 Timothy E. O’Connor
Timothy E. O’Connor Reid Shaw1
Reid Shaw1 Rafael Madero-Marroquin
Rafael Madero-Marroquin Gregory W. Roloff
Gregory W. Roloff- 1Department of Medicine, Loyola University Medical Center, Maywood, IL, United States
- 2Section of Hematology/Oncology, The University of Chicago, Chicago, IL, United States
In recent years, advances in genetics and the integration of clinical-grade next-generation sequencing (NGS) assays into patient care have facilitated broader recognition of hereditary hematopoietic malignancy (HHM) among clinicians, in addition to the identification and characterization of novel HHM syndromes. Studies on genetic risk distribution within affected families and unique considerations of HHM biology represent exciting areas of translational research. More recently, data are now emerging pertaining to unique aspects of clinical management of malignancies arising in the context of pathogenic germline mutations, with particular emphasis on chemotherapy responsiveness. In this article, we explore considerations surrounding allogeneic transplantation in the context of HHMs. We review pre- and post-transplant patient implications, including genetic testing donor selection and donor-derived malignancies. Additionally, we consider the limited data that exist regarding the use of transplantation in HHMs and safeguards that might be pursued to mitigate transplant-related toxicities.
Introduction
It is now well understood that inherited pathogenic mutations contribute to about 10% of newly diagnosed solid tumors (1). The prototypical familial cancer syndrome described by Li and Fraumeni in the 1960s involving neoplasia of the breast, bone, brain, and adrenal glands jumpstarted the nascent field of cancer genetics (2). The first case of hereditary hematopoietic malignancy was detailed in 1922 and ultimately published in series by Gunz and colleagues in 1975. Their description spanned 909 families, identifying 72 individuals with leukemia who had one or more relatives who were also affected with leukemia (3). In 1999, the autosomal dominant familial platelet disorder with propensity to develop acute myeloid leukemia (now known as RUNX1 Familial Platelet Disorder with Associated Myeloid Malignancies, RUNX1-FPDMM) was the first HHM to have its molecular basis elucidated, driven by heterozygous pathogenic RUNX1 variants (4). More than a dozen genes have been subsequently identified as direct drivers of HHMs, with multiple others identified as conferring risk of blood cancer as one phenotypic element within broader solid tumor predisposition syndromes (5, 6).
Although understandably anxiety provoking, the identification of a familial cancer syndrome can also prove empowering to patients. First, it provides knowledge to guide the evaluation of seemingly unaffected family members of the index patient via cascade testing. Because some variants confer risk to more than one tumor type, verifying the presence of a pathogenic germline variant streamlines opportunities for screening and surveillance of other malignancies and disease phenotypes (6). Precision medicine treatment approaches have also emerged and entered the standard of care for common tumor types, such as the use of PARP-inhibitor therapy in germline BRCA-mutant breast cancer (7). Unlike solid tumors, in HHMs the mutation status of family members may influence decision making for the care of the index patient due to considerations of donor source when hematopoietic cell transplant (HCT) is pursued. Additionally, distinct biology of HHM syndromes also influences transplant conditioning regimens and strategies for toxicity mitigation in a vulnerable patient population. In this article, we review high-yield HHM syndromes that are relevant to HCT and consider the place for HCT in disease management strategies. An overview of HHM testing is provided and data regarding HCT outcomes in established HHM syndromes are presented.
Overview of select HHMs
Currently, pathogenic germline variants in DDX41 mutations represent the most common cause of HHMs, representing approximately 2.4% of patients in a large cohort study of 1385 individuals with AML or MDS (8, 9). Development of myeloid malignancy in patients with germline DDX41 mutations occurs at an age that approximates the median age of onset for de novo/sporadic AML/MDS. Most affected individuals demonstrate normal blood counts well into adulthood, suggesting that DDX41 haploinsufficiency is sufficient to support baseline hematopoiesis (9). However, loss of heterozygosity leads to the development of cytopenias and macrocytosis at a mean age of 66 years (range 50-85 years) and hematologic malignancy shortly thereafter with a mean age at diagnosis of 69 years (range 36-88 years. Myeloid malignancy associated with DDX41 variants is felt to be highly treatment responsive. In 20 patients with MDS/AML treated with chemotherapy (n=9) or azacitidine (n=11), response rates were 100% and 73%, respectively, and a median survival of 5.2 years was observed (8). Another study among 3132 unrelated adult patients with myeloid malignancies identified 28 individuals (20/28 male) with germline DDX41 variants diagnosed with AML described chronic clinical trajectories (mean duration 11.2 months) during which patients with cytopenias were monitored before AML onset. When diagnosed, 69% of patients had acquired a second hit (somatic) DDX41 variant and most cases had a normal karyotype (75%) with bone marrow hypocellularity (93%) (10). Recently, in a large, multinational analysis of myeloid neoplasia in 9082 patients, 346 patients with pathogenic/likely pathogenic germline and/or somatic DDX41 variants were identified. Statistically, it was calculated that about 80% of myeloid malignancy with germline risk contribution identified in the study population was attributable to DDX41 mutations alone. Risk variants were enriched within Japanese patients by 10-fold when compared to those from other geographic areas. The lifetime risk of acquiring myeloid malignancy in the population by age 90 was 49%. Moreover, co-mutational status, both at diagnosis and at time of disease progression did not affect clinical outcomes, even in the case of multi-hit TP53 variants, suggesting that DDX41-driven myeloid malignancies comprise a unique subset of myeloid malignancy with distinct molecular biology (11).
Biallelic CEBPA mutations are found in approximately 10% of patients with AML. It is estimated that as many as ten percent of these (~1% total cases) may represent familial variants (12). Familial CEBPA-mutated AML was first described in 2004, with three family members harboring identical N-terminal germline mutations (13). AML development generally occurs when secondarily acquired C-terminal mutations arise, typically at an average age of onset of 24.5 years (range 2-46 years) and with affected kindreds demonstrating >90% penetrance for AML diagnosis (13–15). Due to high chemotherapy responsiveness, familial CEBPA-mutated AML generally portends a favorable prognosis in comparison to cases with sporadic mutations, with 10-year survival rates of 67%, superior to spontaneous bi-allelic (54%) and single CEBPA mutations (29%) (15). However, as discussed more below, recurrent leukemic episodes are common and consideration of allogeneic replacement of a stem cell pool harboring pathogenic germline variants should be considered once remission is achieved.
Autosomal dominant mutations in GATA2 lead to the well-described GATA2 deficiency syndrome. Although the clinical phenotype can vary, the syndrome harbors several notable phenotypic features that must be recognized by hematologists. First, flow cytometric profiling of affected patients often demonstrates substantial reduction or near absence or B-lymphocytes, NK cells, and monocytes, leading to immunodeficiency (16, 17). Recurrent infections manifest throughout late childhood and adolescence. Disseminated nontuberculous mycobacterial, invasive fungal, human papillomavirus infections should warrant dedicated genetics investigation. Additionally, limb lymphedema, deafness, and pulmonary proteinosis also contribute to disease phenotype. This severe immunodeficiency, which typically presents in adolescence, leads to a variety of disseminated infections, including nontuberculous mycobacterial, invasive fungal, and disseminated human papilloma virus infections (18). Finally, GATA2 deficiency confers risk to the development of myeloid malignancy, typically in the form of MDS/AML (19). HHMs driven by germline GATA2 mutations generally have poor overall survival when analyzed by germline variants alone, but this affect appears to be equalized when accounting for karytotypic/cytogenetic features (20).
Familial platelet disorder with predisposition to myeloid malignancy (FPDMM), sometimes referred to as Thrombocytopenia 2, was first described in 1985 and molecularly characterized in 1999 as the result of pathogenic variants in the transcription factor RUNX1 (4). A wide spectrum of variants has been described, including multiexon or whole gene deletions, frameshifts, missense and nonsense mutations. Strikingly, clonal hematopoiesis has been observed in a majority of asymptomatic RUNX1 germline variant carriers, even before age 50. The lifetime risk of developing leukemia is estimated to be ~35% (21, 22). RUNX1-mutated AML is associated with dismal relapse-free and overall survival when compared to controls with RUNX1 wild type status (23). Short telomere syndromes (STS) are characterized by accelerated aging, and associated with mutations in genes that maintain the “molecular clock,” including TERT, TERC, and DKC1 (24–26). The loss of function of these genes lead to a variable phenotypic presentation that includes non-hematopoietic phenotypes including idiopathic pulmonary fibrosis (27), premature graying of hair, cryptogenic cirrhosis, and hematologic consequences including aplastic anemia, MDS, and AML (28, 29). Because the clinical phenotype of STS involve multisystem disorders, clinical surveillance is imperative and patients benefit from involvement of multispecialty teams with experience in cancer risk assessment (Figure 1) (30).
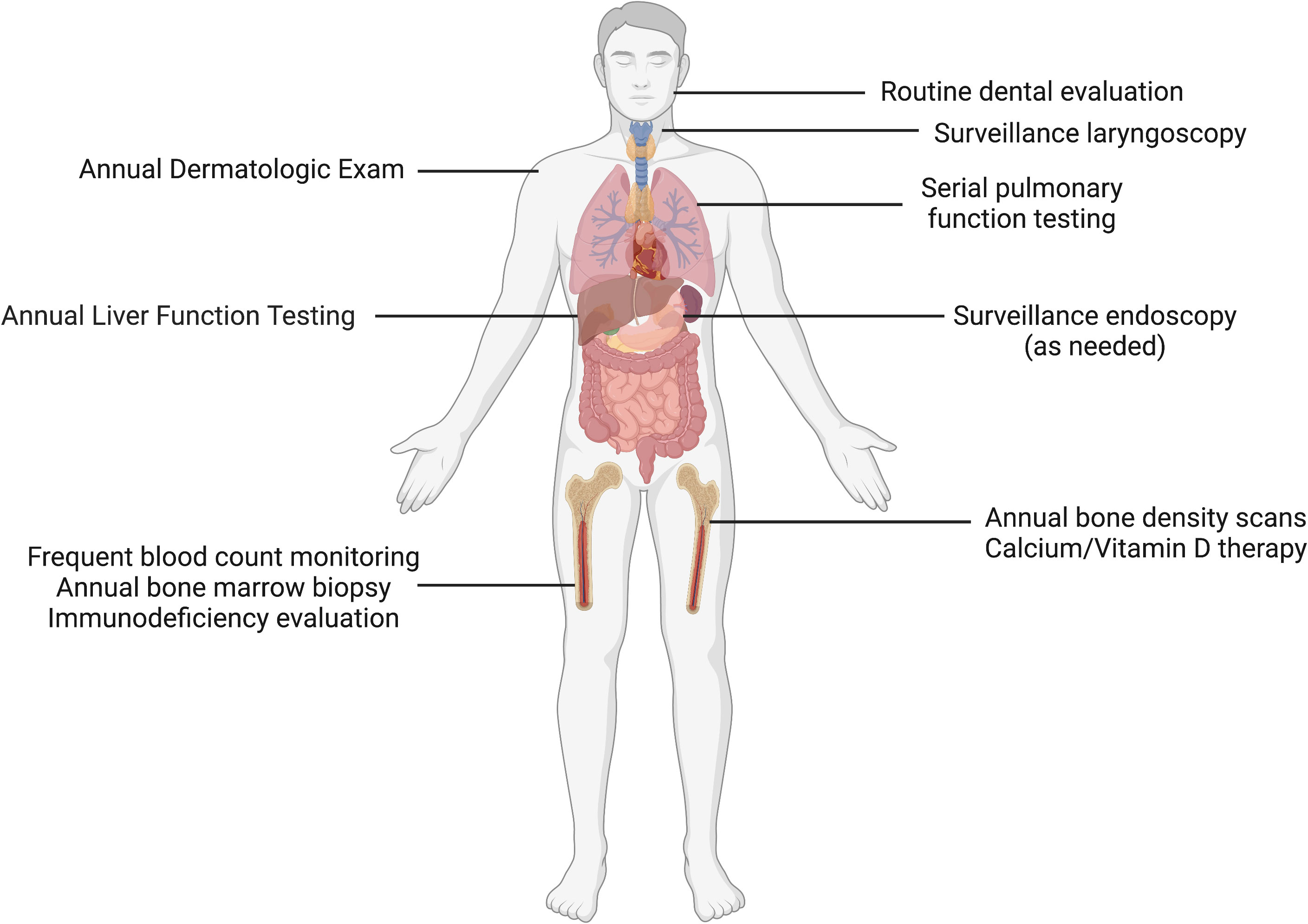
Figure 1 Clinical surveillance in short telomere syndromes. Patients with short telomere syndromes are at highest risk for bone marrow failure, pulmonary fibrosis, liver disease and solid cancers. Surveillance strategies implemented within a multidisciplinary cancer risk program are aimed to identify characteristic disease phenotypes and facilitate timely intervention. These abnormalities include such as cytopenias/bone marrow failure, interstitial lung disease, cryptogenic cirrhosis, and squamous cell carcinomas, especially of the tongue. Image made with BioRender.com.
Unlike familial CEBPA-mutated AML, wherein AML development appears to be the principal clinical feature, other inherited cancer syndromes associated with HHMs and hematopoietic phenotypes also confer solid tumor risk. As mentioned above, the first hereditary cancer syndrome described by Li and Fraumeni detailed kindreds with sarcoma, leukemia, and cancers of the brain, breast, and adrenal gland (2). Hypodiploid ALL is the most common hematopoietic malignancy reported to be associated with Li-Fraumeni syndrome (LFS), although cases of AML and chronic myeloid malignancies such as myelodysplastic syndromes and chronic myeloid leukemia have also been described. Unfortunately, large datasets with carefully annotated clinical trajectories for hematopoietic malignancy in Li-Fraumeni syndrome (LFS) patients remain scarce. In a series of 7 LFS patients who received care for hematopoietic malignancy at MD Anderson Cancer Center (31), five patients underwent HCT. Only one of these five patients, an individual with B-cell ALL, remained in durable remission at least one year after transplantation. Five of the seven patients had malignancies that were classified as therapy-related myeloid neoplasms and experienced dismal outcomes (31). This LFS experience underscores the importance of hematopoietic malignancy risk assessment in patients who have received prior cancer therapy, with particular emphasis on patients with inherited cancer predisposition syndromes. As Churpek and colleagues have identified previously undiagnosed germline mutations in 10/47 (21%) of breast cancer patients who developed therapy-related leukemia, it is important to recognize that the discovery of an inherited predisposition syndrome may not occur until subsequent malignancies develop after an initial cancer diagnosis (32).
Further evidence towards characterizing solid tumor manifestations of HHM-associated germline syndromes is emerging. For example, in seminal studies that established germline variants in DDX41 as HHM-associated genes, 6/33 (18%) individuals with blood cancers had received prior solid tumor diagnoses, with prostate cancer being the most common (3/33, 9%) (8, 9, 33). However, isolated solid tumors in germline variant carriers were also reported in the absence of hematopoietic malignancy, and it is not known whether solid tumor incidence is higher in patients with germline DDX41 variants than would be expected in the general population (5). Table 1 describes cardinal hematopoietic and non-hematopoietic clinical features of the above-discussed HHMs, and summarizes published HCT experiences.
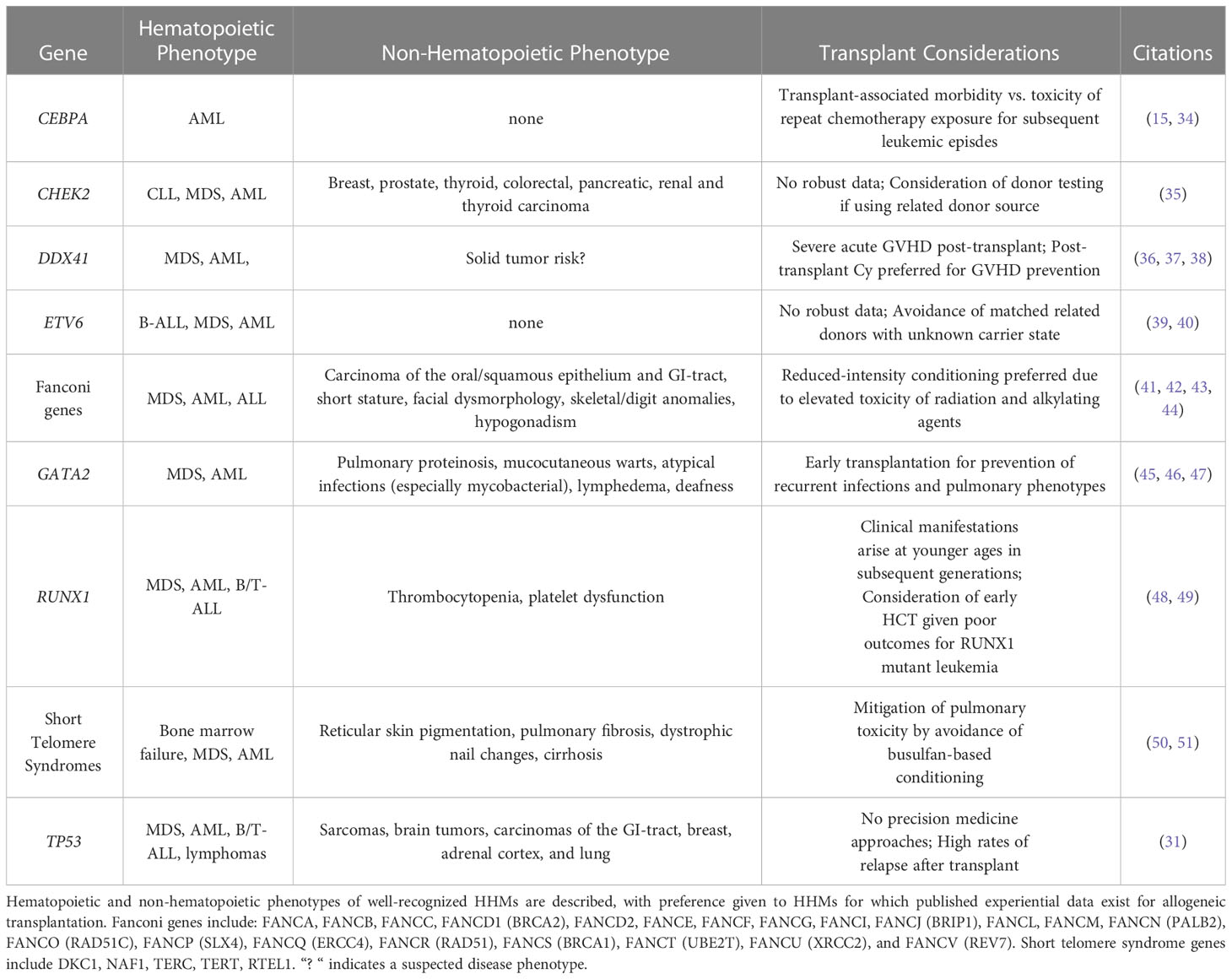
Table 1 High-yield characteristics of hereditary hematopoietic malignancies and stem cell transplant considerations.
Patient-level implications pre- and post-transplant
As mentioned above, proper HHM evaluation, ideally concurrent with initial workup of hematologic malignancy, provides both diagnostic and clinically actionable information that may prove highly relevant to the patient’s course. Expert protocols and recommendations guiding HHM evaluation have been described previously (52, 53). Although it has been established that germline variants may be inferred from tumor-only somatic variant panel testing (54, 55), a proper HHM workup involves sampling non-tumor tissue. Skin biopsies are preferred, as other sources such as saliva may be contaminated with WBC harboring somatic variants and thereby obscuring variant adjudication (56). Due to increasing awareness of HHMs amongst patients and providers, commercial options offering genetic testing specifically for hereditary blood cancer evaluation have been developed and are currently marketed to patients. However, a high degree of heterogeneity exists within the commercial HHM testing market. In addition to differences in cost, turnaround time and variant validation methods, important genes are missing from commercial many commercial panels and companies accept a wide range of tissues as representative germline samples, including peripheral blood and/or bone marrow specimens (57). Furthermore, most panels are not routinely updated in a timely manner to reflect evolving literature (58).
Donor cell leukemia (DCL) is a catastrophic yet often avoidable consequence of allogeneic transplant whereby pathogenic donor clones expand in the recipient and contribute to apparent “relapses.” This phenomenon may more accurately be described as donor-derived hematopoietic malignancy since the initial neoplasm is not truly relapsing from a dormant state, but rather, a new hematologic cancer is propagating. Prior to advent of commercially available NGS, cytogenetic testing was used to identify donor cell leukemias (DCL) as a complication of allogeneic HSCT (59). Early series estimated the incidence of DCL was initially estimated to be as high as 5% (60). However, it is now thought of as a rare complication of allogeneic stem cell transplantation, with multiple studies suggesting an incidence below 1% (61–63). Despite the limited incidence, overall outcomes of reported DCL cohorts have suggested a poor prognosis with a high mortality rate after diagnosis (61, 64). Cases of donor cell MDS has also been reported after matched-related donor transplant, suggesting the possibility of germline variant carriage in the family member ultimately used as a stem cell source (65, 66). Some reports for which donor follow-up was available have also described the clinical trajectory of hematopoietic malignancy development in the donor after donor-derived relapse in the recipient (67). Outcomes for donor cell MDS, while sparsely reported, also appear to be poor, with published institutional experiences underscoring the importance of proceeding to HCT where possible (68).
Several mechanisms have been suspected to enable leukemic transformation of donor-derived cells (61, 69). Instances of germline mutations present in the hematopoietic cells of apparently healthy donors are well-described (70–72). Due to the potentially elevated risk of developing DCL in recipients of cells with mutated germline predisposition alleles, screening and consideration for exclusion of these donor cells has been suggested (62). Donor screening is of particular importance when using related donors and when there is high probability for HHM, for which ample suspicion and appropriate evaluation is critical before allograft (73). One barrier to comprehensive donor HHM might stem concerns regarding turnaround time of NGS (typically several weeks) and the possible need to delay of HCT. However, if a pathogenic variant has been identified in the transplant recipient, panel-based NGS in a donor candidate is likely unnecessary. We prefer direct single-gene testing for the gene in question from the peripheral blood. Although peripheral blood should not be the source of germline sampling in a patient with hematopoietic malignancy, it is appropriate for evaluation of healthy donors.
There are also ethical challenges arising from donor DNA sequencing after allogeneic HSCT. These considerations are of importance in cases of DCL, where donor germline variants can be detected and potentially carry significant clinical implications for the donor (74, 75). Moreover, variants of uncertain significance can also complicate the interpretation of the results. These scenarios raise ethical questions regarding test result disclosure to the donor. These issues are further exacerbated in cases of unrelated donors, where communication with the donor is limited, and in cases involving cord blood HSCT, where the donor is of minor age (76). If pursued, communication with these donors is likely best facilitated by involvement of agencies that support transplant programs (National Marrow Donor Program, DKMS).
HHM transplant evidence and outcomes
Patients with HHMs receive similar therapies to those with de novo disease often because the underlying causative germline variant is not discovered until after treatment has been initiated (5, 77). STS discussed above, develop due to germline mutations in telomeric regulators that affect telomere structure and function. Patients who develop bone marrow failure or MDS often go on to receive HCT (78). Selection of an appropriate chemotherapy conditioning regimen is important for STS due to a noted propensity for affected patients to develop organ failure as part of their underlying genetic syndrome. In STS, these non-hematologic disease manifestations include pulmonary fibrosis and cirrhosis (50, 79, 80). For example, in patients with dyskeratosis congenita (DC), high rates of death due to pulmonary toxicity have been observed in patients who received busulfan-based conditioning regimens. Fludarabine-containing reduced intensity conditioning regimens are becoming standard of care for these patients, having been shown to be safe and effective without accelerating pulmonary damage (51, 81).
Another example of a precision-medicine guided approach to HCT in patients with HHM pertains to mitigation of post-transplant graft-versus-host disease. Patients with germline DDX41 germline variant have been shown to have higher rates of both severe (stage 3-4) acute graft-versus-host disease (GVHD) (38%) and moderate to severe chronic GVHD (33%) compared to those without any identified P/LP germline variant (9% and 10%, respectively) (36). GVHD prophylaxis with cyclophosphamide has been shown to prevent development of GVHD in patients with DDX41 germline variants. For example, among 15 patients with P/LP germline DDX41 variants who underwent HSCT, but did not receive post-HSCT cyclophosphamide, 7 deaths occurred, 4 of which were due to severe GVHD. Five patients with P/LP germline DDX41 variants who received post-HSCT cyclophosphamide were completely free of GVHD and alive at the time of analysis (36). Though the mechanism for cyclophosphamide-mediated GVHD amelioration has not been delineated, DDX41 contributes to STING signaling and may lead to aberrant immune activation after transplant (82, 83).
As mentioned above, GATA2 deficiency, stemming from pathogenic variants in GATA2, is comprised of a syndrome characterized by life-threatening opportunistic infections with non-tuberculous mycobacteria (NTM), fungal infections, or human papillomavirus (HPV) infections and a propensity to progress to MDS, CMML, and AML (17–19). Because of immunodeficiency in multiple cellular compartments, the pursuit of HCT in GATA2 deficiency is aimed at addressing both bone marrow hematopoietic stem cells predisposed to developing myeloid malignancy, in addition to restoring functional immune compartments and avoiding deadly consequences of recurrent pulmonary infections. Accordingly, HCT has been shown to be highly effective at preventing recurrent NTM, with zero patients experiencing recurrence of disease following transplant in a 14-patient cohort treated at the National Institutes of Health (45). Risk for post-HCT HPV infections, however, does not appear to be fully addressed by transplantation, with about 50% of patients showing persistent mucocutaneous warts (84). Furthermore, pulmonary complications, including pulmonary hypertension and alveolar proteinosis appear to be substantially mitigated with HCT (85).
Although HCT is an effective means at mitigating, or even reversing clinical phenotypes associated of GATA2 deficiency, the optimal timing of HCT, and choice of conditioning regimen, donor sources, and GVHD prophylaxis remain areas of active investigation. With the potential to mitigate or avoid future syndromic disease manifestations, preemptive HCT in patients with GATA2 deficiency represents an attractive strategy. From 2013 to 2020, 59 patients underwent HCT with busulfan-based conditioning at the National Institutes of Health. Patients were eligible for transplant if they had at least one episode of a life-threatening opportunistic infection, with almost half of patients having received a transplant due to an established diagnosis of MDS or the presence of cytogenetic changes in the absence of overt morphologic disease, particularly monosomy 7. Overall survival (OS) and event-free survival (EFS) at 4 years were 85.1% and 82.1% respectively (86). It is important to note that malignant myeloid progression is possible after HCT, given the malignant clones can propagate in the setting of MDS. Because GATA2-deficient bone marrow stem cells are at a proliferative disadvantage (45), nonmyeloablative conditioning regimens have also been pursued. These regimens have largely consisted of fludarabine, alemtuzumab and busulfan, which have been shown to be an effective strategy. No grade III-IV acute GVHD was observed in patients who received haploidentical related donor HSCTs and prophylaxis with post-transplantation cyclophosphamide followed by tacrolimus/mycophenolate. Ninety-six percent of these patients had complete reversal of the hematologic disease phenotype by one-year post-transplant (86).
HCT has also been shown to be an effective strategy for familial CEBPA-mutated AML. This syndrome is associated with high penetrance and a young age of AML onset without development of preceding myelodysplasia. Patients typically experience complete response to induction chemotherapy with standard regimens (15). Indeed, the rate of achieving a first CR in a seminal study of 24 individuals spanning 10 families of familial AML was 91% (n = 21/23). Disease recurrence was common, however, with the cumulative incidence of relapse being 56% at 10 years. Although recurrent leukemic episodes remain generally responsive to chemotherapy, treatment-related cardiotoxicity has been observed. Of the 7 patients who died following recurrence of disease, 71% (n = 5) were in remission. One patient had treatment-related complications (cardiotoxicity) and 3 patients had transplant-related complications (GVHD, post-transplant lymphoma, and infection). It is difficult to balance the risk of transplant-related morbidity/mortality with the risk of organic and infectious complications due to cyclical treatment of recurrent leukemic episodes. Preemptive HCT in known variant carriers has been proposed, but data are lacking to widely support empiric HCT vs. proceeding to allograft after appropriate donor evaluation once remission has been achieved (Figure 2) (87).
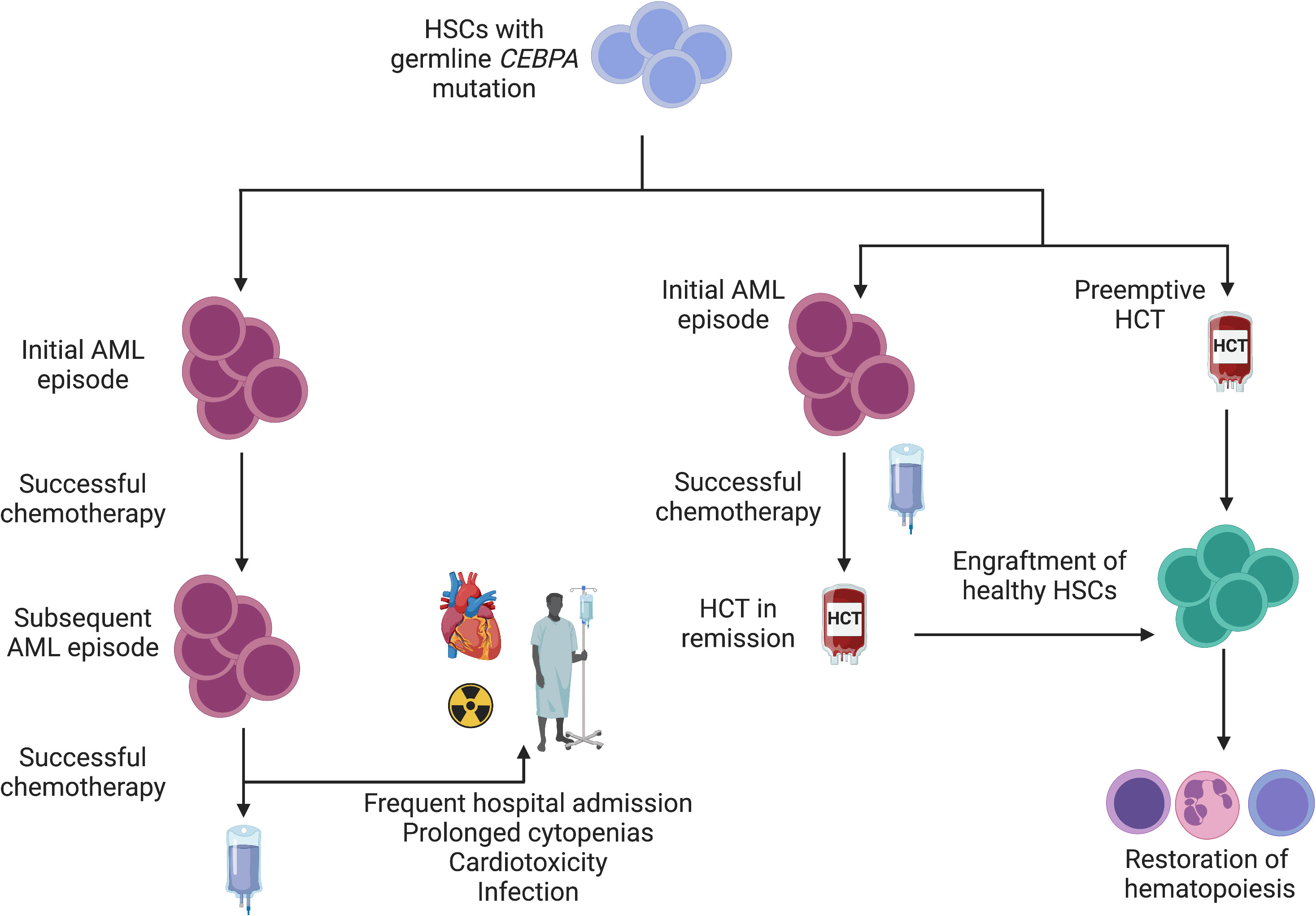
Figure 2 Interventions for familial CEBPA-mutated AML. Given a young age at diagnosis and a high likelihood of AML development among variant carriers, a schematic for management is depicted. Individuals who wish to avoid HCT or for whom HCT is not feasible may achieve disease control with chemotherapy alone. Failure to correct a mutated stem cell pool (left) sets the stage for subsequent leukemic episodes. Although long latency periods between subsequent episodes have been observed (15), repeated exposure to cytotoxic chemotherapy confers risk of infection and organ toxicity. In families with highly penetrant disease (right), empiric HCT is reasonable to mitigate AML development and ensure normal hematopoiesis. Alternatively, HCT can be pursued after initial remission is achieved following an initial AML episode.
Conclusions
The study of HHMs has advanced substantially in recent years due to progress in NGS technologies, their clinical integration, and cost accessibility. These technological achievements have enabled physicians and scientists to partner with motivated patients and uncover novel HHM genes, define mechanistic principles that underlie the stepwise progression towards overt malignancy, define the presence or absence of precursor states, and characterize clinical trajectories. Future research in the field will be aimed at optimizing protocols for clinical surveillance in unaffected variant carriers. As clinical integration of NGS in has dramatically increased the number of HHM patients identified, secondary referrals for their family members have also increased the number of clinically unaffected variant carriers seeking consultation. Thoughtful intervention in high-risk HHM variant carriers is also likely to be an area of exciting investigation. Decision-making models to define the optimal time for intervention are needed. Preemptive HCT remains an attractive option for patients with HHM variants associated with deleterious systemic clinical phenotypes (GATA2 deficiency, for example), but non-relapse mortality remains a serious obstacle towards more uniform deployment of potentially curative strategies to replace deleterious stem cell populations. Furthermore, as gene therapy approaches advance, less invasive means for correcting pathogenic variants may become feasible.
Author contributions
GR conceived the article. TO’C, RS, RM-M, and GR researched and wrote the manuscript. All authors edited the manuscript. GR created figures included in publication. All authors contributed to the article and approved the submitted version.
Acknowledgments
The authors acknowledge patients affected by HHMs and their families for participation in critical research on the topic.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Garber JE, Offit K. Hereditary cancer predisposition syndromes. J Clin Oncol (2005) 23(2):276–92. doi: 10.1200/JCO.2005.10.042
2. Li FP, Fraumeni JF Jr. Soft-tissue sarcomas, breast cancer, and other neoplasms. a familial syndrome? Ann Intern Med (1969) 71(4):747–52. doi: 10.7326/0003-4819-71-4-747
3. Gunz FW, Gunz JP, Veale AM, Chapman CJ, Houston IB. Familial leukaemia: a study of 909 families. Scand J Haematol (1975) 15(2):117–31. doi: 10.1111/j.1600-0609.1975.tb01063.x
4. Song WJ, Sullivan MG, Legare RD, Hutchings S, Tan X, Kufrin D, et al. Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia. Nat Genet (1999) 23(2):166–75. doi: 10.1038/13793
5. Roloff GW, Drazer MW, Godley LA. Inherited susceptibility to hematopoietic malignancies in the era of precision oncology. JCO Precis Oncol (2021) 5:107–22. doi: 10.1200/PO.20.00387
6. Tawana K, Drazer MW, Churpek JE. Universal genetic testing for inherited susceptibility in children and adults with myelodysplastic syndrome and acute myeloid leukemia: are we there yet? Leukemia (2018) 32(7):1482–92. doi: 10.1038/s41375-018-0051-y
7. Robson M, Im SA, Senkus E, Xu B, Domchek SM, Masuda N, et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med (2017) 377(6):523–33. doi: 10.1056/NEJMoa1706450
8. Sebert M, Passet M, Raimbault A, Rahme R, Raffoux E, Sicre de Fontbrune F, et al. Germline DDX41 mutations define a significant entity within adult MDS/AML patients. Blood (2019) 134(17):1441–4. doi: 10.1182/blood.2019000909
9. Lewinsohn M, Brown AL, Weinel LM, Phung C, Rafidi G, Lee MK, et al. Novel germ line DDX41 mutations define families with a lower age of MDS/AML onset and lymphoid malignancies. Blood (2016) 127(8):1017–23. doi: 10.1182/blood-2015-10-676098
10. Li P, White T, Xie W, Cui W, Peker D, Zeng G, et al. AML with germline DDX41 variants is a clinicopathologically distinct entity with an indolent clinical course and favorable outcome. Leukemia (2022) 36(3):664–74. doi: 10.1038/s41375-021-01404-0
11. Makishima H, Saiki R, Nannya Y, Korotev S, Gurnari C, Takeda J, et al. Germ line DDX41 mutations define a unique subtype of myeloid neoplasms. Blood (2023) 141(5):534–49. doi: 10.1182/blood.2022018221
12. Pabst T, Eyholzer M, Haefliger S, Schardt J, Mueller BU. Somatic CEBPA mutations are a frequent second event in families with germline CEBPA mutations and familial acute myeloid leukemia. J Clin Oncol (2008) 26(31):5088–93. doi: 10.1200/JCO.2008.16.5563
13. Smith ML, Cavenagh JD, Lister TA, Fitzgibbon J. Mutation of CEBPA in familial acute myeloid leukemia. N Engl J Med (2004) 351(23):2403–7. doi: 10.1056/NEJMoa041331
14. Sellick GS, Spendlove HE, Catovsky D, Pritchard-Jones K, Houlston RS. Further evidence that germline CEBPA mutations cause dominant inheritance of acute myeloid leukaemia. Leukemia (2005) 19(7):1276–8. doi: 10.1038/sj.leu.2403788
15. Tawana K, Wang J, Renneville A, Bodor C, Hills R, Loveday C, et al. Disease evolution and outcomes in familial AML with germline CEBPA mutations. Blood (2015) 126(10):1214–23. doi: 10.1182/blood-2015-05-647172
16. Bigley V, Haniffa M, Doulatov S, Wang XN, Dickinson R, McGovern N, et al. The human syndrome of dendritic cell, monocyte, b and NK lymphoid deficiency. J Exp Med (2011) 208(2):227–34. doi: 10.1084/jem.20101459
17. Dickinson RE, Griffin H, Bigley V, Reynard LN, Hussain R, Haniffa M, et al. Exome sequencing identifies GATA-2 mutation as the cause of dendritic cell, monocyte, b and NK lymphoid deficiency. Blood (2011) 118(10):2656–8. doi: 10.1182/blood-2011-06-360313
18. Hsu AP, Sampaio EP, Khan J, Calvo KR, Lemieux JE, Patel SY, et al. Mutations in GATA2 are associated with the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome. Blood (2011) 118(10):2653–5. doi: 10.1182/blood-2011-05-356352
19. Hahn CN, Chong CE, Carmichael CL, Wilkins EJ, Brautigan PJ, Li XC, et al. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat Genet (2011) 43(10):1012–7. doi: 10.1038/ng.913
20. Wlodarski MW, Hirabayashi S, Pastor V, Stary J, Hasle H, Masetti R, et al. Prevalence, clinical characteristics, and prognosis of GATA2-related myelodysplastic syndromes in children and adolescents. Blood (2016) 127(11):1387–97. doi: 10.1182/blood-2015-09-669937
21. Speck NA, Gilliland DG. Core-binding factors in haematopoiesis and leukaemia. Nat Rev Cancer (2002) 2(7):502–13. doi: 10.1038/nrc840
22. Michaud J, Simpson KM, Escher R, Buchet-Poyau K, Beissbarth T, Carmichael C, et al. Integrative analysis of RUNX1 downstream pathways and target genes. BMC Genomics (2008) 9:363. doi: 10.1186/1471-2164-9-363
23. Greif PA, Konstandin NP, Metzeler KH, Herold T, Pasalic Z, Ksienzyk B, et al. RUNX1 mutations in cytogenetically normal acute myeloid leukemia are associated with a poor prognosis and up-regulation of lymphoid genes. Haematologica (2012) 97(12):1909–15. doi: 10.3324/haematol.2012.064667
24. Cohen SB, Graham ME, Lovrecz GO, Bache N, Robinson PJ, Reddel RR. Protein composition of catalytically active human telomerase from immortal cells. Science (2007) 315(5820):1850–3. doi: 10.1126/science.1138596
25. Vulliamy T, Marrone A, Goldman F, Dearlove A, Bessler M, Mason PJ, et al. The RNA component of telomerase is mutated in autosomal dominant dyskeratosis congenita. Nature (2001) 413(6854):432–5. doi: 10.1038/35096585
26. Garcia CK, Wright WE, Shay JW. Human diseases of telomerase dysfunction: insights into tissue aging. Nucleic Acids Res (2007) 35(22):7406–16. doi: 10.1093/nar/gkm644
27. Alder JK, Chen JJ, Lancaster L, Danoff S, Su SC, Cogan JD, et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci U S A. (2008) 105(35):13051–6. doi: 10.1073/pnas.0804280105
28. Yamaguchi H, Calado RT, Ly H, Kajigaya S, Baerlocher GM, Chanock SJ, et al. Mutations in TERT, the gene for telomerase reverse transcriptase, in aplastic anemia. N Engl J Med (2005) 352(14):1413–24. doi: 10.1056/NEJMoa042980
29. Knight SW, Heiss NS, Vulliamy TJ, Greschner S, Stavrides G, Pai GS, et al. X-Linked dyskeratosis congenita is predominantly caused by missense mutations in the DKC1 gene. Am J Hum Genet (1999) 65(1):50–8. doi: 10.1086/302446
30. Niewisch MR, Savage SA. An update on the biology and management of dyskeratosis congenita and related telomere biology disorders. Expert Rev Hematol (2019) 12(12):1037–52. doi: 10.1080/17474086.2019.1662720
31. Swaminathan M, Bannon SA, Routbort M, Naqvi K, Kadia TM, Takahashi K, et al. Hematologic malignancies and Li-fraumeni syndrome. Cold Spring Harb Mol Case Stud (2019) 5(1):a003210. doi: 10.1101/mcs.a003210
32. Churpek JE, Marquez R, Neistadt B, Claussen K, Lee MK, Churpek MM, et al. Inherited mutations in cancer susceptibility genes are common among survivors of breast cancer who develop therapy-related leukemia. Cancer (2016) 122(2):304–11. doi: 10.1002/cncr.29615
33. Bannon SA, Routbort MJ, Montalban-Bravo G, Mehta RS, Jelloul FZ, Takahashi K, et al. Next-generation sequencing of DDX41 in myeloid neoplasms leads to increased detection of germline alterations. Front Oncol (2020) 10:582213. doi: 10.3389/fonc.2020.582213
34. Tawana K, Rio-Machin A, Preudhomme C, Fitzgibbon J. Familial CEBPA-mutated acute myeloid leukemia. Semin Hematol (2017) 54(2):87–93. doi: 10.1053/j.seminhematol.2017.04.001
35. Stubbins RJ, Korotev S, Godley LA. Germline CHEK2 and ATM variants in myeloid and other hematopoietic malignancies. Curr Hematol Malig Rep (2022) 17(4):94–104. doi: 10.1007/s11899-022-00663-7
36. Saygin C, Roloff G, Hahn CN, Chhetri R, Gill S, Elmariah H, et al. Allogeneic hematopoietic stem cell transplant outcomes in adults with inherited myeloid malignancies. Blood Adv (2023) 7(4):549–54. doi: 10.1182/bloodadvances.2022008172
37. Duployez N, Largeaud L, Duchmann M, Kim R, Rieunier J, Lambert J, et al. Prognostic impact of DDX41 germline mutations in intensively treated acute myeloid leukemia patients: an ALFA-FILO study. Blood (2022) 140(7):756–68. doi: 10.1182/blood.2021015328
38. Makishima H, Bowman TV, Godley LA. DDX41-associated susceptibility to myeloid neoplasms. Blood (2023) 141(13):1544–52. doi: 10.1182/blood.2022017715
39. Zhang MY, Churpek JE, Keel SB, Walsh T, Lee MK, Loeb KR, et al. Germline ETV6 mutations in familial thrombocytopenia and hematologic malignancy. Nat Genet (2015) 47(2):180–5. doi: 10.1038/ng.3177
40. Feurstein S, Godley LA. Germline ETV6 mutations and predisposition to hematological malignancies. Int J Hematol (2017) 106(2):189–95. doi: 10.1007/s12185-017-2259-4
41. Ebens CL, MacMillan ML, Wagner JE. Hematopoietic cell transplantation in fanconi anemia: current evidence, challenges and recommendations. Expert Rev Hematol (2017) 10(1):81–97. doi: 10.1080/17474086.2016.1268048
42. Peffault de Latour R, Soulier J. How I treat MDS and AML in fanconi anemia. Blood (2016) 127(24):2971–9. doi: 10.1182/blood-2016-01-583625
43. Rosenberg PS, Greene MH, Alter BP. Cancer incidence in persons with fanconi anemia. Blood (2003) 101(3):822–6. doi: 10.1182/blood-2002-05-1498
44. Anur P, Friedman DN, Sklar C, Oeffinger K, Castiel M, Kearney J, et al. Late effects in patients with fanconi anemia following allogeneic hematopoietic stem cell transplantation from alternative donors. Bone Marrow Transplant (2016) 51(7):938–44. doi: 10.1038/bmt.2016.32
45. Grossman J, Cuellar-Rodriguez J, Gea-Banacloche J, Zerbe C, Calvo K, Hughes T, et al. Nonmyeloablative allogeneic hematopoietic stem cell transplantation for GATA2 deficiency. Biol Blood Marrow Transplant (2014) 20(12):1940–8. doi: 10.1016/j.bbmt.2014.08.004
46. Cuellar-Rodriguez J, Gea-Banacloche J, Freeman AF, Hsu AP, Zerbe CS, Calvo KR, et al. Successful allogeneic hematopoietic stem cell transplantation for GATA2 deficiency. Blood (2011) 118(13):3715–20. doi: 10.1182/blood-2011-06-365049
47. Tholouli E, Sturgess K, Dickinson RE, Gennery A, Cant AJ, Jackson G, et al. In vivo T-depleted reduced-intensity transplantation for GATA2-related immune dysfunction. Blood (2018) 131(12):1383–7. doi: 10.1182/blood-2017-10-811489
48. Waidhauser J, Labopin M, Esteve J, Kroger N, Cornelissen J, Gedde-Dahl T, et al. Allogeneic stem cell transplantation for AML patients with RUNX1 mutation in first complete remission: a study on behalf of the acute leukemia working party of the EBMT. Bone Marrow Transplant (2021) 56(10):2445–53. doi: 10.1038/s41409-021-01322-w
49. Godley LA. Inherited predisposition to acute myeloid leukemia. Semin Hematol (2014) 51(4):306–21. doi: 10.1053/j.seminhematol.2014.08.001
50. Rocha V, Devergie A, Socie G, Ribaud P, Esperou H, Parquet N, et al. Unusual complications after bone marrow transplantation for dyskeratosis congenita. Br J Haematol (1998) 103(1):243–8. doi: 10.1046/j.1365-2141.1998.00949.x
51. Dietz AC, Orchard PJ, Baker KS, Giller RH, Savage SA, Alter BP, et al. Disease-specific hematopoietic cell transplantation: nonmyeloablative conditioning regimen for dyskeratosis congenita. Bone Marrow Transplant (2011) 46(1):98–104. doi: 10.1038/bmt.2010.65
52. Draze MW, Feurstein S, West AH, Jones MF, Churpek JE, Godley LA.University of Chicago hematopoietic malignancies cancer risk t. how I diagnose and manage individuals at risk for inherited myeloid malignancies. Blood (2016) 128(14):1800–13. doi: 10.1182/blood-2016-05-670240
53. Tawana K, Brown AL, Churpek JE. Integrating germline variant assessment into routine clinical practice for myelodysplastic syndrome and acute myeloid leukaemia: current strategies and challenges. Br J Haematol (2022) 196(6):1293–310. doi: 10.1111/bjh.17855
54. Drazer MW, Kadri S, Sukhanova M, Patil SA, West AH, Feurstein S, et al. Prognostic tumor sequencing panels frequently identify germ line variants associated with hereditary hematopoietic malignancies. Blood Adv (2018) 2(2):146–50. doi: 10.1182/bloodadvances.2017013037
55. Kraft IL, Godley LA. Identifying potential germline variants from sequencing hematopoietic malignancies. Blood (2020) 136(22):2498–506. doi: 10.1182/blood.2020006910
56. Godley LA. Inherited predisposition to myeloid malignancies. Blood Adv (2019) 3(17):2688. doi: 10.1182/bloodadvances.2019000172
57. Roloff GW, Godley LA, Drazer MW. Assessment of technical heterogeneity among diagnostic tests to detect germline risk variants for hematopoietic malignancies. Genet Med (2021) 23(1):211–4. doi: 10.1038/s41436-020-0934-y
58. Roloff GW, Shaw R, O'Connor TE, Hathaway F, Drazer MW. Stagnation in quality of next-generation sequencing assays for the diagnosis of hereditary hematopoietic malignancies. J Genet Couns (2023) 00:1–6. doi: 10.1002/jgc4.1672
59. Fialkow PJ, Thomas ED, Bryant JI, Neiman PE. Leukaemic transformation of engrafted human marrow cells in vivo. Lancet (1971) 1(7693):251–5. doi: 10.1016/S0140-6736(71)90998-6
60. Boyd CN, Ramberg RC, Thomas ED. The incidence of recurrence of leukemia in donor cells after allogeneic bone marrow transplantation. Leuk Res (1982) 6(6):833–7. doi: 10.1016/0145-2126(82)90067-4
61. Suarez-Gonzalez J, Martinez-Laperche C, Kwon M, Balsalobre P, Carbonell D, Chicano M, et al. Donor cell-derived hematologic neoplasms after hematopoietic stem cell transplantation: a systematic review. Biol Blood Marrow Transplant (2018) 24(7):1505–13. doi: 10.1016/j.bbmt.2018.01.033
62. Gibson CJ, Kim HT, Zhao L, Murdock HM, Hambley B, Ogata A, et al. Donor clonal hematopoiesis and recipient outcomes after transplantation. J Clin Oncol (2022) 40(2):189–201. doi: 10.1200/JCO.21.02286
63. Kato M, Yamashita T, Suzuki R, Matsumoto K, Nishimori H, Takahashi S, et al. Donor cell-derived hematological malignancy: a survey by the Japan society for hematopoietic cell transplantation. Leukemia (2016) 30(8):1742–5. doi: 10.1038/leu.2016.23
64. Engel N, Rovo A, Badoglio M, Labopin M, Basak GW, Beguin Y, et al. European Experience and risk factor analysis of donor cell-derived leukaemias/MDS following haematopoietic cell transplantation. Leukemia (2019) 33(2):508–17. doi: 10.1038/s41375-018-0218-6
65. Farina M, Bernardi S, Gandolfi L, Zanaglio C, Morello E, Turra A, et al. Case report: late onset of myelodysplastic syndrome from donor progenitor cells after allogeneic stem cell transplantation. which lessons can we draw from the reported case? Front Oncol (2020) 10:564521. doi: 10.3389/fonc.2020.564521
66. Komrokji R, Ifthikharuddin JJ, Felgar RE, Abboud CN, Wedow LA, Connaughton A, et al. Donor cell myelodysplastic syndrome after allogeneic stem cell transplantation responding to donor lymphocyte infusion: case report and literature review. Am J Hematol (2004) 76(4):389–94. doi: 10.1002/ajh.20111
67. Orciuolo E, Azzara A, Bandini G, Galimberti S, Bonifazi F, Fazzi R, et al. Contemporaneous appearance, 18 years after allogeneic bone marrow transplantation, of myelodysplastic syndrome in the patient and the donor. Bone Marrow Transplant (2004) 33(8):859–61. doi: 10.1038/sj.bmt.1704417
68. Dietz AC, DeFor TE, Brunstein CG, Wagner JE Jr. Donor-derived myelodysplastic syndrome and acute leukaemia after allogeneic haematopoietic stem cell transplantation: incidence, natural history and treatment response. Br J Haematol (2014) 166(2):209–12. doi: 10.1111/bjh.12847
69. Wiseman DH. Donor cell leukemia: a review. Biol Blood Marrow Transplant (2011) 17(6):771–89. doi: 10.1016/j.bbmt.2010.10.010
70. Kobayashi S, Kobayashi A, Osawa Y, Nagao S, Takano K, Okada Y, et al. Donor cell leukemia arising from preleukemic clones with a novel germline DDX41 mutation after allogenic hematopoietic stem cell transplantation. Leukemia (2017) 31(4):1020–2. doi: 10.1038/leu.2017.44
71. Berger G, van den Berg E, Sikkema-Raddatz B, Abbott KM, Sinke RJ, Bungener LB, et al. Re-emergence of acute myeloid leukemia in donor cells following allogeneic transplantation in a family with a germline DDX41 mutation. Leukemia (2017) 31(2):520–2. doi: 10.1038/leu.2016.310
72. Xiao H, Shi J, Luo Y, Tan Y, He J, Xie W, et al. First report of multiple CEBPA mutations contributing to donor origin of leukemia relapse after allogeneic hematopoietic stem cell transplantation. Blood (2011) 117(19):5257–60. doi: 10.1182/blood-2010-12-326322
73. Churpek JE, Artz A, Bishop M, Liu H, Godley LA. Correspondence regarding the consensus statement from the worldwide network for blood and marrow transplantation standing committee on donor issues. Biol Blood Marrow Transplant (2016) 22(1):183–4. doi: 10.1016/j.bbmt.2015.10.008
74. Marwa B, Krueger J, Stephenson EA, Davidson S, Allan D, Knoppers B, et al. Ethical and analytic challenges with genomic sequencing of relapsed hematologic malignancies following allogeneic hematopoietic stem-cell transplantation. JCO Precis Oncol (2021) 5:1339–47. doi: 10.1200/PO.20.00489
75. Godley LA, Shimamura A. Genetic predisposition to hematologic malignancies: management and surveillance. Blood (2017) 130(4):424–32. doi: 10.1182/blood-2017-02-735290
76. Crow J, Youens K, Michalowski S, Perrine G, Emhart C, Johnson F, et al. Donor cell leukemia in umbilical cord blood transplant patients: a case study and literature review highlighting the importance of molecular engraftment analysis. J Mol Diagn (2010) 12(4):530–7. doi: 10.2353/jmoldx.2010.090215
77. Ahmadmehrabi K, Haque AR, Aleem A, Griffiths EA, Roloff GW. Targeted therapies for the evolving molecular landscape of acute myeloid leukemia. Cancers (Basel). (2021) 13(18). doi: 10.3390/cancers13184646
78. Tummala H, Walne A, Dokal I. The biology and management of dyskeratosis congenita and related disorders of telomeres. Expert Rev Hematol (2022) 15(8):685–96. doi: 10.1080/17474086.2022.2108784
79. de la Fuente J, Dokal I. Dyskeratosis congenita: advances in the understanding of the telomerase defect and the role of stem cell transplantation. Pediatr Transplant (2007) 11(6):584–94. doi: 10.1111/j.1399-3046.2007.00721.x
80. Dror Y, Freedman MH, Leaker M, Verbeek J, Armstrong CA, Saunders FE, et al. Low-intensity hematopoietic stem-cell transplantation across human leucocyte antigen barriers in dyskeratosis congenita. Bone Marrow Transplant (2003) 31(10):847–50. doi: 10.1038/sj.bmt.1703931
81. Bhoopalan SV, Wlodarski M, Reiss U, Triplett B, Sharma A. Reduced-intensity conditioning-based hematopoietic cell transplantation for dyskeratosis congenita: single-center experience and literature review. Pediatr Blood Cancer (2021) 68(10):e29177. doi: 10.1002/pbc.29177
82. Barreyro L, Chlon TM, Starczynowski DT. Chronic immune response dysregulation in MDS pathogenesis. Blood (2018) 132(15):1553–60. doi: 10.1182/blood-2018-03-784116
83. Chlon TM, Stepanchick E, Hershberger CE, Daniels NJ, Hueneman KM, Kuenzi Davis A, et al. Germline DDX41 mutations cause ineffective hematopoiesis and myelodysplasia. Cell Stem Cell (2021) 28(11):1966–81 e6. doi: 10.1016/j.stem.2021.08.004
84. Parta M, Cole K, Avila D, Duncan L, Baird K, Schuver BB, et al. Hematopoietic cell transplantation and outcomes related to human papillomavirus disease in GATA2 deficiency. Transplant Cell Ther (2021) 27(5):435 e1– e11. doi: 10.1016/j.jtct.2020.12.028
85. Marciano BE, Olivier KN, Folio LR, Zerbe CS, Hsu AP, Freeman AF, et al. Pulmonary manifestations of GATA2 deficiency. Chest (2021) 160(4):1350–9. doi: 10.1016/j.chest.2021.05.046
86. Nichols-Vinueza DX, Parta M, Shah NN, Cuellar-Rodriguez JM, Bauer TR Jr., West RR, et al. Donor source and post-transplantation cyclophosphamide influence outcome in allogeneic stem cell transplantation for GATA2 deficiency. Br J Haematol (2022) 196(1):169–78. doi: 10.1111/bjh.17840
87. Hamilton KV, Maese L, Marron JM, Pulsipher MA, Porter CC, Nichols KE. Stopping leukemia in its tracks: should preemptive hematopoietic stem-cell transplantation be offered to patients at increased genetic risk for acute myeloid leukemia? J Clin Oncol (2019) 37(24):2098–104. doi: 10.1200/JCO.19.00181
Keywords: hereditary hematopoietic malignancy, AML, MDS, genetic testing, stem cell transplant
Citation: O’Connor TE, Shaw R, Madero-Marroquin R and Roloff GW (2023) Clinical considerations at the intersection of hematopoietic cell transplantation and hereditary hematopoietic malignancy. Front. Oncol. 13:1180439. doi: 10.3389/fonc.2023.1180439
Received: 06 March 2023; Accepted: 28 April 2023;
Published: 12 May 2023.
Edited by:
Claire Homan, University of South Australia, AustraliaReviewed by:
Simona Bernardi, University of Brescia, ItalyYifan Pang, Levine Cancer Institute, United States
Copyright © 2023 O’Connor, Shaw, Madero-Marroquin and Roloff. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gregory W. Roloff, Z3JlZ29yeS5yb2xvZmZAdWNob3NwaXRhbHMuZWR1