 Kristina Zalevskaja
Kristina Zalevskaja Jukka-Pekka Mecklin3,4
Jukka-Pekka Mecklin3,4 Toni T. Seppälä
Toni T. Seppälä- 1Applied Tumor Genomics Research Program, Research Programs Unit, University of Helsinki, Helsinki, Finland
- 2Department of Gastrointestinal Surgery, Helsinki University Central Hospital, Helsinki, Finland
- 3Faculty of Sport and Health Sciences, University of Jyväskylä, Jyväskylä, Finland
- 4Department of Education and Research, Jyväskylä Hospital Nova, Jyväskylä, Finland
- 5Faculty of Medicine and Health Technology and Tays Cancer Centre, University of Tampere, Tampere, Finland
Introduction: Patients with Lynch syndrome (LS) have an increased lifetime risk of pancreatic cancer (PC) and biliary tract cancer (BTC). These cancers have a notoriously pessimistic prognosis due to late diagnosis and limited therapeutic options. There are limited data based on small cohorts reviewing PC and BTC in LS patients.
Methods: In this retrospective study of the Lynch Syndrome Registry of Finland (LSRFi), records of genetically verified LS patients diagnosed with PC or BTC between 1982 and 2020 were analyzed.
Results: Thirty-nine patients were included: tumor(s) were in the pancreas in 26 patients, in the biliary tract in 10, and in the ampulla of Vater in three. A pathogenic germline variant was found in MLH1 in 33 of 39 patients. Twenty-six patients with 28 tumors located in the pancreas were identified: 23 pancreatic ductal adenocarcinomas (PDACs) and five neuroendocrine tumors (NETs). The median age at diagnosis of PC was 64 years (range of 38–81). In PC, the 5-year overall survival (OS) rate was 20%, and in PDAC, it was 13.6%. Ten patients with BTC were diagnosed: two intrahepatic, five perihilar, two distal extrahepatic cholangiocarcinomas, and one gallbladder carcinoma. Eight patients were male, and the median age at diagnosis was 54 years (range of 34–82). The 5-year OS rate for BTC was 30%. Metachronous tumors were diagnosed in 28 patients (70%). Colorectal cancer was the most common metachronous tumor, diagnosed in 20 patients (51%), and diagnosed prior to PC or BTC in all cases. Curative surgery was attempted on 17 of 39 patients. For 30 patients (91%), the cause of death was PC or BTC; two patients died from another LS-associated cancer, and one died from a stroke.
Conclusion: Although the survival of LS patients with PC or BTC is better than in sporadic cancers, it is still poor and may be reflected by the relatively higher surgical resectability accounted for by the earlier age of onset. More studies on analyses of the molecular and immune profile, screening, and management of LS-associated pancreaticobiliary cancers are warranted.
1 Introduction
Lynch syndrome (LS), previously known as hereditary nonpolyposis colorectal cancer (HNPCC), is an autosomal dominant disorder caused by pathogenic germline variants in one of the DNA mismatch repair (MMR) genes, MLH1, MSH2, MSH6, or PMS2, or by deletions in the EPCAM gene (1–3). It is the most common hereditary cancer syndrome, with a prevalence estimated as high as 1 in 279 (4). Pathogenic MMR variant carriers have a high lifetime risk of developing colorectal and endometrial cancers and an increased risk of developing gastric, ovarian, urothelial, pancreatic, biliary tract, small bowel, prostate, breast, brain, and skin cancers, depending on the gene affected (5). LS-associated cancers usually display MMR deficiency (dMMR) that leads to microsatellite instability (MSI) in the tumors.
Increased risk of pancreatic cancer (PC) in LS carriers was first observed by Lynch et al. in 1985 (6). In 1992, Mecklin et al. described 11 LS patients with biliary tract cancer (BTC), suggesting an association between BTC and LS (7). Since then, numerous retrospective studies and one review have confirmed an increased incidence of PCs and BTCs in LS patients (8–12). The Prospective Lynch Syndrome Database (PLSD) report has shown different lifetime risks for PC depending on the germline mutation variant (5).
LS-associated colorectal, gynecological, and gastric cancers have a better prognosis than sporadic cancers (5, 13–16). Unfortunately, PC and BTC remain aggressive and have a poor prognosis. As of lately, immune checkpoint inhibitor therapy has been an exciting development in the treatment of solid tumors with MSI and dMMR, including PC and BTC, with promising results (17–20). However, there are limited data based only on small cohorts reviewing pancreatic and biliary tract malignancies in LS patients.
In this article, we present the largest cohort of LS patients with PC and BTC to date and characterize their clinical features.
2 Methods
2.1 Study cohort
We retrospectively reviewed the medical records of patients in the Lynch Syndrome Registry of Finland (LSRFi) who were diagnosed with pancreatic or biliary tract malignant tumors between 1982 and 2020. The nationwide registry, established in 1982, includes, at present, 1,800 verified pathogenic variant carriers from 400 families and contains clinicopathological information on all cancers of registered individuals. The data have been regularly cross-checked against the Finnish national cancer registry.
This multicenter retrospective study was approved by the national authority for registry research (Findata), waiving the requirement for informed consent to use data obtained from medical records.
2.2 Survival analyses
Overall survival (OS) was defined as the time from diagnosis until death from any cause or the last date of confirmed survival. OS was analyzed in R using the Kaplan–Meier method (21).
2.3 Pathological classification
Pancreatic adenocarcinomas (PDACs) were graded according to histopathological WHO criteria (22). Pancreatic neuroendocrine tumors (NETs) were classified according to the WHO 2020 classification for pancreatic neuroendocrine neoplasms (23). Biliary tract tumors were classified according to system based on their anatomical location and categorized as intrahepatic, perihilar, or distal cholangiocarcinomas (24). According to the WHO classification of digestive system tumors, adenocarcinomas of the ampulla of Vater are histologically closer to the small intestine but anatomically near the pancreas and biliary tract (22). Therefore, this rare type of cancer was included in the cohort but analyzed separately.
3 Results
3.1 Patient characteristics
Forty LS patients were diagnosed with PC or BTC or ampullary cancer between 1982 and 2020 (Table 1). Among them, tumors in 26 patients were in the pancreas, 10 in the biliary tract, and three in the ampulla of Vater. One patient was excluded from the study due to non-pancreatic and non-biliary histology. A pathogenic germline variant of MLH1 was detected in 33 patients, MSH2 in five, and MSH6 in one patient. MLH1 variant carriers had 21 out of 26 PCs, nine out of 10 BTC, and all three ampullary cancers (Figure 1).
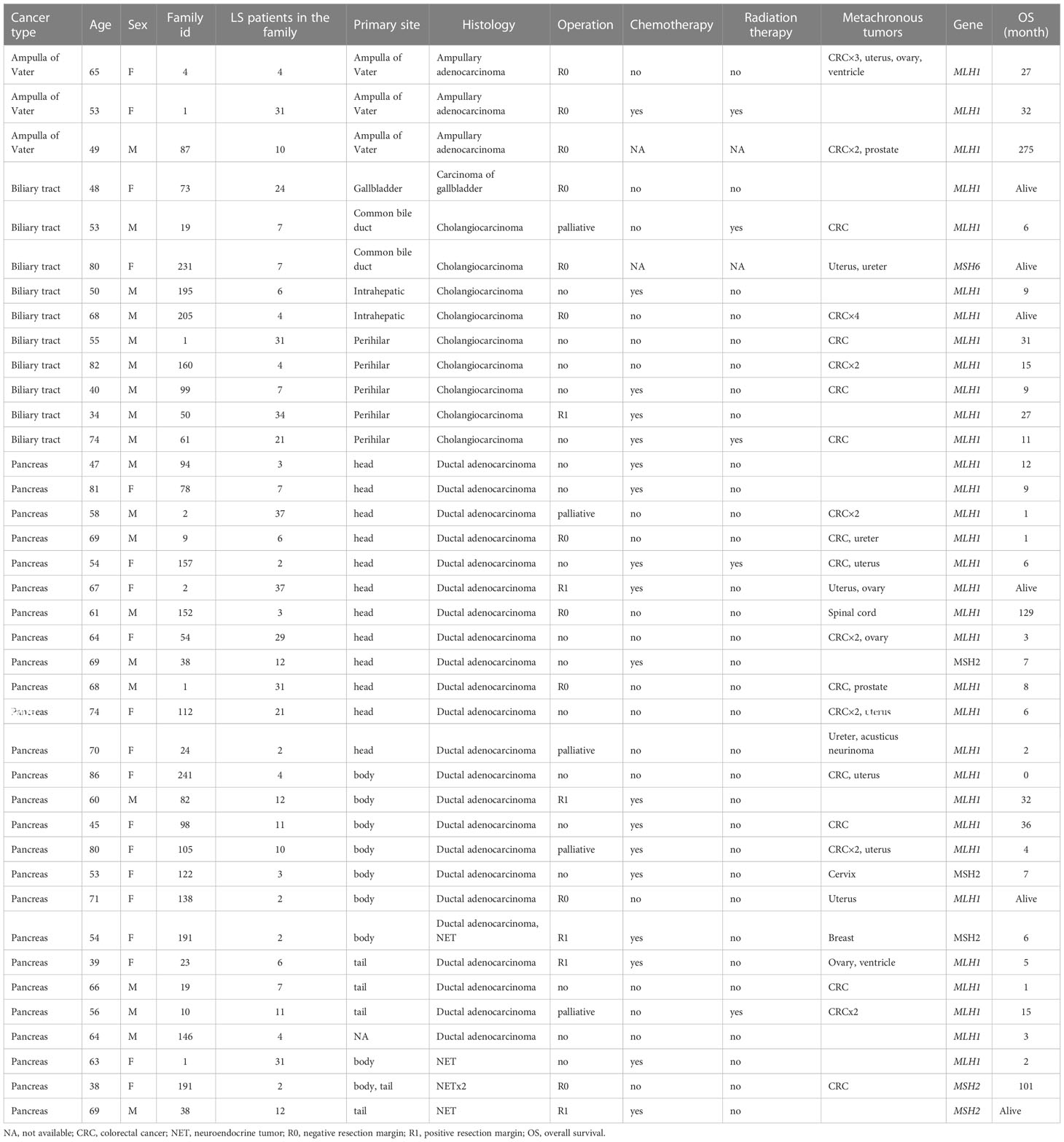
Table 1 Clinicopathological characteristics of PC, BTC, and ampullary cancer in LS patients.
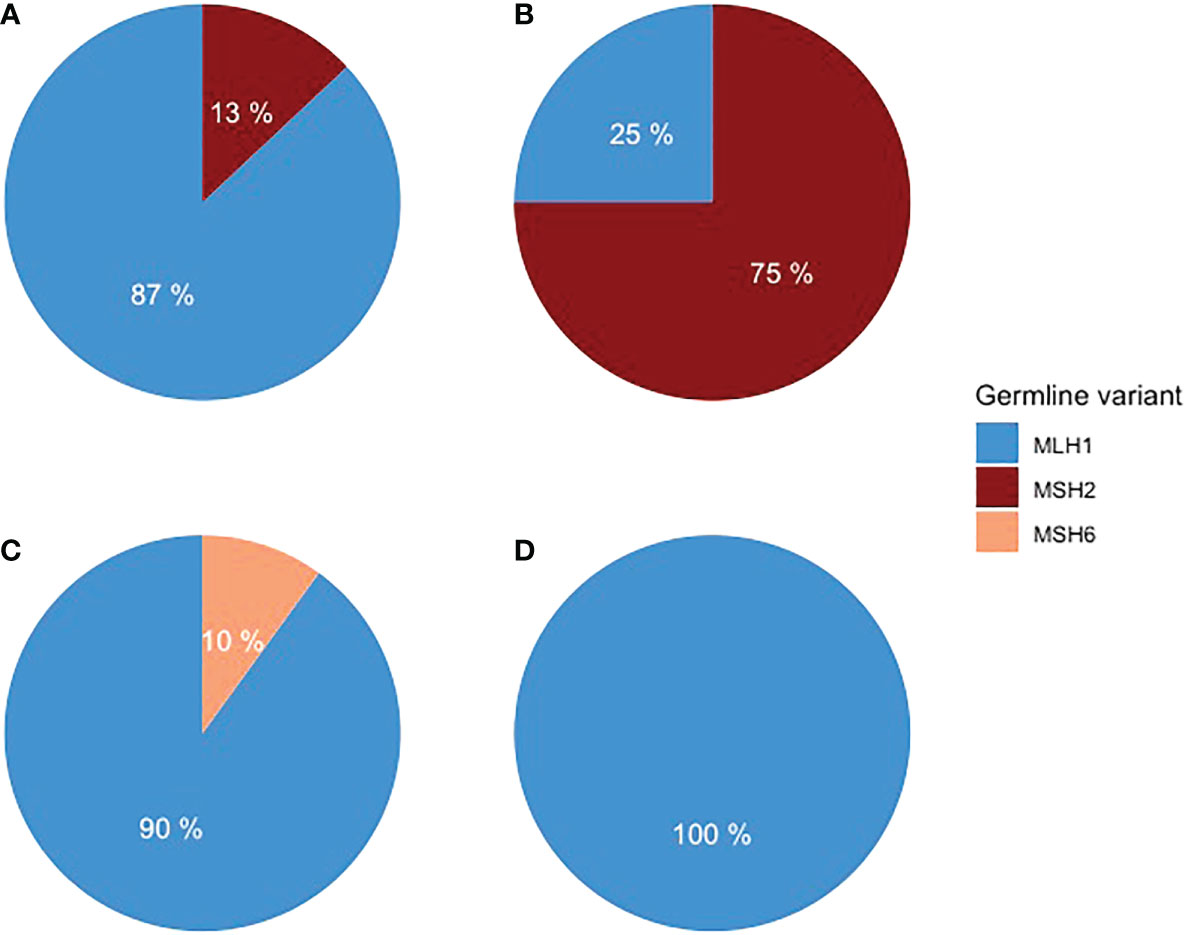
Figure 1 Distribution of germline variants in (A) PDAC, (B) pancreatic NET, (C) BTC, and (D) ampullary adenocarcinoma.
LS was diagnosed prior to PC or BTC in 22 patients and simultaneously in 14 patients. Each patient’s family in LSRFi and the number of LS-diagnosed patients in this family are presented in Table 1. Five families had more than one family member diagnosed with PC or BTC.
In 26 patients with 28 tumors located in the pancreas, 23 were PDAC and five were NETs. Fifteen were female, and the median age at diagnosis was 64 years (range of 38–81). The distribution of anatomical locations of PC was head 12, body 10, and tail five (one location was not recorded). Germline variants of MLH1 were detected in 21 patients and MSH2 in five patients. One patient had simultaneously PDAC and NET, and one patient had two NETs. In both patients, multiple endocrine neoplasia type 1 (MEN1) syndrome was additionally diagnosed. The diagnosis of pancreatic malignancies in 20 patients was based on clinical, radiological, and pathology reports, but in six patients, it was based on clinical and radiological findings only.
Of the 10 BTC patients, two were intrahepatic, five perihilar, two distal extrahepatic cholangiocarcinomas, and one gallbladder carcinoma. Eight were male, and the median age at diagnosis was 54 years (ranging from 34 to 82). Germline variants of MLH1 were detected in nine patients and of MSH6 in one patient. The BTC diagnosis was based on a pathology report in seven patients and on computer tomography in three patients. The patient with gallbladder carcinoma was primarily operated on due to pain caused by gallstones but received an unexpected diagnosis of gallbladder carcinoma. The treatment was later completed with liver segment II–IV resections with R0 margins.
Adenocarcinoma of the ampulla of Vater was diagnosed in three patients, all MLH1 carriers. Two were women, and the median age at diagnosis was 53 years (ranging from 49 to 65). As all three patients underwent surgery, the diagnosis was verified by a pathological report.
3.2 Metachronous tumors
Metachronous tumors were diagnosed in 28 (72%) patients (median number 1; range of 0–6). Colorectal cancer was the most common, diagnosed in 20 patients, and endometrial cancer in eight patients. In all cases, colorectal cancer and endometrial cancer were diagnosed prior to PC or BTC.
3.3 Treatment and survival
Curative surgery was attempted in 17 (43.5%) patients and treatment with non-curative intent, life-prolonging or palliative therapy, was provided to 22 patients (Figure 2).
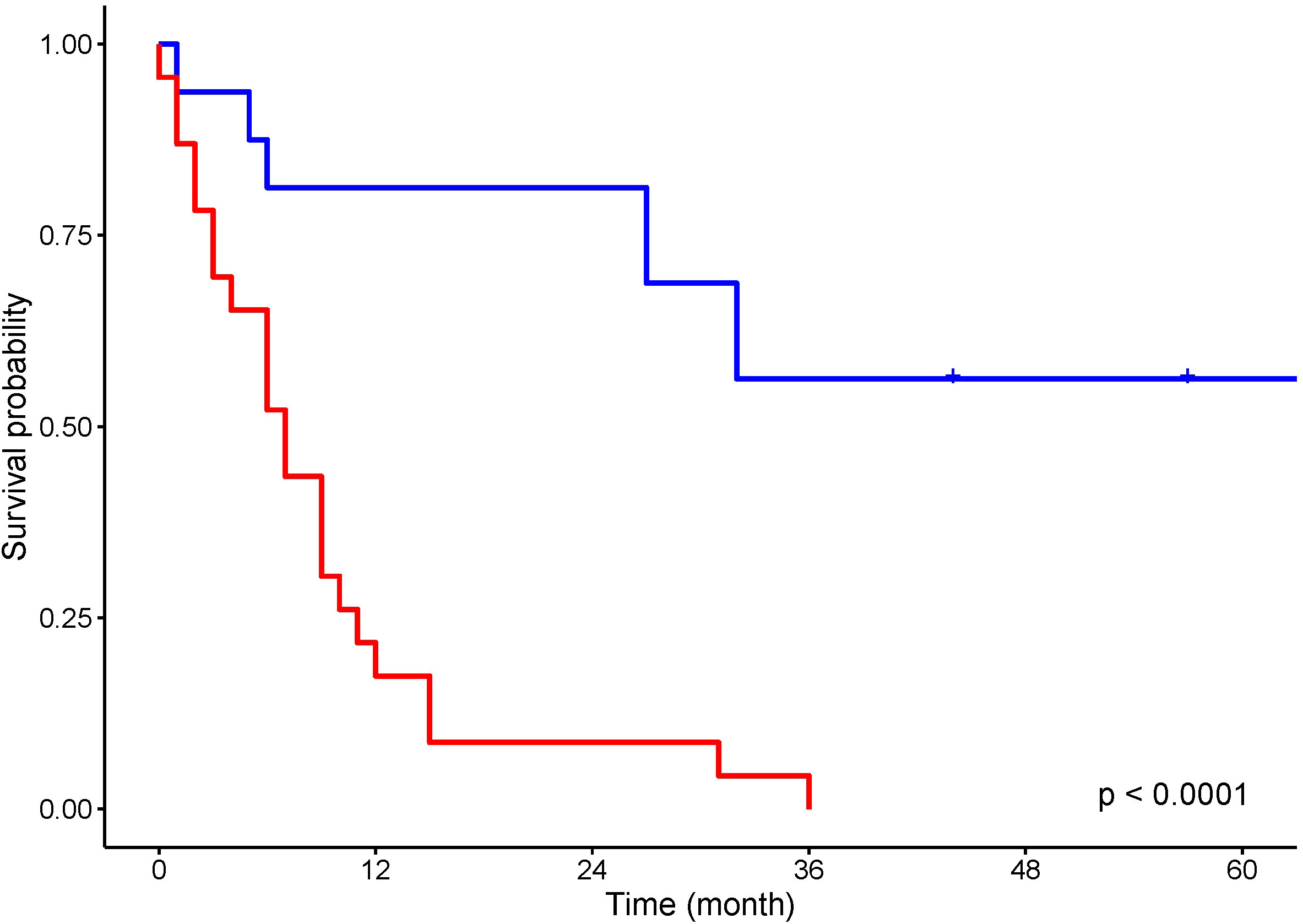
Figure 2 Kaplan–Meier analysis for overall survival of patients treated with curative intent (blue) and non curative intent (red).
Ten out of 26 patients with PC underwent surgery with curative intent. Five patients had surgical resection with a negative resection margin. Five patients’ resection margins were positive, and adjuvant chemotherapy was administered to these patients. In LS patients with PC who were treated with curative intent, the 5-year survival rate was 50%. In the 10 LS patients with PDAC resected with curative intent, the 5-year survival rate was 38%. Seven patients were treated with chemotherapy and one patient with chemoradiation therapy. One patient received additional immunotherapy with pembrolizumab for MLH1/PMS2-deficient PDAC. Eight patients were provided with symptomatic treatment. In 16 patients who were not treated with curative intent, the median OS was 5 months. For all patients with PC, the 5-year OS rate was 20%, and for those with PDAC, it was 13.6% (Figure 3A). One patient died from pneumonia after a palliative operation within 72 h and was excluded from survival calculations. Endoscopic stent placement was performed in eight cases and percutaneous transhepatic drainage in one.
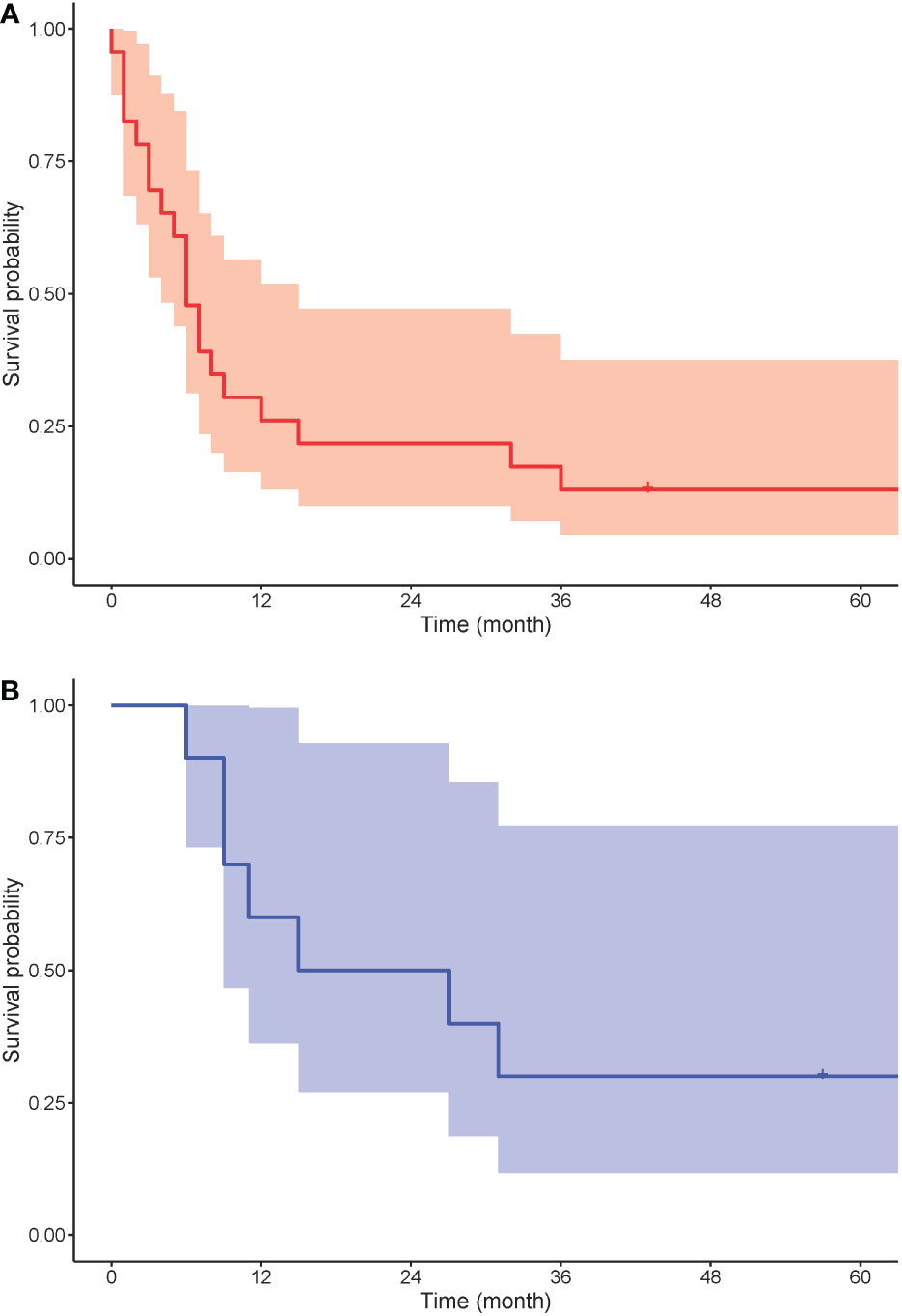
Figure 3 Kaplan-Meier survival analysis for overall survival of (A) PDAC (red) and (B) BTC (blue).
Four patients with BTC underwent surgical resection with curative intent. Three patients’ resection margin was negative. One patient’s resection margin was positive, and adjuvant chemotherapy was administered. Among these four patients with BTC who were operated on with curative intent, the 5-year survival rate was 75%. Two patients were treated with chemotherapy, and one with chemoradiation therapy. Three patients were treated symptomatically. In six patients who were not treated with curative intent, the median OS was 10 months. For LS patients with BTC, the 5-year OS rate was 30% (Figure 3B). Endoscopic stent placement was performed in three cases, and percutaneous transhepatic drainage was placed in four cases.
All three patients with adenocarcinomas of the ampulla of Vater underwent curative surgery with a negative resection margin. Still two patients died of recurrence of ampullary cancer within 1.5 years and one from small bowel cancer.
Thirty-three patients (85%) out of 39 were deceased. For thirty patients (91%), the cause of death was PC, BTC, or ampullary cancer. Two patients died from another LS-associated cancer and one patient died from a stroke. An overview of clinical characteristics and treatment is presented in Table 2.
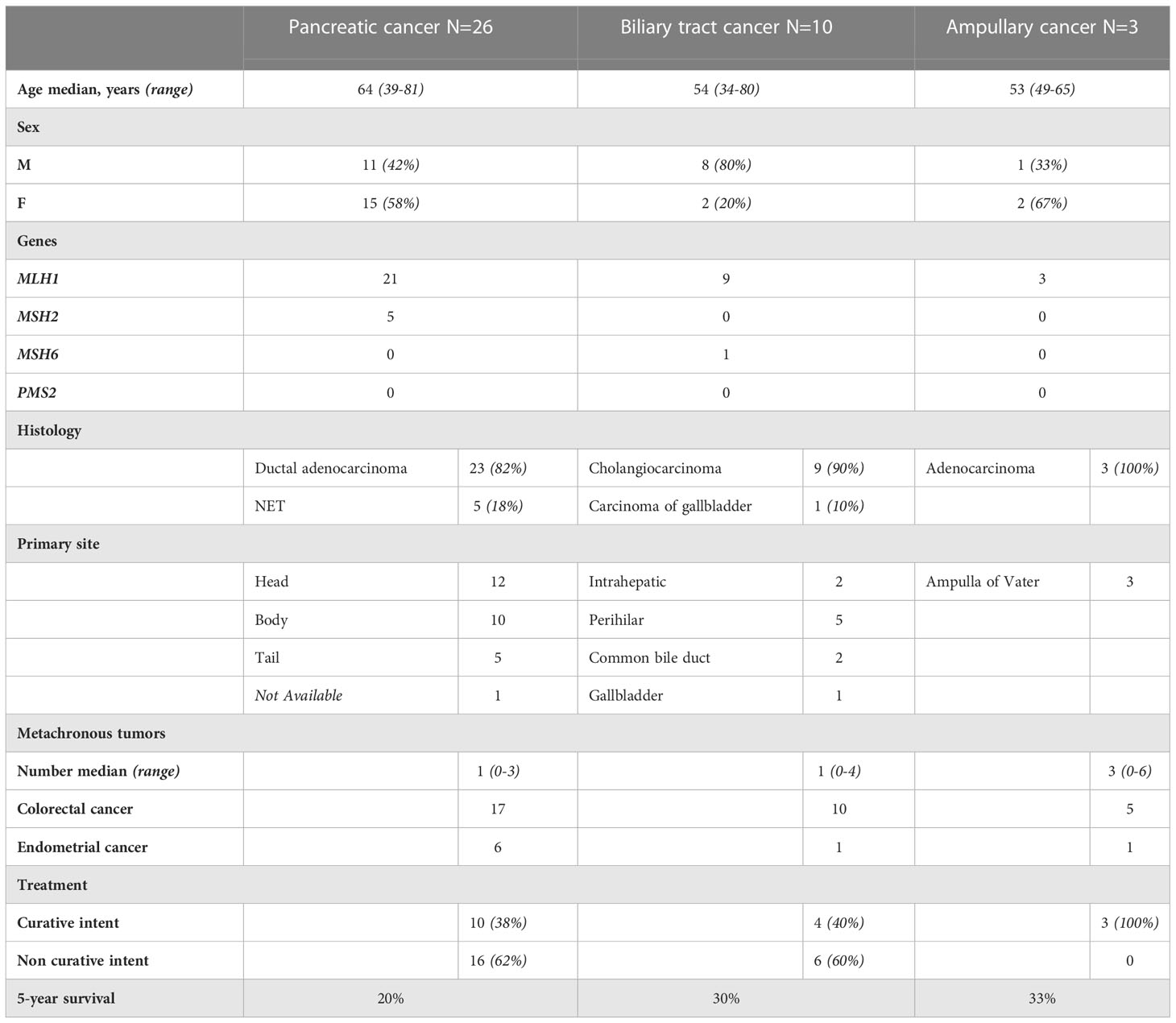
Table 2 Overview of clinical characteristics and treatment in LS patients with pancreatic, biliary tract and ampullary cancer.
4 Discussion
Sporadic PCs and BTCs have a dismal prognosis due to being asymptomatic in the early stages, resulting in a late diagnosis. Treatment options are limited and lack effectiveness. The five-year survival rate is only 10% for both cancer types in the United States (25, 26). LS patients have an increased lifetime risk of developing PC and BTC. Analysis of data from the Prospective Lynch Syndrome Database (PLSD) has shown the cumulative risk at 75 years of age for PC is 6.2%, 0.5%, and 1.4%, and for BTC is 3.7%, 1.7%, and 0%, respectively, for carriers of MLH1, MSH2, and MSH6 germline variants (5). Our retrospective study supports pathogenic MLH1 germline variant carriers being overrepresented among LS patients with PC and BTC.
We did not detect any pancreaticobiliary cancers in PMS2 carriers, although the number of identified PMS2 families is low in Finland. No clear evidence of an increased risk of PC or BTC has been shown in PMS2 pathogenic variant carriers, even in the larger series (27). In a study by Møller et al., none of the 124 PMS2 pathogenic variant carriers were diagnosed with PC or BTC (5). Hu et al. reported three PMS2 carriers with PDAC. Two of these patients developed MMR-proficient PDAC (28). Ando et al. performed immunohistochemistry (IHC) analysis on 116 operated BTC patients, identifying two PMS2 germline variant carriers, both microsatellite stable (MSS) (29). These findings suggest that PDAC and BTC in PMS2 germline mutation carriers might be sporadic. The cautionary tale of Wang et al. raises the importance of routine tumor testing for both MMR deficiency and MSI to detect patients who might have a better chance of responding to immunotherapy (30). Hendifar et al. presented a case report underscoring the importance of testing every cancer in LS patients for MMR, as not all of them might respond to immunotherapy (31).
The incidence of sporadic PC and BTC increases with age and the median age at the diagnosis is 70 years (32). In the current study, LS patients with PC or BTC were younger, resembling the early age phenomenon which is typical for all LS associated cancers. Also, PC was diagnosed equally in females and males. The small sample size of this study does not allow definitive conclusions drawn, but the latest PLSD report did report substantial sex difference in upper gastrointestinal cancers with 22% in male MLH1 carriers by 75 years versus 11% in female MLH1 carriers (33).
Three-quarters of LS patients with PC or BTC had metachronous tumors. Colorectal and endometrial cancers were diagnosed in all cases prior to PC or BTC. These findings suggest that most, but not all, LS patients develop PC or BTC later in life after the more common primary cancers. A personal cancer history of LS carriers over 60 years of age may serve as an indicator for healthcare to stay alert for unspecific symptoms the upper gastrointestinal cancers may induce. On the other hand, a quarter of the patients did not have a previous cancer history, and PC or BTC was their first malignancy.
Most sporadic PCs are in the head of the pancreas. PC in the body or tail has a worse prognosis compared to pancreatic head cancer due to remaining asymptomatic for a longer period, resulting in late diagnoses (34, 35). Takamizawa et al. described the anatomical location of the PC in six LS patients, identifying five of the six PCs as being in the body and the tail of the pancreas (36). In our study, PC was located equally often in the head, the body, and the tail of the pancreas. Our series suggests that no distinct primary anatomical site is more prevalent. BTC was found in all parts of the biliary tract, as also previously shown by Cloyd et al. (11).
Surgical resection is the only curative treatment for PC and BTC. Curative surgery can be performed in 10%–20% of sporadic PC cases and in 20% of sporadic BTC cases (37, 38). In this study, curative surgical resection was performed twice as often, resulting in better overall survival outcomes. This might be explained by the fact that half of the cases already had an LS diagnosis and participated in regular surveillance. In Finland, surveillance for PC and BTC in LS patients is symptom-based, as European guidelines for LS recommend (13). In practice, it means LS patients are educated about symptoms they might encounter and are encouraged to contact secondary and tertiary healthcare providers with expertise in LS if they experience any “red flag” symptoms.
Immune modulation therapy with checkpoint inhibition is a new promising option for LS-associated cancer types with poor prognosis, as histology-agnostic FDA approval for any dMMR or MSI solid cancers is in place (18). Pancreaticobiliary cancers are often deemed unresectable, but a proper molecular pathological examination revealing MSI with even some response to checkpoint inhibition may convert an inoperable case back to operable. However, good biopsies for histology might be difficult to get, and especially known LS carriers should be referred to experienced centers with high volumes of endoscopic ultrasound-guided biopsies to avoid false-negative biopsies for dMMR and MSI due to poor sample quality. It is especially important to not suffice with imaging or cytology-informed diagnosis alone, but a diagnostic biopsy for dMMR or MSI testing must be obtained in all cases with known or suspected LS.
This study has several limitations, such as a retrospective design, a small sample size, and, in some cases, a lack of pathological verification of cancer. Even though the small sample size of this cohort limits the power of statistical analysis, it is still the largest series reported to date. Although it seems that the survival of LS patients with PC or BTC is better than in sporadic cancers, it is still poor. The relatively higher surgical resectability may be accounted for by selection bias due to the earlier age of onset.
To conclude, there is a growing need for molecular and immune profiling of LS-associated PDAC and BTC to clarify the suitability of these cancers with an extremely poor prognosis for immune or any other upcoming therapy.
Data availability statement
The original contributions presented in the study are included in the article/supplementary materials. Further inquiries can be directed to the corresponding authors.
Ethics statement
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
Author contributions
KZ: Data curation, formal analysis, writing—original draft. J-PM: Conceptualization, supervision, writing—review and editing. TS: Supervision, writing—review and editing. All authors contributed to the article and approved the submitted version.
Funding
The research of the group was funded by the Jane and Aatos Erkko Foundation, Cancer Foundation Finland, Sigrid Juselius Foundation, Emil Aaltonen Foundation, Relander Foundation, Academy of Finland, iCAN Digital Precision Medicine flagship, and the State research funding.
Acknowledgments
The authors would like to acknowledge Maija Röntynen and Kirsi Pylvänäinen for help with the Lynch Syndrome Registry of Finland (LSRFi). We would like to thank Erdogan Pekcan Erkan for proofreading the article.
Conflict of interest
TS reports consultation fees from Boehringer Ingelheim Finland and Amgen Finland and being a co-owner and CEO of Healthfund Finland.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Lynch H, Lynch P, Lanspa S, Snyder C, Lynch J, Boland C. Review of the lynch syndrome: history, molecular genetics, screening, differential diagnosis, and medicolegal ramifications. Clin Genet (2009) 76(1):1–18. doi: 10.1111/j.1399-0004.2009.01230.x
2. Ligtenberg MJL, Kuiper RP, Chan TL, Goossens M, Hebeda KM, Voorendt M, et al. Heritable somatic methylation and inactivation of MSH2 in families with lynch syndrome due to deletion of the 3′ exons of TACSTD1. Nat Genet (2009) 41(1):112–7. doi: 10.1038/ng.283
3. Kovacs ME, Papp J, Szentirmay Z, Otto S, Olah E. Deletions removing the last exon of TACSTD1 constitute a distinct class of mutations predisposing to lynch syndrome. Hum Mutation. (2009) 30(2):197–203. doi: 10.1002/humu.20942
4. Win AK, Jenkins MA, Dowty JG, Antoniou AC, Lee A, Giles GG, et al. Prevalence and penetrance of major genes and polygenes for colorectal cancer. Cancer Epidemiol Biomarkers Prev (2017) 26(3):404–12. doi: 10.1158/1055-9965.EPI-16-0693
5. Møller P, Seppälä TT, Bernstein I, Holinski-Feder E, Sala P, Evans DG, et al. Cancer risk and survival in path_MMR carriers by gene and gender up to 75 years of age: a report from the prospective lynch syndrome database. Gut (2018) 67(7):1306–16. doi: 10.1136/gutjnl-2017-314057
6. Lynch HT, Voorhees GJ, Lanspa SJ, McGreevy PS, Lynch JF. Pancreatic carcinoma and hereditary nonpolyposis colorectal cancer: a family study(1985). Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1977116/ (Accessed April 6, 2022).
7. Mecklin JP, Järvinen HJ, Virolainen M. The association between cholangiocarcinoma and hereditary nonpolyposis colorectal carcinoma. Cancer (1992) 69(5):1112–4. doi: 10.1002/cncr.2820690508
8. Kastrinos F, Stoffel EM, Balmaña J, Steyerberg EW, Mercado R, Syngal S. Phenotype comparison of MLH1 and MSH2 mutation carriers in a cohort of 1,914 individuals undergoing clinical genetic testing in the united states. Cancer Epidemiology Biomarkers Prev (2008) 17(8):2044–51. doi: 10.1158/1055-9965.EPI-08-0301
9. Geary J, Sasieni P, Houlston R, Izatt L, Eeles R, Payne SJ, et al. Gene-related cancer spectrum in families with hereditary non-polyposis colorectal cancer (HNPCC). Familial Cancer. (2008) 7(2):163–72. doi: 10.1007/s10689-007-9164-6
10. Borelli I, Casalis Cavalchini GC, Del Peschio S, Micheletti M, Venesio T, Sarottoet I, et al. A founder MLH1 mutation in lynch syndrome families from piedmont, Italy, is associated with an increased risk of pancreatic tumours and diverse immunohistochemical patterns. Fam Cancer. (2014) 13(3):401–13. doi: 10.1007/s10689-014-9726-3
11. Cloyd JM, Chun YS, Ikoma N, Vauthey JN, Aloia TA, Cuddy A, et al. Clinical and genetic implications of DNA mismatch repair deficiency in biliary tract cancers associated with lynch syndrome. J Gastrointest Cancer. (2018) 49(1):93–6. doi: 10.1007/s12029-017-0040-9
12. Bujanda L, Herreros-Villanueva M. Pancreatic cancer in lynch syndrome patients. J Cancer. (2017) 8(18):3667–74. doi: 10.7150/jca.20750
13. Seppälä TT, Latchford A, Negoi I, Sampaio Soares A, Jimenez-Rodriguez R, Sanchez-Guillén L, et al. European Guidelines from the EHTG and ESCP for lynch syndrome: an updated third edition of the mallorca guidelines based on gene and gender. Br J Surgery. (2021) 108(5):484–98. doi: 10.1002/bjs.11902
14. Crosbie EJ, Ryan NAJ, Arends MJ, Bosse T, Burn J, Corneset JM, et al. The Manchester international consensus group recommendations for the management of gynecological cancers in lynch syndrome. Genet Med (2019) 21(10):2390–400. doi: 10.1038/s41436-019-0489-y
15. Ryan N a. J, Evans DG, Green K, Crosbie EJ. Pathological features and clinical behavior of lynch syndrome-associated ovarian cancer. Gynecologic Oncol (2017) 144(3):491. doi: 10.1016/j.ygyno.2017.01.005
16. Møller P, Seppälä T, Bernstein I, Holinski-Feder E, Sala P, Evans DG, et al. Cancer incidence and survival in lynch syndrome patients receiving colonoscopic and gynaecological surveillance: first report from the prospective lynch syndrome database. Gut (2017) 66(3):464–72. doi: 10.1136/gutjnl-2015-309675
17. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch-repair deficiency predicts response of solid tumors to PD-1 blockade. Science (2017) 357(6349):409–13. doi: 10.1126/science.aan6733
18. Marcus L, Lemery SJ, Keegan P, Pazdur R. FDA Approval summary: Pembrolizumab for the treatment of microsatellite instability-high solid tumors. Clin Cancer Res (2019) 25(13):3753–8. doi: 10.1158/1078-0432.CCR-18-4070
19. Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz H-J, Morse MA, et al. Nivolumab in patients with metastatic DNA mismatch repair deficient/microsatellite instability–high colorectal cancer (CheckMate 142): results of an open-label, multicentre, phase 2 study. Lancet Oncol (2017) 18(9):1182–91. doi: 10.1016/S1470-2045(17)30422-9
20. Zeng FL, Chen JF. Application of immune checkpoint inhibitors in the treatment of cholangiocarcinoma. Technol Cancer Res Treat (2021) 20:15330338211039952. doi: 10.1177/15330338211039952
21. R: The r project for statistical computing . Available at: https://www.r-project.org/ (Accessed May 5, 2022).
22. Publication of WHO classification of tumours, 5th edition, volume 1: Digestive system tumours . IARC. Available at: https://www.iarc.who.int/news-events/publication-of-who-classification-of-tumours-5th-edition-volume-1-digestive-system-tumours/ (Accessed May 5, 2022).
23. Khanna L, Prasad SR, Sunnapwar A, Kondapaneni S, Dasyam A, Tammisetti VS, et al. Pancreatic neuroendocrine neoplasms: 2020 update on pathologic and imaging findings and classification. RadioGraphics (2020) 40(5):1240–62. doi: 10.1148/rg.2020200025
24. Kendall T, Verheij J, Gaudio E, Evert M, Guido M, Goeppert B, et al. Anatomical, histomorphological and molecular classification of cholangiocarcinoma. Liver Int (2019) 39(S1):7–18. doi: 10.1111/liv.14093
25. Pancreatic cancer - statistics (2012). Cancer.Net. Available at: https://www.cancer.net/cancer-types/pancreatic-cancer/statistics (Accessed October 20, 2022).
26. Bile duct cancer (Cholangiocarcinoma) - statistics (2012). Cancer.Net. Available at: https://www.cancer.net/cancer-types/bile-duct-cancer-cholangiocarcinoma/statistics (Accessed October 20, 2022).
27. ten Broeke SW, van der Klift HM, Tops CMJ, Aretz S, Bernstein I, Buchanan DD, et al. Cancer risks for PMS2-associated lynch syndrome. JCO (2018) 36(29):2961–8. doi: 10.1200/JCO.2018.78.4777
28. Hu ZI, Shia J, Stadler ZK, Varghese AM, Capanu M, Salo-Mullen E, et al. Evaluating mismatch repair deficiency in pancreatic adenocarcinoma: Challenges and recommendations. Clin Cancer Res (2018) 24(6):1326–36. doi: 10.1158/1078-0432.CCR-17-3099
29. Ando Y, Kumamoto K, Matsukawa H, RYOU Ishikawa R, Suto H, Oshima M, et al. Low prevalence of biliary tract cancer with defective mismatch repair genes in a Japanese hospital-based population. Oncol Lett (2022) 23(1):4. doi: 10.3892/ol.2021.13122
30. Wang Y, Cuggia A, Pacis A, Boileau J-C, Marcuset VA, Gao ZH, et al. Pancreatic cancer progression in a patient with lynch syndrome receiving immunotherapy: A cautionary tale. J Natl Compr Cancer Network. (2021) 19(8):883–7. doi: 10.6004/jnccn.2021.7049
31. Hendifar AE, Larson BK, Rojansky R, Guan M, Gong J, Veronica Placencio V, et al. Pancreatic cancer ‘mismatch’ in lynch syndrome. BMJ Open Gastroenterol (2019) 6(1):e000274. doi: 10.1136/bmjgast-2019-000274
32. Wang H, Liu J, Xia G, Lei S, Huang X, Huang X. Survival of pancreatic cancer patients is negatively correlated with age at diagnosis: a population-based retrospective study. Sci Rep (2020) 10(1):7048. doi: 10.1038/s41598-020-64068-3
33. Dominguez-Valentin M, Sampson JR, Seppälä TT, ten Broeke SW, Plazzer J-P, Nakken S, et al. Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: findings from the prospective lynch syndrome database. Genet Med (2020) 22(1):15–25. doi: 10.1038/s41436-019-0596-9
34. Artinyan A, Soriano PA, Prendergast C, Low T, Ellenhorn JDI, Kim J. The anatomic location of pancreatic cancer is a prognostic factor for survival. HPB (Oxford). (2008) 10(5):371–6. doi: 10.1080/13651820802291233
35. Lee M, Kwon W, Kim H, Byun Y, Han Y, Kanget JS, et al. The role of location of tumor in the prognosis of the pancreatic cancer. Cancers (Basel). (2020) 12(8):2036. doi: 10.3390/cancers12082036
36. Takamizawa S, Morizane C, Tanabe N, Maruki Y, Kondo S, Susumu Hijioka S, et al. Clinical characteristics of pancreatic and biliary tract cancers associated with lynch syndrome. J Hepatobiliary Pancreat Sci (2022) 29(3):377–84. doi: 10.1002/jhbp.1063
37. Cillo U, Fondevila C, Donadon M, Gringeri E, Mocchegiani F, Schlitt HJ, et al. Surgery for cholangiocarcinoma. Liver Int (2019) 39(Suppl 1):143–55. doi: 10.1111/liv.14089
Keywords: Lynch syndrome (LS), hereditary nonpolyposis colon cancer (HNPCC), pancreatic cancer, biliary tract cancer, microsatellite instability (MSI)
Citation: Zalevskaja K, Mecklin J-P and Seppälä TT (2023) Clinical characteristics of pancreatic and biliary tract cancers in Lynch syndrome: A retrospective analysis from the Finnish National Lynch Syndrome Research Registry. Front. Oncol. 13:1123901. doi: 10.3389/fonc.2023.1123901
Received: 14 December 2022; Accepted: 18 January 2023;
Published: 01 February 2023.
Edited by:
Jianxun J. Song, Texas A&M Health Science Center, United StatesReviewed by:
Jiaqiao Fan, Dalian Medical University, ChinaAnil Kumar, Texas A&M Health Science Center, United States
Copyright © 2023 Zalevskaja, Mecklin and Seppälä. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kristina Zalevskaja, a3Jpc3RpbmEuemFsZXZza2FqYUBoZWxzaW5raS5maQ==; Toni T. Seppälä, dG9uaS5zZXBwYWxhQGhlbHNpbmtpLmZp