential Expression of a High Number of ATP Binding Cassette Transporters in Both Normal and Leukemic CD34+CD38-Cells. Leukemia (2006) 20(4):750–4. doi: 10.1038/sj.leu.2404131
96. Cole SPC. Targeting Multidrug Resistance Protein 1 (MRP1, ABCC1): Past, Present, and Future. Annu Rev Pharmacol Toxicol (2014) 54(1):95–117. doi: 10.1146/annurev-pharmtox-011613-135959
97. Fukuda Y, Wang Y, Lian S, Lynch J, Nagai S, Fanshawe B, et al. Upregulated Heme Biosynthesis, an Exploitable Vulnerability in MYCN-Driven Leukemogenesis. JCI Insight (2017) 2(15):e92409. doi: 10.1172/jci.insight.92409
98. Zhou S, Schuetz JD, Bunting KD, Colapietro A-M, Sampath J, Morris JJ, et al. The ABC Transporter Bcrp1/ABCG2 is Expressed in a Wide Variety of Stem Cells and is a Molecular Determinant of the Side-Population Phenotype. Nat Med (2001) 7(9):1028–34. doi: 10.1038/nm0901-1028
99. van den Heuvel-Eibrink MM, van der Holt B, Burnett AK, Knauf WU, Fey MF, Verhoef GE, et al. CD34-Related Coexpression of MDR1 and BCRP Indicates a Clinically Resistant Phenotype in Patients With Acute Myeloid Leukemia (AML) of Older Age. Ann Hematol (2007) 86(5):329–37. doi: 10.1007/s00277-007-0269-7
100. Robinson AN, Tebase BG, Francone SC, Huff LM, Kozlowski H, Cossari D, et al. Coexpression of ABCB1 and ABCG2 in a Cell Line Model Reveals Both Independent and Additive Transporter Function. Drug Metab Dispos (2019) 47(7):715–23. doi: 10.1124/dmd.118.086181
101. Klener P, Sovilj D, Renesova N, Andera L. BH3 Mimetics in Hematologic Malignancies. Int J Mol Sci (2021) 22(18):10157. doi: 10.3390/ijms221810157
102. Campos L, Rouault J, Sabido O, Oriol P, Roubi N, Vasselon C, et al. High Expression of Bcl-2 Protein in Acute Myeloid Leukemia Cells is Associated With Poor Response to Chemotherapy. Blood (1993) 81(11):3091–6. doi: 10.1182/blood.v81.11.3091.3091
103. Karakas T, Maurer U, Weidmann E, Miething CC, Hoelzer D, Bergmann L. High Expression of Bcl-2 mRNA as a Determinant of Poor Prognosis in Acute Myeloid Leukemia. Ann Oncol (1998) 9(2):159–65. doi: 10.1023/a:1008255511404
104. Garciaz S, Saillard C, Hicheri Y, Hospital M-A, Vey N. Venetoclax in Acute Myeloid Leukemia: Molecular Basis, Evidences for Preclinical and Clinical Efficacy and Strategies to Target Resistance. Cancers (2021) 13(22):5608. doi: 10.3390/cancers13225608
105. Kaufmann SH, Karp JE, Svingen PA, Krajewski S, Burke PJ, Gore SD, et al. Elevated Expression of the Apoptotic Regulator Mcl-1 at the Time of Leukemic Relapse. Blood (1998) 91(3):991–1000. doi: 10.1182/blood.v91.3.991
106. Pei S, Pollyea DA, Gustafson A, Stevens BM, Minhajuddin M, Fu R, et al. Monocytic Subclones Confer Resistance to Venetoclax-Based Therapy in Patients With Acute Myeloid Leukemia. Cancer Discov (2020) 10(4):536–51. doi: 10.1158/2159-8290.cd-19-0710
107. Ramsey HE, Fischer MA, Lee T, Gorska AE, Arrate MP, Fuller L, et al. A Novel MCL1 Inhibitor Combined With Venetoclax Rescues Venetoclax-Resistant Acute Myelogenous Leukemia. Cancer Discov (2018) 8(12):1566–81. doi: 10.1158/2159-8290.cd-18-0140
108. Cidado J, Boiko S, Proia T, Ferguson D, Criscione SW, San Martin M, et al. AZD4573 Is a Highly Selective CDK9 Inhibitor That Suppresses MCL-1 and Induces Apoptosis in Hematologic Cancer Cells. Clin Cancer Res (2020) 26(4):922–34. doi: 10.1158/1078-0432.ccr-19-1853
109. Khoo KH, Verma CS, Lane DP. Drugging the P53 Pathway: Understanding the Route to Clinical Efficacy. Nat Rev Drug Discov (2014) 13(3):217–36. doi: 10.1038/nrd4236
110. Rücker FG, Schlenk RF, Bullinger L, Kayser S, Teleanu V, Kett H, et al. TP53 Alterations in Acute Myeloid Leukemia With Complex Karyotype Correlate With Specific Copy Number Alterations, Monosomal Karyotype, and Dismal Outcome. Blood (2012) 119(9):2114–21. doi: 10.1182/blood-2011-08-375758
111. Nechiporuk T, Kurtz SE, Nikolova O, Liu TT, Jones CL, D’Alessandro A, et al. The TP53 Apoptotic Network Is a Primary Mediator of Resistance to BCL2 Inhibition in AML Cells. Cancer Discov (2019) 9(7):910–25. doi: 10.1158/2159-8290.Cd-19-0125
112. Wattel E, Preudhomme C, Hecquet B, Vanrumbeke M, Quesnel B, Dervite I, et al. P53 Mutations are Associated With Resistance to Chemotherapy and Short Survival in Hematologic Malignancies. Blood (1994) 84(9):3148–57. doi: 10.1182/blood.v84.9.3148.3148
113. Kuo YH, Qi J, Cook GJ. Regain Control of P53: Targeting Leukemia Stem Cells by Isoform-Specific HDAC Inhibition. Exp Hematol (2016) 44(5):315–21. doi: 10.1016/j.exphem.2016.02.007
114. Weisberg E, Halilovic E, Cooke VG, Nonami A, Ren T, Sanda T, et al. Inhibition of Wild-Type P53-Expressing AML by the Novel Small Molecule HDM2 Inhibitor Cgm097. Mol Cancer Ther (2015) 14(10):2249–59. doi: 10.1158/1535-7163.mct-15-0429
115. Pan R, Ruvolo V, Mu H, Leverson JD, Nichols G, Reed JC, et al. Synthetic Lethality of Combined Bcl-2 Inhibition and P53 Activation in AML: Mechanisms and Superior Antileukemic Efficacy. Cancer Cell (2017) 32(6):748–60.e6. doi: 10.1016/j.ccell.2017.11.003
116. De Haan G, Lazare SS. Aging of Hematopoietic Stem Cells. Blood (2018) 131(5):479–87. doi: 10.1182/blood-2017-06-746412
117. Xiao Y, Zou P, Wang J, Song H, Zou J, Liu LB. Lower Phosphorylation of P38 MAPK Blocks the Oxidative Stress-Induced Senescence in Myeloid Leukemic CD34(+)CD38(-) Cells. J Huazhong Univ Sci Technol Med Sci (2012) 32(3):328–33. doi: 10.1007/s11596-012-0057-z
118. Wajapeyee N, Wang S-Z, Serra RW, Solomon PD, Nagarajan A, Zhu X, et al. Senescence Induction in Human Fibroblasts and Hematopoietic Progenitors by Leukemogenic Fusion Proteins. Blood (2010) 115(24):5057–60. doi: 10.1182/blood-2009-09-245928
119. Peng DY, Wang HF, Li L, Ma X, Chen Y, Zhou H, et al. miR-34c-5p Promotes Eradication of Acute Myeloid Leukemia Stem Cells by Inducing Senescence Through Selective RAB27B Targeting to Inhibit Exosome Shedding. Leukemia (2018) 32(5):1180–8. doi: 10.1038/s41375-018-0015-2
120. Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, et al. Cellular Senescence: Defining a Path Forward. Cell (2019) 179(4):813–27. doi: 10.1016/j.cell.2019.10.005
121. Haferlach C, Dicker F, Herholz H, Schnittger S, Kern W, Haferlach T. Mutations of the TP53 Gene in Acute Myeloid Leukemia are Strongly Associated With a Complex Aberrant Karyotype. Leukemia (2008) 22(8):1539–41. doi: 10.1038/leu.2008.143
122. Barbosa K, Li S, Adams PD, Deshpande AJ. The Role of TP53 in Acute Myeloid Leukemia: Challenges and Opportunities. Genes Chromosomes Cancer (2019) 58(12):875–88. doi: 10.1002/gcc.22796
123. Nabinger SC, Chen S, Gao R, Yao C, Kobayashi M, Vemula S, et al. Mutant P53 Enhances Leukemia-Initiating Cell Self-Renewal to Promote Leukemia Development. Leukemia (2019) 33(6):1535–9. doi: 10.1038/s41375-019-0377-0
124. Woods D, Turchi JJ. Chemotherapy Induced DNA Damage Response. Cancer Biol Ther (2013) 14(5):379–89. doi: 10.4161/cbt.23761
125. Park Y, Gerson SL. DNA Repair Defects in Stem Cell Function and Aging. Annu Rev Med (2005) 56(1):495–508. doi: 10.1146/annurev.med.56.082103.104546
126. Popp HD, Naumann N, Brendel S, Henzler T, Weiss C, Hofmann WK, et al. Increase of DNA Damage and Alteration of the DNA Damage Response in Myelodysplastic Syndromes and Acute Myeloid Leukemias. Leuk Res (2017) 57:112–8. doi: 10.1016/j.leukres.2017.03.011
127. Xu RD, Yu SY, Zhu D, Huang XP, Xu YQ, Lao YM, et al. hCINAP Regulates the DNA-Damage Response and Mediates the Resistance of Acute Myelocytic Leukemia Cells to Therapy. Nat Commun (2019) 10:3812. doi: 10.1038/s41467-019-11795-5
128. Delia D, Mizutani S. The DNA Damage Response Pathway in Normal Hematopoiesis and Malignancies. Int J Hematol (2017) 106(3):328–34. doi: 10.1007/s12185-017-2300-7
129. Duy C, Li M, Teater M, Meydan C, Garrett-Bakelman FE, Lee TC, et al. Chemotherapy Induces Senescence-Like Resilient Cells Capable of Initiating AML Recurrence. Cancer Discov (2021) 11(6):1542–61. doi: 10.1158/2159-8290.Cd-20-1375
130. Bose S, Zhang C, Le A. Glucose Metabolism in Cancer: The Warburg Effect and Beyond. Springer International Publishing (2021) 1311:3–15. doi: 10.1007/978-3-030-65768-0_1
131. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science (2009) 324(5930):1029–33. doi: 10.1126/science.1160809
132. Chen W-L, Wang J-H, Zhao A-H, Xu X, Wang Y-H, Chen T-L, et al. A Distinct Glucose Metabolism Signature of Acute Myeloid Leukemia With Prognostic Value. Blood (2014) 124(10):1645–54. doi: 10.1182/blood-2014-02-554204
133. Takubo K, Nagamatsu G, Kobayashi CI, Nakamura-Ishizu A, Kobayashi H, Ikeda E, et al. Regulation of Glycolysis by Pdk Functions as a Metabolic Checkpoint for Cell Cycle Quiescence in Hematopoietic Stem Cells. Cell Stem Cell (2013) 12(1):49–61. doi: 10.1016/j.stem.2012.10.011
134. Sriskanthadevan S, Jeyaraju DV, Chung TE, Prabha S, Xu W, Skrtic M, et al. AML Cells Have Low Spare Reserve Capacity in Their Respiratory Chain That Renders Them Susceptible to Oxidative Metabolic Stress. Blood (2015) 125(13):2120–30. doi: 10.1182/blood-2014-08-594408
135. Jones CL, Stevens BM, D’Alessandro A, Reisz JA, Culp-Hill R, Nemkov T, et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell (2018) 34(5):724–740.e4. doi: 10.1016/j.ccell.2018.10.005
136. Jones CL, Stevens BM, Pollyea DA, Culp-Hill R, Reisz JA, Nemkov T, et al. Nicotinamide Metabolism Mediates Resistance to Venetoclax in Relapsed Acute Myeloid Leukemia Stem Cells. Cell Stem Cell (2020) 27(5):748–764.e4. doi: 10.1016/j.stem.2020.07.021
137. Caenepeel S, Karen R, Belmontes B, Verlinsky A, Tan H, Yang YJ, et al. Discovery and Preclinical Evaluation of AMG 397, a Potent, Selective and Orally Bioavailable MCL1 Inhibitor. Cancer Res (2020) 80(16):6218. doi: 10.1158/1538-7445.Am2020-6218
138. Stevens BM, Jones CL, Pollyea DA, Culp-Hill R, D’Alessandro A, Winters A, et al. Fatty Acid Metabolism Underlies Venetoclax Resistance in Acute Myeloid Leukemia Stem Cells. Nat Cancer (2020) 1(12):1176–87. doi: 10.1038/s43018-020-00126-z
139. Ye HB, Adane B, Khan N, Sullivan T, Minhajuddin M, Gasparetto M, et al. Leukemic Stem Cells Evade Chemotherapy by Metabolic Adaptation to an Adipose Tissue Niche. Cell Stem Cell (2016) 19(1):23–37. doi: 10.1016/j.stem.2016.06.001
140. Tabe Y, Yamamoto S, Saitoh K, Sekihara K, Monma N, Ikeo K, et al. Bone Marrow Adipocytes Facilitate Fatty Acid Oxidation Activating AMPK and a Transcriptional Network Supporting Survival of Acute Monocytic Leukemia Cells. Cancer Res (2017) 77(6):1453–64. doi: 10.1158/0008-5472.can-16-1645
141. Powell JA, Wallington-Beddoe CT, Pitson SM. Targeting Sphingosine Kinase 1 in Acute Myeloid Leukemia: Translation to Clinic. Int J Hematol Oncol (2017) 6(2):31–4. doi: 10.2217/ijh-2017-0011
142. Xie SZ, Kaufmann KB, Wang W, Chan-Seng-Yue M, Gan OI, Laurenti E, et al. Sphingosine-1-Phosphate Receptor 3 Potentiates Inflammatory Programs in Normal and Leukemia Stem Cells to Promote Differentiation. Blood Cancer Discov (2021) 2(1):32–53. doi: 10.1158/2643-3230.BCD-20-0155
143. Im AP, Sehgal AR, Carroll MP, Smith BD, Tefferi A, Johnson DE, et al. DNMT3A and IDH Mutations in Acute Myeloid Leukemia and Other Myeloid Malignancies: Associations With Prognosis and Potential Treatment Strategies. Leukemia (2014) 28(9):1774–83. doi: 10.1038/leu.2014.124
144. DiNardo CD, Schuh AC, Stein EM, Montesinos P, Wei AH, de Botton S, et al. Enasidenib Plus Azacitidine Versus Azacitidine Alone in Patients With Newly Diagnosed, Mutant-IDH2 Acute Myeloid Leukaemia (AG221-AML-005): A Single-Arm, Phase 1b and Randomised, Phase 2 Trial. Lancet Oncol (2021) 22(11):1597–608. doi: 10.1016/s1470-2045(21)00494-0
145. Stein EM, Dinardo CD, Pollyea DA, Fathi AT, Roboz GJ, Altman JK, et al. Enasidenib in Mutant IDH2 Relapsed or Refractory Acute Myeloid Leukemia. Blood (2017) 130(6):722–31. doi: 10.1182/blood-2017-04-779405
146. Dinardo CD, Stein EM, De Botton S, Roboz GJ, Altman JK, Mims AS, et al. Durable Remissions With Ivosidenib Inidh1-Mutated Relapsed or Refractory AML. N Engl J Med (2018) 378(25):2386–98. doi: 10.1056/nejmoa1716984
147. Dinardo CD, Stein AS, Stein EM, Fathi AT, Frankfurt O, Schuh AC, et al. Mutant Isocitrate Dehydrogenase 1 Inhibitor Ivosidenib in Combination With Azacitidine for Newly Diagnosed Acute Myeloid Leukemia. J Clin Oncol (2021) 39(1):57–65. doi: 10.1200/jco.20.01632
148. Amatangelo MD, Quek L, Shih A, Stein EM, Roshal M, David MD, et al. Enasidenib Induces Acute Myeloid Leukemia Cell Differentiation to Promote Clinical Response. Blood (2017) 130(6):732–41. doi: 10.1182/blood-2017-04-779447
149. Choe S, Wang H, Dinardo CD, Stein EM, De Botton S, Roboz GJ, et al. Molecular Mechanisms Mediating Relapse Following Ivosidenib Monotherapy in IDH1-Mutant Relapsed or Refractory AML. Blood Adv (2020) 4(9):1894–905. doi: 10.1182/bloodadvances.2020001503
150. Yang X, Wong MPM, Ng RK. Aberrant DNA Methylation in Acute Myeloid Leukemia and Its Clinical Implications. Int J Mol Sci (2019) 20(18):4576. doi: 10.3390/ijms20184576
151. Liu XL, Liu HQ, Li J, Mao CY, He JT, Zhao X. Role of Epigenetic in Leukemia: From Mechanism to Therapy. Chem Biol Interact (2020) 317:108963. doi: 10.1016/j.cbi.2020.108963
152. Sato H, Wheat JC, Steidl U, Ito K. DNMT3A and TeT2 in the Pre-Leukemic Phase of Hematopoietic Disorders. Front Oncol (2016) 6:187. doi: 10.3389/fonc.2016.00187
153. Garg S, Reyes-Palomares A, He L, Bergeron A, Lavallée V-P, Lemieux S, et al. Hepatic Leukemia Factor is a Novel Leukemic Stem Cell Regulator in DNMT3A, NPM1, and FLT3-ITD Triple-Mutated AML. Blood (2019) 134(3):263–76. doi: 10.1182/blood.2018862383
154. Weissmann S, Alpermann T, Grossmann V, Kowarsch A, Nadarajah N, Eder C, et al. Landscape of TET2 Mutations in Acute Myeloid Leukemia. Leukemia (2012) 26(5):934–42. doi: 10.1038/leu.2011.326
155. Zhou KG, Zhou M, Cheng L, Chen X, Wang XM, Chu YJ, et al. Loss of MBD2 Attenuates MLL-AF9-Driven Leukemogenesis by Suppressing the Leukemic Cell Cycle via CDKN1C. Oncogenesis (2021) 10(11):79. doi: 10.1038/s41389-021-00366-3
156. Jung N, Dai B, Gentles AJ, Majeti R, Feinberg AP. An LSC Epigenetic Signature is Largely Mutation Independent and Implicates the HOXA Cluster in AML Pathogenesis. Nat Commun (2015) 6:8489. doi: 10.1038/ncomms9489
157. Vu LP, Pickering BF, Cheng Y, Zaccara S, Nguyen D, Minuesa G, et al. The N6-Methyladenosine (M6a)-Forming Enzyme METTL3 Controls Myeloid Differentiation of Normal Hematopoietic and Leukemia Cells. Nat Med (2017) 23(11):1369–76. doi: 10.1038/nm.4416
158. Barbieri I, Tzelepis K, Pandolfini L, Shi J, Millán-Zambrano G, Robson SC, et al. Promoter-Bound METTL3 Maintains Myeloid Leukaemia by M6a-Dependent Translation Control. Nature (2017) 552(7683):126–31. doi: 10.1038/nature24678
159. Yankova E, Blackaby W, Albertella M, Rak J, De Braekeleer E, Tsagkogeorga G, et al. Small-Molecule Inhibition of METTL3 as a Strategy Against Myeloid Leukaemia. Nature (2021) 593(7860):597–601. doi: 10.1038/s41586-021-03536-w
160. Weng HY, Huang HL, Wu HZ, Qin X, Zhao BXS, Dong L, et al. METTL14 Inhibits Hematopoietic Stem/Progenitor Differentiation and Promotes Leukemogenesis via mRNA M(6)A Modification. Cell Stem Cell (2018) 22(2):191–205.e9. doi: 10.1016/j.stem.2017.11.016
161. Su R, Dong L, Li CY, Nachtergaele S, Wunderlich M, Qing Y, et al. R-2hg Exhibits Anti-Tumor Activity by Targeting FTO/m(6)A/MYC/CEBPA Signaling. Cell (2018) 172(1-2):90–105.e23. doi: 10.1016/j.cell.2017.11.031
162. Su R, Dong L, Li YC, Gao M, Han L, Wunderlich M, et al. Targeting FTO Suppresses Cancer Stem Cell Maintenance and Immune Evasion. Cancer Cell (2020) 38(1):79–96.e11. doi: 10.1016/j.ccell.2020.04.017
163. Shen C, Sheng Y, Zhu AC, Robinson S, Jiang X, Dong L, et al. RNA Demethylase ALKBH5 Selectively Promotes Tumorigenesis and Cancer Stem Cell Self-Renewal in Acute Myeloid Leukemia. Cell Stem Cell (2020) 27(1):64–80.e9. doi: 10.1016/j.stem.2020.04.009
164. Wang JZ, Li YC, Wang PP, Han GQ, Zhang TT, Chang JW, et al. Leukemogenic Chromatin Alterations Promote AML Leukemia Stem Cells via a KDM4C-ALKBH5-AXL Signaling Axis. Cell Stem Cell (2020) 27(1):81–97.e8. doi: 10.1016/j.stem.2020.04.001
165. Paris J, Morgan M, Campos J, Spencer GJ, Shmakova A, Ivanova I, et al. Targeting the RNA M6a Reader YTHDF2 Selectively Compromises Cancer Stem Cells in Acute Myeloid Leukemia. Cell Stem Cell (2019) 25(1):137–48.e6. doi: 10.1016/j.stem.2019.03.021
166. Cheng YM, Xie W, Pickering BF, Chu KL, Savino AM, Yang XJ, et al. N-6-Methyladenosine on mRNA Facilitates a Phase-Separated Nuclear Body That Suppresses Myeloid Leukemic Differentiation. Cancer Cell (2021) 39(7):958–972.e8. doi: 10.1016/j.ccell.2021.04.017
167. Nguyen AT, Taranova O, He J, Zhang Y. DOT1L, the H3K79 Methyltransferase, is Required for MLL-AF9–mediated Leukemogenesis. Blood (2011) 117(25):6912–22. doi: 10.1182/blood-2011-02-334359
168. Harris WJ, Huang X, Lynch JT, Spencer GJ, Hitchin JR, Li YY, et al. The Histone Demethylase KDM1A Sustains the Oncogenic Potential of MLL-AF9 Leukemia Stem Cells. Cancer Cell (2012) 21(4):473–87. doi: 10.1016/j.ccr.2012.03.014
169. Pedicona F, Casado P, Hijazi M, Gribben JG, Rouault-Pierre K, Cutillas PR, et al. Targeting the Lysine-Specific Demethylase 1 Rewires Kinase Networks and Primes Leukemia Cells for Kinase Inhibitor Treatment. Sci Signal (2022) 15(730):eabl7989. doi: 10.1126/scisignal.abl7989
170. Rau RE, Rodriguez BA, Luo M, Jeong M, Rosen A, Rogers JH, et al. DOT1L as a Therapeutic Target for the Treatment of DNMT3A-Mutant Acute Myeloid Leukemia. Blood (2016) 128(7):971–81. doi: 10.1182/blood-2015-11-684225
171. Yang C, Wang K, Liang QL, Tian TT, Zhong ZF. Role of NSD1 as Potential Therapeutic Target in Tumor. Pharmacol Res (2021) 173:105888. doi: 10.1016/j.phrs.2021.105888
172. Huang H, Howard CA, Zari S, Cho HJ, Shukla S, Li H, et al. Covalent Inhibition of NSD1 Histone Methyltransferase. Nat Chem Biol (2020) 16(12):1403–10. doi: 10.1038/s41589-020-0626-6
173. Mohanty S, Jyotsana N, Sharma A, Othman B, Kloos A, Mandhania M, et al. Targeted Inhibition of the NUP98-NSD1 Fusion Oncogene in AML. Blood (2019) 134:2545. doi: 10.1182/blood-2019-123709
174. Margueron R, Reinberg D. The Polycomb Complex PRC2 and its Mark in Life. Nature (2011) 469(7330):343–9. doi: 10.1038/nature09784
175. Göllner S, Oellerich T, Agrawal-Singh S, Schenk T, Klein H-U, Rohde C, et al. Loss of the Histone Methyltransferase EZH2 Induces Resistance to Multiple Drugs in Acute Myeloid Leukemia. Nat Med (2017) 23(1):69–78. doi: 10.1038/nm.4247
176. Basheer F, Giotopoulos G, Meduri E, Yun H, Mazan M, Sasca D, et al. Contrasting Requirements During Disease Evolution Identify EZH2 as a Therapeutic Target in AML. J Exp Med (2019) 216(4):966–81. doi: 10.1084/jem.20181276
177. Zhou J, Bi C, Cheong L-L, Mahara S, Liu S-C, Tay K-G, et al. The Histone Methyltransferase Inhibitor, DZNep, Up-Regulates TXNIP, Increases ROS Production, and Targets Leukemia Cells in AML. Blood (2011) 118(10):2830–9. doi: 10.1182/blood-2010-07-294827
178. Illiano M, Conte M, Salzillo A, Ragone A, Spina A, Nebbioso A, et al. The KDM Inhibitor GSKJ4 Triggers CREB Downregulation via a Protein Kinase A and Proteasome-Dependent Mechanism in Human Acute Myeloid Leukemia Cells. Front Oncol (2020) 10:799. doi: 10.3389/fonc.2020.00799
179. Mallaney C, Ostrander EL, Celik H, Kramer AC, Martens A, Kothari A, et al. Kdm6b Regulates Context-Dependent Hematopoietic Stem Cell Self-Renewal and Leukemogenesis. Leukemia (2019) 33(10):2506–21. doi: 10.1038/s41375-019-0462-4
180. Macpherson L, Anokye J, Yeung MM, Lam EYN, Chan Y-C, Weng C-F, et al. HBO1 is Required for the Maintenance of Leukaemia Stem Cells. Nature (2020) 577(7789):266–70. doi: 10.1038/s41586-019-1835-6
181. Zhang J, Gao XF, Yu L. Roles of Histone Deacetylases in Acute Myeloid Leukemia With Fusion Proteins. Front Oncol (2021) 11:741746. doi: 10.3389/fonc.2021.741746
182. Moschoi R, Imbert V, Nebout M, Chiche J, Mary D, Prebet T, et al. Protective Mitochondrial Transfer From Bone Marrow Stromal Cells to Acute Myeloid Leukemic Cells During Chemotherapy. Blood (2016) 128(2):253–64. doi: 10.1182/blood-2015-07-655860
183. Forte D, Garcia-Fernandez M, Sanchez-Aguilera A, Stavropoulou V, Fielding C, Martin-Perez D, et al. Bone Marrow Mesenchymal Stem Cells Support Acute Myeloid Leukemia Bioenergetics and Enhance Antioxidant Defense and Escape From Chemotherapy. Cell Metab (2020) 32(5):829–843.e9. doi: 10.1016/j.cmet.2020.09.001
184. Kamga PT, Bassi G, Cassaro A, Midolo M, Di Trapani M, Gatti A, et al. Notch Signalling Drives Bone Marrow Stromal Cell-Mediated Chemoresistance in Acute Myeloid Leukemia. Oncotarget (2016) 7(16):21713–27. doi: 10.18632/oncotarget.7964
185. Abdul-Aziz AM, Sun Y, Hellmich C, Marlein CR, Mistry J, Forde E, et al. Acute Myeloid Leukemia Induces Protumoral P16ink4a-Driven Senescence in the Bone Marrow Microenvironment. Blood (2019) 133(5):446–56. doi: 10.1182/blood-2018-04-845420
186. Kagoya Y, Yoshimi A, Kataoka K, Nakagawa M, Kumano K, Arai S, et al. Positive Feedback Between NF-κb and TNF-α Promotes Leukemia-Initiating Cell Capacity. J Clin Invest (2014) 124(2):528–42. doi: 10.1172/jci68101
187. Barreyro L, Will B, Bartholdy B, Zhou L, Todorova TI, Stanley RF, et al. Overexpression of IL-1 Receptor Accessory Protein in Stem and Progenitor Cells and Outcome Correlation in AML and MDS. Blood (2012) 120(6):1290–8. doi: 10.1182/blood-2012-01-404699
188. Wang ZD, Chen JH, Wang MZ, Zhang LL, Yu L. One Stone, Two Birds: The Roles of Tim-3 in Acute Myeloid Leukemia. Front Immunol (2021) 12:618710. doi: 10.3389/fimmu.2021.618710
189. Binder S, Luciano M, Horejs-Hoeck J. The Cytokine Network in Acute Myeloid Leukemia (AML): A Focus on Pro- and Anti-Inflammatory Mediators. Cytokine Growth Factor Rev (2018) 43:8–15. doi: 10.1016/j.cytogfr.2018.08.004
190. Mirfakhraie R, Noorazar L, Mohammadian M, Hajifathali A, Gholizadeh M, Salimi M, et al. Treatment Failure in Acute Myeloid Leukemia: Focus on the Role of Extracellular Vesicles. Leuk Res (2022) 112:106751. doi: 10.1016/j.leukres.2021.106751
191. Lyu T, Wang Y, Li D, Yang H, Qin B, Zhang W, et al. Exosomes From BM-MSCs Promote Acute Myeloid Leukemia Cell Proliferation, Invasion and Chemoresistance via Upregulation of S100A4. Exp Hematol Oncol (2021) 10(1):24. doi: 10.1186/s40164-021-00220-7
192. Chen TT, Zhang GZ, Kong LZ, Xu SJ, Wang Y, Dong M. Leukemia-Derived Exosomes Induced IL-8 Production in Bone Marrow Stromal Cells to Protect the Leukemia Cells Against Chemotherapy. Life Sci (2019) 221:187–95. doi: 10.1016/j.lfs.2019.02.003
193. Bouvy C, Wannez A, Laloy J, Chatelain C, Dogne JM. Transfer of Multidrug Resistance Among Acute Myeloid Leukemia Cells via Extracellular Vesicles and Their microRNA Cargo. Leuk Res (2017) 62:70–6. doi: 10.1016/j.leukres.2017.09.014
194. Fang ZG, Wang XZ, Wu JY, Xiao RZ, Liu JJ. High Serum Extracellular Vesicle miR-10b Expression Predicts Poor Prognosis in Patients With Acute Myeloid Leukemia. Cancer Biomarkers (2020) 27(1):1–9. doi: 10.3233/cbm-190211
195. Sharifi H, Shafiee A, Molavi G, Razi E, Mousavi N, Sarvizadeh M, et al. Leukemia-Derived Exosomes: Bringing Oncogenic Signals to Blood Cells. J Cell Biochem (2019) 120(10):16307–15. doi: 10.1002/jcb.29018
196. Huan J, Hornick NI, Shurtleff MJ, Skinner AM, Goloviznina NA, Roberts CT, et al. RNA Trafficking by Acute Myelogenous Leukemia Exosomes. Cancer Res (2013) 73(2):918–29. doi: 10.1158/0008-5472.Can-12-2184
197. Hong C-S, Sharma P, Yerneni SS, Simms P, Jackson EK, Whiteside TL, et al. Circulating Exosomes Carrying an Immunosuppressive Cargo Interfere With Cellular Immunotherapy in Acute Myeloid Leukemia. Sci Rep (2017) 7(1):14684. doi: 10.1038/s41598-017-14661-w
198. Morita K, Wang F, Jahn K, Hu T, Tanaka T, Sasaki Y, et al. Clonal Evolution of Acute Myeloid Leukemia Revealed by High-Throughput Single-Cell Genomics. Nat Commun (2020) 11(1):5327. doi: 10.1038/s41467-020-19119-8
Keywords: acute myeloid leukemia, leukemia stem cells, drug resistance, resistant mechanisms, LSCs remolding, clonal heterogeneity
Citation: Niu J, Peng D and Liu L (2022) Drug Resistance Mechanisms of Acute Myeloid Leukemia Stem Cells. Front. Oncol. 12:896426. doi: 10.3389/fonc.2022.896426
Received: 15 March 2022; Accepted: 06 June 2022;
Published: 05 July 2022.
Edited by:
Pengxu Qian, Zhejiang University, ChinaReviewed by:
Pengcheng Shi, Southern Medical University, ChinaYajing Chu, Chinese Academy of Medical Sciences and Peking Union Medical College, China
Copyright © 2022 Niu, Peng and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lingbo Liu, bGluZ2JvbGl1NjRAcXEuY29t
†These authors have contributed equally to this work