 Xiangchen Hu
Xiangchen Hu Zhe Wang2†
Zhe Wang2†- 1Department of General Surgery, Shengjing Hospital of China Medical University, Shenyang, China
- 2Department of Pathology, Shengjing Hospital of China Medical University, Shenyang, China
- 3Medical Research Center, Shengjing Hospital of China Medical University, Shenyang, China
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal tumors of the gastrointestinal tract. At present, surgery is the first-line treatment for primary resectable GISTs; however, the recurrence rate is high. Imatinib mesylate (IM) is an effective first-line drug used for the treatment of unresectable or metastatic recurrent GISTs. More than 80% of patients with GISTs show significantly improved 5-year survival after treatment; however, approximately 50% of patients develop drug resistance after 2 years of IM treatment. Therefore, an in-depth research is urgently needed to reveal the mechanisms of secondary resistance to IM in patients with GISTs and to develop new therapeutic targets and regimens to improve their long-term prognoses. In this review, research on the mechanisms of secondary resistance to IM conducted in the last 5 years is discussed and summarized from the aspects of abnormal energy metabolism, gene mutations, non-coding RNA, and key proteins. Studies have shown that different drug-resistance mechanism networks are closely linked and interconnected. However, the influence of these drug-resistance mechanisms has not been compared. The combined inhibition of drug-resistance mechanisms with IM therapy and the combined inhibition of multiple drug-resistance mechanisms are expected to become new therapeutic options in the treatment of GISTs. In addition, implementing individualized therapies based on the identification of resistance mechanisms will provide new adjuvant treatment options for patients with IM-resistant GISTs, thereby delaying the progression of GISTs. Previous studies provide theoretical support for solving the problems of drug-resistance mechanisms. However, most studies on drug-resistance mechanisms are still in the research stage. Further clinical studies are needed to confirm the safety and efficacy of the inhibition of drug-resistance mechanisms as a potential therapeutic target.
1 Introduction
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal tumors of the digestive tract, originating from gastrointestinal pacemaker cells (interstitial cells of Cajal, ICC) or related stem cells (1, 2). GISTs are typically driven by mutations of the receptor tyrosine kinase oncogene (C-KIT) or the platelet-derived growth factor receptor α (PDGFRα), which account for more than 80% and 5%–10% of all cases of GIST, respectively (3–5). GISTs without KIT or PDGFRα mutations are known as wild-type GISTs (WT-GISTs), which account for 10%–15% of all cases of adult GISTs and up to 85% of all cases of pediatric GISTs (6–8). In this category, 20%–40% are characterized by the loss of succinate dehydrogenase complex (SDH-deficient GISTs), approximately 15% carry BRAF/RAS or NF1 mutations, and the remainder is referred to as KIT/PDGFRA/SDH/RAS-P WT-GISTs (or quadruple WT-GISTs) (9, 10). A careful examination for germline mutations is of great significance for all patients with WT-GISTs (11). Analyses conducted using tissue microarrays have shown that the DOG1 gene is relatively specifically expressed in GISTs, regardless of the KIT or PDGFRA mutation status (12). A monoclonal antibody against DOG1 has been proven to be a highly sensitive specific marker for the diagnosis of GISTs, and its sensitivity is higher than that of KIT (13).
Imatinib mesylate (IM) is a selective tyrosine kinase inhibitor (TKI) that targets KIT and PDGFRα for the treatment of unresectable or metastatic GISTs, which significantly improves the 5-year survival of patients (14, 15). The efficacy of IM varies among different KIT and PDGFRA mutation types, depending on the exons involved (16, 17). Approximately 14% of patients with GISTs are initially resistant to IM (18), whereas approximately 50% of patients develop resistance after 2 years of treatment, the so-called secondary resistance to IM (19). Therefore, it is important to clarify the mechanisms of secondary resistance to IM through research and develop new therapeutic targets and regimens to improve the long-term prognoses of patients with GISTs. A recent study demonstrated that IM specifically increases the expression of the complex II (SDHB) protein in oxidative phosphorylation (OXPHOS) proteins by downregulating miR-483-3p (Figure 1A). This study demonstrated the molecular mechanism of increased OXPHOS protein expression induced by IM and confirmed the biological role of miR-483-3p in regulating energy metabolism after IM treatment (20).This review will focus on the discussion and summary of research on the mechanisms of secondary resistance to IM conducted in the last 5 years from the aspects of abnormal gene mutation, energy metabolism, non-coding RNA, and key proteins.
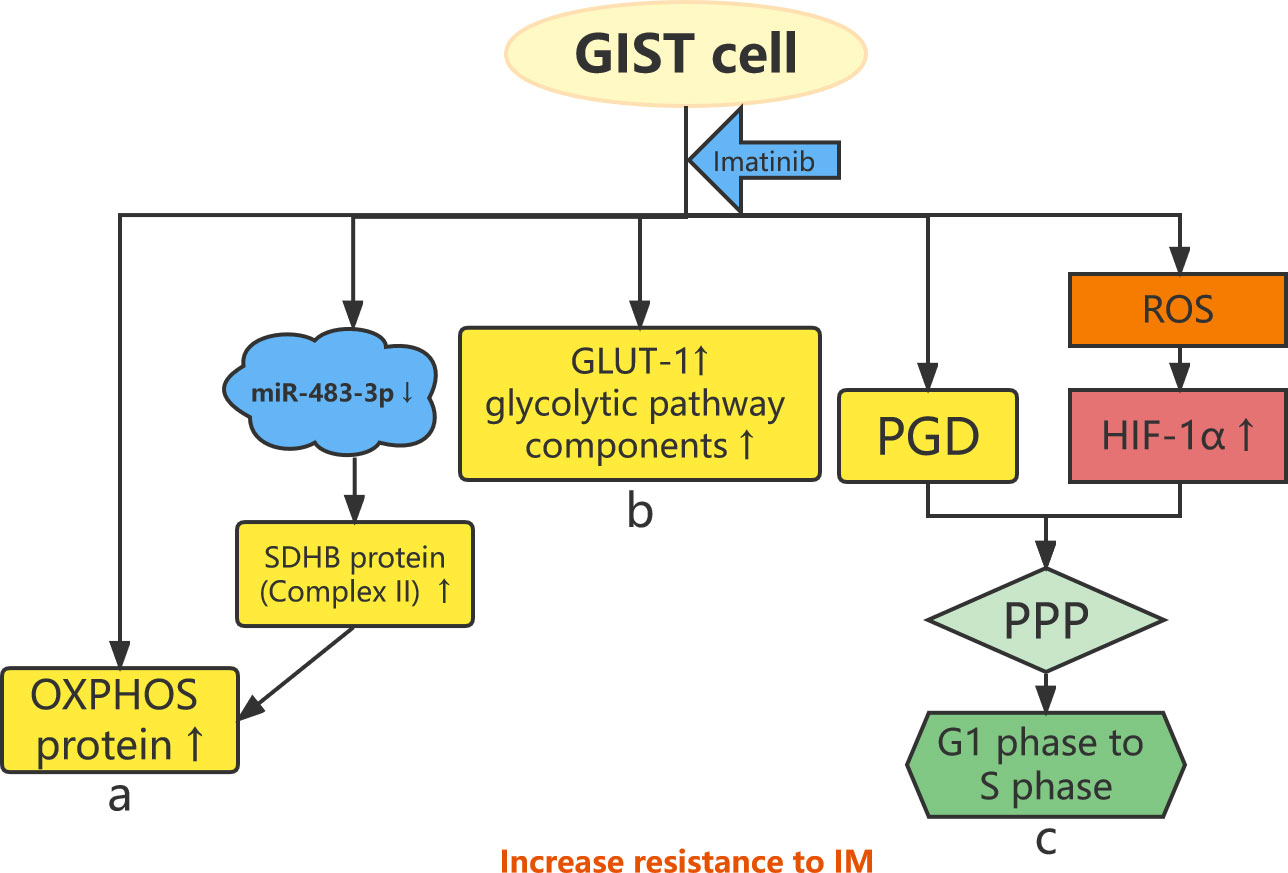
Figure 1 Abnormal energy metabolism and resistance to imatinib. (A) OXPHOS protein expression is increased in IM-resistant GIST cells, and IM specifically increases the expression of complex II (SDHB) protein by downregulating miR-483-3p. (B) GLUT-1 and glycolytic pathway components increase in IM-resistant GIST cells. (C) The HIF-1α–PGD–PPP axis and IM-induced ROS stimulate GIST cells from the G1 phase to the S phase, leading to drug resistance.
2 Mechanisms of secondary resistance to imatinib
2.1 Gene mutation and resistance to imatinib
Secondary KIT and PDGFRA mutations are the main causes of secondary resistance to IM in non–wild-type GIST (21, 22). In most cases of GIST, secondary KIT mutations reactivate KIT downstream signaling pathways, such as the PI3K/AKT/mTOR pathway, and continue to drive GIST proliferation and survival, leading to acquired IM resistance (23–27). KIT T670I is one of the most common types of secondary KIT mutations (24). Cassier et al. found that PDGFRA exon 18 D842V gene subtype mutation is associated with primary resistance to IM (16). Secondary PDGFRA mutations are less common in IM-resistant GISTs than secondary KIT mutations (28, 29). Secondary KIT mutations or PDGFRA mutations do not occur in wild-type IM-resistant GISTs (30).
In a previous study of 210 Chinese patients with IM-resistant GIST who underwent next-generation sequencing for the identification and characterization of secondary KIT mutations, the results showed that 63.81% of the patients had mutations on exon 13, 4.76% had mutations on exon 14, and 31.43% had mutations on exon 17. All secondary KIT mutations were missense mutations, mostly located in the kinase domain (31). Zhao et al. obtained consistent results in an analysis of the distribution of the most common Kit mutation forms in 2,273 Chinese patients with GIST. The results showed that KIT exon 13 V654A and exon 17 N822K were the most common secondary mutations in GISTs with primary mutations in exon 11 (32). These two secondary KIT mutations induce resistance to IM by activating the PI3K/AKT/mTOR pathway (33). Inhibition of PI3K induces massive apoptosis in IM-resistant GISTs (34). Interestingly, KIT is overactivated in IM-resistant GISTs with secondary KIT mutations; however, the expression levels are not significantly increased. Secondary PDGFRA mutations are mostly located in exon 18 (24, 35).
In addition to secondary KIT and PDGFRA mutations, several additional genetic mutations have been associated with secondary resistance to IM in GISTs. Additional mutations of RB1, SMARCB1, and MAX (myc-related protein) are important causes of resistance to IM. Notably, GISTs caused by different gene mutations show different clinicopathological characteristics (31). Genome-scale CRISPR-Cas9 knockout (GeCKO) screening classifies TP53 and SOCS6 as candidate genes for resistance to IM owing to their presence in multiple signaling pathways, such as the apoptosis pathway, Wnt signaling pathway, and JAK-STAT signaling access (36).
Identifying the abovementioned types of gene mutation is a key supplement to the existing GIST risk assessment model. In addition, the discovery of new potential candidate therapeutic targets for different genetic mutations will be beneficial in delaying the progression of GISTs. Individualized therapy based on the identification of types of genetic mutation will also provide new adjuvant treatment options for patients with IM-resistant GIST. Furthermore, the type of gene mutation may be used as a biomarker to help identify patients who can benefit more from adjuvant therapy and to predict the risk of recurrence of GISTs.
2.2 Abnormal energy metabolism and resistance to imatinib
An important feature of cancer cells is abnormal energy metabolism, which is characterized by strong aerobic glycolysis and reduced mitochondrial energy metabolism. This feature is called the Warburg effect (37). Metabolic reprogramming of cancer cells sets the stage for rapid growth and metastasis (38, 39). Drug-resistant cancer cell subsets depend on the enhancement of mitochondrial function and OXPHOS (40, 41). Moreover, the metabolic adaptation of cancer cells to the toxic effects of targeted drugs contributes to drug resistance (42–45). GIST cells exhibit high levels of glucose uptake and aerobic glycolytic activity, and metabolic reprogramming induced by IM stress enhances mitochondrial function and OXPHOS (46).
IM alters the metabolic phenotype of GISTs (46) and increases the expression of several OXPHOS proteins, including complexes II, III, and V (40). Huang et al. found that IM-resistant GIST cells show increased OXPHOS protein expression compared with IM-sensitive GIST cells (Figure 1A) (40). In addition, IM-resistant GIST cells show higher OXPHOS levels and glycolysis rates than IM-sensitive cells and are more susceptible to glycolysis inhibition. Inhibition of OXPHOS increases the sensitivity of GISTs to IM. OXPHOS protein expression is increased in IM-sensitive GIST cells after IM treatment but not in IM-resistant GIST cells (47). Notably, there is a heterogeneity of metabolic phenotypes in IM-resistant GIST (40). Glucose transporter 1 (GLUT-1) is a key component of the glycolytic pathway and is associated with secondary resistance to IM in GIST cells. IM downregulates the expression of GLUT-1 and the glycolytic pathway components hexokinase 2, pyruvate kinase M2, and lactate dehydrogenase in IM-sensitive GIST cell lines. In contrast, the expression of GLUT-1 and these glycolytic pathway components increases after the treatment of IM-resistant GIST cell lines using IM (Figure 1B). This indicates that IM-resistant GIST cells have a higher glycolysis rate than IM-sensitive GIST cells (48).
Following chronic IM induction, energy metabolism in GIST cells shifts from the tricarboxylic acid cycle to the pentose phosphate pathway (PPP) (47). On one hand, the expression of phosphate glucose dehydrogenase (PGD), one of the rate-limiting enzymes of the PPP, is significantly upregulated in IM-resistant GIST cell lines. Overexpression of PGD promotes GIST cell proliferation and inhibits cell apoptosis. On the other hand, the level of hypoxia-inducible factor 1α (HIF-1α) is elevated under prolonged stimulation of reactive oxygen species generated by IM (47). HIF-1α leads to changes in metabolic pathways as follows: the HIF-1α–PGD–PPP axis stimulates GIST cells from the G1 phase to the S phase, inhibits GIST cell apoptosis through metabolic reprogramming, and ultimately leads to IM resistance (Figure 1C) (47).
Most of the research viewpoints on energy metabolism in GIST cells have reached a consensus, which provides a theoretical basis for overcoming resistance to IM from the perspective of abnormal energy metabolism. Therapy involving the inhibition of the energy metabolism pathway combined with IM, such as VLX600 combined with IM and WZB117 combined with IM, requires further preclinical validation (46, 48).
2.3 Non-coding RNAs and resistance to imatinib
2.3.1 Long non-coding RNAs
Long non-coding RNAs (lncRNAs) are transcripts longer than 200 nucleotides with no or limited protein-coding capacity (49–51). LncRNAs play key roles in several important biological processes, including regulation of epigene expression, as well as transcriptional and posttranscriptional regulation (52). Numerous studies have demonstrated that lncRNAs play key regulatory roles in the disease course of human cancers, including cancer cell proliferation, apoptosis, and drug resistance (53–55). In addition, recent studies have shown that lncRNAs can modulate the sensitivity of patients to anticancer drugs and thus have the potential to be therapeutic targets in the treatment of drug-resistant tumors (56, 57). Moreover, lncRNAs may promote the progression and metastasis of GISTs, and the expression of many lncRNAs in primary GIST tissue differs from that in recurrent GIST tissue (58, 59). Furthermore, lncRNAs are associated with secondary resistance to IM in GISTs, and the resistance mechanisms are mostly related to signaling pathways (60–63). LncRNAs, such as the HOX antisense intergenic RNA (HOTAIR), can also promote IM resistance by activating autophagy in GIST cells (62).
The lncRNA coiled-coil domain-containing 26 (CCDC26), located on chromosome 8q24.21, is a retinoic acid–dependent regulator of myeloid differentiation, also known as RAM (64). CCDC26 interacts with C-KIT and regulates its transcription. In addition, CCDC26 downregulates the expression of c-Kit in GISTs, whereas CCDC26 knockout induces IM resistance in GIST cells by upregulating the expression of C-KIT (Figure 2A) (60). CCDC26 knockout also upregulates the expression of insulin-like growth factor 1 receptor (IGF-1R) (Figure 2A). IGF-1R induces drug resistance by participating in the apoptosis pathway (Figure 2C), whereas inhibition of IGF-1R reverses CCDC26 knockout–induced drug resistance (59, 65, 66). These findings suggest that treatment targeting the CCDC26 or CCDC26-IGF-1R axis may improve sensitivity to IM in patients with IM-resistant GISTs.
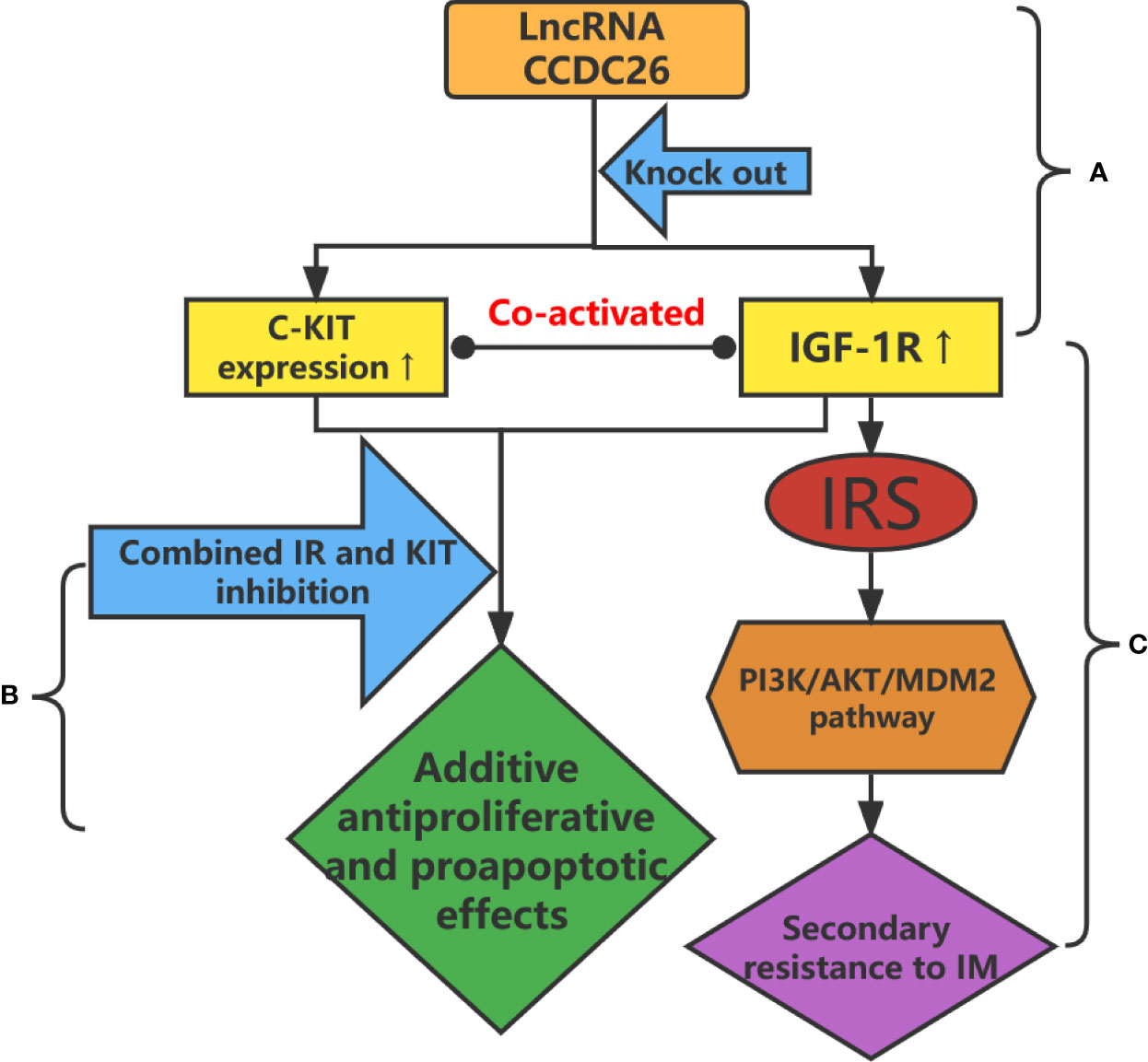
Figure 2 (A) CCDC26 knockout upregulates the expression of C-KIT and IGF-1R in IM-resistant GISTs. (B) Additive antiproliferative and proapoptotic effects are obtained after the combined inhibition of IR and KIT in IM-resistant GIST cells. (C) Upregulation of IGF-1R leads to drug resistance through the PI3K/AKT/MDM2 signaling pathway.
Yan et al. identified a set of dysregulated lncRNAs in IM-resistant GISTs using chip technology and found that lncRNAJC6-2 is associated with the HIF-1α pathway, which links lncRNAs to energy metabolism (61). Using high-throughput RNA sequencing, Shao et al. found that GIST samples express 40% of all annotated lncRNAs in humans. Notably, the number of downregulated lncRNA expressions was greater than the number of upregulated expressions, irrespective of the presence or absence of resistance to IM. The expression of RP11616M22.7 is significantly increased in IM-resistant samples than that in non-resistant samples and is closely related to the Hippo pathway. Overexpression of RP11-616M22.7 induces resistance to IM in GIST cells, whereas RP11616M22.7 gene knockout enhances IM resistance in GIST cells both in vitro and in vivo (62).
Some studies have demonstrated that the expression and dysregulation of lncRNAs are more cancer-specific than those of protein-coding genes (67). Therefore, specific lncRNAs in GISTs are likely to be involved in unique biological functions related to treatment and drug resistance. With an in-depth exploration of specific lncRNAs and their mechanisms, our understanding of the non-coding transcriptome of GISTs will become more comprehensive, which will, in turn, accelerate the development of new effective therapeutic targets.
2.3.2. MicroRNAs
MicroRNAs (MiRNAs) are 22-nucleotide non-coding small ribonucleic acids that control tumor cell growth by regulating the expression of multiple gene products and the function of cellular pathways (68). MiRNAs play important roles in the pathogenesis, invasion, and drug resistance of tumors and are thus identified as targets for cancer diagnosis, therapy, and prognosis (69–72). Akçakaya et al. analyzed miRNA expression profiles to study the miRNA expression signatures associated with response to IM and KIT mutation status in patients with GIST. They found that miR-125a-5p and its target gene, tyrosine-protein phosphatase non-receptor type 18 (PTPN18), play important roles in IM resistance. The mechanism behind this is that overexpression of miR-125a-5p downregulates the level of PTPN18 expression in GISTs and promotes resistance to IM (73). Subsequent studies demonstrated that the effects of miR-125a-5p and PTPN18 on IM resistance are mediated through phosphorylated FAK levels (Figure 3) (28). By comparing two groups of IM-resistant GIST samples with and without secondary mutations, Amirnasr et al. detected 22 significantly differentially expressed miRNAs and almost completely separated the two groups of samples. Three of these miRNAs, namely, miR-92a-3p, miR-99a-5p, and miR-101-3p, are potential effectors of IM resistance. This suggests that the distribution of miRNA biomarkers may be related to the presence of secondary mutations (74). Zhang et al. used the microarray data preserved by Akçakaya et al. to identify five key miRNAs in the lncRNA–miRNA target gene regulatory network, confirming that overexpression of miR-28-5p and miR-125a-5p is significantly related to secondary resistance to IM (75). Kou et al. studied the miRNA expression profiles in the serums of patients with GIST and found that the levels of miR-518e-5p and miR-548e in the serums of the patients in the IM-resistant group were significantly higher than those of the patients in the IM-sensitive and healthy control groups. This indicates that the serum level of miR-518e-5p can distinguish IM-resistant patients from IM-sensitive patients or healthy individuals (76).
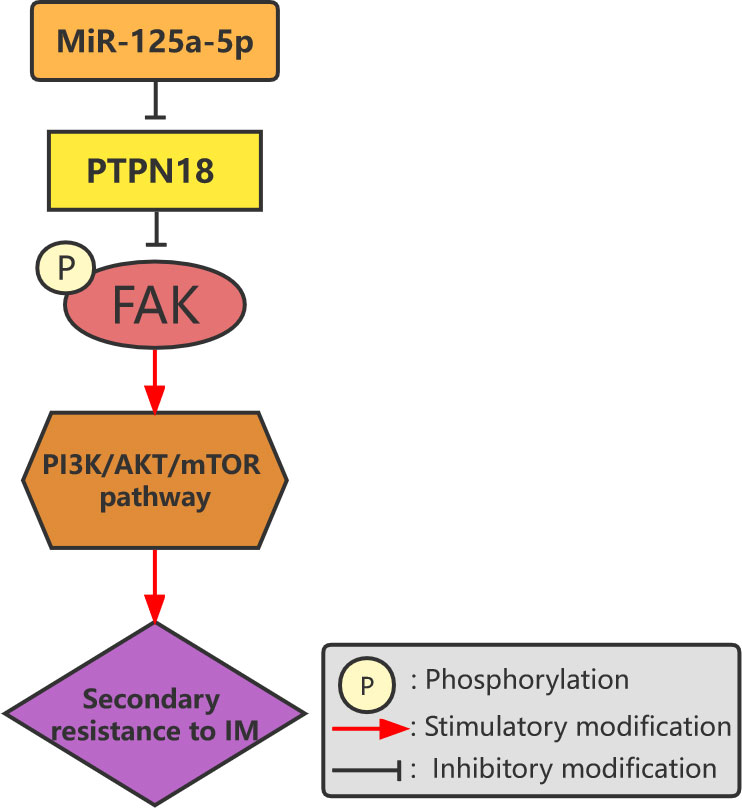
Figure 3 Overexpression of miR-125a-5p downregulates the expression of PTPN18 and promotes IM resistance in GISTs mediated by phosphorylated FAK levels.
Studies have demonstrated that miRNAs can regulate resistance to chemotherapy by inducing autophagy in GIST cells (77, 78). Chen et al. found that miR-30a sensitizes GIST cells to IM by inhibiting autophagy and confirmed that the autophagy marker Beclin-1 is a target gene of miR-30a (79). Zhang et al. found that HOTAIR targeting the autophagy-related protein 2 homolog B inhibitor miR-130a promotes resistance to IM by upregulating the level of autophagy (63).
Information regarding most miRNAs associated with secondary resistance to IM is still in the discovery stage; thus, the resistance mechanisms need to be studied further. Because miRNAs are closely related to the pathogenesis, invasion, metastasis, and drug resistance of tumors, research ideas should be broadened rather than limited to one aspect. Several studies have confirmed that lncRNAs can regulate other non-coding RNAs, especially miRNAs, and that miRNAs also have regulatory effects on lncRNAs (80, 81). Therefore, improving the regulatory network of miRNAs and lncRNAs in IM-resistant GISTs is also a promising research direction.
2.4. Several key proteins and resistance to imatinib
From a protein perspective, approximately 10% of KIT-positive GISTs lose the expression of KIT oncoproteins and become resistant to TKIs owing to the transition to a KIT-independent state (KIT-negative) during TKI treatment (82). Tu et al. found that Axl in TKIs is highly expressed in KIT-negative GISTs and that Axl gene knockout or silencing can inhibit the proliferation of KIT-negative GISTs. This information provides a new perspective regarding the Axl/P53 signaling axis as a therapeutic target for a subset of KIT-negative GISTs (83).
Cyclin D1 can regulate the cell cycle through the activation of the cyclin-dependent kinase (CDK), activation of transcription factors, RAD51 co-regulation of DNA repair, and activation of the AMPK-LKB1 signaling pathway (84). Cyclin D1 is highly expressed in each KIT-independent GIST cell subline. In addition, inhibition of cyclin D1 has antiproliferative and proapoptotic effects in KIT-independent GISTs, which are associated with Rb activation and p27 upregulation. Notably, PRKCQ is a negative regulator of cyclin D1 expression, whereas the Jun and Hippo pathway effector molecules YAP and TAZ are positive regulators of cyclin D1 expression. The PRKCQ, Jun, and Hippo pathways synergistically regulate cyclin D1 expression in GISTs (85). Using GeCKO screening, (85) found that CDK1 is highly expressed in advanced and IM-resistant GISTs in three patient cohorts. CDK1 is the founding member of the CDK family (86). It can promote the proliferation and progression of GISTs by binding to substrate protein kinase B (Akt) and regulating its phosphorylation (87). In most solid tumors, Aurora kinase A (AURKA) promotes cell cycle progression by regulating cell cycle checkpoints (88). A clinical analysis has demonstrated that AURKA can be an independent prognostic factor for GISTs. In addition, experiments have shown that overexpression of AURKA can promote the proliferation of GIST-T1 cells, inhibit cell apoptosis, and enhance the resistance of cells to IM (89).
Several multidrug transporters play key roles in secondary drug resistance by regulating drug concentrations in tumor cells. Multidrug resistance–related protein 1 (MRP1) is one of the major multidrug transporters (90). Intracellular IM level plays an important role in the development of IM resistance in patients with chronic myeloid leukemia (91). Studies have confirmed that MRP1 and breast cancer resistance protein are highly expressed in IM-resistant GIST cell lines and that IM-resistant patients with GIST show significantly lower intracellular IM levels than IM-sensitive patients (92). This suggests that drug transporters may play an important role in IM resistance. Xu et al. proposed the following mechanism for this: the methyltransferase METTL3 mediates 6-methyladenosine (M6A) to modify the 5’end non-coding region of the multidrug transporter MRP1 mRNA and promotes the translation of MRP1 mRNA, leading to drug resistance in GISTs (93). M6A is a common mRNA modification that regulates mRNA stability, splicing, and translation (94, 95). These findings suggest that drug transporters may be potential therapeutic targets in the treatment of IM-resistant GISTs.
The insulin receptor (IR) is a member of the tyrosine kinase family, including homologous types 1 and 2 (IGF-1R and IGF-2R) (96). IR and IGF-1/2R play important roles in energy metabolism and cell growth, division, and differentiation (97). Chen et al. showed that IR and Kit are co-activated in IM-resistant GIST cells and biopsy samples but not in IM-sensitive GIST cells (Figure 2A). They also found that additive antiproliferative and proapoptotic effects were obtained after the combined inhibition of IR and KIT in IM-resistant GIST cells (Figure 2B) (98). Thus, the inactivation of IR increases the sensitivity of resistant cells to IM, suggesting that the combined inhibition of IR and KIT is a promising therapeutic strategy in the treatment of IM-resistant GISTs.
Serrano-Candelas et al. found that the linker molecule SH3-binding protein 2 (SH3BP2) is expressed in non–wild-type GISTs. SH3BP2 is involved in the regulation of the expression and cellular activity of KIT and PDGFRA in GISTs. They also found that silencing of SH3BP2 is accompanied by downregulation of oncogenic KIT and PDGFRA and significant promotion of apoptosis in IM-sensitive and resistant GIST cells (99).
The relationship between various key proteins and IM resistance mechanisms is intricate and interconnected. However, there is no clear comparison of the role of each protein network in the mechanism of resistance to IM. Targeted therapy that involves a single protein network may not solve the problem of secondary resistance to IM. The combined inhibition of multiple protein networks may become a new research direction for the treatment of IM-resistant GISTs.
2.5. Mutation and other gene aberrations and resistance to imatinib
2.5.1. Oncogenic KIT signaling on the Golgi apparatus
The Golgi apparatus may serve as a platform for oncogenic KIT signaling (100, 101). Moreover, oncogenic KIT signaling on the Golgi apparatus is essential for the autonomous proliferation of GIST cells (101). In IM-resistant GISTs with secondary KIT mutations, oncogenic KIT signaling is predominantly localized to the Golgi apparatus (100). This KIT activates the PI3K/AKT/mTOR pathway, MEK-Erk pathway, and signal transducer and activator of transcription 5 (Figure 4) (100). Blocking KIT biosynthetic transport from the endoplasmic reticulum to the Golgi apparatus suppresses oncogenic signaling, suggesting that Kit autophosphorylation is spatiotemporally regulated (100, 101). In an analysis of this mechanism, Obata et al. discovered a biosynthetic protein, 2-methylcopropylamide (M-COPA; also known as “AMF-26”), which blocks the transport of KIT from the endoplasmic reticulum to the Golgi apparatus by inhibiting the autophosphorylation of KIT at Y703/Y721/Y730/Y936 and ultimately inhibits oncogenic KIT signaling (Figure 4) (101). M-COPA inhibits the activation of Kit kinase domain mutants, thereby inhibiting the proliferation of IM-resistant GISTs (101). A novel heat shock protein 90 inhibitor, TAS-116, also inhibits the growth of drug-resistant cells and induces their apoptosis by reducing KIT autophosphorylation in the Golgi apparatus (102). Notably, the effect of TAS-116 has been validated in an animal study conducted using a xenograft mouse model (102).
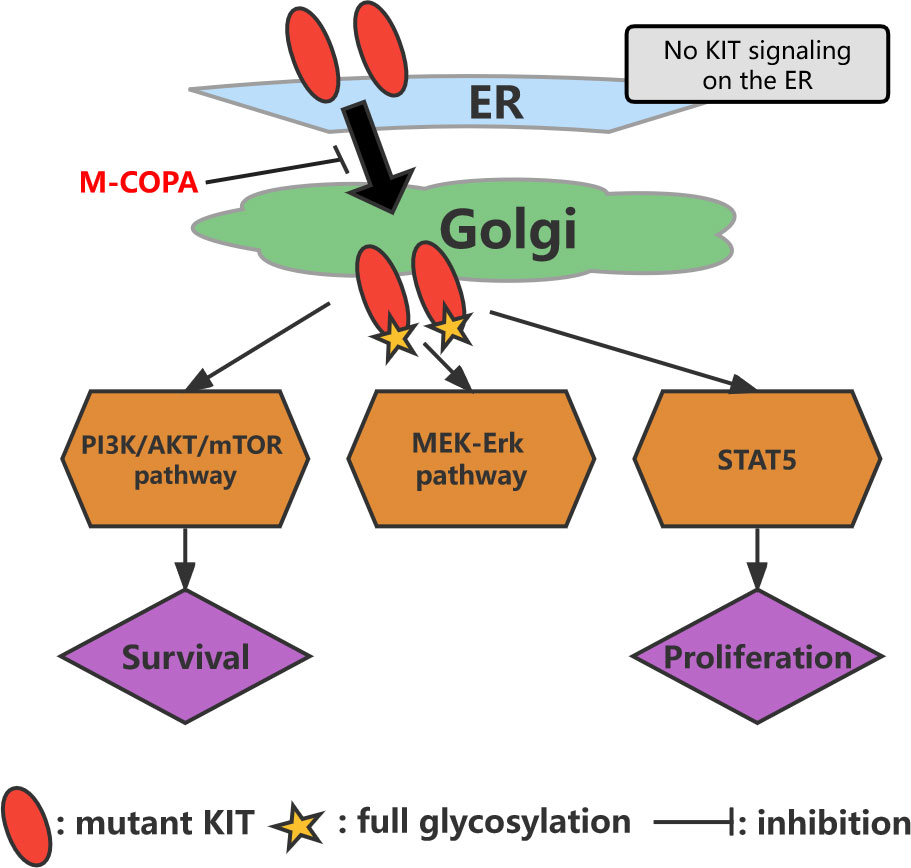
Figure 4 Model of oncogenic KIT signaling on intracellular compartments in GISTs. KIT is normally transported from the endoplasmic reticulum to the Golgi apparatus, followed by full glycosylation. After reacting with the Golgi apparatus, KIT can activate the PI3K/AKT/mTOR pathway, MEK-Erk pathway, and STAT5. M-COPA inhibits oncogenic signaling by blocking the transport of KIT from the endoplasmic reticulum to the Golgi because KIT activates downstream molecules only on the Golgi apparatus.
Oncogenic KIT signaling on the Golgi apparatus provides new insights into not only the pathogenesis of KIT but also the treatment of IM-resistant GISTs that express mutant KIT. However, further studies are needed to confirm the clinical efficacy of drug therapies that target this carcinogenic signal.
2.5.2. KITlow cell subsets
KITlow cell subsets may be a cell bank that mediates the progression and recurrence of GISTs (103). Bardsley et al. detected a precursor cell of ICCs in the stomach wall of a mouse that possesses stem cell properties, including the ability to self-renew and differentiate into mature ICCs. This ICC precursor cell–derived cell line was able to spontaneously transform to form GIST-like tumors. Notably, the expression of Kit in this ICC precursor cell was lower than that in mature ICC precursor cells (104, 105).
Inherently, IM-resistant CD34 KITlow cells are a distinct subset of GIST cells. KITlow cells have stronger replication ability and clonogenic potential than KITHigh cell subsets. This subpopulation has tumor stem cell–like expression characteristics and behaviors and can self-renew and differentiate into IM-sensitive CD34 KITHigh progeny. Notably, TKI treatment results in the enrichment of this KITlow cell subset, which may be mediated by cell-associated transcription factors (OCT4 and NANOG) (103). The KITlow cell subset represents a novel mechanism of primary resistance to TKIs and a targetable subpopulation in the treatment of GISTs. This provides valuable therapeutic ideas for overcoming the persistence and recurrence of GISTs after TKI therapy.
3 Conclusions
In this review, the findings of studies on mechanisms of secondary resistance conducted over the last 5 years are summarized from the aspects of abnormal energy metabolism, gene mutations, non-coding RNA, and key proteins. These previous studies provide theoretical support for solving the problem of the mechanism of resistance to IM. However, the available data on most drug-resistance mechanisms are still in the research stage. Further clinical studies are needed to confirm the safety and efficacy of utilizing drug-resistance mechanisms as potential therapeutic targets.
Addressing the problem of secondary resistance to IM has always been the key to improving the treatment outcomes and prognoses of patients with GISTs. Different resistance mechanisms are closely linked and interact with each other; thus, using a single resistance mechanism as a therapeutic target should be avoided. The combined inhibition of drug-resistance mechanisms with IM therapy and the combined inhibition of multiple drug-resistance mechanisms are expected to become new options in the treatment of GISTs. Implementing individualized therapy based on the identification of resistance mechanisms will provide new adjuvant treatment options for patients with IM-resistant GISTs, thereby delaying the progression of GISTs.
Author contributions
Conceptualization: ZW. Article collection and analysis: YK, XH, PS, and QZ. Manuscript writing: XH and QZ. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (No. 81601692), the Technology Research from the Department of Education of Liaoning Province (No. JCZR2020013), and the 345 Talent Project of Shengjing hospital of China Medical University.
Acknowledgments
The authors would like to thank all the reviewers who participated in the review and also Editage (www.editage.cn) for its linguistic assistance during the preparation of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Joensuu H, Fletcher C, Dimitrijevic S, Silberman S, Roberts P, Demetri G. Management of malignant gastrointestinal stromal tumours. Lancet Oncol (2002) 3(11):655–64. doi: 10.1016/S1470-2045(02)00899-9
2. Miettinen M, Lasota J. Gastrointestinal stromal tumors: pathology and prognosis at different sites. Semin Diagn Pathol (2006) 23(2):70–83. doi: 10.1053/j.semdp.2006.09.001
3. Rubin BP, Heinrich MC, Corless CL. Gastrointestinal stromal tumour. Lancet (London England) (2007) 369(9574):1731–41. doi: 10.1016/S0140-6736(07)60780-6
4. Joensuu H, Hohenberger P, Corless CL. Gastrointestinal stromal tumour. Lancet (London England) (2013) 382(9896):973–83. doi: 10.1016/S0140-6736(13)60106-3
5. Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Sci (New York NY) (1998) 279(5350):577–80. doi: 10.1126/science.279.5350.577
6. Wozniak A, Gebreyohannes YK, Debiec-Rychter M, Schöffski P. New targets and therapies for gastrointestinal stromal tumors. Expert Rev Anticancer Ther (2017) 17(12):1117–29. doi: 10.1080/14737140.2017.1400386
7. Janeway KA, Liegl B, Harlow A, Le C, Perez-Atayde A, Kozakewich H, et al. Pediatric KIT wild-type and platelet-derived growth factor receptor alpha-wild-type gastrointestinal stromal tumors share KIT activation but not mechanisms of genetic progression with adult gastrointestinal stromal tumors. Cancer Res (2007) 67(19):9084–8. doi: 10.1158/0008-5472.CAN-07-1938
8. Pappo AS, Janeway KA. Pediatric gastrointestinal stromal tumors. Hematol/oncol Clinics North America (2009) 23(1):15–34, vii. doi: 10.1016/j.hoc.2008.11.005
9. Urbini M, Astolfi A, Indio V, Nannini M, Schipani A, Bacalini MG, et al. Gene duplication, rather than epigenetic changes, drives FGF4 overexpression in KIT/PDGFRA/SDH/RAS-p WT GIST. Sci Rep (2020) 10(1):19829. doi: 10.1038/s41598-020-76519-y
10. Astolfi A, Indio V, Nannini M, Saponara M, Schipani A, De Leo A, et al. Targeted deep sequencing uncovers cryptic KIT mutations in KIT/PDGFRA/SDH/RAS-p wild-type GIST. Front Oncol (2020) 10:504. doi: 10.3389/fonc.2020.00504
11. Kays JK, Sohn JD, Kim BJ, Goze K, Koniaris LG. Approach to wild-type gastrointestinal stromal tumors. Trans Gastroenterol Hepatol (2018) 3:92. doi: 10.21037/tgh.2018.10.13
12. West RB, Corless CL, Chen X, Rubin BP, Subramanian S, Montgomery K, et al. The novel marker, DOG1, is expressed ubiquitously in gastrointestinal stromal tumors irrespective of KIT or PDGFRA mutation status. Am J Pathol (2004) 165(1):107–13. doi: 10.1016/S0002-9440(10)63279-8
13. Espinosa I, Lee CH, Kim MK, Rouse BT, Subramanian S, Montgomery K, et al. A novel monoclonal antibody against DOG1 is a sensitive and specific marker for gastrointestinal stromal tumors. Am J Surg Pathol (2008) 32(2):210–8. doi: 10.1097/PAS.0b013e3181238cec
14. Joensuu H, Eriksson M, Sundby Hall K, Reichardt A, Hermes B, Schütte J, et al. Survival outcomes associated with 3 years vs 1 year of adjuvant imatinib for patients with high-risk gastrointestinal stromal tumors: An analysis of a randomized clinical trial after 10-year follow-up. JAMA Oncol (2020) 6(8):1241–6. doi: 10.1001/jamaoncol.2020.2091
15. Casali PG, Blay JY, Abecassis N, Bajpai J, Bauer S, Biagini R, et al. Gastrointestinal stromal tumours: ESMO-EURACAN-GENTURIS clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol (2022) 33(1):20–33. doi: 10.1016/j.annonc.2021.09.005
16. Cassier PA, Fumagalli E, Rutkowski P, Schöffski P, Van Glabbeke M, Debiec-Rychter M, et al. Outcome of patients with platelet-derived growth factor receptor alpha-mutated gastrointestinal stromal tumors in the tyrosine kinase inhibitor era. Clin Cancer Res (2012) 18(16):4458–64. doi: 10.1158/1078-0432.CCR-11-3025
17. Szucs Z, Thway K, Fisher C, Bulusu R, Constantinidou A, Benson C, et al. Molecular subtypes of gastrointestinal stromal tumors and their prognostic and therapeutic implications. Future Oncol (London England) (2017) 13(1):93–107. doi: 10.2217/fon-2016-0192
18. Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. New Engl J Med (2002) 347(7):472–80. doi: 10.1056/NEJMoa020461
19. Raut CP, Espat NJ, Maki RG, Araujo DM, Trent J, Williams TF, et al. Efficacy and tolerability of 5-year adjuvant imatinib treatment for patients with resected intermediate- or high-risk primary gastrointestinal stromal tumor: The PERSIST-5 clinical trial. JAMA Oncol (2018) 4(12):e184060. doi: 10.1001/jamaoncol.2018.4060
20. Huang WK, Shi H, Akçakaya P, Zeljic K, Gangaev A, Caramuta S, et al. Imatinib regulates miR-483-3p and mitochondrial respiratory complexes in gastrointestinal stromal tumors. Int J Mol Sci (2021) 22(19):10600. doi: 10.3390/ijms221910600
21. Wang WL, Conley A, Reynoso D, Nolden L, Lazar AJ, George S, et al. Mechanisms of resistance to imatinib and sunitinib in gastrointestinal stromal tumor. Cancer chemother Pharmacol (2011) 67 Suppl 1:S15–24. doi: 10.1007/s00280-010-1513-8
22. Antonescu C. Gastrointestinal stromal tumors. Curr topics Microbiol Immunol (2012) 355:41–57. doi: 10.1007/82_2011_161
23. Van Glabbeke M, Verweij J, Casali PG, Le Cesne A, Hohenberger P, Ray-Coquard I, et al. Initial and late resistance to imatinib in advanced gastrointestinal stromal tumors are predicted by different prognostic factors: a European organisation for research and treatment of cancer-Italian sarcoma group-Australasian gastrointestinal trials group study. J Clin Oncol (2005) 23(24):5795–804. doi: 10.1200/JCO.2005.11.601
24. Antonescu CR, Besmer P, Guo T, Arkun K, Hom G, Koryotowski B, et al. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res (2005) 11(11):4182–90. doi: 10.1158/1078-0432.CCR-04-2245
25. Wardelmann E, Merkelbach-Bruse S, Pauls K, Thomas N, Schildhaus HU, Heinicke T, et al. Polyclonal evolution of multiple secondary KIT mutations in gastrointestinal stromal tumors under treatment with imatinib mesylate. Clin Cancer Res (2006) 12(6):1743–9. doi: 10.1158/1078-0432.CCR-05-1211
26. Desai J, Shankar S, Heinrich MC, Fletcher JA, Fletcher CD, Manola J, et al. Clonal evolution of resistance to imatinib in patients with metastatic gastrointestinal stromal tumors. Clin Cancer Res (2007) 13(18 Pt 1):5398–405. doi: 10.1158/1078-0432.CCR-06-0858
27. Liegl B, Kepten I, Le C, Zhu M, Demetri GD, Heinrich MC, et al. Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J Pathol (2008) 216(1):64–74. doi: 10.1002/path.2382
28. Huang WK, Akçakaya P, Gangaev A, Lee L, Zeljic K, Hajeri P, et al. miR-125a-5p regulation increases phosphorylation of FAK that contributes to imatinib resistance in gastrointestinal stromal tumors. Exp Cell Res (2018) 371(1):287–96. doi: 10.1016/j.yexcr.2018.08.028
29. Lim KH, Huang MJ, Chen LT, Wang TE, Liu CL, Chang CS, et al. Molecular analysis of secondary kinase mutations in imatinib-resistant gastrointestinal stromal tumors. Med Oncol (Northwood London England) (2008) 25(2):207–13. doi: 10.1007/s12032-007-9014-2
30. Kang W, Zhu C, Yu J, Ye X, Ma Z. KIT gene mutations in gastrointestinal stromal tumor. Front biosci (Landmark edition) (2015) 20(6):919–26. doi: 10.2741/4346
31. Du J, Wang S, Wang R, Wang SY, Han Q, Xu HT, et al. Identifying secondary mutations in Chinese patients with imatinib-resistant gastrointestinal stromal tumors (GISTs) by next generation sequencing (NGS). Pathol Oncol res: POR (2020) 26(1):91–100. doi: 10.1007/s12253-019-00770-6
32. Zhao Q, Zhang C, Qi C, Yang J, Chen Y, Ge S, et al. Preclinical model-based evaluation of imatinib resistance induced by KIT mutations and its overcoming strategies in gastrointestinal stromal tumor (GIST). Am J Trans Res (2021) 13(12):13608–24.
33. Wang CM, Huang K, Zhou Y, Du CY, Ye YW, Fu H, et al. Molecular mechanisms of secondary imatinib resistance in patients with gastrointestinal stromal tumors. J Cancer Res Clin Oncol (2010) 136(7):1065–71. doi: 10.1007/s00432-009-0753-7
34. Bauer S, Duensing A, Demetri GD, Fletcher JA. KIT oncogenic signaling mechanisms in imatinib-resistant gastrointestinal stromal tumor: PI3-kinase/AKT is a crucial survival pathway. Oncogene (2007) 26(54):7560–8. doi: 10.1038/sj.onc.1210558
35. Heinrich MC, Corless CL, Blanke CD, Demetri GD, Joensuu H, Roberts PJ, et al. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol (2006) 24(29):4764–74. doi: 10.1200/JCO.2006.06.2265
36. Cao J, Wei J, Yang P, Zhang T, Chen Z, He F, et al. Genome-scale CRISPR-Cas9 knockout screening in gastrointestinal stromal tumor with imatinib resistance. Mol Cancer (2018) 17(1):121. doi: 10.1186/s12943-018-0865-2
37. Warburg O. On respiratory impairment in cancer cells. Sci (New York NY) (1956) 124(3215):269–70. doi: 10.1126/science.124.3215.269
38. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the warburg effect: the metabolic requirements of cell proliferation. Sci (New York NY) (2009) 324(5930):1029–33. doi: 10.1126/science.1160809
39. DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab (2008) 7(1):11–20. doi: 10.1016/j.cmet.2007.10.002
40. Huang WK, Gao J, Chen Z, Shi H, Yuan J, Cui HL, et al. Heterogeneity of metabolic vulnerability in imatinib -resistant gastrointestinal stromal tumor. Cells (2020) 9(6):1333. doi: 10.3390/cells9061333
41. Faubert B, Solmonson A, DeBerardinis RJ. Metabolic reprogramming and cancer progression. Sci (New York NY) (2020) 368(6487):eaaw5473. doi: 10.1126/science.aaw5473
42. Roesch A, Vultur A, Bogeski I, Wang H, Zimmermann KM, Speicher D, et al. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer Cell (2013) 23(6):811–25. doi: 10.1016/j.ccr.2013.05.003
43. Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sánchez N, Marchesini M, et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature (2014) 514(7524):628–32. doi: 10.1038/nature13611
44. Yoshida GJ. Metabolic reprogramming: the emerging concept and associated therapeutic strategies. J Exp Clin Cancer Res (2015) 34:111. doi: 10.1186/s13046-015-0221-y
45. Zhang G, Frederick DT, Wu L, Wei Z, Krepler C, Srinivasan S, et al. Targeting mitochondrial biogenesis to overcome drug resistance to MAPK inhibitors. J Clin Invest (2016) 126(5):1834–56. doi: 10.1172/JCI82661
46. Vitiello GA, Medina BD, Zeng S, Bowler TG, Zhang JQ, Loo JK, et al. Mitochondrial inhibition augments the efficacy of imatinib by resetting the metabolic phenotype of gastrointestinal stromal tumor. Clin Cancer Res (2018) 24(4):972–84. doi: 10.1158/1078-0432.CCR-17-2697
47. Xu K, He Z, Chen M, Wang N, Zhang D, Yang L, et al. HIF-1α regulates cellular metabolism, and imatinib resistance by targeting phosphogluconate dehydrogenase in gastrointestinal stromal tumors. Cell Death Dis (2020) 11(7):586. doi: 10.1038/s41419-020-02768-4
48. Shima T, Taniguchi K, Tokumaru Y, Inomata Y, Arima J, Lee SW, et al. Glucose transporter−1 inhibition overcomes imatinib resistance in gastrointestinal stromal tumor cells. Oncol Rep (2022) 47(1):7. doi: 10.3892/or.2021.8218
49. Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell (2011) 43(6):904–14. doi: 10.1016/j.molcel.2011.08.018
50. Marchese FP, Raimondi I, Huarte M. The multidimensional mechanisms of long noncoding RNA function. Genome Biol (2017) 18(1):206. doi: 10.1186/s13059-017-1348-2
51. Zhang J, Li Z, Liu L, Wang Q, Li S, Chen D, et al. Long noncoding RNA TSLNC8 is a tumor suppressor that inactivates the interleukin-6/STAT3 signaling pathway. Hepatol (Baltimore Md) (2018) 67(1):171–87. doi: 10.1002/hep.29405
52. Kung JT, Colognori D, Lee JT. Long noncoding RNAs: past, present, and future. Genetics (2013) 193(3):651–69. doi: 10.1534/genetics.112.146704
53. Liu XH, Sun M, Nie FQ, Ge YB, Zhang EB, Yin DD, et al. Lnc RNA HOTAIR functions as a competing endogenous RNA to regulate HER2 expression by sponging miR-331-3p in gastric cancer. Mol Cancer (2014) 13:92. doi: 10.1186/1476-4598-13-92
54. Loewen G, Jayawickramarajah J, Zhuo Y, Shan B. Functions of lncRNA HOTAIR in lung cancer. J Hematol Oncol (2014) 7:90. doi: 10.1186/s13045-014-0090-4
55. Anastasiadou E, Jacob LS, Slack FJ. Non-coding RNA networks in cancer. Nat Rev Cancer (2018) 18(1):5–18. doi: 10.1038/nrc.2017.99
56. Pichler M, Rodriguez-Aguayo C, Nam SY, Dragomir MP, Bayraktar R, Anfossi S, et al. Therapeutic potential of FLANC, a novel primate-specific long non-coding RNA in colorectal cancer. Gut (2020) 69(10):1818–31. doi: 10.1136/gutjnl-2019-318903
57. Shen S, Wang J, Zheng B, Tao Y, Li M, Wang Y, et al. LINC01714 enhances gemcitabine sensitivity by modulating FOXO3 phosphorylation in cholangiocarcinoma. Mol Ther Nucleic Acids (2020) 19:446–57. doi: 10.1016/j.omtn.2019.11.028
58. Niinuma T, Suzuki H, Nojima M, Nosho K, Yamamoto H, Takamaru H, et al. Upregulation of miR-196a and HOTAIR drive malignant character in gastrointestinal stromal tumors. Cancer Res (2012) 72(5):1126–36. doi: 10.1158/0008-5472.CAN-11-1803
59. Yan J, Chen D, Chen X, Sun X, Dong Q, Hu C, et al. Downregulation of lncRNA CCDC26 contributes to imatinib resistance in human gastrointestinal stromal tumors through IGF-1R upregulation. Braz J Med Biol Res (2019) 52(6):e8399. doi: 10.1590/1414-431x20198399
60. Cao K, Li M, Miao J, Lu X, Kang X, Zhu H, et al. CCDC26 knockdown enhances resistance of gastrointestinal stromal tumor cells to imatinib by interacting with c-KIT. Am J Trans Res (2018) 10(1):274–82.
61. Yan J, Chen D, Chen X, Sun X, Dong Q, Du Z, et al. Identification of imatinib-resistant long non-coding RNAs in gastrointestinal stromal tumors. Oncol Lett (2019) 17(2):2283–95. doi: 10.3892/ol.2018.9821
62. Shao Y, Lian S, Zheng J, Tong H, Wang J, Xu J, et al. RP11-616M22.7 recapitulates imatinib resistance in gastrointestinal stromal tumor. Mol Ther Nucleic Acids (2021) 25:264–76. doi: 10.1016/j.omtn.2021.05.017
63. Zhang J, Chen K, Tang Y, Luan X, Zheng X, Lu X, et al. LncRNA-HOTAIR activates autophagy and promotes the imatinib resistance of gastrointestinal stromal tumor cells through a mechanism involving the miR-130a/ATG2B pathway. Cell Death Dis (2021) 12(4):367. doi: 10.1038/s41419-021-03650-7
64. Hirano T, Ike F, Murata T, Obata Y, Utiyama H, Yokoyama KK. Genes encoded within 8q24 on the amplicon of a large extrachromosomal element are selectively repressed during the terminal differentiation of HL-60 cells. Mutat Res (2008) 640(1-2):97–106. doi: 10.1016/j.mrfmmm.2007.12.008
65. Li X, Liu H, Wang J, Qin J, Bai Z, Chi B, et al. Curcumol induces cell cycle arrest and apoptosis by inhibiting IGF-1R/PI3K/Akt signaling pathway in human nasopharyngeal carcinoma CNE-2 cells. Phytother Res (2018) 32(11):2214–25. doi: 10.1002/ptr.6158
66. Zhang Y, Yan N, Wang X, Chang Y, Wang Y. MiR-129-5p regulates cell proliferation and apoptosis via IGF-1R/Src/ERK/Egr-1 pathway in RA-fibroblast-like synoviocytes. Biosci Rep (2019) 39(12):BSR20192009. doi: 10.1042/BSR20192009
67. Yan X, Hu Z, Feng Y, Hu X, Yuan J, Zhao SD, et al. Comprehensive genomic characterization of long non-coding RNAs across human cancers. Cancer Cell (2015) 28(4):529–40. doi: 10.1016/j.ccell.2015.09.006
68. Zhang J, Cheng J, Zeng Z, Wang Y, Li X, Xie Q, et al. Comprehensive profiling of novel microRNA-9 targets and a tumor suppressor role of microRNA-9 via targeting IGF2BP1 in hepatocellular carcinoma. Oncotarget (2015) 6(39):42040–52. doi: 10.18632/oncotarget.5969
69. Subramanian S, Lui WO, Lee CH, Espinosa I, Nielsen TO, Heinrich MC, et al. MicroRNA expression signature of human sarcomas. Oncogene (2008) 27(14):2015–26. doi: 10.1038/sj.onc.1210836
70. Sarkar FH, Li Y, Wang Z, Kong D, Ali S. Implication of microRNAs in drug resistance for designing novel cancer therapy. Drug resist updates: Rev commentaries antimicrobial Anticancer chemother (2010) 13(3):57–66. doi: 10.1016/j.drup.2010.02.001
71. Almeida MI, Nicoloso MS, Zeng L, Ivan C, Spizzo R, Gafà R, et al. Strand-specific miR-28-5p and miR-28-3p have distinct effects in colorectal cancer cells. Gastroenterology (2012) 142(4):886–96.e9. doi: 10.1053/j.gastro.2011.12.047
72. Zheng Q, Chen C, Guan H, Kang W, Yu C. Prognostic role of microRNAs in human gastrointestinal cancer: A systematic review and meta-analysis. Oncotarget (2017) 8(28):46611–23. doi: 10.18632/oncotarget.16679
73. Akçakaya P, Caramuta S, Åhlen J, Ghaderi M, Berglund E, Östman A, et al. microRNA expression signatures of gastrointestinal stromal tumours: associations with imatinib resistance and patient outcome. Br J Cancer (2014) 111(11):2091–102. doi: 10.1038/bjc.2014.548
74. Amirnasr A, Gits CMM, van Kuijk PF, Smid M, Vriends ALM, Rutkowski P, et al. Molecular comparison of imatinib-naïve and resistant gastrointestinal stromal tumors: Differentially expressed microRNAs and mRNAs. Cancers (2019) 11(6):882. doi: 10.3390/cancers11060882
75. Zhang Z, Jiang NY, Guan RY, Zhu YK, Jiang FQ, Piao D. Identification of critical microRNAs in gastrointestinal stromal tumor patients treated with imatinib. Neoplasma (2018) 65(5):683–92. doi: 10.4149/neo_2018_170906N575
76. Kou Y, Yang R, Wang Q. Serum miR-518e-5p is a potential biomarker for secondary imatinib-resistant gastrointestinal stromal tumor. J Biosci (2018) 43(5):1015–23. doi: 10.1007/s12038-018-9805-y
77. Gupta A, Roy S, Lazar AJ, Wang WL, McAuliffe JC, Reynoso D, et al. Autophagy inhibition and antimalarials promote cell death in gastrointestinal stromal tumor (GIST). Proc Natl Acad Sci United States America (2010) 107(32):14333–8. doi: 10.1073/pnas.1000248107
78. Jiang B, Xue M, Xu D, Song J, Zhu S. Down-regulated lncRNA HOTAIR alleviates polycystic ovaries syndrome in rats by reducing expression of insulin-like growth factor 1 via microRNA-130a. J Cell Mol Med (2020) 24(1):451–64. doi: 10.1111/jcmm.14753
79. Chen W, Li Z, Liu H, Jiang S, Wang G, Sun L, et al. MicroRNA-30a targets BECLIN-1 to inactivate autophagy and sensitizes gastrointestinal stromal tumor cells to imatinib. Cell Death Dis (2020) 11(3):198. doi: 10.1038/s41419-020-2390-7
80. Jalali S, Bhartiya D, Lalwani MK, Sivasubbu S, Scaria V. Systematic transcriptome wide analysis of lncRNA-miRNA interactions. PloS One (2013) 8(2):e53823. doi: 10.1371/journal.pone.0053823
81. Awan FM, Naz A, Obaid A, Ikram A, Ali A, Ahmad J, et al. MicroRNA pharmacogenomics based integrated model of miR-17-92 cluster in sorafenib resistant HCC cells reveals a strategy to forestall drug resistance. Sci Rep (2017) 7(1):11448. doi: 10.1038/s41598-017-11943-1
82. Debiec-Rychter M, Cools J, Dumez H, Sciot R, Stul M, Mentens N, et al. Mechanisms of resistance to imatinib mesylate in gastrointestinal stromal tumors and activity of the PKC412 inhibitor against imatinib-resistant mutants. Gastroenterology (2005) 128(2):270–9. doi: 10.1053/j.gastro.2004.11.020
83. Tu Y, Zuo R, Ni N, Eilers G, Wu D, Pei Y, et al. Activated tyrosine kinases in gastrointestinal stromal tumor with loss of KIT oncoprotein expression. Cell Cycle (Georgetown Tex) (2018) 17(23):2577–92. doi: 10.1080/15384101.2018.1553335
84. Casimiro MC, Di Sante G, Di Rocco A, Loro E, Pupo C, Pestell TG, et al. Cyclin D1 restrains oncogene-induced autophagy by regulating the AMPK-LKB1 signaling axis. Cancer Res (2017) 77(13):3391–405. doi: 10.1158/0008-5472.CAN-16-0425
85. Ou WB, Ni N, Zuo R, Zhuang W, Zhu M, Kyriazoglou A, et al. Cyclin D1 is a mediator of gastrointestinal stromal tumor KIT-independence. Oncogene (2019) 38(39):6615–29. doi: 10.1038/s41388-019-0894-3
86. Otto T, Sicinski P. Cell cycle proteins as promising targets in cancer therapy. Nat Rev Cancer (2017) 17(2):93–115. doi: 10.1038/nrc.2016.138
87. Lu X, Pang Y, Cao H, Liu X, Tu L, Shen Y, et al. Integrated screens identify CDK1 as a therapeutic target in advanced gastrointestinal stromal tumors. Cancer Res (2021) 81(9):2481–94. doi: 10.1158/0008-5472.CAN-20-3580
88. Yan M, Wang C, He B, Yang M, Tong M, Long Z, et al. Aurora-a kinase: A potent oncogene and target for cancer therapy. Med Res Rev (2016) 36(6):1036–79. doi: 10.1002/med.21399
89. Cheng X, Wang J, Lu S, Fan W, Wang W. Aurora kinase a (AURKA) promotes the progression and imatinib resistance of advanced gastrointestinal stromal tumors. Cancer Cell Int (2021) 21(1):407. doi: 10.1186/s12935-021-02111-7
90. Fletcher JI, Williams RT, Henderson MJ, Norris MD, Haber M. ABC Transporters as mediators of drug resistance and contributors to cancer cell biology. Drug resist updates: Rev commentaries antimicrobial Anticancer chemother (2016) 26:1–9. doi: 10.1016/j.drup.2016.03.001
91. Widmer N, Rumpold H, Untergasser G, Fayet A, Buclin T, Decosterd LA. Resistance reversal by RNAi silencing of MDR1 in CML cells associated with increase in imatinib intracellular levels. Leukemia (2007) 21(7):1561–2. doi: 10.1038/sj.leu.2404671
92. Zhang Q, Li Z, Xu K, Qian Y, Chen M, Sun L, et al. Intracellular concentration and transporters in imatinib resistance of gastrointestinal stromal tumor. Scand J Gastroenterol (2019) 54(2):220–6. doi: 10.1080/00365521.2019.1577488
93. Xu K, Zhang Q, Chen M, Li B, Wang N, Li C, et al. N(6)-methyladenosine modification regulates imatinib resistance of gastrointestinal stromal tumor by enhancing the expression of multidrug transporter MRP1. Cancer Lett (2022) 530:85–99. doi: 10.1016/j.canlet.2022.01.008
94. Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, et al. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell (2015) 161(6):1388–99. doi: 10.1016/j.cell.2015.05.014
95. Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature (2014) 505(7481):117–20. doi: 10.1038/nature12730
96. Arcaro A. Targeting the insulin-like growth factor-1 receptor in human cancer. Front Pharmacol (2013) 4:30. doi: 10.3389/fphar.2013.00030
97. Menting JG, Whittaker J, Margetts MB, Whittaker LJ, Kong GK, Smith BJ, et al. How insulin engages its primary binding site on the insulin receptor. Nature (2013) 493(7431):241–5. doi: 10.1038/nature11781
98. Chen W, Kuang Y, Qiu HB, Cao Z, Tu Y, Sheng Q, et al. Dual targeting of insulin receptor and KIT in imatinib-resistant gastrointestinal stromal tumors. Cancer Res (2017) 77(18):5107–17. doi: 10.1158/0008-5472.CAN-17-0917
99. Serrano-Candelas E, Ainsua-Enrich E, Navinés-Ferrer A, Rodrigues P, García-Valverde A, Bazzocco S, et al. Silencing of adaptor protein SH3BP2 reduces KIT/PDGFRA receptors expression and impairs gastrointestinal stromal tumors growth. Mol Oncol (2018) 12(8):1383–97. doi: 10.1002/1878-0261.12332
100. Obata Y, Horikawa K, Takahashi T, Akieda Y, Tsujimoto M, Fletcher JA, et al. Oncogenic signaling by kit tyrosine kinase occurs selectively on the golgi apparatus in gastrointestinal stromal tumors. Oncogene (2017) 36(26):3661–72. doi: 10.1038/onc.2016.519
101. Obata Y, Horikawa K, Shiina I, Takahashi T, Murata T, Tasaki Y, et al. Oncogenic kit signalling on the golgi is suppressed by blocking secretory trafficking with m-COPA in gastrointestinal stromal tumours. Cancer Lett (2018) 415:1–10. doi: 10.1016/j.canlet.2017.11.032
102. Saito Y, Takahashi T, Obata Y, Nishida T, Ohkubo S, Nakagawa F, et al. TAS-116 inhibits oncogenic KIT signalling on the golgi in both imatinib-naïve and imatinib-resistant gastrointestinal stromal tumours. Br J Cancer (2020) 122(5):658–67. doi: 10.1038/s41416-019-0688-y
103. Banerjee S, Yoon H, Ting S, Tang CM, Yebra M, Wenzel AT, et al. KIT(low) cells mediate imatinib resistance in gastrointestinal stromal tumor. Mol Cancer Ther (2021) 20(10):2035–48. doi: 10.1158/1535-7163.MCT-20-0973
104. Bardsley MR, Horváth VJ, Asuzu DT, Lorincz A, Redelman D, Hayashi Y, et al. Kitlow stem cells cause resistance to kit/platelet-derived growth factor alpha inhibitors in murine gastrointestinal stromal tumors. Gastroenterology (2010) 139(3):942–52. doi: 10.1053/j.gastro.2010.05.083
Keywords: gastrointestinal stromal tumor, imatinib, secondary imatinib resistance, drug-resistance mechanism, therapeutic targets
Citation: Hu X, Wang Z, Su P, Zhang Q and Kou Y (2022) Advances in the research of the mechanism of secondary resistance to imatinib in gastrointestinal stromal tumors. Front. Oncol. 12:933248. doi: 10.3389/fonc.2022.933248
Received: 30 April 2022; Accepted: 18 August 2022;
Published: 06 September 2022.
Edited by:
Emilio Francesco Giunta, Università degli Studi della Campania Luigi Vanvitelli, ItalyReviewed by:
Alessandra Merlini, University of Turin, ItalyEmanuela Bostjancic, University of Ljubljana, Slovenia
Copyright © 2022 Hu, Wang, Su, Zhang and Kou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Youwei Kou, a291eXdAc2otaG9zcGl0YWwub3Jn
†These authors share first authorship