 Chiara Agnoletto
Chiara Agnoletto Stefano Volinia
Stefano Volinia- 1Rete Oncologica Veneta (ROV), Veneto Institute of Oncology IOV-IRCCS, Padua, Italy
- 2Laboratorio per le Tecnologie delle Terapie Avanzate (LTTA), Department of Translational Medicine, University of Ferrara, Ferrara, Italy
- 3Biological and Chemical Research Centre (CNBCh UW), University of Warsaw, Warsaw, Poland
- 4Center of New Technologies, University of Warsaw, Warsaw, Poland
Circulating tumor cells (CTCs) represent a subset of heterogeneous cells, which, once released from a tumor site, have the potential to give rise to metastasis in secondary sites. Recent research focused on the attempt to detect and characterize these rare cells in the circulation, and advancements in defining their molecular profile have been reported in diverse tumor species, with potential implications for clinical applications. Of note, metabolic alterations, involving mitochondria, have been implicated in the metastatic process, as key determinants in the transition of tumor cells to a mesenchymal or stemness-like phenotype, in drug resistance, and in induction of apoptosis. This review aimed to briefly analyse the most recent knowledge relative to mitochondria dysfunction in CTCs, and to envision implications of altered mitochondria in CTCs for a potential utility in clinics.
Liquid biopsy for tumor diagnostics
Cancer metastasis occurs through a series of sequential steps, which include epithelial mesenchymal transition (EMT) of primary tumor cells into tumor-initiating cells (TICs) and their intravasation into the bloodstream as circulating tumor cells (1), and subsequent extravasation at distant sites, with generation of metastasis upon mesenchymal epithelial transition (MET) (2–4). CTCs have been detected in the majority of patients with tumors, they have been proven to be heterogeneous and a subgroup of them represents cancer stem-like cells (CSLCs) or TICs (5, 6).
Recent progress in the identification of cancer biomarkers opened a new field of cancer diagnostics (7). Due to the non-invasive nature of liquid biopsy, repeated sampling and testing of blood have been performed for the accurate early disease detection and monitoring of treatment responses (8). In clinics, enumeration and phenotyping of CTCs have been proven as novel biomarkers to estimate the risk for metastatic relapse or disease progression in various tumors (9). CTCs have been detected in 30-50% of patients with metastatic colon, prostate and/or breast cancer (9–11). High CTC levels, as enumerated with the CellSearch™ assay, which specifically detects tumour cells with epithelial phenotype, defined as 4’,6-diamidino-2-phenylindole (DAPI)+, CK+, CD45- cells, are associated with poor clinical outcome, i.e. shorter progression free survival (PFS) and overall survival (OS), in breast, colorectal, and prostate cancers (3, 9, 10, 12–18). Advanced cancer patients with high CTC counts even after systemic therapy have poor clinical outcome, and elevation of CTC levels during follow-up predicts a high risk of progression (10, 19–21). The prognostic potential of CTCs for monitoring metastasis or the efficacy of chemotherapy has been reported: in metastatic breast cancer, persistently elevated CTC levels after one cycle of treatment correlates with a poor prognosis (9), while a reduction in CTC levels indicates improved prognosis (22–24).
Further, detection of CTCs might represent an alternative approach for early diagnosis (25). Technical advances have assessed the feasibility of detecting and profiling CTCs at the very early steps of tumor invasion (26). Indeed, CTCs have been detected in 10% of CRC precancers (adenomas) and in 3% of patients with chronic obstructive pulmonary disease (COPD), who have an elevated risk of developing lung cancer 1–4 years before CT screening of lung nodules (27, 28). In this contest, tissue-specific transcriptome profiling of single CTCs might address in verifying the location of the occult lesion, to select the appropriate imaging or diagnostic methodology (29).
As an alternative liquid biopsy, cell-free (cf) circulating DNA is continuously released by clonal tumor cells into the circulation (25). Alterations in cfDNA can be identified through ultra-deep NGS even at very low frequency (<1%), and have been used for early detection of recurrence in several tumors (e.g. colorectal cancer, pancreatic cancer, neuroblastoma) and, recently, also of premalignant lung and bladder disorders (27, 30, 31). At present, different platform technologies provide sufficient sensitivity in circulating tumor (ct) DNA detection to identify lung tumor patients relapsing within a year of subclonal detection, and for precision screening in cervical premalignancy (31, 32). However, methodologies require to be improved for detection and profiling of ctDNA, and measurements need higher sensitivity and specificity, by defining quantitative thresholds to avoid overdiagnosis. A relevant biological limitation is the amount of ctDNA recovered from early-stage cancer patients, even if less than 0.1% of ctDNA in plasma has been detected by digital droplet PCR or NGS methods (33). Enrichment steps have to be performed, based on biological properties or physics. Confounding results due to cancer-associated mutations not restricted to tumor patients and the presence of clonal alterations in blood cells due to aging and clonal hematopoiesis both represent critical issues (34, 35). Genomic driver alterations of tumors in BRAF, RAS, EGFR, HER2, FGFR3, PIK3CA, TP53, CDKN2A, and NF1/2 genes can also be identified in non tumor specimens. Additionally, determining the tissue of origin of the neoplastic lesion can be extremely challenging (27). Thus, limitations for clinical utility of ctDNA analysis are evident.
Mitochondria dysfunction in cancer: minimal integrity point
An altered metabolism is a distinct feature of tumorigenesis, and the metabolic profile in tumour cells represents a crucial step for their survival. Tumor cells depend strongly on both enhanced glycolysis, the pentose phosphate pathway, and glutaminolysis, as a result of dysfunctional mitochondria, to efficiently respond to energetics requirements (36–39). Yet, tumour cells require a minimal functional mitochondrial pool, in order to produce sufficient amount of biosynthetic precursors (40–42).
Mitochondria act at multiple levels and coordinate several biological processes, with generation of reactive oxygen species (ROS), release of oncoproteins and oncometabolites, modulation of calcium homeostasis and autophagic processes, cell death, and metabolism (43, 44). Mitochondria integrity is a central checkpoint for cancer cells (36), as they actively participate in plasticity of tumour cells and act on several mechanisms to address environmental conditions.
Despite the mitochondrion constitutes a key actor, the dualities of its function in tumour metastasis and therapy resistance have only recently been depicted, with opposite effects on both processes. Through the analysis of large-scale data sets from The Cancer Genome Atlas (TCGA), the underpinning genetic determinants of these changes have been identified, which are orchestrated by oncogenes and tumor suppressors (45). Further, tumors share a subset of metabolic gene signatures independent of their tissue of origin, and upregulate genes that encode for glycolysis and nucleotide biosynthesis enzymes, with important implications for cancer diagnosis and patient stratification (46, 47). Experimental data suggest that mitochondrial dysfunction can reach a threshold where it turns to opposite effects for tumour cells, and a fine regulation of mitochondrial function is required to drive tumorigenesis. Coherently recent evidence indicates that the metabolic phenotype of cancer varies at different disease stages, and contribute to tumor progression (48, 49). Thus, stage-specific metabolic traits have been identified in prostate, breast, renal, and lung tumors (50–53), and transcriptional analysis of 21 tumor types collected by the TCGA confirmed the strict association of inhibition in genes of mitochondrial metabolism with the presence of an EMT gene signature, which is linked to tumor initiation, invasion, and metastasis, and poor clinical outcome (54). In accordance, complementary studies have confirmed that mutations of enzymes from the TCA cycle, SDH and FH, are linked to EMT and invasive phenotype in pheochromocytoma and paraganglioma and renal cancer (55, 56). Similarly, decreased mtDNA content, which is associated with bioenergetics defects, linked with poor patient prognosis in several tumours (57).
Yet, defects in mitochondrial function have been reported to reduce tumour aggressiveness, as demonstrated in renal oncocytoma, with an aberrant accumulation of dysfunctional mitochondria inhibiting the autophagic machinery (58). Consistent, inhibition of autophagy leads to mitochondrial dysfunction and reprograms tumor fate toward benign neoplasms (59).
The hypothesis is that the reduction in mitochondrial function could be progressively advantageous for tumour cells until a ‘minimal integrity point’, below which this alteration becomes deleteriuos.
The determinants of the metabolic adaptations during dissemination and metastasis are only partially defined. Overall, cancer cells that detach from the primary tumor experience oxidative stress, and thus activate mitochondrial antioxidant networks to eventually metastasize. Coherently, in human specimens of prostate cancer the peroxisome proliferator- activated receptor gamma coactivator 1 alpha (PGC1α), which is the master transcriptional regulator of mitochondrial oxidative metabolism, is downmodulated (50). The role of mitochondrial dysfunction in promoting metastasis is further confirmed by a partial inhibition of mitochondrial respiratory chain due to rotenone, with induction of cell migration and clonogenicity in vitro and lung metastasis in vivo (60). Finally, mtDNA mutations affecting complex I support breast cancer metastasis in vivo via deregulation of NAD+/NADH and activation of autophagy (61).
Despite these consistent lines of evidence, an increased mitochondrial oxidative phosphorylation genes function was detected in CTCs from orthotopically implanted breast cancer mice, while in distant metastases expression of PGC1α was increased, suggesting that tumor-specific reprogramming might occur during metastasis, thus reconciling apparent discrepant data from literature (62). Of note, differential use of pyruvate in the mitochondria has been recently demonstrated to dictate the site of metastasis in breast cancer (63, 64).
Last, several results support a role for metabolic adaptation specifically mediated by activated mitochondrial function as a key determinant of therapy resistance (48). As an explicative example, resistance to mitogen-activated protein kinase (MAPK) inhibitors in BRafV600-driven melanoma is associated with increased mtDNA content and oxidative phosphorylation (65), while inhibition of BRafV600 induces an oxidative phosphorylation switch activated by PGC1α (66, 67), enhancing the detoxification capacities of these cell.
To date, numerous drugs have been proposed to modulate different functions of mitochondria for tumour therapy, which have been reviewed elsewhere (68, 69). Briefly, these strategies aim to compensate alterations in all relevant mitochondrial activities, i.e. bioenergetics, signaling, and biosynthetic functions (68, 69). At present, most drugs have been tested for antitumour activity in clinical trials (68). Moreover, the strict interconnection between mitochondrial metabolism and core cellular checkpoints reveals the potential of targeting mitochondrial activity in combination therapies (48). However, the variety of therapeutic targets and the ability of cells to adapt and compensate need to be considered. Mitochondrial metabolism is heterogeneous within and between tumors (70), as evidenced in the diverse responsiveness to antiangiogenic therapies (71–73), suggesting that the efficacy of anticancer therapy may depend on the adaptive metabolic capacity of tumour cells. Furthermore, cancer-initiating and therapy-resistant cells present a more oxidative metabolic program, thus the emergence of therapy-resistant cancer clones could rely on the newly acquired metabolic state, while this metabolic plasticity can be therapeutically exploited through the combination of standard and antimetabolic therapies (48).
In this article, our objective is to review the most recent evidence in support of a role for mitochondria dysfunction in circulating tumor cells behaviour, with the attempt to eventually reconcile apparent discordant results, and envision their implications for a potential utility in clinics.
Potential impact of mtDNA mutations in CTCs
Mitochondrial DNA (mtDNA), with its mutations and polymorphisms, has only recently acquired novel attention in tumor research. Yet, mitochondrial genetics in cancer has been neglected for a long time. Only recent large-scale sequencing efforts and clinical studies have highlighted the prevalence of mutations in mtDNA and their potential roles in tumorigenesis (74, 75). Human mtDNA is maternally inherited, with several mtDNA copies per mitochondrion and hundreds of mitochondria per cell, and encodes 37 genes, which include 22 transfer RNAs, 2 ribosomal RNAs and 13 protein subunits of the electron transport chain (ETC) complexes and ATP synthase (mtOXPHOS proteins) (75, 76). mtDNA is highly polymorphic due to a mutation rate an order of magnitude higher than the nuclear genome. Further, functional variants can be beneficial or deleterious depending on the context. A subset of mtDNA variants have been reported to cause minimal adaptive changes in OXPHOS, with modulation of multiple mitochondrial functions including stress, autophagy, and oncogenic responses to environment (77–79). Moreover, mtDNA can also influence the inflammasome, innate immunity, IL-1b and NFkB inflammatory pathways, and T-cell immune surveillance (79, 80).
Several examples in literature reported that a nearly total loss of mtDNA copy number in vitro and in vivo results in subtle or temporally delayed effects on mitochondrial function (81). mtDNA is subjected to the phenomenon of heteroplasmy, i.e. the existence of diverse subsets of mtDNA molecules into a given cell, due to the multi-copy nature of mtDNA. Mitochondrial DNA heterogeneity occurs frequently and is an important concept for the development of mitochondrial dysfunction (82). The availability of great datasets, such as the International Cancer Genome Consortium (ICGC) and the TCGA, demonstrated that ~60% of all solid tumors present at least one mtDNA mutation (83, 84). A great majority of mutations are at high levels of heteroplasmy, with a minority of tumours achieving near-complete mutation homoplasmy, thus indicating that dysregulation of mitochondrial function via mtDNA mutation is a feature of tumour. Also, in general, oncocytic tumours with high heteroplasmy of mtDNA mutations, and significant mitochondrial dysfunction, are benign, non-aggressive, low proliferating lesions (85). Recent clinical and genetic studies pointed to mtDNA mutations as potential drivers or phenotypic modifiers of prostate and thyroid cancers (86, 87), yet a definitive experimental evidence of mtDNA mutations as a key driver event in tumorigenesis is lacking.
The heteroplasmic mtDNA genotype is continuously remodelled during successive cytokinesis, thus several genotypes with diverse oncogenic potential are generated among tissues within the same individual over time (77).
The effect of mtDNA haplotype in tumor predisposition and development has only recently been confirmed, as discussed in details in a recent review on the importance of mtDNA alterations to drive precision prevention trials (88–91). Inherited missense alterations, potentially extremely deleterious, in mtDNA genes, such as ND6 (NADH dehydrogenase subunit 6) and COI (cytochrome oxidase subunit I), which code for subunits of OXPHOS complexes I and IV, have been associated with risks of tumors (77, 78, 92–95). Such alterations in mtDNA are heteroplasmic and frequently lethal if exceeding a biochemical threshold, depending on several criteria, among which the type of tissue (93–96). In mitosis milder mtDNA polymorphisms can shift to become predominantly enriched within individual cells, potentially contributing to neoplastic transformation. The importance of this phenomenon for cancer predisposition has been demonstrated as the mtDNA complex I ND5 m.12425delA frameshift mutation, inherited as a germline mutation and transmitted at lower heteroplasmy levels (5–10% mutant), shifted to homoplasmic mutation exclusively in nasopharyngeal tumor cells and correlated with lack of the ND6 subunit (94). Genetic or pharmacologic (metformin) disruption of mitochondrial respiration increased autophagy and prevented cancer development in a mouse model of Li-Fraumeni syndrome. On the other hand, in a pilot study of Li-Fraumeni patients, metformin decreased mitochondrial activity while activating a cell-signaling event which is known to lead to rhabdomyosarcoma development (97). Of great relevance for clinical applications, nuclear DNA germline mutations influence mitochondrial genomic instability for cancer predisposition, as described e.g. for the nuclear genes BRCA1, SUV3, SOD (36, 74, 88, 98). Accumulating evidence suggests that mtDNA mutations may also contribute to cancer cell development, tissue invasion and metastasis. Indeed mtDNA variations, such as deletions, point mutations and copy number differences, are associated with several cancer types (99). In breast cancer, a compromised mitochondrial function, due to mtDNA mutations and low mtDNA copy number, has been associated with increased metastasis and poor prognosis (78, 100); also low mtDNA copy number promotes metastasis by inducing EMT via mitochondrial retrograde signaling (101). In addition, cells with compromised mitochondrial integrity rapidly progress to malignancy (74, 99), and clonal expansion of mutant mtDNA species was reported in 27–80% (average 54%) of malignant tumor samples (102).
Despite this evidence, it has been reported that mitochondria of tumor cells are functional and perform oxidative phosphorylation. This concept further supports the notion of a minimal integrity point determining the relevance of a defective mitochondrial function on tumorigenesis. As a proof, targeted depletion of mitochondrial DNA can reduce tumorigenic potential in vivo (36). A recent paper demonstrated the effects of complete mitochondrial DNA deletion on the ability of tumors to metastasize in vivo (40). Normal cells contain both discrete and networked mitochondria each with multiple mtDNA copies. Melanoma and breast carcinoma cells completely deprived of mtDNA, named ρ0 cells, upon injection intravenously in syngeneic murine models, have delayed tumor growth (40, 41). Of note, cells derived from primary tumors originating from ρ0 cells, and their circulating and metastatic counterparts, acquired a partial mitochondrial network, and progressively recovered a full respiratory function; this effect was associated with stepwise assembly of mitochondrial electron transport chain complexes and correlated with tumorigenicity (40). The acquisition of a full mitochondrial competence is dependent from horizontal mtDNA transfer, consistent with previous in vitro results (103, 104). A crucial step of full respiration recovery is associated with the assembly of the respirasome and ETC complex II (CII), in accordance with the requirement for efficient OXPHOS in metastatic dissemination (62). Consistent with these observations, autophagy is activated in CTCs when respiration is partially restored, in order to eliminate dysfunctional mitochondria. In accordance, higher levels of TFAM, a critical factor for replication, transcription, and packaging of mtDNA (105) and OPA1 (106) have been observed, while mitochondria-to-nucleus retrograde signaling eventually restores both mtDNA distribution and respiratory function (40). A minimum level of mtDNA damage is needed to initiate intercellular transfer of functional mitochondria. An independent confirmation has been reported in human glioblastoma cells (107). Thus, tumor cells deprived of mtDNA can acquire mtDNA of host origin, resulting in stepwise recovery of respiration from primary to metastatic tumor cells, with the crucial role of the complete assembly of the respirasome and CII (40).
As previously outlined, polymorphic sites are distributed along the complete mitochondrial genome. The genetic diversity of mtDNA in blood is strongly associated with tumor and may serve as a diagnostic marker. The small size (16,569 bp) of mtDNA is especially suitable for the accurate assessment of such profiles and the association of genetic heterogeneity, rather than specific mutations, with cancer, together with its clonal expansion, high copy number and high mutation rate (77). In a recent study, a significant depletion of mtDNA has been reported for several types of tumors, such as bladder, breast, kidney, and liver cancer (57), thus the identification of specific mutant variants in tested blood is quite difficult. Assessment of heterogeneity profiles of intra-host mtDNA variants from blood has been proven to overcome the identification of specific mutations for diagnostic detection of HCC (108). Consensus sequences of mtDNA differ between tumor and blood from ~ 58% of patients, while most tumor-specific variants (99.4%) were present in less than 5% of HCC patient, limiting their use as general cancer markers (108). In contrast, accurate estimation of heterogeneity can be performed at a moderate sequencing depth, thus providing a more reliable source of cancer-specific markers (108). Thus, a strong genetic signal consisting in a intra-host mtDNA profile has been documented, despite the presence of tumor-specific mutant mtDNA species at a very low concentration in plasma. However, at present the strict HCC specificity of the classifier has not been confirmed (108).
Effect of dysfunctional mitochondria in CTCs
The deregulation of cellular energetics is a hallmark of tumor cells, with enhanced glycolysis, pentose phosphate pathway, and glutaminolysis, as a result of altered mitochondrial function (37, 38). In order to dissect the metastasis-related deregulation of metabolic genes in CTCs, a panel of genes have been analyzed in prostate cancer cell lines with different metastatic capacities (109). Eight metabolic genes were differentially expressed in metastatic cell lines, HK2, PDP2, G6PD, PGK1, PHKA1, PYGL, PDK1, and PKM2 (109), with a confirmed and remarkable association between their functions and the metastatic capacity of tumor cells (110–112). Of clinical relevance, the identified genes were detected in the CTCs of 54 clinical samples. Of note, two key enzymes of glycolysis and the pentose phosphate pathway, respectively, PGK1 and G6PD, were determined as efficacious markers for CTCs metabolic analysis (109). Further, PGK1/G6PD-marked hypermetabolic CTCs (GM+CTCs, i.e. DAPI+CD45−PGK1/G6PD+ cells) potentially represent a more accurate marker than EMT-CTCs for the diagnosis of metastasis in prostate cancer patients (109). Indeed, increased GM+CTCs level was associated with advanced tumor stage and metastasis (P < 0.05), and presented higher AUCs of the ROC curve (0.780) in the discrimination of metastatic patients than the EMT CTCs subtypes (E-CTCs 0.729, H-CTCs 0.741, and M-CTCs 0.648) (109).
In breast tumor patients CTC exhibited enhanced mitochondria biogenesis and respiration, with higher expression levels of genes associated with mitochondrial biogenesis (PGC-1α, PGC-1β, NRF1, and ERRα) and oxidative phosphorylation (Cox5b, Cox4i, ATPsynth, CytC) (62). CTCs were largely quiescent and specifically upregulated PGC-1α, and presented a more aerobic metabolism compared to both primary and metastatic tumors (62). These effects were proven to be mediated by PGC-1α (62). Of clinical relevance, high PGC-1α expression was detected in over 80% of CTC from IDC patients with lung metastases, confirming its association with distant metastasis and poor outcome (62). Thus, some invasive and migratory properties of tumor cells are dependent on mitochondrial respiration and PGC-1α is a potential target for therapeutic intervention. A dynamic shifts in the metabolic program of tumor cells facilitates diverse steps in cancer progression and metastasis, and mitochondrial biogenesis and respiration induced by PGC-1α is essential for functional motility of cancer cells and metastasis (62). The reversible shift in patterns of metabolic gene expression is synergistically coupled with genes frequently associated with EMT and acquisition of enhanced migratory and invasive properties of tumor cells (62). Several reports proved the mutual regulation of metabolic genes by EMT and vice versa, through both in vivo and in vitro experiments, thus synergistically promoting cancer metastasis (113–115). Altering mitochondrial function also determines survival and acquisition of cancer stem cell properties, in part via retrograde mitochondria-nucleus signaling (116). Mitochondrial activity and ROS detoxification are critical for cancer cell viability (117), and ensure cancer cell survival detaching from basement membrane (118). Further, E-cadherin expression is an important determinant of metastatic potential in metastatic lung nodules and CTCs in breast cancer (119), consistent with the loss of its expression due to EMT occurring frequently during tumor metastasis. E-cadherin activation inhibits metastasis at multiple stages, including the accumulation of CTCs from the primary tumor and the extravasation of tumor cells from the vasculature (119). Activating mAbs increased the frequency of apoptosis in CTCs and tumor cells in metastatic nodules, through upmodulation of Bax mRNA expression, and downmodulation of Bcl-xL mRNA expression (119). Overall these data reconcile with the notion that tumor-specific reprogramming might occur during sequential stage in tumorigenesis and in the metastatic cascade, as outlined previously in the manuscript (47, 48).
In a recent paper, testing the presence of CTC in peripheral blood of patients with renal cell carcinoma (RCC) undergoing surgery, authors reported a difference of the mitochondrial network between CTCs and basophils, monocytes and neutrophils, as evaluated by mitochondria staining, was observed (120). RCC is a highly invasive tumor, and patients respond poorly to chemotherapy, even in combination with immunotherapy (121, 122). Further, early detection of RCC remains a significant challenge. Predictive markers of response still lack in clinical practice. Testing CTCs profiles by gene expression analysis of the targetable genes may improve RCC therapy outcomes. Multiregional sequencing of RCC tumors and metastatic tissues evidenced the high intra-tumoral heterogeneity with respect to adjacent normal kidney tissue (123). The frequent lack of epithelial antigens and concomitant EMT in RCC tumor cells (124) often compromise CTC capture. The size-based isolation of CTCs by using the ISET (isolation by size of epithelial tumor cells) filtration method in combination with mitochondria staining allows to differentiate non-hematopoietic cells in the peripheral blood and define CTC subgroups possibly associated with metastatic potential, confirming that an altered mitochondrial network is relevant in determining the metastatic phenotype of CTCs (120). A relevant drawback in this study is the lack of data related to differences in mitochondrial network observed between CTCs and leukocytes, if related to mitochondrial volume or structure, and, especially, if a correlation with specific CTC subsets was conceivable.
ROS function in circulation-related stresses in CTCs
Epithelial–mesenchymal transitioned CTCs enter into the vasculature, due to loose mosaic vessels and remodelling of extracellular matrix (ECM) (125, 126). Intravascularly, these metastasis-initiating CTCs need to maintain survival under anoikis, immune attack, and severe shear stress. Although the great majority of CTCs die into the circulation (127), 0.1% of CTCs survive as disseminated cells and eventually relapse (128). Into the circulation tumor cells respond to mechanical forces, and the role of the fluid microenvironment in metastasis has been recently proven, both as interstitial flow (~ 0.1 dyn/cm2), blood (1–30 dyn/cm2)/lymphatic circulation (~ 0.64 dyn/cm2), and target organ-specific fluid microenvironments (129). Of clinical relevance, the interstitial flow determines the direction of tumor cell metastasis to specific organs (130). One of the most crucial mechanical forces is generated by liquid flowing on the cell surface, i.e. the laminar shear stress (LSS) (129, 131). The laminar shear stress regulates the survival and function of normal cells, such as endothelial cells, osteoblasts, and circulating hematopoietic embryonic stem cells (132–135), and also promotes metastatic potential and anoikis resistance in breast CTCs (136, 137).
In vivo and in vitro evidence has proven that conversion from epithelial tumor cells into CSLCs/TICs can occur within blood vessels, due exclusively to hydrodynamic shear stress experienced during systemic circulation, without additional requirement for growth factors or a hypoxic stromal niche (138). A recent study has demonstrated that the fluid shear stress (0.05 dyne/cm2) in the interstitium can promote cancer motility through modulating the Yes-associated protein (YAP1)-related ROCK-LIMK-cofilin signaling pathway (139).
Adhesion to the ECM helps to maintain normal tissue architecture, and loss of anchorage activate a programmed cell death termed ‘anoikis’ (140). Anoikis is due to cell detachment from the ECM and prevents anchorage-independent tumor cell growth. CTCs need to acquire resistance to anoikis to survive in the circulatory system, where cells encounter the fluid shear stress. Resistance to anoikis is a hallmark of malignant tumor cells, and both the dynamic ECM network (141, 142) and hypoxic conditions (143) have been proven to promote anoikis resistance and increase survival in epithelial and carcinoma cells. Dissecting the molecular mechanisms that protect tumor cells from undergoing anoikis is critical, and novel strategies to target CTCs within the circulation could reduce their metastatic potential. Cancer cells acquire anoikis resistance via several mechanisms and signaling molecules including phosphoinositide 3‐kinase (PI3K)– protein kinase B (Akt) (144), Ras–extracellular signal regulated kinases (ERK) (145), Jun‐ N‐terminal kinase (146), mitogen‐activated protein–extracellular signal‐regulated kinase (147), and integrins (148). In addition, tumor microenvironments can also contribute to anoikis resistance in cancer cells by altering matrix rigidity, increasing oxidative stress, and depriving cells of adequate oxygen supply (149–151). An increased ROS level has been reported in CTCs (118). ROS have been confirmed to be crucial regulators of cell adhesion (8), and attachment of CTCs to the lining of the microvasculature is a crucial step for cancer cell extravasation and metastasis generation (152, 153). A high ROS level is associated with enhanced invasiveness and metastasis in hepatocellular carcinoma (HCC) (154). In pancreatic cancer and melanoma, acquisition of anoikis resistance protects cells from apoptosis, and promotes cell invasion and metastatic potential through the phosphorylation of STAT3 at Tyr705 (155). Anoikis is a highly complex multistep process, and both acquisition of apoptosis resistance and autophagy promote epithelial cell survival during anoikis (156).
Signaling activated by EMT‐related transcription factors constitutively activate specific signals in metastasis, including evasion of anoikis, with enhanced stemness and clonogenic features of cancer cells (157). The importance of EMT in metastasis is doubtful and recently discussed in several studies (158–160). Even the most mesenchymal states are not irreversibly committed (161), and a lot of studies support the notion that the metastatic potential greatly correlates with an intermediate EMT state (54, 125, 162–164). Yet, distinct hybrid phenotypes states determine the invasive, metastatic, and differentiation characteristics of tumor cells, with implications in tumor heterogeneity, invasion, metastasis, and resistance to therapy. Fluid shear stress experienced in systemic circulation can lead to specific acquisition of MSC-like potential in breast tumor CTCs, that promotes EMT, and acquisition of CSLCs/TIC potential (138). Such effects were proven to be mediated by promoting conversion of CD24middle/CD44high/CD133middle/CXCR4low/ALDH1low primary patient epithelial tumor cells into CD24low/CD44low/CD133high/CXCR4high/ALDH1high cancer stem-like cells (CSLCs), with plasticity and self-renewal capacity (138). This activation is dependent on ROS/NO generation, and suppression of extracellular signal-related kinase (ERK)/glycogen synthase kinase (GSK) 3β, an analogous mechanism operating in embryonic stem cells to prevent their differentiation while promoting self-renewal (138). Briefly, activated stress-responsive signaling pathways induces the transition from tumor cells to more highly invasive TIC (138).
Tumor cells detached from the ECM, upon invasion through the basement membrane, and entering into the circulatory system, encounter blood flow‐induced low shear stress (LSS; 2 dyn/cm2), which induces expression of Caveolin‐1 (Cav‐1), a 22‐kDa integral membrane protein. Cav‐1 has been proven to induce breast cancer cell motility, invadopodia formation, and metastasis via the PI3K–Akt–mechanistic target of rapamycin signaling pathway (141). In addition, LSS protects breast tumor cells from anoikis under anchorage‐ independent conditions via a Cav‐1‐ dependent signaling pathway, by inhibiting Cav‐1‐dependent extrinsic and intrinsic apoptotic crosstalk signaling (136). Indeed, LSS‐induced dissociation of Cav‐1–Fas inhibited the generation of the death‐inducing signaling complex, caspase‐8 activation, with inhibition of the extrinsic apoptosis signaling pathway (136). Likewise, LSS blocked the mitochondrial pathway through promotion of integrin β1–focal adhesion kinase‐mediated multicellular aggregation, suppression of truncated BID translocation, inactivation of caspase‐8 and mediated crosstalk between the extrinsic and intrinsic apoptotic pathways, which in turn inactivate Bcl‐2 and Bcl‐xL, thus preventing mitochondrial membrane permeabilization through Bax oligomerization (136). Accordingly, depletion of Cav‐1 restored sensitivity to anoikis. Also, upon LSS a significant decrease in Beclin‐1 has been observed, thus autophagy might be another regulator of LSS‐induced anoikis resistance (136). These data underline a novel role for flow‐induced shear stress in the regulation of anoikis in neoplastic cells, indicating that LSS‐induced anoikis resistance is a critical mechanism that increases tumor malignancy (136). In human lung carcinoma cells, ROS prevent Cav‐1 ubiquitination and degeneration (149). Also, a recent study demonstrated that Cav‐1 is involved in anoikis resistance in human lung cancer cells through regulation of myeloid cell leukemia 1 (Mcl‐1) by interacting with Mcl‐1 and preventing it from degradation (165). Overall, these results may lead to potential therapeutic strategies targeting Cav-1 and modulating the tumor microenvironment (136).
The effect of cyclic laminar shear stress (LSS) has been recently studied in vitro on CTCs of colorectal tumor (CRC) (166). Suspended tumor cells with a CK8+/CD45−/DAPI+ phenotype actively responded to LSS by activating the expression of atonal bHLH transcription factor 8 (ATOH8), a fluid mechanosensor, with key roles in intravascular survival and metabolism plasticity (166). Molecules, with the capability of sensing and translating mechanical forces, can transform physical stimulation into biological signals (167). As a new LSS-response molecule, ATOH8 is induced by 10 dyn/cm2 LSS in endothelial cells, and is also involved in angiogenesis, skeletal muscle formation, and embryonic development (168–170). ATOH8 expression among tumors is heterogeneous, and its role as a tumor suppressor or tumor promoter is still controversial. ATOH8 could inhibit stem cell features of hepatocellular carcinoma cells (171) and malignant phenotypes of nasopharyngeal carcinoma (172), while promoting cell proliferation and inhibiting apoptosis in CRC cells (173). In CRC it exerts a tumor promoting effect and is associated with hematogenous metastasis and poor prognosis in patients (166). Specifically, ATOH8 was upregulated in CTCs via activation of VEGFR2/AKT signalling pathway mediated by LSS induced VEGF release in vitro and in vivo (166). ATOH8 transcriptionally activated HK2-mediated glycolysis and inhibited cell death pathway in CTCs, thus mediating the intravascular survival of colorectal tumor cells in the circulation, and ultimately providing a novel potential target for the prevention and treatment of hematogenous metastasis in CRC (166, 174) Metabolism and cell survival are inextricably linked, and cancer cells can switch between different metabolic states to respond to adverse conditions such as metabolic stress, anoikis, and mechanical stress (131, 175). ATOH8 overexpression could promote CRC CTCs migration, invasion, anoikis resistance, facilitating CTC survival (166). HK2 is one of the key enzymes of glycolysis, participating in the regulation of cancer cell metabolism and death, and its overexpression is significantly positively correlated with CRC recurrence (176). HK2 can support cell survival via promoting glycolysis and reducing overabundant ROS or generating HK2-VDAC complex with inhibition of mitochondria-mediated apoptosis. As expected, in ATOH8-overexpressing CRC cells ROS level were down-regulated while mitochondrial HK2 was up-regulated (166). Briefly, a VEGF-VEGFR2-AKT signal axis in CRC m-CTCs contributes to the high expression of ATOH8 and ultimately promotes CTC survival in the fluid microenvironment upon LSS exposure (166).
Mitochondria and drug resistance in CTCs
CTCs may respond differently to chemotherapies compared to primary tumor cells. CTCs isolated from advanced breast cancer patients are more resistant to the DNA-damaging and pro-apoptotic effects of chemotherapy than tumor cells attached to the ECM (177). Of note, the response of CTCs to chemotherapy has a prognostic significance. Apoptosis in CTCs correlates with systemic chemotherapeutic response and disease progression upon therapy (177).
Chemotherapy activates the intrinsic pathway of apoptosis (178, 179). The anti-apoptotic Bcl-2 proteins are expressed on the mitochondrial membrane and prevent apoptotic cell death upon directly binding to pro-death Bax and Bak. ROS both induce the mitochondrial anti-apoptotic proteins via activation of the transcription factors NF-kB, Nrf-2, and HIF-1a (180, 181), and reduce the expression of pro-apoptotic proteins via the ERK/MAPK and PI3K/Akt pathways (182). As a proof of evidence for therapeutic strategies, caspase 3 activation and Bcl-xL depletion are correlated with a decreased number of CTCs and metastasis (119).The interactions of certain Bcl-2 proteins occur at the BH3 domains (178), and BH3 profiling measures the relative interactions of pro- and anti-apoptotic proteins to determine whether a tumor cell is near the threshold to activate apoptosis through mitochondrial outer membrane permeabilization (178, 179). Coherently, BH3 profiling acts as a metabolic signature and can predict response to chemotherapy and resistance to targeted therapy (183, 184). For example, a tumor with high level of functional Bcl-xL, an anti-apoptotic protein, may be resistant to therapy and BH3 profiling should correlate with lack of treatment efficacy (179). A recent trail, registered at Clinicaltrials.gov. (NCT03223662) has been set with the primary objective to determine whether a metabolomic signature or BH3 profiling of pre-neoadjuvant tumor biopsy correlates with the outcome of pathological complete response (pCR) after neoadjuvant chemoradiotherapy for patients with esophageal adenocarcinoma or squamous cell carcinoma, to serve as a basis for precision-based, personalized strategies for future treatment (185). Mitochondrial priming is dynamic, therefore, its threshold for apoptosis can be decreased by selecting tumor specific therapies. Stratification of patients based on whether pCR occurs may identify metabolomic signatures associated with response. Furthermore, future trials will be based on altering the mitochondrial threshold for apoptosis to increase the susceptibility to standard therapeutics.
Progression into the cell cycle, CD40L-NF-kB–mediated Bcl-xL upregulation, downmodulation in the expression of the proapoptotic Bim, Bax, and Bak proteins (186) and ultimately a decrease in mitochondrial priming and drug resistance have been recently confirmed in Mantle cell lymphoma (MCL) by using an ex vivo model (187). Using BH3 profiling, the central role of microenvironment-dependent signaling has been confirmed, with sequestration of the BH3-only activator Bim by Bcl-xL proteins at the mitochondrial level (187). Further, anti-CD20 antibody OBN Obinutuzumab efficiently counteract overexpression of Bcl-xL through NF-kB inhibition and loss of mitochondrial priming and drug sensitivity (187); consistently, OBN has demonstrated promising clinical activity with increased progression-free survival (PFS) in combination with bendamustine (188). The combined use of ibrutinib, which mediates indirect Bcl-xL down-modulation upon BTK-dependent binding, and venetoclax may improve clinical responses with more efficiency and less toxicity than the current standard of care. At present, an ongoing Obinutuzumab, GDC-0199 Plus Ibrutinib in Relapsed/Refractory Mantle Cell Lymphoma Patients (OAsIs) Trial for MCL patients (OBN, ibrutinib, and venetoclax, www.nationalclinicaltrials.gov,#NCT02558816) has been designed to determine in vivo efficacy through a selective approach targeting the lymphoma niche (187).
Induction of apoptosis is a common effect of several drugs. B-cell lymphoma 2 (Bcl2) expression is associated with resistance to apoptosis, and might be a marker for relative therapeutic resistance. Serial apoptosis monitoring might provide insight into resistance to a therapeutic regime. Bcl-2 should thus represent a biomarker of biological and clinical interest (189–191). A pilot study has been performed on metastatic breast cancer (MBC) patients with the aim to estimate Bcl-2 expression and apoptosis in CTCs after initiation of a new therapy, in order to assess the therapeutic efficacy (192). At baseline, apoptosis inversely correlated with CTC number and modestly with Bcl-2 positive CTC. As expected, higher CTC levels at baseline or first follow-up were associated with worse prognosis (9) and provide novel observations for Bcl-2 expression and apoptosis in CTCs. After one cycle of therapy in patients with elevated CTC, higher levels of CTC apoptosis were associated with worse prognosis, while higher CTC-Bcl-2 levels correlated with decreased apoptosis and superior PFS, resembling that for patients without elevated CTC (192). Actually the cohort consisted of a relatively small number of patients. In a recent paper, in patients with ER positive breast cancer who received adjuvant endocrine therapy, Bcl-2 predicted favourable outcomes, although its presence has been associated with worse prognosis (193–195). Since some patients had ER positive disease while others were ER negative, and some received endocrine therapy while others received chemotherapy, the association of CTC-Bcl-2 with outcomes may be quite heterogeneous. In summary, the results from this pilot study confirmed the prognostic significance of CTC at baseline and first follow-up for patients with metastatic breast cancer (192, 196). Further studies are needed to incorporate these assays into larger, more definitive trials as well as standard clinical practice.
CTCs have been reported as prognostic in all stages of breast cancer. A novel strategy for the isolation and expression profiling of pure populations of CTCs based on immunomagnetic capture and fluorescence-activated cell sorting (IE/FACS) has been described (197). Unsupervised hierarchical clustering revealed that CTC profiles clustered with more aggressive subtypes of primary breast tumors, with downregulated apoptosis, the relative absence of immune-related signals and down-modulation of ribosomes (197), relative to peripheral blood, suggesting a relatively quiescent state in circulation (198). As expected, CTCs from MBC had significantly higher risk of recurrence scores than primary tumors (197).
Recently a higher expression of Bcl2 and reduced expression of acetyl coenzyme A carboxylase-1 (ACC1) in tumors associated with CTC clusters (CTCcls) positivity has been reported (199). CTCcls represent a unique subset with higher metastatic potential and resistance to chemotherapy compared to single CTCs (5, 200). Of clinical relevance, patients with early-stage/locally-advanced BC have higher median CTCcl counts compared to patients with metastatic tumor (201). Further, CTCcls have a clinical prognostic value both at baseline and after treatment in terms of PFS and OS in cancer patients (9, 202, 203). Of note, a CTCcl+ gene signature consisting of 54 upregulated genes has been identified, significantly associated with poor relapse-free survival (RFS) in 360 patients with basal-like BC advancements (199), confirming Bcl2 expression as a poor prognostic factor in triple negative breast cancer (TNBC) patients, especially in the absence of adjuvant therapy (204). Also, Bcl-2 is upmodulated in CTC of xenograft models of TNBC (199), and correlated with higher levels of adhesion molecules including E-selectin, ICAM-1, and VCAM-1 (205).
Of clinical relevance, despite a subset of CTC consists of circulating cancer stem cells with high tumorigenic potential (206), CTCs are resistant to chemotherapy through mechanisms different from those activated in CSCs (207, 208). Stress induced in CTC, upon loss of ECM attachment, determines either anoikis-associated apoptosis (209) or generation of elevated levels of ROS (118, 210), with mild DNA damage and pre-activation of DNA checkpoints (211), as recapitulated in in vitro experiments. As a result, the DNA damage repair is efficiently activated upon chemotherapy, while inhibiting ROS production dramatically reduces the efficiency of post-chemotherapy DNA damage repair. Different from CTCs, breast CSCs had lower levels of ROS as compared with non-CSCs (212). A DNA damage repair response in cancer cells has been confirmed to determine tumor resistance to several DNA-damaging therapies, including anthracyclines and platinums (213). ATM/Chk2 and ATR/Chk1, which are two major kinase signaling pathways involved in the canonical DNA damage response network, are pre-activated in CTCs, and cause cell cycle arrest (177). Activation of checkpoint kinases represents an important mechanism limiting chemotherapeutic efficacy (213, 214). Thus, several Chk1/Chk2 inhibitors, including XL-844, AZD7762, and PF00477736, which potentiate the effects of DNA-damaging therapies by abrogating DNA damage-induced cell cycle arrest, have entered clinical trials for cancer therapy in combination with chemotherapeutic drugs (215). In accordance, these agents sensitize resistant CTCs to chemotherapy in vitro, with reduction of the number of CTCs and inhibition of lung and liver metastasis in xenograft models (177).
Coherently, exposure to cytotoxic/oxidative stress mediates a switch of CTC to a less proliferative but more drug-resistant phenotype (216). CTC in women with advanced oestrogen receptor (ER)-positive/human epidermal growth factor receptor 2 (HER2)-negative breast cancer acquire a HER2-positive phenotype after multiple courses of therapy (217, 218). While primary breast cancer is highly sensitive to HER2-targeted therapy, the clinical significance of acquired HER2 heterogeneity in metastatic tumor has been only recently analyzed (216). Cultured CTC isolated from women with ER+/HER2− primary tumors, 84% of whom had acquired HER2 expression, consisted of discrete subpopulations: a more proliferative HER2+ CTC subset, not addicted to HER2, consistent with activation of multiple signalling pathways, and a HER2− CTC subset, resistant to cytotoxic chemotherapy, while sensitive to Notch inhibition, due to activation of Notch and DNA damage pathways (216). Treatment of HER2+ CTCs with low doses of docetaxel or induction of oxidative stress induced rapid shifts from HER2+ to HER2−, thus modulating the HER2+/HER2− interconversion (216). HER2+ and HER2− CTCs interconverted spontaneously, and had comparable tumor initiating potential (216). Simultaneous treatment with paclitaxel and Notch inhibitors determined suppression of tumorigenesis in orthotopic CTC-derived tumor models (216). Together, these results point to distinct interconverting phenotypes within CTC, contributing to progression of breast cancer and acquisition of drug resistance.
Last, a number of studies have highlighted the role of tumor microenvironment in promoting tumor metastasis (219). In addition to circulating tumor cells, increased levels of viable circulating endothelial cells (CEC) are also released from primary tumors in patients with progressive disease (220). In head and neck cancer patients tumor-associated CEC express significantly higher level of Bcl-2, that is directly correlated with metastatic status, since they co-migrated with tumor cells to lung (221). CECs expressing Bcl-2 in the patient blood samples might be originating from tumor microvasculature and their binding to tumor cells induces a marked increase in Src and FAK activation in tumor cells, with anchorage independent survival (222), inhibition of both apoptosis, through regulating Bim, and anoikis, through regulating BAD (223, 224). Endothelial cells overexpressing Bcl-2 (EC-Bcl-2) expressed significantly higher levels of E-selectin and exhibited enhanced tumor cell binding (205). In addition, tumor cells bound to EC-Bcl-2 showed significantly higher anoikis resistance that was mediated by the Src-FAK signaling pathway (205). Furthermore, SCID mice coinjected with tumor cells and EC-Bcl-2 showed significantly higher lung metastasis (205). These results demonstrated a novel role for tumor-associated endothelial cells in protecting tumor cells from anoikis and chaperoning the tumor cells to distal sites.
Discussion
In the present review, we discussed the most recent research describing mitochondria alterations in the context of CTCs and their main effects on tumorigenesis and metastasis, and on CTCs behaviour, as summarised in Figure 1 and Table 1. In literature there are relatively few data on this argument, with some main limitations related predominantly to the low detection rate and difficulties in isolation of viable CTCs from circulation.
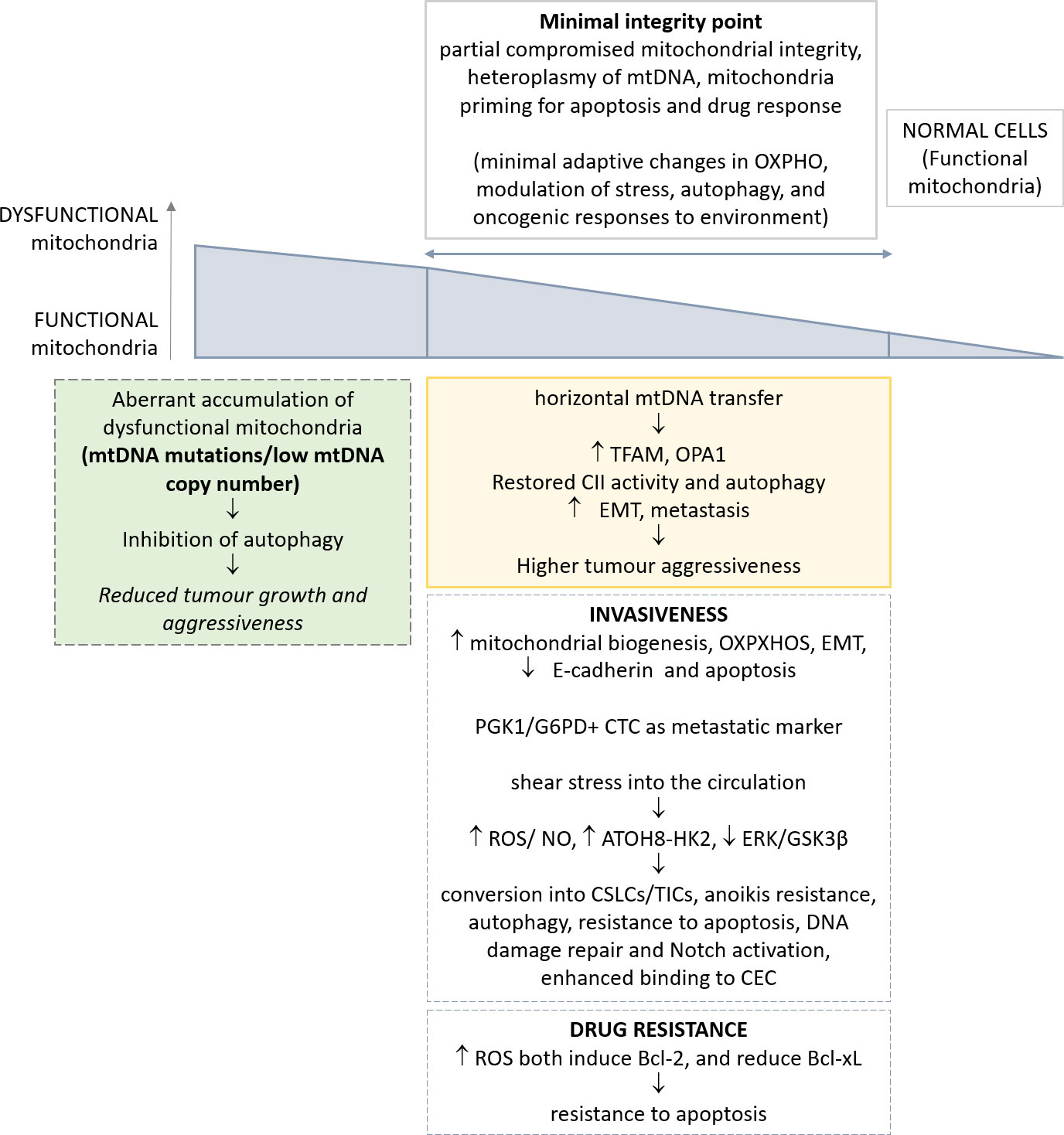
Figure 1 Schematic representation of the most relevant alterations reported in mitochondria, and their implications in CTC phenotype. The genetic and metabolic plasticity of tumour cells and the identification of a minimal integrity point threshold in mitochondria permit greater oncogenic/metastatic potential in CTCs. The effects of such adaptations in CTCs are depicted.
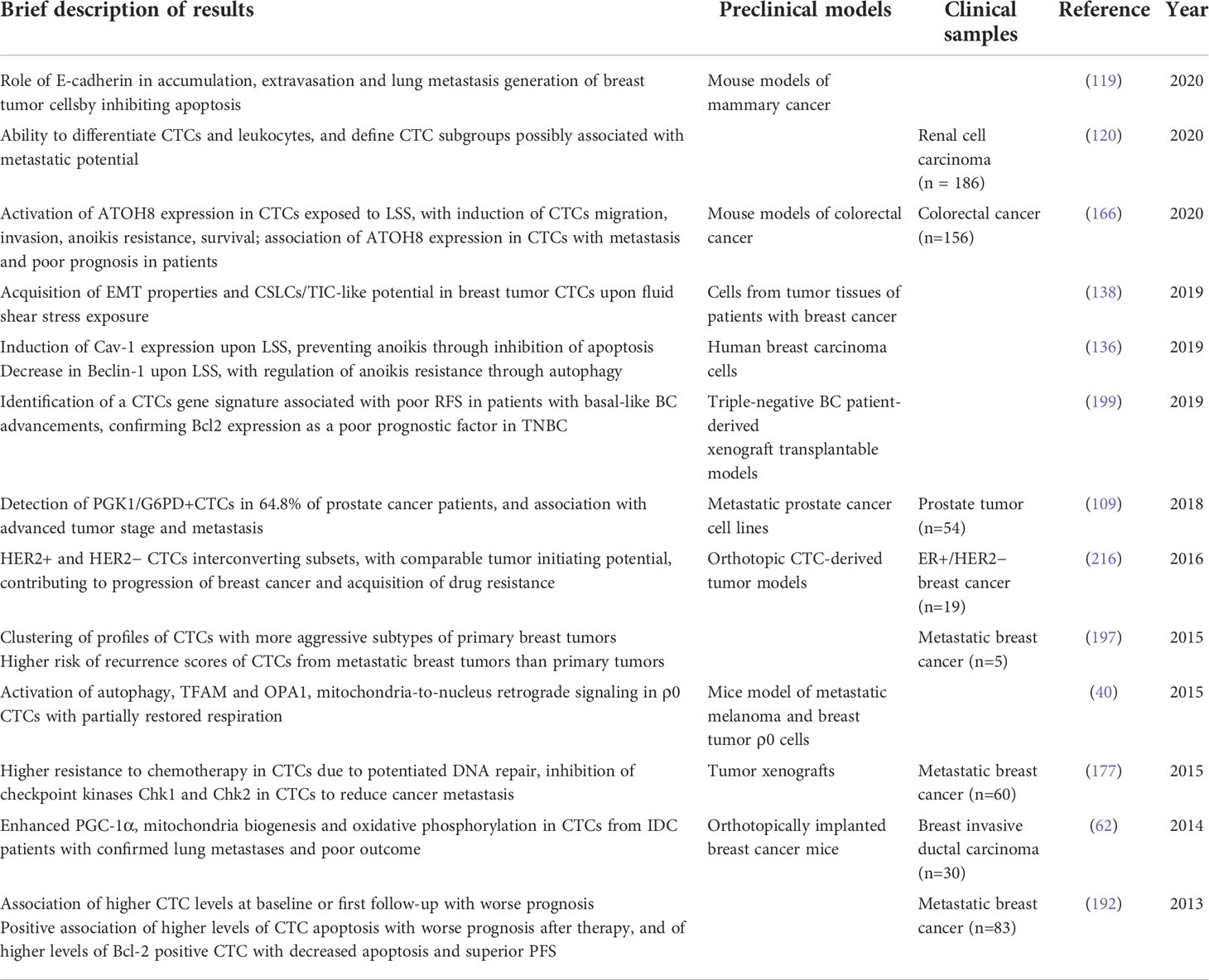
Table 1 Most relevant data on the implications of mitochondria alterations identified in CTC, as discussed in the review.
Thus, most reports relate to a selection of tumor types, lacking a comprehensive overview of the significance of mitochondria dysfunctions in CTC and their role in mediating tumor metastasis. Despite this shortcut, a wealth of information still demonstrates a role for mitochondria in neoplastic transformation, and suggests a potential clinical use, for both diagnostic and therapeutic purposes.
A novel concept, outlined in the present manuscript, and supported by the most recent advancement on mitochondria dysfunction in tumor, is related with the high plasticity of these organelles. Altogether, the heteroplasmy of mtDNA, the identification of a minimal integrity point for mitochondria functionality, and, finally, the mitochondria priming in apoptosis and drug response influence the adaptive capability of these organelles to the requirement of cancer cells over time. Indeed, many apparently controversial reports from literature can be eventually reconciled, allowing for diverse tissues of origin and the different stages of cells throughout tumorigenesis and metastasis. Such adaptive responses of mitochondria render apparently difficult to depict a mode of intervention for their effective drug modulation.
Nevertheless, the interest in dissecting the mechanisms through which mitochondria might participate in determining CTC responses to microenvironment and dictating their metastatic potential, is obvious, both to the research community and clinicians. In consideration of the current evolution rate in CTCs isolation and profiling in a growing panel of tumors (e.g. mesenchymal versus epithelial tumors), we are expecting relevant advancement on this argument in the coming years. The possibility of detecting and more extensively characterizing mitochondria alterations in CTCs will allow to obtain more robust and direct proofs of the still incomplete data presented in this review.
Author contributions
All authors participated in conceptualization, writing, review and editing the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The research was funded by Rete Oncologica Veneta (ROV).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell (2014) 14:275–91. doi: 10.1016/j.stem.2014.02.006
2. Alix-Panabières C, Pantel K. Challenges in circulating tumour cell research. Nat Rev Cancer (2014) 14:623–31. doi: 10.1038/nrc3820
3. Cristofanilli M, Pierga J-Y, Reuben J, Rademaker A, Davis AA, Peeters DJ, et al. The clinical use of circulating tumor cells (CTCs) enumeration for staging of metastatic breast cancer (MBC): International expert consensus paper. Crit Rev Oncol Hematol (2019) 134:39–45. doi: 10.1016/j.critrevonc.2018.12.004
4. Deutsch TM, Stefanovic S, Feisst M, Fischer C, Riedel F, Fremd C, et al. Cut-off analysis of CTC change under systemic therapy for defining early therapy response in metastatic breast cancer. Cancers (Basel) (2020) 12:e1055. doi: 10.3390/cancers12041055
5. Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell (2014) 158:1110–22. doi: 10.1016/j.cell.2014.07.013
6. Yu M, Bardia A, Wittner BS, Stott SL, Smas ME, Ting DT, et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science (2013) 339:580–4. doi: 10.1126/science.1228522
7. Larrea E, Sole C, Manterola L, Goicoechea I, Armesto M, Arestin M, et al. New concepts in cancer biomarkers: Circulating MiRNAs in liquid biopsies. Int J Mol Sci (2016) 17:e627. doi: 10.3390/ijms17050627
9. Cristofanilli M, Budd GT, Ellis MJ, Stopeck A, Matera J, Miller MC, et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N Engl J Med (2004) 351:781–91. doi: 10.1056/NEJMoa040766
10. de Bono JS, Scher HI, Montgomery RB, Parker C, Miller MC, Tissing H, et al. Circulating tumor cells predict survival benefit from treatment in metastatic castration-resistant prostate cancer. Clin Cancer Res (2008) 14:6302–9. doi: 10.1158/1078-0432.CCR-08-0872
11. Cohen SJ, Punt CJA, Iannotti N, Saidman BH, Sabbath KD, Gabrail NY, et al. Prognostic significance of circulating tumor cells in patients with metastatic colorectal cancer. Ann Oncol (2009) 20:1223–9. doi: 10.1093/annonc/mdn786
12. Budd GT, Cristofanilli M, Ellis MJ, Stopeck A, Borden E, Miller MC, et al. Circulating tumor cells versus imaging–predicting overall survival in metastatic breast cancer. Clin Cancer Res (2006) 12:6403–9. doi: 10.1158/1078-0432.CCR-05-1769
13. Cohen SJ, Punt CJA, Iannotti N, Saidman BH, Sabbath KD, Gabrail NY, et al. Relationship of circulating tumor cells to tumor response, progression-free survival, and overall survival in patients with metastatic colorectal cancer. J Clin Oncol (2008) 26:3213–21. doi: 10.1200/JCO.2007.15.8923
14. Cristofanilli M, Hayes DF, Budd GT, Ellis MJ, Stopeck A, Reuben JM, et al. Circulating tumor cells: A novel prognostic factor for newly diagnosed metastatic breast cancer. J Clin Oncol (2005) 23:1420–30. doi: 10.1200/JCO.2005.08.140
15. Hayes DF, Cristofanilli M, Budd GT, Ellis MJ, Stopeck A, Miller MC, et al. Circulating tumor cells at each follow-up time point during therapy of metastatic breast cancer patients predict progression-free and overall survival. Clin Cancer Res (2006) 12:4218–24. doi: 10.1158/1078-0432.CCR-05-2821
16. Liu MC, Shields PG, Warren RD, Cohen P, Wilkinson M, Ottaviano YL, et al. Circulating tumor cells: A useful predictor of treatment efficacy in metastatic breast cancer. J Clin Oncol (2009) 27:5153–9. doi: 10.1200/JCO.2008.20.6664
17. Bidard F-C, Peeters DJ, Fehm T, Nolé F, Gisbert-Criado R, Mavroudis D, et al. Clinical validity of circulating tumour cells in patients with metastatic breast cancer: A pooled analysis of individual patient data. Lancet Oncol (2014) 15:406–14. doi: 10.1016/S1470-2045(14)70069-5
18. Riebensahm C, Joosse SA, Mohme M, Hanssen A, Matschke J, Goy Y, et al. Clonality of circulating tumor cells in breast cancer brain metastasis patients. Breast Cancer Res (2019) 21:101. doi: 10.1186/s13058-019-1184-2
19. De Giorgi U, Valero V, Rohren E, Dawood S, Ueno NT, Miller MC, et al. Circulating tumor cells and [18F]Fluorodeoxyglucose positron emission Tomography/Computed tomography for outcome prediction in metastatic breast cancer. J Clin Oncol (2009) 27:3303–11. doi: 10.1200/JCO.2008.19.4423
20. Negin BP, Cohen SJ. Circulating tumor cells in colorectal cancer: Past, present, and future challenges. Curr Treat Options Oncol (2010) 11:1–13. doi: 10.1007/s11864-010-0115-3
21. Krebs MG, Sloane R, Priest L, Lancashire L, Hou J-M, Greystoke A, et al. Evaluation and prognostic significance of circulating tumor cells in patients with non-Small-Cell lung cancer. J Clin Oncol (2011) 29:1556–63. doi: 10.1200/JCO.2010.28.7045
22. Smerage JB, Barlow WE, Hortobagyi GN, Winer EP, Leyland-Jones B, Srkalovic G, et al. Circulating tumor cells and response to chemotherapy in metastatic breast cancer: SWOG S0500. J Clin Oncol (2014) 32:3483–9. doi: 10.1200/JCO.2014.56.2561
23. Gennari A, Foca F, Zamarchi R, Rocca A, Amadori D, De Censi A, et al. Insulin-like growth factor-1 receptor (IGF-1R) expression on circulating tumor cells (CTCs) and metastatic breast cancer outcome: Results from the TransMYME trial. Breast Cancer Res Treat (2020) 181:61–8. doi: 10.1007/s10549-020-05596-4
24. Papadaki MA, Koutsopoulos AV, Tsoulfas PG, Lagoudaki E, Aggouraki D, Monastirioti A, et al. Clinical relevance of immune checkpoints on circulating tumor cells in breast cancer. Cancers (Basel) (2020) 12:e376. doi: 10.3390/cancers12020376
25. Haber DA, Velculescu VE. Blood-based analyses of cancer: Circulating tumor cells and circulating tumor DNA. Cancer Discov (2014) 4:650–61. doi: 10.1158/2159-8290.CD-13-1014
26. Pantel K. Blood-based analysis of circulating cell-free DNA and tumor cells for early cancer detection. PloS Med (2016) 13:e1002205. doi: 10.1371/journal.pmed.1002205
27. Spira A, Yurgelun MB, Alexandrov L, Rao A, Bejar R, Polyak K, et al. Precancer atlas to drive precision prevention trials. Cancer Res (2017) 77:1510–41. doi: 10.1158/0008-5472.CAN-16-2346
28. Ilie M, Hofman V, Long-Mira E, Selva E, Vignaud J-M, Padovani B, et al. “Sentinel” circulating tumor cells allow early diagnosis of lung cancer in patients with chronic obstructive pulmonary disease. PloS One (2014) 9:e111597. doi: 10.1371/journal.pone.0111597
29. Gorges TM, Kuske A, Röck K, Mauermann O, Müller V, Peine S, et al. Accession of tumor heterogeneity by multiplex transcriptome profiling of single circulating tumor cells. Clin Chem (2016) 62:1504–15. doi: 10.1373/clinchem.2016.260299
30. Birkenkamp-Demtröder K, Nordentoft I, Christensen E, Høyer S, Reinert T, Vang S, et al. Genomic alterations in liquid biopsies from patients with bladder cancer. Eur Urol (2016) 70:75–82. doi: 10.1016/j.eururo.2016.01.007
31. Izumchenko E, Chang X, Brait M, Fertig E, Kagohara LT, Bedi A, et al. Targeted sequencing reveals clonal genetic changes in the progression of early lung neoplasms and paired circulating DNA. Nat Commun (2015) 6:8258. doi: 10.1038/ncomms9258
32. Guerrero-Preston R, Valle BL, Jedlicka A, Turaga N, Folawiyo O, Pirini F, et al. Molecular triage of premalignant lesions in liquid-based cervical cytology and circulating cell-free DNA from urine, using a panel of methylated human papilloma virus and host genes. Cancer Prev Res (Phila) (2016) 9:915–24. doi: 10.1158/1940-6207.CAPR-16-0138
33. Newman AM, Lovejoy AF, Klass DM, Kurtz DM, Chabon JJ, Scherer F, et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat Biotechnol (2016) 34:547–55. doi: 10.1038/nbt.3520
34. Forshew T, Murtaza M, Parkinson C, Gale D, Tsui DWY, Kaper F, et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med (2012) 4:136ra68. doi: 10.1126/scitranslmed.3003726
35. Fernandez-Cuesta L, Perdomo S, Avogbe PH, Leblay N, Delhomme TM, Gaborieau V, et al. Identification of circulating tumor DNA for the early detection of small-cell lung cancer. EBioMedicine (2016) 10:117–23. doi: 10.1016/j.ebiom.2016.06.032
36. Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab (2016) 23:27–47. doi: 10.1016/j.cmet.2015.12.006
37. Weber GF. Time and circumstances: Cancer cell metabolism at various stages of disease progression. Front Oncol (2016) 6:257. doi: 10.3389/fonc.2016.00257
38. Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
39. Mullen AR, Hu Z, Shi X, Jiang L, Boroughs LK, Kovacs Z, et al. Oxidation of alpha-ketoglutarate is required for reductive carboxylation in cancer cells with mitochondrial defects. Cell Rep (2014) 7:1679–90. doi: 10.1016/j.celrep.2014.04.037
40. Tan AS, Baty JW, Dong L-F, Bezawork-Geleta A, Endaya B, Goodwin J, et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab (2015) 21:81–94. doi: 10.1016/j.cmet.2014.12.003
41. Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, et al. Mitochondrial metabolism and ROS generation are essential for kras-mediated tumorigenicity. Proc Natl Acad Sci USA (2010) 107:8788–93. doi: 10.1073/pnas.1003428107
42. Bajzikova M, Kovarova J, Coelho AR, Boukalova S, Oh S, Rohlenova K, et al. Reactivation of dihydroorotate dehydrogenase-driven pyrimidine biosynthesis restores tumor growth of respiration-deficient cancer cells. Cell Metab (2019) 29:399–416.e10. doi: 10.1016/j.cmet.2018.10.014
43. Weinberg SE, Chandel NS. Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol (2015) 11:9–15. doi: 10.1038/nchembio.1712
44. Porporato PE, Filigheddu N, Pedro J.M.B.-S., Kroemer G, Galluzzi L. Mitochondrial metabolism and cancer. Cell Res (2018) 28:265–80. doi: 10.1038/cr.2017.155
45. Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer (2011) 11:85–95. doi: 10.1038/nrc2981
46. Hu J, Locasale JW, Bielas JH, O’Sullivan J, Sheahan K, Cantley LC, et al. Heterogeneity of tumor-induced gene expression changes in the human metabolic network. Nat Biotechnol (2013) 31:522–9. doi: 10.1038/nbt.2530
47. Gaude E, Frezza C. Tissue-specific and convergent metabolic transformation of cancer correlates with metastatic potential and patient survival. Nat Commun (2016) 7:13041. doi: 10.1038/ncomms13041
48. Valcarcel-Jimenez L, Gaude E, Torrano V, Frezza C, Carracedo A. Mitochondrial metabolism: Yin and yang for tumor progression. Trends Endocrinol Metab (2017) 28:748–57. doi: 10.1016/j.tem.2017.06.004
49. Lipinski KA, Barber LJ, Davies MN, Ashenden M, Sottoriva A, Gerlinger M. Cancer evolution and the limits of predictability in precision cancer medicine. Trends Cancer (2016) 2:49–63. doi: 10.1016/j.trecan.2015.11.003
50. Torrano V, Valcarcel-Jimenez L, Cortazar AR, Liu X, Urosevic J, Castillo-Martin M, et al. The metabolic Co-regulator PGC1α suppresses prostate cancer metastasis. Nat Cell Biol (2016) 18:645–56. doi: 10.1038/ncb3357
51. Denkert C, Bucher E, Hilvo M, Salek R, Orešič M, Griffin J, et al. Metabolomics of human breast cancer: New approaches for tumor typing and biomarker discovery. Genome Med (2012) 4:37. doi: 10.1186/gm336
52. Hakimi AA, Reznik E, Lee C-H, Creighton CJ, Brannon AR, Luna A, et al. An integrated metabolic atlas of clear cell renal cell carcinoma. Cancer Cell (2016) 29:104–16. doi: 10.1016/j.ccell.2015.12.004
53. Kerr EM, Gaude E, Turrell FK, Frezza C, Martins CP. Mutant kras copy number defines metabolic reprogramming and therapeutic susceptibilities. Nature (2016) 531:110–3. doi: 10.1038/nature16967
54. Nieto MA, Huang RY-J, Jackson RA, Thiery JP. EMT: 2016. Cell (2016) 166:21–45. doi: 10.1016/j.cell.2016.06.028
55. Loriot C, Burnichon N, Gadessaud N, Vescovo L, Amar L, Libé R, et al. Epithelial to mesenchymal transition is activated in metastatic pheochromocytomas and paragangliomas caused by SDHB gene mutations. J Clin Endocrinol Metab (2012) 97:E954–962. doi: 10.1210/jc.2011-3437
56. Sciacovelli M, Gonçalves E, Johnson TI, Zecchini VR, da Costa ASH, Gaude E, et al. Fumarate is an epigenetic modifier that elicits epithelial-to-Mesenchymal transition. Nature (2016) 537:544–7. doi: 10.1038/nature19353
57. Reznik E, Miller ML, Şenbabaoğlu Y, Riaz N, Sarungbam J, Tickoo SK, et al. Mitochondrial DNA copy number variation across human cancers. Elife (2016) 5:e10769. doi: 10.7554/eLife.10769
58. Joshi S, Tolkunov D, Aviv H, Hakimi AA, Yao M, Hsieh JJ, et al. The genomic landscape of renal oncocytoma identifies a metabolic barrier to tumorigenesis. Cell Rep (2015) 13:1895–908. doi: 10.1016/j.celrep.2015.10.059
59. Guo JY, Chen H-Y, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, et al. Activated ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev (2011) 25:460–70. doi: 10.1101/gad.2016311
60. Porporato PE, Payen VL, Pérez-Escuredo J, De Saedeleer CJ, Danhier P, Copetti T, et al. A mitochondrial switch promotes tumor metastasis. Cell Rep (2014) 8:754–66. doi: 10.1016/j.celrep.2014.06.043
61. Santidrian AF, Matsuno-Yagi A, Ritland M, Seo BB, LeBoeuf SE, Gay LJ, et al. Mitochondrial complex I activity and NAD+/NADH balance regulate breast cancer progression. J Clin Invest (2013) 123:1068–81. doi: 10.1172/JCI64264
62. LeBleu VS, O’Connell JT, Gonzalez Herrera KN, Wikman H, Pantel K, Haigis MC, et al. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol (2014) 16(10):992–15. doi: 10.1038/ncb3039
63. Christen S, Lorendeau D, Schmieder R, Broekaert D, Metzger K, Veys K, et al. Breast cancer-derived lung metastases show increased pyruvate carboxylase-dependent anaplerosis. Cell Rep (2016) 17:837–48. doi: 10.1016/j.celrep.2016.09.042
64. Dupuy F, Tabariès S, Andrzejewski S, Dong Z, Blagih J, Annis MG, et al. PDK1-dependent metabolic reprogramming dictates metastatic potential in breast cancer. Cell Metab (2015) 22:577–89. doi: 10.1016/j.cmet.2015.08.007
65. Zhang G, Frederick DT, Wu L, Wei Z, Krepler C, Srinivasan S, et al. Targeting mitochondrial biogenesis to overcome drug resistance to MAPK inhibitors. J Clin Invest (2016) 126:1834–56. doi: 10.1172/JCI82661
66. Vazquez F, Lim J-H, Chim H, Bhalla K, Girnun G, Pierce K, et al. PGC1α expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell (2013) 23:287–301. doi: 10.1016/j.ccr.2012.11.020
67. Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, et al. Oncogenic BRAF regulates oxidative metabolism via PGC1α and MITF. Cancer Cell (2013) 23:302–15. doi: 10.1016/j.ccr.2013.02.003
68. Missiroli S, Perrone M, Genovese I, Pinton P, Giorgi C. Cancer metabolism and mitochondria: Finding novel mechanisms to fight tumours. EBioMedicine (2020) 59:102943. doi: 10.1016/j.ebiom.2020.102943
69. Fiorillo M, Ózsvári B, Sotgia F, Lisanti MP. High ATP production fuels cancer drug resistance and metastasis: Implications for mitochondrial ATP depletion therapy. Front Oncol (2021) 11:740720. doi: 10.3389/fonc.2021.740720
70. Hensley CT, Faubert B, Yuan Q, Lev-Cohain N, Jin E, Kim J, et al. Metabolic heterogeneity in human lung tumors. Cell (2016) 164:681–94. doi: 10.1016/j.cell.2015.12.034
71. Allen E, Miéville P, Warren CM, Saghafinia S, Li L, Peng M-W, et al. Metabolic symbiosis enables adaptive resistance to anti-angiogenic therapy that is dependent on MTOR signaling. Cell Rep (2016) 15:1144–60. doi: 10.1016/j.celrep.2016.04.029
72. Jiménez-Valerio G, Martínez-Lozano M, Bassani N, Vidal A, Ochoa-de-Olza M, Suárez C, et al. Resistance to antiangiogenic therapies by metabolic symbiosis in renal cell carcinoma PDX models and patients. Cell Rep (2016) 15:1134–43. doi: 10.1016/j.celrep.2016.04.015
73. Pisarsky L, Bill R, Fagiani E, Dimeloe S, Goosen RW, Hagmann J, et al. Targeting metabolic symbiosis to overcome resistance to anti-angiogenic therapy. Cell Rep (2016) 15:1161–74. doi: 10.1016/j.celrep.2016.04.028
75. Gammage PA, Frezza C. Mitochondrial DNA: The overlooked oncogenome? BMC Biol (2019) 17:53. doi: 10.1186/s12915-019-0668-y
76. Taylor RW, Turnbull DM. Mitochondrial DNA mutations in human disease. Nat Rev Genet (2005) 6:389–402. doi: 10.1038/nrg1606
77. Wallace DC. Mitochondrial DNA variation in human radiation and disease. Cell (2015) 163:33–8. doi: 10.1016/j.cell.2015.08.067
78. Petros JA, Baumann AK, Ruiz-Pesini E, Amin MB, Sun CQ, Hall J, et al. MtDNA mutations increase tumorigenicity in prostate cancer. Proc Natl Acad Sci U.S.A. (2005) 102:719–24. doi: 10.1073/pnas.0408894102
79. Zong W-X, Rabinowitz JD, White E. Mitochondria and cancer. Mol Cell (2016) 61:667–76. doi: 10.1016/j.molcel.2016.02.011
80. Banoth B, Cassel SL. Mitochondria in innate immune signaling. Transl Res (2018) 202:52–68. doi: 10.1016/j.trsl.2018.07.014
81. Jazayeri M, Andreyev A, Will Y, Ward M, Anderson CM, Clevenger W. Inducible expression of a dominant negative DNA polymerase-gamma depletes mitochondrial DNA and produces a Rho0 phenotype. J Biol Chem (2003) 278:9823–30. doi: 10.1074/jbc.m211730200
82. Stewart JB, Chinnery PF. The dynamics of mitochondrial DNA heteroplasmy: Implications for human health and disease. Nat Rev Genet (2015) 16:530–42. doi: 10.1038/nrg3966
83. Yuan Y, Ju YS, Kim Y, Li J, Wang Y, Yoon CJ, et al. Comprehensive molecular characterization of mitochondrial genomes in human cancers. Nat Genet (2020) 52:342–52. doi: 10.1038/s41588-019-0557-x
84. Stewart JB, Alaei-Mahabadi B, Sabarinathan R, Samuelsson T, Gorodkin J, Gustafsson CM, et al. Simultaneous DNA and RNA mapping of somatic mitochondrial mutations across diverse human cancers. PloS Genet (2015) 11:e1005333. doi: 10.1371/journal.pgen.1005333
85. Gasparre G, Romeo G, Rugolo M, Porcelli AM. Learning from oncocytic tumors: Why choose inefficient mitochondria? Biochim Biophys Acta (2011) 1807:633–42. doi: 10.1016/j.bbabio.2010.08.006
86. Gopal RK, Calvo SE, Shih AR, Chaves FL, McGuone D, Mick E, et al. Early loss of mitochondrial complex I and rewiring of glutathione metabolism in renal oncocytoma. Proc Natl Acad Sci USA (2018) 115:E6283–90. doi: 10.1073/pnas.1711888115
87. Hopkins JF, Sabelnykova VY, Weischenfeldt J, Simon R, Aguiar JA, Alkallas R, et al. Mitochondrial mutations drive prostate cancer aggression. Nat Commun (2017) 8:656. doi: 10.1038/s41467-017-00377-y
88. Feeley KP, Bray AW, Westbrook DG, Johnson LW, Kesterson RA, Ballinger SW, et al. Mitochondrial genetics regulate breast cancer tumorigenicity and metastatic potential. Cancer Res (2015) 75:4429–36. doi: 10.1158/0008-5472.CAN-15-0074
89. Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science (2005) 308:1909–11. doi: 10.1126/science.1106653
90. Goh J, Enns L, Fatemie S, Hopkins H, Morton J, Pettan-Brewer C, et al. Mitochondrial targeted catalase suppresses invasive breast cancer in mice. BMC Cancer (2011) 11:191. doi: 10.1186/1471-2407-11-191
91. Latorre-Pellicer A, Moreno-Loshuertos R, Lechuga-Vieco AV, Sánchez-Cabo F, Torroja C, Acín-Pérez R, et al. Mitochondrial and nuclear DNA matching shapes metabolism and healthy ageing. Nature (2016) 535:561–5. doi: 10.1038/nature18618
92. Liu VWS, Wang Y, Yang H-J, Tsang PCK, Ng T-Y, Wong L-C, et al. Mitochondrial DNA variant 16189T>C is associated with susceptibility to endometrial cancer. Hum Mutat (2003) 22:173–4. doi: 10.1002/humu.10244
93. Williams SB, Ye Y, Huang M, Chang DW, Kamat AM, Pu X, et al. Mitochondrial DNA content as risk factor for bladder cancer and its association with mitochondrial DNA polymorphisms. Cancer Prev Res (Phila) (2015) 8:607–13. doi: 10.1158/1940-6207.CAPR-14-0414
94. Gasparre G, Iommarini L, Porcelli AM, Lang M, Ferri GG, Kurelac I, et al. An inherited mitochondrial DNA disruptive mutation shifts to homoplasmy in oncocytic tumor cells. Hum Mutat (2009) 30:391–6. doi: 10.1002/humu.20870
95. Picard M, Zhang J, Hancock S, Derbeneva O, Golhar R, Golik P, et al. Progressive increase in MtDNA 3243A>G heteroplasmy causes abrupt transcriptional reprogramming. Proc Natl Acad Sci USA (2014) 111:E4033–4042. doi: 10.1073/pnas.1414028111
96. Hahn A, Zuryn S. Mitochondrial genome (MtDNA) mutations that generate reactive oxygen species. Antioxidants (Basel) (2019) 8:E392. doi: 10.3390/antiox8090392
97. Wang P-Y, Li J, Walcott FL, Kang J-G, Starost MF, Talagala SL, et al. Inhibiting mitochondrial respiration prevents cancer in a mouse model of Li-fraumeni syndrome. J Clin Invest (2017) 127:132–6. doi: 10.1172/JCI88668
98. Chen P-L, Chen C-F, Chen Y, Guo XE, Huang C-K, Shew J-Y, et al. Mitochondrial genome instability resulting from SUV3 haploinsufficiency leads to tumorigenesis and shortened lifespan. Oncogene (2013) 32:1193–201. doi: 10.1038/onc.2012.120
99. van Gisbergen MW, Voets AM, Starmans MHW, de Coo IFM, Yadak R, Hoffmann RF, et al. How do changes in the MtDNA and mitochondrial dysfunction influence cancer and cancer therapy? challenges, opportunities and models. Mutat Res Rev Mutat Res (2015) 764:16–30. doi: 10.1016/j.mrrev.2015.01.001
100. Tseng L-M, Yin P-H, Chi C-W, Hsu C-Y, Wu C-W, Lee L-M, et al. Mitochondrial DNA mutations and mitochondrial DNA depletion in breast cancer. Genes Chromosomes Cancer (2006) 45:629–38. doi: 10.1002/gcc.20326
101. Guha M, Avadhani NG. Mitochondrial retrograde signaling at the crossroads of tumor bioenergetics, genetics and epigenetics. Mitochondrion (2013) 13:577–91. doi: 10.1016/j.mito.2013.08.007
102. Khaidakov M, Shmookler Reis RJ. Possibility of selection against MtDNA mutations in tumors. Mol Cancer (2005) 4:36. doi: 10.1186/1476-4598-4-36
103. Spees JL, Olson SD, Whitney MJ, Prockop DJ. Mitochondrial transfer between cells can rescue aerobic respiration. Proc Natl Acad Sci USA (2006) 103:1283–8. doi: 10.1073/pnas.0510511103
104. Dong L-F, Kovarova J, Bajzikova M, Bezawork-Geleta A, Svec D, Endaya B, et al. Horizontal transfer of whole mitochondria restores tumorigenic potential in mitochondrial DNA-deficient cancer cells. Elife (2017) 6:e22187. doi: 10.7554/eLife.22187
105. Kukat C, Larsson N-G. MtDNA makes a U-turn for the mitochondrial nucleoid. Trends Cell Biol (2013) 23:457–63. doi: 10.1016/j.tcb.2013.04.009
106. Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana-Cabrera R, Corrado M, et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell (2013) 155:160–71. doi: 10.1016/j.cell.2013.08.032
107. Dickinson A, Yeung KY, Donoghue J, Baker MJ, Kelly RD, McKenzie M, et al. The regulation of mitochondrial DNA copy number in glioblastoma cells. Cell Death Differ (2013) 20:1644–53. doi: 10.1038/cdd.2013.115
108. Campo DS, Nayak V, Srinivasamoorthy G, Khudyakov Y. Entropy of mitochondrial DNA circulating in blood is associated with hepatocellular carcinoma. BMC Med Genomics (2019) 12:74. doi: 10.1186/s12920-019-0506-7
109. Chen J, Cao S, Situ B, Zhong J, Hu Y, Li S, et al. Metabolic reprogramming-based characterization of circulating tumor cells in prostate cancer. J Exp Clin Cancer Res (2018) 37:127. doi: 10.1186/s13046-018-0789-0
110. Favaro E, Bensaad K, Chong MG, Tennant DA, Ferguson DJP, Snell C, et al. Glucose utilization via glycogen phosphorylase sustains proliferation and prevents premature senescence in cancer cells. Cell Metab (2012) 16:751–64. doi: 10.1016/j.cmet.2012.10.017
111. Kamarajugadda S, Stemboroski L, Cai Q, Simpson NE, Nayak S, Tan M, et al. Glucose oxidation modulates anoikis and tumor metastasis. Mol Cell Biol (2012) 32:1893–907. doi: 10.1128/MCB.06248-11
112. Koukourakis MI, Kalamida D, Mitrakas AG, Liousia M, Pouliliou S, Sivridis E, et al. Metabolic cooperation between Co-cultured lung cancer cells and lung fibroblasts. Lab Invest (2017) 97:1321–31. doi: 10.1038/labinvest.2017.79
113. Wu Y-H, Lee Y-H, Shih H-Y, Chen S-H, Cheng Y-C, Tsun-Yee Chiu D. Glucose-6-Phosphate dehydrogenase is indispensable in embryonic development by modulation of epithelial-mesenchymal transition via the NOX/Smad3/MiR-200b axis. Cell Death Dis (2018) 9:10. doi: 10.1038/s41419-017-0005-8
114. Yang W, Xia Y, Ji H, Zheng Y, Liang J, Huang W, et al. Nuclear PKM2 regulates β-catenin transactivation upon EGFR activation. Nature (2011) 480:118–22. doi: 10.1038/nature10598
115. Yang L, Hou Y, Yuan J, Tang S, Zhang H, Zhu Q, et al. Twist promotes reprogramming of glucose metabolism in breast cancer cells through PI3K/AKT and P53 signaling pathways. Oncotarget (2015) 6:25755–69. doi: 10.18632/oncotarget.4697
116. Jones AWE, Yao Z, Vicencio JM, Karkucinska-Wieckowska A, Szabadkai G. PGC-1 family coactivators and cell fate: Roles in cancer, neurodegeneration, cardiovascular disease and retrograde mitochondria-nucleus signalling. Mitochondrion (2012) 12:86–99. doi: 10.1016/j.mito.2011.09.009
117. Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer (2011) 11:325–37. doi: 10.1038/nrc3038
118. Schafer ZT, Grassian AR, Song L, Jiang Z, Gerhart-Hines Z, Irie HY, et al. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature (2009) 461:109–13. doi: 10.1038/nature08268
119. Na T-Y, Schecterson L, Mendonsa AM, Gumbiner BM. The functional activity of e-cadherin controls tumor cell metastasis at multiple steps. Proc Natl Acad Sci USA (2020) 117:5931–7. doi: 10.1073/pnas.1918167117
120. Klezl P, Pospisilova E, Kolostova K, Sonsky J, Maly O, Grill R, et al. Detection of circulating tumor cells in renal cell carcinoma: Disease stage correlation and molecular characterization. J Clin Med (2020) 9:E1372. doi: 10.3390/jcm9051372
121. Motzer RJ, Russo P. Systemic therapy for renal cell carcinoma. J Urol (2000) 163:408–17. doi: 10.1016/S0022-5347(05)67889-5
122. Gore ME, Griffin CL, Hancock B, Patel PM, Pyle L, Aitchison M, et al. Interferon Alfa-2a versus combination therapy with interferon Alfa-2a, interleukin-2, and fluorouracil in patients with untreated metastatic renal cell carcinoma (MRC RE04/EORTC GU 30012): An open-label randomised trial. Lancet (2010) 375:641–8. doi: 10.1016/S0140-6736(09)61921-8
123. Gerlinger M, Horswell S, Larkin J, Rowan AJ, Salm MP, Varela I, et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet (2014) 46:225–33. doi: 10.1038/ng.2891
124. Harten SK, Shukla D, Barod R, Hergovich A, Balda MS, Matter K, et al. Regulation of renal epithelial tight junctions by the von hippel-lindau tumor suppressor gene involves occludin and claudin 1 and is independent of e-cadherin. Mol Biol Cell (2009) 20:1089–101. doi: 10.1091/mbc.e08-06-0566
125. Lambert AW, Pattabiraman DR, Weinberg RA. Emerging biological principles of metastasis. Cell (2017) 168:670–91. doi: 10.1016/j.cell.2016.11.037
126. Kwan TT, Bardia A, Spring LM, Giobbie-Hurder A, Kalinich M, Dubash T, et al. A digital RNA signature of circulating tumor cells predicting early therapeutic response in localized and metastatic breast cancer. Cancer Discov (2018) 8:1286–99. doi: 10.1158/2159-8290.CD-18-0432
127. Massagué J, Obenauf AC. Metastatic colonization by circulating tumour cells. Nature (2016) 529:298–306. doi: 10.1038/nature17038
128. Krebs MG, Hou J-M, Ward TH, Blackhall FH, Dive C. Circulating tumour cells: Their utility in cancer management and predicting outcomes. Ther Adv Med Oncol (2010) 2:351–65. doi: 10.1177/1758834010378414
129. Huang Q, Hu X, He W, Zhao Y, Hao S, Wu Q, et al. Fluid shear stress and tumor metastasis. Am J Cancer Res (2018) 8:763–77.
130. Polacheck WJ, Charest JL, Kamm RD. Interstitial flow influences direction of tumor cell migration through competing mechanisms. Proc Natl Acad Sci USA (2011) 108:11115–20. doi: 10.1073/pnas.1103581108
131. Zhao Y, Liu Y, Lin L, Huang Q, He W, Zhang S, et al. The LncRNA MACC1-AS1 promotes gastric cancer cell metabolic plasticity via AMPK/Lin28 mediated MRNA stability of MACC1. Mol Cancer (2018) 17:69. doi: 10.1186/s12943-018-0820-2
132. Sathanoori R, Bryl-Gorecka P, Müller CE, Erb L, Weisman GA, Olde B, et al. P2Y2 receptor modulates shear stress-induced cell alignment and actin stress fibers in human umbilical vein endothelial cells. Cell Mol Life Sci (2017) 74:731–46. doi: 10.1007/s00018-016-2365-0
133. Delaine-Smith RM, Sittichokechaiwut A, Reilly GC. Primary cilia respond to fluid shear stress and mediate flow-induced calcium deposition in osteoblasts. FASEB J (2014) 28:430–9. doi: 10.1096/fj.13-231894
134. Toh Y-C, Voldman J. Fluid shear stress primes mouse embryonic stem cells for differentiation in a self-renewing environment via heparan sulfate proteoglycans transduction. FASEB J (2011) 25:1208–17. doi: 10.1096/fj.10-168971
135. Adamo L, Naveiras O, Wenzel PL, McKinney-Freeman S, Mack PJ, Gracia-Sancho J, et al. Biomechanical forces promote embryonic haematopoiesis. Nature (2009) 459:1131–5. doi: 10.1038/nature08073
136. Li S, Chen Y, Zhang Y, Jiang X, Jiang Y, Qin X, et al. Shear stress promotes anoikis resistance of cancer cells via caveolin-1-Dependent extrinsic and intrinsic apoptotic pathways. J Cell Physiol (2019) 234:3730–43. doi: 10.1002/jcp.27149
137. Triantafillu UL, Park S, Klaassen NL, Raddatz AD, Kim Y. Fluid shear stress induces cancer stem cell-like phenotype in MCF7 breast cancer cell line without inducing epithelial to mesenchymal transition. Int J Oncol (2017) 50:993–1001. doi: 10.3892/ijo.2017.3865
138. Choi HY, Yang G-M, Dayem AA, Saha SK, Kim K, Yoo Y, et al. Hydrodynamic shear stress promotes epithelial-mesenchymal transition by downregulating ERK and GSK3β activities. Breast Cancer Res (2019) 21:6. doi: 10.1186/s13058-018-1071-2
139. Lee HJ, Diaz MF, Price KM, Ozuna JA, Zhang S, Sevick-Muraca EM, et al. Fluid shear stress activates YAP1 to promote cancer cell motility. Nat Commun (2017) 8:14122. doi: 10.1038/ncomms14122
141. Kodama K, Ishii G, Miyamoto S, Goya M, Zhang S-C, Sangai T, et al. Laminin 5 expression protects against anoikis at aerogenous spread and lepidic growth of human lung adenocarcinoma. Int J Cancer (2005) 116:876–84. doi: 10.1002/ijc.21136
142. Kamarajan P, Kapila YL. An altered fibronectin matrix induces anoikis of human squamous cell carcinoma cells by suppressing integrin alpha v levels and phosphorylation of FAK and ERK. Apoptosis (2007) 12:2221–31. doi: 10.1007/s10495-007-0138-9
143. Whelan KA, Caldwell SA, Shahriari KS, Jackson SR, Franchetti LD, Johannes GJ, et al. Hypoxia suppression of bim and bmf blocks anoikis and luminal clearing during mammary morphogenesis. Mol Biol Cell (2010) 21:3829–37. doi: 10.1091/mbc.E10-04-0353
144. de Sousa Mesquita AP, de Araújo Lopes S, Pernambuco Filho PCA, Nader HB, Lopes CC. Acquisition of anoikis resistance promotes alterations in the Ras/ERK and PI3K/Akt signaling pathways and matrix remodeling in endothelial cells. Apoptosis (2017) 22:1116–37. doi: 10.1007/s10495-017-1392-0
145. Zugasti O, Rul W, Roux P, Peyssonnaux C, Eychene A, Franke TF, et al. Raf-MEK-Erk cascade in anoikis is controlled by Rac1 and Cdc42. via Akt. Mol Cell Biol (2001) 21:6706–17. doi: 10.1128/MCB.21.19.6706-6717.2001
146. Chen Y-J, Kuo C-D, Tsai Y-M, Yu C-C, Wang G-S, Liao H-F. Norcantharidin induces anoikis through jun-N-Terminal kinase activation in CT26 colorectal cancer cells. Anticancer Drugs (2008) 19:55–64. doi: 10.1097/CAD.0b013e3282f18826
147. Fukazawa H, Noguchi K, Murakami Y, Uehara Y. Mitogen-activated Protein/Extracellular signal-regulated kinase kinase (MEK) inhibitors restore anoikis sensitivity in human breast cancer cell lines with a constitutively activated extracellular-regulated kinase (ERK) pathway. Mol Cancer Ther (2002) 1:303–9.
148. Morozevich GE, Kozlova NI, Susova OY, Karalkin PA, Berman AE. Implication of α2β1 integrin in anoikis of MCF-7 human breast carcinoma cells. Biochem (Mosc) (2015) 80:97–103. doi: 10.1134/S0006297915010113
149. Rungtabnapa P, Nimmannit U, Halim H, Rojanasakul Y, Chanvorachote P. Hydrogen peroxide inhibits non-small cell lung cancer cell anoikis through the inhibition of caveolin-1 degradation. Am J Physiol Cell Physiol (2011) 300:C235–245. doi: 10.1152/ajpcell.00249.2010
150. Tang X, Kuhlenschmidt TB, Li Q, Ali S, Lezmi S, Chen H, et al. Mechanically-induced colon cancer cell population shows increased metastatic potential. Mol Cancer (2014) 13:131. doi: 10.1186/1476-4598-13-131
151. Rohwer N, Welzel M, Daskalow K, Pfander D, Wiedenmann B, Detjen K, et al. Hypoxia-inducible factor 1alpha mediates anoikis resistance via suppression of Alpha5 integrin. Cancer Res (2008) 68:10113–20. doi: 10.1158/0008-5472.CAN-08-1839
152. Nguyen DX, Bos PD, Massagué J. Metastasis: From dissemination to organ-specific colonization. Nat Rev Cancer (2009) 9:274–84. doi: 10.1038/nrc2622
153. Li J, King MR. Adhesion receptors as therapeutic targets for circulating tumor cells. Front Oncol (2012) 2:79. doi: 10.3389/fonc.2012.00079
154. Ren T, Zhang H, Wang J, Zhu J, Jin M, Wu Y, et al. MCU-dependent mitochondrial Ca2+ inhibits NAD+/SIRT3/SOD2 pathway to promote ROS production and metastasis of HCC cells. Oncogene (2017) 36:5897–909. doi: 10.1038/onc.2017.167
155. Fofaria NM, Srivastava SK. STAT3 induces anoikis resistance, promotes cell invasion and metastatic potential in pancreatic cancer cells. Carcinogenesis (2015) 36:142–50. doi: 10.1093/carcin/bgu233
156. Fung C, Lock R, Gao S, Salas E, Debnath J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol Biol Cell (2008) 19:797–806. doi: 10.1091/mbc.e07-10-1092
157. Taddei ML, Giannoni E, Morandi A, Ippolito L, Ramazzotti M, Callari M, et al. Mesenchymal to amoeboid transition is associated with stem-like features of melanoma cells. Cell Commun Signal (2014) 12:24. doi: 10.1186/1478-811X-12-24
158. Gao D, Joshi N, Choi H, Ryu S, Hahn M, Catena R, et al. Myeloid progenitor cells in the premetastatic lung promote metastases by inducing mesenchymal to epithelial transition. Cancer Res (2012) 72:1384–94. doi: 10.1158/0008-5472.CAN-11-2905
159. Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong STC, et al. Epithelial-to-Mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature (2015) 527:472–6. doi: 10.1038/nature15748
160. Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, et al. Epithelial-to-Mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature (2015) 527:525–30. doi: 10.1038/nature16064
161. Ye X, Weinberg RA. Epithelial-mesenchymal plasticity: A central regulator of cancer progression. Trends Cell Biol (2015) 25:675–86. doi: 10.1016/j.tcb.2015.07.012
162. Pastushenko I, Brisebarre A, Sifrim A, Fioramonti M, Revenco T, Boumahdi S, et al. Identification of the tumour transition states occurring during EMT. Nature (2018) 556:463–8. doi: 10.1038/s41586-018-0040-3
163. Chaffer CL, Weinberg RAA. Perspective on cancer cell metastasis. Science (2011) 331:1559–64. doi: 10.1126/science.1203543
164. Tsai JH, Yang J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev (2013) 27:2192–206. doi: 10.1101/gad.225334.113
165. Lloyd PG. Caveolin-1, antiapoptosis signaling, and anchorage-independent cell growth. focus on “Caveolin-1 regulates mcl-1 stability and anoikis in lung carcinoma cells”. Am J Physiol Cell Physiol (2012) 302:C1282–1283. doi: 10.1152/ajpcell.00075.2012
166. Huang Q, Li S, Hu X, Sun M, Wu Q, Dai H, et al. Shear stress activates ATOH8 via autocrine VEGF promoting glycolysis dependent-survival of colorectal cancer cells in the circulation. J Exp Clin Cancer Res (2020) 39:25. doi: 10.1186/s13046-020-1533-0
167. Lim C-G, Jang J, Kim C. Cellular machinery for sensing mechanical force. BMB Rep (2018) 51:623–9. doi: 10.5483/BMBRep.2018.51.12.237
168. Fang F, Wasserman SM, Torres-Vazquez J, Weinstein B, Cao F, Li Z, et al. The role of Hath6, a newly identified shear-Stress-Responsive transcription factor, in endothelial cell differentiation and function. J Cell Sci (2014) 127:1428–40. doi: 10.1242/jcs.136358
169. Güttsches A-K, Balakrishnan-Renuka A, Kley RA, Tegenthoff M, Brand-Saberi B, Vorgerd M. ATOH8: A novel marker in human muscle fiber regeneration. Histochem Cell Biol (2015) 143:443–52. doi: 10.1007/s00418-014-1299-6
170. Wang B, Balakrishnan-Renuka A, Napirei M, Theiss C, Brand-Saberi B. Spatiotemporal expression of Math6 during mouse embryonic development. Histochem Cell Biol (2015) 143:575–82. doi: 10.1007/s00418-014-1305-z
171. Zhang Y, Tang B, Song J, Yu S, Li Y, Su H, et al. Lnc-PDZD7 contributes to stemness properties and chemosensitivity in hepatocellular carcinoma through EZH2-mediated ATOH8 transcriptional repression. J Exp Clin Cancer Res (2019) 38:92. doi: 10.1186/s13046-019-1106-2
172. Wang Z, Xie J, Yan M, Wang J, Wang X, Zhang J, et al. Downregulation of ATOH8 induced by EBV-encoded LMP1 contributes to the malignant phenotype of nasopharyngeal carcinoma. Oncotarget (2016) 7:26765–79. doi: 10.18632/oncotarget.8503
173. Ye M, He Y, Lin H, Yang S, Zhou Y, Zhou L, et al. High expression of atonal homolog 8 predicts a poor clinical outcome in patients with colorectal cancer and contributes to tumor progression. Oncol Rep (2017) 37:2955–63. doi: 10.3892/or.2017.5554
174. Nakajima H, Yamamoto K, Agarwala S, Terai K, Fukui H, Fukuhara S, et al. Flow-dependent endothelial YAP regulation contributes to vessel maintenance. Dev Cell (2017) 40:523–536.e6. doi: 10.1016/j.devcel.2017.02.019
175. Boroughs LK, DeBerardinis RJ. Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol (2015) 17:351–9. doi: 10.1038/ncb3124
176. Xiong X, Wen Y-A, Mitov MI, C Oaks M, Miyamoto S, Gao T. PHLPP regulates hexokinase 2-dependent glucose metabolism in colon cancer cells. Cell Death Discovery (2017) 3:16103. doi: 10.1038/cddiscovery.2016.103
177. Gong C, Liu B, Yao Y, Qu S, Luo W, Tan W, et al. Potentiated DNA damage response in circulating breast tumor cells confers resistance to chemotherapy. J Biol Chem (2015) 290:14811–25. doi: 10.1074/jbc.M115.652628
178. Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, Armstrong SA, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell (2006) 9:351–65. doi: 10.1016/j.ccr.2006.03.027
179. Ni Chonghaile T, Sarosiek KA, Vo T-T, Ryan JA, Tammareddi A, Moore VDG, et al. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science (2011) 334:1129–33. doi: 10.1126/science.1206727
180. Chen N, Chen X, Huang R, Zeng H, Gong J, Meng W, et al. BCL-XL is a target gene regulated by hypoxia-inducible factor-1{alpha}. J Biol Chem (2009) 284:10004–12. doi: 10.1074/jbc.M805997200
181. Niture SK, Jaiswal AK. Nrf2-induced antiapoptotic bcl-XL protein enhances cell survival and drug resistance. Free Radic Biol Med (2013) 57:119–31. doi: 10.1016/j.freeradbiomed.2012.12.014
182. Son Y, Cheong Y-K, Kim N-H, Chung H-T, Kang DG, Pae H-O. Mitogen-activated protein kinases and reactive oxygen species: How can ROS activate MAPK pathways? J Signal Transduct (2011) 2011:792639. doi: 10.1155/2011/792639
183. Spratlin JL, Serkova NJ, Eckhardt SG. Clinical applications of metabolomics in oncology: A review. Clin Cancer Res (2009) 15:431–40. doi: 10.1158/1078-0432.CCR-08-1059
184. Montero J, Sarosiek KA, DeAngelo JD, Maertens O, Ryan J, Ercan D, et al. Drug-induced death signaling strategy rapidly predicts cancer response to chemotherapy. Cell (2015) 160:977–89. doi: 10.1016/j.cell.2015.01.042
185. Taylor Ripley R, Surman DR, Diggs LP, Trepel JB, Lee M-J, Ryan J, et al. Metabolomic and BH3 profiling of esophageal cancers: Novel assessment methods for precision therapy. BMC Gastroenterol (2018) 18:94. doi: 10.1186/s12876-018-0823-x
186. Lwin T, Lin J, Choi YS, Zhang X, Moscinski LC, Wright KL, et al. Follicular dendritic cell-dependent drug resistance of non-Hodgkin lymphoma involves cell adhesion-mediated bim down-regulation through induction of MicroRNA-181a. Blood (2010) 116:5228–36. doi: 10.1182/blood-2010-03-275925
187. Chiron D, Bellanger C, Papin A, Tessoulin B, Dousset C, Maiga S, et al. Rational targeted therapies to overcome microenvironment-dependent expansion of mantle cell lymphoma. Blood (2016) 128:2808–18. doi: 10.1182/blood-2016-06-720490
188. Sehn LH, Chua N, Mayer J, Dueck G, Trněný M, Bouabdallah K, et al. Obinutuzumab plus bendamustine versus bendamustine monotherapy in patients with rituximab-refractory indolent non-Hodgkin lymphoma (GADOLIN): A randomised, controlled, open-label, multicentre, phase 3 trial. Lancet Oncol (2016) 17:1081–93. doi: 10.1016/S1470-2045(16)30097-3
189. Riethdorf S, Müller V, Zhang L, Rau T, Loibl S, Komor M, et al. Detection and HER2 expression of circulating tumor cells: Prospective monitoring in breast cancer patients treated in the neoadjuvant GeparQuattro trial. Clin Cancer Res (2010) 16:2634–45. doi: 10.1158/1078-0432.CCR-09-2042
190. Meng S, Tripathy D, Shete S, Ashfaq R, Haley B, Perkins S, et al. HER-2 gene amplification can be acquired as breast cancer progresses. Proc Natl Acad Sci U.S.A. (2004) 101:9393–8. doi: 10.1073/pnas.0402993101
191. Meng S, Tripathy D, Shete S, Ashfaq R, Saboorian H, Haley B, et al. UPAR and HER-2 gene status in individual breast cancer cells from blood and tissues. Proc Natl Acad Sci USA (2006) 103:17361–5. doi: 10.1073/pnas.0608113103
192. Smerage JB, Budd GT, Doyle GV, Brown M, Paoletti C, Muniz M, et al. Monitoring apoptosis and bcl-2 on circulating tumor cells in patients with metastatic breast cancer. Mol Oncol (2013) 7:680–92. doi: 10.1016/j.molonc.2013.02.013
193. Albain KS, Barlow WE, Shak S, Hortobagyi GN, Livingston RB, Yeh I-T, et al. Prognostic and predictive value of the 21-gene recurrence score assay in postmenopausal women with node-positive, oestrogen-Receptor-Positive breast cancer on chemotherapy: A retrospective analysis of a randomised trial. Lancet Oncol (2010) 11:55–65. doi: 10.1016/S1470-2045(09)70314-6
194. Callagy GM, Webber MJ, Pharoah PDP, Caldas C. Meta-analysis confirms BCL2 is an independent prognostic marker in breast cancer. BMC Cancer (2008) 8:153. doi: 10.1186/1471-2407-8-153
195. Dowsett M, Cuzick J, Wale C, Forbes J, Mallon EA, Salter J, et al. Prediction of risk of distant recurrence using the 21-gene recurrence score in node-negative and node-positive postmenopausal patients with breast cancer treated with anastrozole or tamoxifen: A TransATAC study. J Clin Oncol (2010) 28:1829–34. doi: 10.1200/JCO.2009.24.4798
196. Hayes DF, Smerage J. Is there a role for circulating tumor cells in the management of breast cancer? Clin Cancer Res (2008) 14:3646–50. doi: 10.1158/1078-0432.CCR-07-4481
197. Lang JE, Scott JH, Wolf DM, Novak P, Punj V, Magbanua MJM, et al. Expression profiling of circulating tumor cells in metastatic breast cancer. Breast Cancer Res Treat (2015) 149:121–31. doi: 10.1007/s10549-014-3215-0
198. Meng S, Tripathy D, Frenkel EP, Shete S, Naftalis EZ, Huth JF, et al. Circulating tumor cells in patients with breast cancer dormancy. Clin Cancer Res (2004) 10:8152–62. doi: 10.1158/1078-0432.CCR-04-1110
199. Thangavel H, De Angelis C, Vasaikar S, Bhat R, Jolly MK, Nagi C, et al. A CTC-Cluster-Specific signature derived from OMICS analysis of patient-derived xenograft tumors predicts outcomes in basal-like breast cancer. J Clin Med (2019) 8:E1772. doi: 10.3390/jcm8111772
200. Hou J-M, Krebs M, Ward T, Sloane R, Priest L, Hughes A, et al. Circulating tumor cells as a window on metastasis biology in lung cancer. Am J Pathol (2011) 178:989–96. doi: 10.1016/j.ajpath.2010.12.003
201. Fina E, Reduzzi C, Motta R, Di Cosimo S, Bianchi G, Martinetti A, et al. Did circulating tumor cells tell us all they could? the missed circulating tumor cell message in breast cancer. Int J Biol Markers (2015) 30:e429–433. doi: 10.5301/jbm.5000166
202. Hayashi N, Nakamura S, Tokuda Y, Shimoda Y, Yagata H, Yoshida A, et al. Prognostic value of HER2-positive circulating tumor cells in patients with metastatic breast cancer. Int J Clin Oncol (2012) 17:96–104. doi: 10.1007/s10147-011-0260-0
203. Wang C, Mu Z, Chervoneva I, Austin L, Ye Z, Rossi G, et al. Longitudinally collected CTCs and CTC-clusters and clinical outcomes of metastatic breast cancer. Breast Cancer Res Treat (2017) 161:83–94. doi: 10.1007/s10549-016-4026-2
204. Ozretic P, Alvir I, Sarcevic B, Vujaskovic Z, Rendic-Miocevic Z, Roguljic A, et al. Apoptosis regulator bcl-2 is an independent prognostic marker for worse overall survival in triple-negative breast cancer patients. Int J Biol Markers (2018) 33:109–15. doi: 10.5301/ijbm.5000291
205. Yadav A, Kumar B, Yu J-G, Old M, Teknos TN, Kumar P. Tumor-associated endothelial cells promote tumor metastasis by chaperoning circulating tumor cells and protecting them from anoikis. PloS One (2015) 10:e0141602. doi: 10.1371/journal.pone.0141602
206. Pilati P, Mocellin S, Bertazza L, Galdi F, Briarava M, Mammano E, et al. Prognostic value of putative circulating cancer stem cells in patients undergoing hepatic resection for colorectal liver metastasis. Ann Surg Oncol (2012) 19:402–8. doi: 10.1245/s10434-011-2132-2
207. Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong C, et al. Let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell (2007) 131:1109–23. doi: 10.1016/j.cell.2007.10.054
208. Hirschmann-Jax C, Foster AE, Wulf GG, Nuchtern JG, Jax TW, Gobel U, et al. Distinct “Side population” of cells with high drug efflux capacity in human tumor cells. Proc Natl Acad Sci U.S.A. (2004) 101:14228–33. doi: 10.1073/pnas.0400067101
209. Zhong X, Rescorla FJ. Cell surface adhesion molecules and adhesion-initiated signaling: Understanding of anoikis resistance mechanisms and therapeutic opportunities. Cell Signal (2012) 24:393–401. doi: 10.1016/j.cellsig.2011.10.005
210. Avivar-Valderas A, Salas E, Bobrovnikova-Marjon E, Diehl JA, Nagi C, Debnath J, et al. PERK integrates autophagy and oxidative stress responses to promote survival during extracellular matrix detachment. Mol Cell Biol (2011) 31:3616–29. doi: 10.1128/MCB.05164-11
211. Rai P. Oxidation in the nucleotide pool, the DNA damage response and cellular senescence: Defective bricks build a defective house. Mutat Res (2010) 703:71–81. doi: 10.1016/j.mrgentox.2010.07.010
212. Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature (2009) 458:780–3. doi: 10.1038/nature07733
213. Tobin LA, Robert C, Nagaria P, Chumsri S, Twaddell W, Ioffe OB, et al. Targeting abnormal DNA repair in therapy-resistant breast cancers. Mol Cancer Res (2012) 10:96–107. doi: 10.1158/1541-7786.MCR-11-0255
214. Dai Y, Grant S. New insights into checkpoint kinase 1 in the DNA damage response signaling network. Clin Cancer Res (2010) 16:376–83. doi: 10.1158/1078-0432.CCR-09-1029
215. Ashwell S, Janetka JW, Zabludoff S. Keeping checkpoint kinases in line: New selective inhibitors in clinical trials. Expert Opin Investig Drugs (2008) 17:1331–40. doi: 10.1517/13543784.17.9.1331
216. Jordan NV, Bardia A, Wittner BS, Benes C, Ligorio M, Zheng Y, et al. HER2 expression identifies dynamic functional states within circulating breast cancer cells. Nature (2016) 537:102–6. doi: 10.1038/nature19328
217. Arteaga CL, Engelman JA. ERBB receptors: From oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell (2014) 25:282–303. doi: 10.1016/j.ccr.2014.02.025
218. Houssami N, Macaskill P, Balleine RL, Bilous M, Pegram MD. HER2 discordance between primary breast cancer and its paired metastasis: Tumor biology or test artefact? insights through meta-analysis. Breast Cancer Res Treat (2011) 129:659–74. doi: 10.1007/s10549-011-1632-x
219. Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer (2009) 9:239–52. doi: 10.1038/nrc2618
220. Beerepoot LV, Mehra N, Vermaat JSP, Zonnenberg BA, Gebbink MFGB, Voest EE. Increased levels of viable circulating endothelial cells are an indicator of progressive disease in cancer patients. Ann Oncol (2004) 15:139–45. doi: 10.1093/annonc/mdh017
221. Kumar P, Ning Y, Polverini PJ. Endothelial cells expressing bcl-2 promotes tumor metastasis by enhancing tumor angiogenesis, blood vessel leakiness and tumor invasion. Lab Invest (2008) 88:740–9. doi: 10.1038/labinvest.2008.46
222. Paoli P, Giannoni E, Chiarugi P. Anoikis molecular pathways and its role in cancer progression. Biochim Biophys Acta (2013) 1833:3481–98. doi: 10.1016/j.bbamcr.2013.06.026
223. Bouchard V, Demers M-J, Thibodeau S, Laquerre V, Fujita N, Tsuruo T, et al. Fak/Src signaling in human intestinal epithelial cell survival and anoikis: Differentiation state-specific uncoupling with the PI3-K/Akt-1 and MEK/Erk pathways. J Cell Physiol (2007) 212:717–28. doi: 10.1002/jcp.21096
Keywords: CTC, mitochondria, ROS, drug resistance, invasiveness
Citation: Agnoletto C and Volinia S (2022) Mitochondria dysfunction in circulating tumor cells. Front. Oncol. 12:947479. doi: 10.3389/fonc.2022.947479
Received: 18 May 2022; Accepted: 11 July 2022;
Published: 04 August 2022.
Edited by:
Hamid Morjani, Université de Reims Champagne-Ardenne, FranceReviewed by:
Gloria Bonuccelli, University of Salford, United KingdomM. Cecilia Caino, University of Colorado Anschutz Medical Campus, United States
Copyright © 2022 Agnoletto and Volinia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chiara Agnoletto, Y2hpYXJhLmFnbm9sZXR0b0Bpb3YudmVuZXRvLml0