 Michael S. Petronek1*
Michael S. Petronek1* Khaliunaa Bayanbold1
Khaliunaa Bayanbold1 Koffi Amegble2Ann M. Tomanek-Chalkley1Bryan G. Allen1
Koffi Amegble2Ann M. Tomanek-Chalkley1Bryan G. Allen1 Douglas R. Spitz1
Douglas R. Spitz1 Charvann K. Bailey2*
Charvann K. Bailey2*- 1Department of Radiation Oncology, Division of Free Radical and Radiation Biology, University of Iowa, Iowa City, IA, United States
- 2Department of Biology, Grinnell College, Grinnell, IA, United States
A distinctive feature of cancer is the upregulation of sirtuin proteins. Sirtuins are class III NAD+-dependent deacetylases involved in cellular processes such as proliferation and protection against oxidative stress. SIRTs 1 and 2 are also overexpressed in several types of cancers including non-small cell lung cancer (NSCLC). Sirtinol, a sirtuin (SIRT) 1 and 2 specific inhibitor, is a recent anti-cancer agent that is cytotoxic against several types of cancers including NSCLC. Thus, sirtuins 1 and 2 represent valuable targets for cancer therapy. Recent studies show that sirtinol functions as a tridentate iron chelator by binding Fe3+ with 3:1 stoichiometry. However, the biological consequences of this function remain unexplored. Consistent with preliminary literature, we show that sirtinol can deplete intracellular labile iron pools in both A549 and H1299 non-small cell lung cancer cells acutely. Interestingly, a temporal adaptive response occurs in A549 cells as sirtinol enhances transferrin receptor stability and represses ferritin heavy chain translation through impaired aconitase activity and apparent IRP1 activation. This effect was not observed in H1299 cells. Holo-transferrin supplementation significantly enhanced colony formation in A549 cells while increasing sirtinol toxicity. This effect was not observed in H1299 cells. The results highlight the fundamental genetic differences that may exist between H1299 and A549 cells and offer a novel mechanism of how sirtinol kills NSCLC cells.
Introduction
Globally, lung cancer is one of the leading causes of cancer deaths. Approximately 80%–85% of all lung cancer diagnoses are non-small cell lung cancer (NSCLC), and the 5-year overall survival remains approximately 28% (1). Such a despondent figure indicates a need for more effective treatment. Historically, treatments for cancer consisted of radiotherapy and chemotherapy, which have had limited success, treating NSCLC (2, 3). More contemporary techniques like targeted therapy allow researchers to exploit distinctive characteristics of cancer cells for more precise treatment (4). One consistent attribute of lung cancer cells is the overexpression of sirtuin 1 and 2 (SIRT1/2) proteins compared to normal human bronchial epithelial cells where enhanced SIRT1/2 expression has been correlated with a poor prognosis in NSCLC patients (5, 6). SIRTs are class III nicotinamide adenine dinucleotide (NAD)+-dependent histone deacetylases that modulate senescence, cell cycle regulation, apoptosis, and oxidative stress response (7). By using NAD+ as a cosubstrate for their modulation of histone deacetylation, targets such as H3K9ac or non-histone targets like Foxo3a and sirtuins are critical for mediating cell survival (8). Current literature reveals that targeted inhibition of SIRT1 and 2 proteins in cancer is beneficial for constraining their growth and defense mechanisms—making them more susceptible to apoptosis.
Sirtinol is a SIRT 1 and 2 pharmacological inhibitor that was discovered during cell-based screening of Saccharomyces cerevisiae yeast by accessing Sir2p inhibition (9). Its potent inhibition of SIRT1 and 2 and has been investigated as an anticancer agent (10). Sirtinol induces senescence and apoptosis in MCF-7 breast cancer cells and H1299 NSCLCs (11, 12). Although sirtinol is not currently a mainline therapeutic used to treat NSCLCs, several studies show its potential for use in the clinic. For example, Fong et al. showed that sirtinol is cytotoxic to NSCLC cells in a dose-dependent manner after 24 and 48 h treatment (13). This study also showed that sirtinol reduces the proliferation of cancer cells by pausing the cell in the G1 phase of the cell cycle. Sirtinol also enhances the cytotoxicity of traditional chemotherapeutic agents like gemcitabine and cisplatin (14, 15), while reducing inflammation in normal epithelial cells, suggesting selective sensitization (16).
Recently, sirtinol was also discovered to be an intracellular iron chelator (17). This observation evolved from evaluating the chemical structure of sirtinol relative to other iron-chelating molecules [e.g., desferrioxamine (DFO) or deferasirox] (7). Sirtinol’s structure contains a 2-hydroxynaphthalenyl moiety that is connected to a benzamide through an aldiminic nitrogen atom (Figures 1A, B). This group can provide a tridentate O – N – O donor moiety that is often observed in other iron-chelating molecules. Crystallography analysis showed that sirtinol can function as a tridentate iron chelator by binding a single Fe3+ atom with 3:1 (sirtinol:Fe3+) stoichiometry to form an octahedral, high-spin (S = 5/2) Fe3+ complex comprised of five donor oxygen atoms and one donor nitrogen atom (Figure 1C) (17). This is a critical finding as iron chelation may serve as an important drug effect associated with the cytotoxicity of sirtinol. However, the biological significance of sirtinol’s iron-chelator function remains unclear. Thus, it was hypothesized that sirtinol can impair iron metabolism in NSCLC cells and this study aimed to evaluate the biological significance of the chelator function of sirtinol in NSCLC.
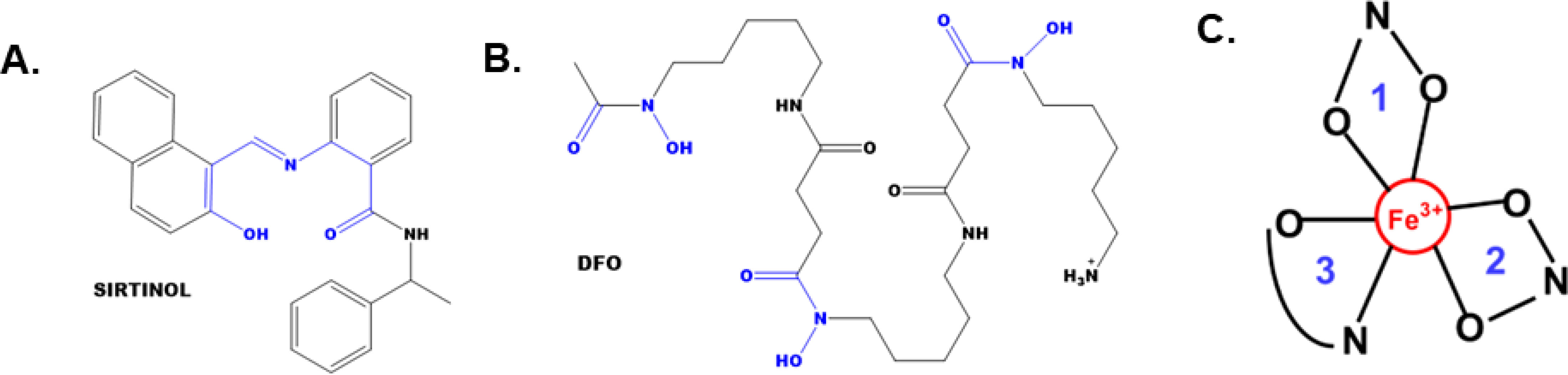
Figure 1 The structures of sirtinol and desferrioxamine (DFO) and its iron coordination sites. (A, B) The structures of sirtinol (A) and DFO (B). The putative iron coordination site within both structures are highlighted in blue. (C) Tridentate, octahedral, high-spin Fe3+ complex formed from the binding of sirtinol to a free Fe atom. Blue numbers represent independent sirtinol molecules bound to the Fe atom.
Iron metabolism is a critical feature of a multitude of cellular functions central to cancer progression (18). It is altered in a wide array of cancer types with cancer cells typically harboring an iron dependency as compared to their normal tissue counterparts. Iron metabolism is altered in human NSCLC tumor tissue with increased hepcidin expression (19). Increased transferrin receptor (TfR) expression has also been observed in human NSCLC tumors (20). These data suggest that NSCLC tumors accumulate iron preferentially. Iron chelation therapy may be an attractive anticancer therapeutic strategy. For example, deferoxamine has recently been shown to selectively induce mitochondrial dysfunction in cancer cells (21).
In this study, A549 and H1299 NSCLC cells were used to evaluate the chelator function of sirtinol. These cells represent two widely used NSCLC in vitro models with varying clinically relevant genetic backgrounds (Table 1). Importantly, A549 cells harbor mutations that have been observed in aggressive lung tumors. STK11 comutations as observed in the A549 cells are relatively frequent with both KRAS (54%) and KEAP1 (27%), where STK11 comutations are associated with worse clinical outcomes (27). Thus, A549 cells provide an in vitro model of an aggressive NSCLC tumor.
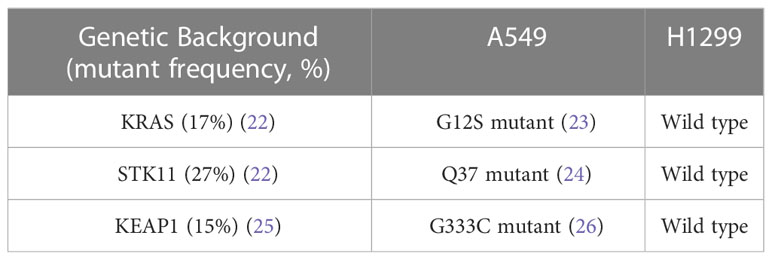
Table 1 Genetic differences in cell lines used in this study.
Results
First, the proposed chelator function was evaluated in this in vitro NSCLC model. Consistent with this function, a 72 h treatment of sirtinol caused decrease in intracellular labile iron in both H1299 and A549 cells (Figure 2). Importantly, the decrease in labile iron by sirtinol is the same effect as observed by DFO. An interesting observation was that this effect was greater in the A549 cells (79.2% decrease) as compared to the H1299 (36.3% decrease). Thus, we have been able to recapitulate the ability of sirtinol to serve as an iron chelator to a differential effect in these genetic subtypes of NSCLC.
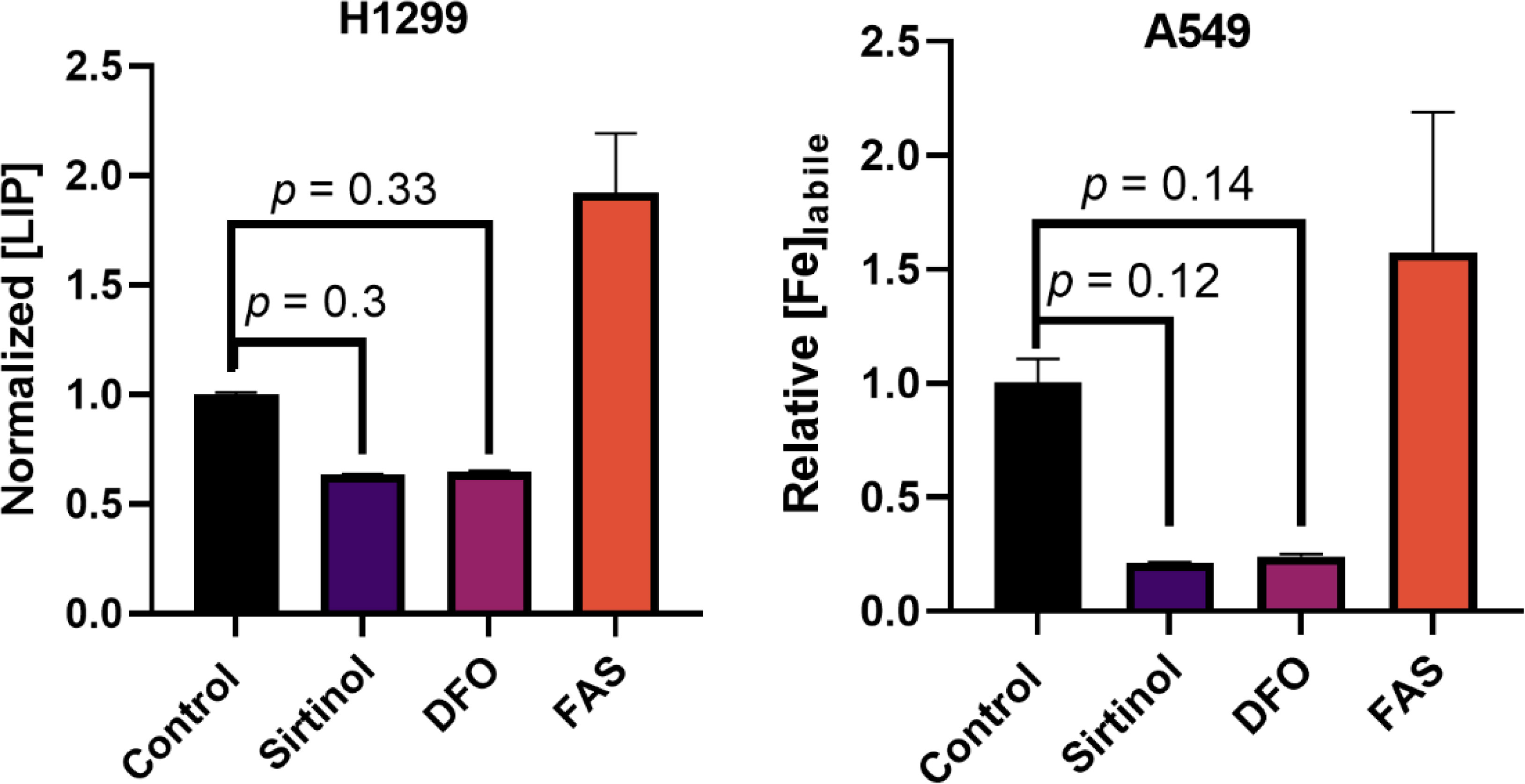
Figure 2 Sirtinol behaves like DFO to chelate iron in non-small cell lung cancer (NSCLC). Effects on labile iron in H1299 and A549 cells by a 50 µM 72 h treatment of sirtinol and DFO using a calcein–AM flow cytometry probe. Conversely, 3 h, 50 µM ferrous ammonium sulfate was used as a positive control. Error bars represent the mean ± SEM of n = 3 replicates. Statistical analysis was done using a one-way ANOVA with a post-hoc Tukey’s test for individual comparisons.
Sirtinol alters iron metabolic features in non-small cell lung cancer cells
Because of the initial validation of the sirtinol iron chelator function, it was hypothesized that this would result in metabolic alterations associated with labile iron regulation. Aconitase was first interrogated as it is an iron-dependent tricarboxylic acid (TCA)-cycle intermediate that utilizes a complete [4Fe-4S]2+ cluster for the isomerization of citrate to isocitrate (28). Under conditions of limited intracellular iron availability, the cluster will be in an incomplete [3Fe-4S]+ form leading to enzyme inactivation (29). In both A549 and H1299 cell lines, a significant decrease in aconitase activity (>50% reduction in enzymatic activity, p< 0.05) was observed (Figure 3A), further indicating that sirtinol’s chelator activity can sequester iron to impair the activity of iron-dependent enzymes.
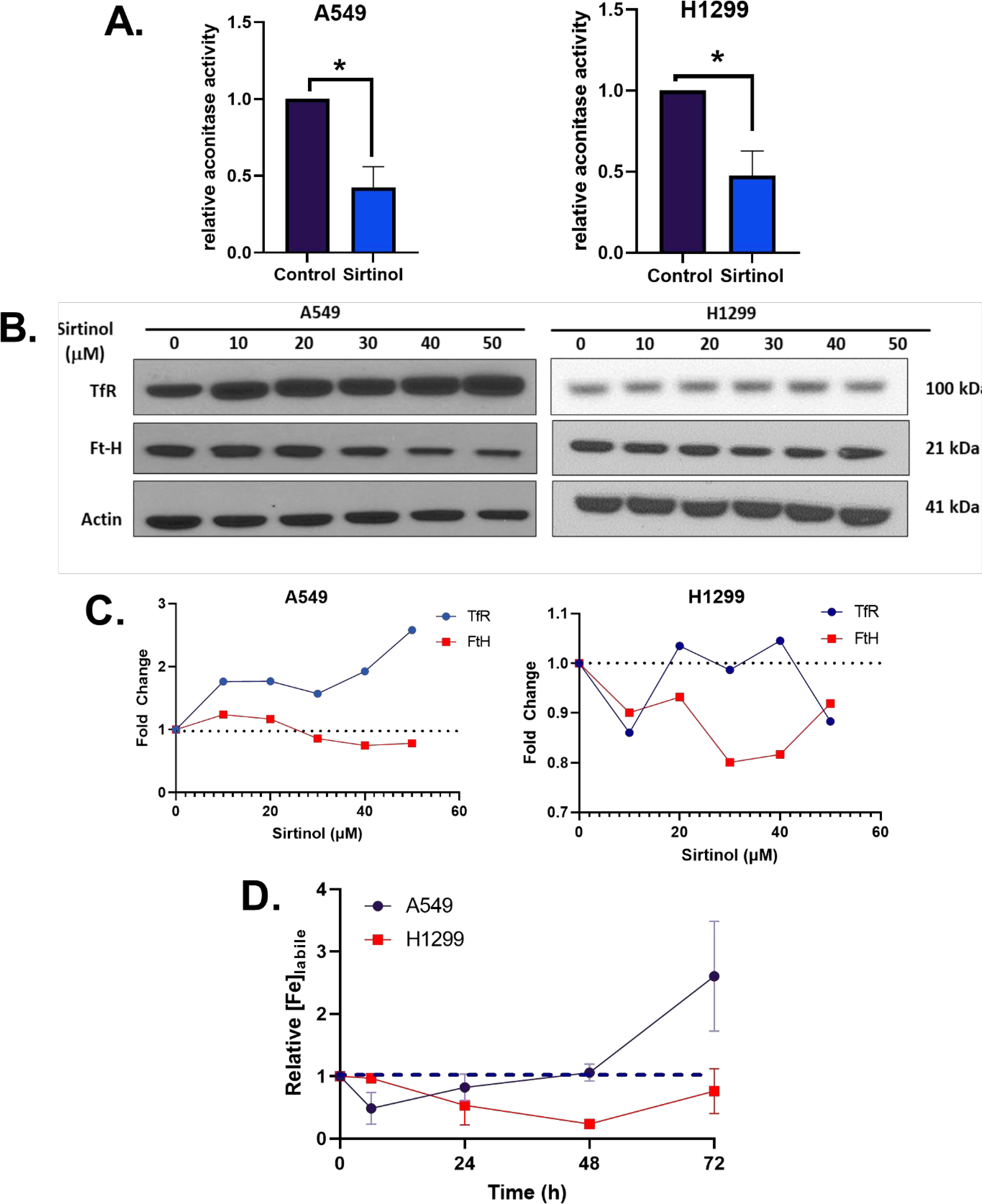
Figure 3 Sirtinol alters iron metabolic features. (A) Relative aconitase activity in A549 and H1299 cells following a 24 h treatment of 50 µM sirtinol. Aconitase activity was evaluated by measuring the rate of appearance of nicotinamide adenine dinucleotide phosphate (NADPH) at 340 nm in the presence of citrate and isocitrate dehydrogenase. Error bars represent the mean ± SEM of n = 3–4 experiments where *p< 0.05 using an unpaired, Welch’s T-test. (B) A549 and H1299 NSCLC cells were treated for 24 h with 0–50 µM sirtinol and then harvested for evaluation of the transferrin receptor (TfR) and ferritin (FtH) expression using a Western blot approach. (C) Western blot quantification TfR and FtH changes in A549 and H1299 cells. (D) A549 and H1299 NSCLC cells were treated for 6, 24, 48, or 72 h with 50 µM sirtinol and then harvested for evaluation of labile iron pool concentrations using a colorimetric ferrozine-based assay. Each timepoint was normalized to an untreated control. Dashed black line indicates baseline measures. Error bars represent mean ± SD from triplicate measures.
Converse to its TCA-cycle function, in an inactive [3Fe-4S]+ form, aconitase can function as iron-responsive protein-1 (IRP1). IRP1 responds to intracellular Fe levels to promote either stabilization or degradation of TfR and ferritin heavy-chain (FtH) mRNAs (30). When there is limited intracellular iron availability, IRP1 binds to iron response elements (IREs) to maintain homeostasis by binding at the 3’ end of TfR mRNA to enhance stability and promote translation (31–33) or the 5’ end of FtH to repress translation (34, 35). Interestingly, a sirtinol concentration–dependent increase in TfR protein levels and FtH repression cells was observed in A549 cells that was not apparent in H1299 cells (Figures 3B, C). This further supports the hypothesis that the chelator function of sirtinol disrupts iron metabolism by limiting intracellular iron availability, although the biological effects appear to present as a cell type–dependent effect. Therefore, it appears that sirtinol may be able to impair the IRP-IRE system through its chelator function; however, this requires further validation. Based on the observed iron metabolic perturbations, the temporal-dependent changes in intracellular iron were interrogated. Using this colorimetric assay, sirtinol was observed to chelate iron in both H1299 and A549 cell lines, as relative labile iron concentrations were decreased in both cell lines acutely (Figure 3D). In A549 cells, the labile iron pool was decreased acutely (within 6 h) while noticeable decreases did not occur in H1299 cells until 24 h sirtinol treatment was used. Interestingly, after the acute decrease in labile iron in A549, there was an apparent adaptive response where labile iron continued to increase above the basal level until 72 h. A similar trend was not observed in the H1299 cells as labile iron was decreased through 48 h until a slight increase from 48 to 72 h occurred but remained below the basal level. These results are consistent with the cell type–specific adaptive response that occurred in the A549 cells. These results further support the iron chelator function of sirtinol, and these observations may represent a fundamental difference in iron metabolic regulation and response to perturbations between NSCLC cell lines with variable genetic backgrounds.
Sirtinol impairs iron-dependent colony formation in A549 cells
Based upon sirtinol’s induced iron-metabolic shifts, the biological relevance of these findings was interrogated by investigating its effects on iron-dependent colony formation. First, the effects of holo-transferrin [hTf; di-ferric transferrin; Tf-(Fe3+)2] on NSCLC plating efficiency were assessed. Interestingly, a significant increase in plating efficiency (40%–60%) was observed in A549 cells following a 24 h supplementation of cell culture media with hTf while there was no effect in H1299 cells (Figure 4A). Thus, A549 cells exhibit a pattern of iron dependency to facilitate colony formation, while H1299 cells do not. Similarly, sirtinol enhanced cell-killing in A549 cells supplemented with hTf but had little to no effect in H1299 cells (Figure 4B). Therefore, the chelator function of sirtinol appears to play a role in sirtinol’s cytotoxicity but is largely context dependent.

Figure 4 Sirtinol impairs iron-dependent colony formation in A549 cells. (A) Plating efficiency (%) of A549 and H1299 cells following a 24 h supplement of 200 µg ml-1 holo-transferrin (hTf). Error bars represent mean ± SD (n = 3) with *p< 0.05 using a paired, two-tailed Welch’s T-test. (B) Clonogenic survival of A549 and H1299 cells treated for 24 h ± sirtinol (50 µM) ± hTf (200 µg ml-1). Error bars represent mean ± SD (n = 3) with *p< 0.05 using a one-way ANOVA test.
Discussion
This study has shown that sirtinol can function as an intracellular iron chelator to induce iron metabolic changes. Importantly, the chelator function of sirtinol has been further validated in NSCLC cells as sirtinol exhibited the same effects on labile iron as DFO. The ability of sirtinol to deplete intracellular labile iron is consistent with the previous literature that sirtinol can function as a tridentate iron chelator (17). Furthermore, sirtinol has been observed to deplete intracellular labile iron acutely leading to an adaptive response; however, the adaptive response is cell line dependent. The adaptive response that we observed, primarily in A549 cells, is consistent with decreased aconitase activity (i.e., enhanced IRP1 activation) leading to increased TfR stability and decreased FtH translation. This is likely due to the sequestering of iron leading to an incomplete [3Fe-4S]+ cluster in aconitase (30). Thus, it can be hypothesized that a disruption in the IRP-IRE system results in an increase in TfR stability leading to increased iron uptake via receptor-mediated endocytosis (36, 37). Meanwhile, the decrease in FtH expression indicates decreased iron storage capacity, as ferritin is the primary iron storage enzyme of the cell (38). Further experiments are required to elucidate the effects of sirtinol on the IRP-IRE system. However, an adaptive response to increase iron uptake and decrease storage would explain the temporally dependent increases in labile iron observed for extended sirtinol treatments in A549 cells. The increase in labile iron at 72 h observed using ferrozine is slightly contradictory to the calcein-AM results. This is likely due to the nature of each individual assay as calcein-AM can bind iron irrespective of its oxidation state but is unable to remove iron from other complexes, while ferrozine is Fe2+-dependent and utilizes ascorbic acid to convert all of the iron in a highly acidic (pH ≈ 4–4.5) solution to convert all of the iron to Fe2+. Thus, the ferrozine results at 72 h may be more reflective of the total iron accumulated within the cells in addition to the iron bound to sirtinol, while the calcein-AM results are more likely to reflect the amount of iron remaining to be chelated after treatment. This hypothesis is based on the notion that sirtinol iron binding permits iron recycling allowing for the iron to be reduced to Fe2+ under the acidic conditions. This would be consistent with previously reported literature that a sirtinol-Fe3+ complex can enhance oxidation of the DCFH fluorescent probe, indicating that this is a redox-active chelator capable of catalyze redox reactions (39). Based on these observations, the iron-chelator capacity of sirtinol previously described chemically appears to have biological importance and is a drug effect that should be considered when evaluating the therapeutic effects of sirtinol (17).
An unexpected discovery is the iron-dependent differences observed between A549 and H1299 NSCLC cells. Consistently, a more pronounced iron-metabolic disruption has been observed in the A549 cells throughout this study. A major, correlative difference between these two cell lines is their KRAS/STK11/KEAP-1 mutational status. A549 cells are KRAS/STK11/KEAP-1 mutant cells resulting in constitutive activation of NRF2 (40, 41) while H1299 cells are not (42). This is of particular interest because KEAP-1 mutant NSCLC cells, such as A549, are particularly aggressive and exhibit therapy resistance as patients with tumors harboring this mutational profile appear to have worse clinical outcomes (42–44). A recent retrospective study of NSCLC patients showed that KEAP-1 was a negative prognostic marker in advanced-stage (stage IIIB–IV) tumors (HR = 1.40, 95% CI: 1.23–1.61, p< 0.001, N = 4,779) (44). Therefore, strategies aimed at enhancing clinical responses in mutant tumors may be of critical importance in managing NSCLC. Interestingly, we have observed that STK11/KRAS/KEAP-1 mutant A549 cells exhibit a pattern of iron-dependent clonogenicity that the chelator function of sirtinol can exploit while the H1299 tumors do not. While this finding is intriguing, it also represents a significant limitation of our study. Because of the concurrent mutations observed in the A549, it is currently unclear to what extent each mutation contributes to the differential iron metabolic regulation observed in these cells. Future studies should be designed to evaluate each of these mutations independently and assess their impacts on iron metabolic regulation.
Conclusions
In summary, it has been observed that the chemical iron-chelator function of sirtinol has biological consequences. It has been shown that sirtinol can chelate iron in NSCLC cells leading to a decrease in labile iron acutely and a cell line–specific adaptive response characterized by a decrease in aconitase activity leading to a shift toward IRP1 activation, enhanced TfR stability, and repressed FtH translation. Intriguingly, KRAS/STK11/KEAP-1 mutant cells exhibit iron-dependent colony formation that can be inhibited by sirtinol, which does not occur in H1299 cells. Overall, it can be concluded that the chelator function of sirtinol has context-dependent biological consequences that may contribute to its toxicity in NSCLC.
Materials and methods
Cell culture
A549 and H1299 cells were grown to 80% confluence before experimentation at 21% O2. Cells were treated with sirtinol prepared in 50mM stocks in DMSO and stored at −80°C for the appropriate concentration and time. For iron supplementation, 10 mg ml-1 of human holo-transferrin (T0665 Sigma-Aldrich, St. Louis, MO) prepared in H2O was added directly to the cell culture media at a concentration of 200 µg ml-1. For experiments testing the combination, both sirtinol and holo-transferrin were added simultaneously. For colony formation assays, cells were treated, washed, and trypsinized. Following trypsinization, cells were counted and plated as single cells in a 6-well dish (≈500 cells per well). Cells were left undisturbed for 7–10 days to allow for colony formation. Colonies were then washed with 70% EtOH for fixation and stained with Coomassie blue. Stained colonies (≥50 cells) were counted under a microscope.
Labile iron pool measures with flow cytometry
Intracellular labile iron pool measures were performed using a Calcein-AM fluorescent dye. Cells were harvested by trypsinization. After cell harvesting, cell pellets were washed in phosphate buffered saline (PBS) and then resuspended in 500 nM Calcein-AM diluted in PBS. Samples were incubated for 15 min at 4% O2 (37°C, 5% CO2). Following incubation, extracellular Calcein-AM was removed by washing with PBS, and cells were resuspended in 1 ml PBS. Following incubation 10,000 cells were analyzed on an LSR II Flow Cytometer (BD Biosciences; λex = 488 nm, λem = 515/20 nm). The labile iron pool was quantified using the following formula:
An inverse normalization was done to approximate the labile iron pool because calcein-AM functions as a “turn-off” probe.
Colorimetric labile iron assay
Labile iron and total iron concentrations were done using a ferrozine-based colorimetric assay. Cells were homogenized in 1X RIPA lysis buffer (Sigma-Aldrich; R0278). Cells were centrifuged at maximum speed for 10 min to remove cell debris, and 100 µl of the supernatant was then diluted 1:1 in ferrozine buffer (5 mM ferrozine, 1.25 M ammonium acetate, and 10 mM ascorbate) and centrifuged again at maximum speed for 10 min to remove any protein aggregates. This step is critical as the acidic nature of the buffer (pH ≈ 4–4.5) will result in protein aggregation that can cause the samples to become cloudy and alter the absorbance profile, resulting in an experimental artifact. Thus, samples with remaining protein aggregates were removed from analysis. The supernatant was then placed in a single well of a clear 96-well plate. Following dilution, the 96-well plate was evaluated for the formation of a Fe2+-ferrozine complex by monitoring the absorbance at 562 nm and Fe concentration was calculated using Beer’s Law:
where A562 is the measured absorbance at 562 nm, ϵ562 is the molar extinction coefficient for a Fe2+-ferrozine complex = 27,900 M-1 cm-1, [Fe] is the calculated Fe concentration (M), and L is the pathlength for 200 µl of liquid ≈ 0.55 cm.
Western blotting
Total protein (25 μg) was electrophoresed on a 4%–20% gradient gel (Bio-Rad) at 150 V for approximately 1.5 h. The separated proteins were transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, CA) and non-specific binding was blocked using 5% non-fat dry milk in PBS-Tween (0.2%) for 1 h at room temperature. The membranes were incubated with primary antibodies (ferritin-heavy chain, 1:1,000 from Abcam, Cambridge, MA, TfR, 1:1,000, Invitrogen, Camarillo, CA) at 4°C overnight. B-actin served as a loading control (1:4,000; Sigma-Aldrich). Following three 5 min TBS-Tween washes, the membranes were probed with secondary antibodies (mouse anti-rabbit; 1:10,000; Sigma-Aldrich, St. Louis, MO) that were conjugated with horseradish peroxidase for 45 min. The washed membranes were incubated with a Super Signal West Pico Chemiluminescent Substrate (Thermo Scientific, Rockford, IL) and exposed to CareStream BioMax MR Film (CareStream Health, Rochester, NY). Quantification of Western blots were performed in ImageJ.
Aconitase activity
Exponentially growing cells were scraped and frozen as dry pellets until assayed for total aconitase activity adapted from as previously described (45). Briefly, cell pellets were resuspended in 50 mM Tris-HCl, pH 7.4 with 0.6 mM MnCl2, and 5 mM Na-citrate and sonicated 3 × 10 s each. Protein was quantified by the Lowry method (46). Aconitase activity was measured as the rate of appearance of NADPH (at 340 nm; Beckman DU 800 spectrophotometer, Brea, CA) for 45 min during the reaction of 200 μg total sample protein with 200 μM NADP+ and 10 U isocitrate dehydrogenase.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
Author contributions
MP and CB performed study designs; MP, CB, KB, KA, and AT-C performed experiments; MP, CB, KB, KA, AT-C, BA, and DS performed data analysis; MP and CB prepared the manuscript; MP, CB, KB, KA, AT-C, BA, and DS contributed to editing. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by NIH grants, P01 CA217797, P01 CA244091, and the Gateway for Cancer Research grant G-17-1500. Core facilities were supported in part by the Carver College of Medicine and the Holden Comprehensive Cancer Center, NIH P30 CA086862.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Author disclaimer
The content is solely the responsibility of the authors and does not represent the views of the National Institutes of Health.
References
1. Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA: A Cancer J Clin (2023) 73:17–48. doi: 10.3322/caac.21763
2. Parashar B, Arora S, Wernicke A. Radiation therapy for early stage lung cancer. Semin interventional Radiol (2013) 30:185–90. doi: 10.1055/s-0033-1342960
3. Zhang J-G, Hong D-F, Zhang C-W, Sun X-D, Wang Z-F, Shi Y, et al. SIRT1 facilitates chemoresistance of pancreatic cancer cells by regulating adaptive response to chemotherapy-induced stress. Cancer Sci (2014) 105(4):445–54. doi: 10.1111/cas.12364
4. Reyes-Habito C, Roh E. Cutaneous reactions to chemotherapeutic drugs and targeted therapy for cancer: part II. targeted therapy. J Am Acad Dermatol (2014) 71:217.e1–217.e11. doi: 10.1016/j.jaad.2014.04.013
5. Zheng M, Hu C, Wu M, Chin Y. Emerging role of SIRT2 in non-small cell lung cancer (Review). Oncol Lett (2021) 22:731. doi: 10.3892/ol.2021.12992
6. Grbesa I, Pajares MJ, Martínez-Terroba E, Agorreta J, Mikecin A-M, Larráyoz M, et al. Expression of sirtuin 1 and 2 is associated with poor prognosis in non-small cell lung cancer patients. PloS One (2015) 10:e0124670. doi: 10.1371/journal.pone.0124670
7. Chalkiadaki A, Guarente L. The multifaceted functions of sirtuins in cancer. Nat Rev Cancer (2015) 15:608–24. doi: 10.1038/nrc3985
8. Wang F, Chan C-H, Chen K, Guan X, Lin H-K, Tong Q, et al. Deacetylation of FOXO3 by SIRT1 or SIRT2 leads to Skp2-mediated FOXO3 ubiquitination and degradation. Oncogene (2012) 31:1546–57. doi: 10.1038/onc.2011.347
9. Bedalov A, Chowdhury S, Simon JA. Chapter nine - biology, chemistry, and pharmacology of sirtuins. In: Marmorstein R, editor. Enzymes of Epigenetics, Part B, Methods in Enzymology, vol. 574 . Academic Press (2016). p. 183–211.
10. Hu J, Jing H, Lin H. Sirtuin inhibitors as anticancer agents. Future Medicinal Chem (2014) 6:945–66. doi: 10.4155/fmc.14.44
11. Wang J, Kim T-H, Ahn MY, Lee J, Jung JH, Choi WS, et al. Sirtinol, a class III HDAC inhibitor, induces apoptotic and autophagic cell death in MCF-7 human breast cancer cells. Int J Oncol (2012) 41:1101–9. doi: 10.3892/ijo.2012.1534
12. Ota H, Tokunaga E, Chang K, Hikasa M, Iijima K, Eto M, et al. Sirt1 inhibitor, sirtinol, induces senescence-like growth arrest with attenuated ras–MAPK signaling in human cancer cells. Oncogene (2006) 25:176–85. doi: 10.1038/sj.onc.1209049
13. Fong Y, Lin Y-C, Wu C-Y, Wang H-MD, Lin L-L, Chou HL, et al. The antiproliferative and apoptotic effects of sirtinol, a sirtuin inhibitor on human lung cancer cells by modulating akt/β-Catenin-Foxo3A axis. Sci World J (2014) 2014:937051. doi: 10.1155/2014/937051
14. Jin KL, Park J-Y, Noh EJ, Hoe KL, Lee JH, Kim J-H, et al. The effect of combined treatment with cisplatin and histone deacetylase inhibitors on HeLa cells. J Gynecol Oncol (2010) 21:262–8. doi: 10.3802/jgo.2010.21.4.262
15. Gong D-J, Zhang J-M, Yu M, Zhuang B, Guo Q-Q. Inhibition of SIRTI combined with gemcitabine therapy for pancreatic carcinoma. Clin Interventions Aging (2013) 8:889–97. doi: 10.2147/CIA.S45064
16. Orecchia A, Scarponi C, Di Felice F, Cesarini E, Avitabile S, Mai A, et al. Sirtinol treatment reduces inflammation in human dermal microvascular endothelial cells. PloS One (2011) 6:e24307. doi: 10.1371/journal.pone.0024307
17. Gautam R, Akam EA, Astashkin AV, Loughrey JJ, Tomat E. Sirtuin inhibitor sirtinol is an intracellular iron chelator. Chem Commun (2015) 51:5104–7. doi: 10.1039/C5CC00829H
18. Petronek MS, Spitz DR, Buettner GR, Allen BG. Linking cancer metabolic dysfunction and genetic instability through the lens of iron metabolism. Cancers (Basel) (2019) 11:1077. doi: 10.3390/cancers11081077
19. Chen Q, Wang L, Ma Y, Wu X, Jin L, Yu F, et al. Increased hepcidin expression in non-small cell lung cancer tissue and serum is associated with clinical stage. Thorac Cancer (2014) 5:14–24. doi: 10.1111/1759-7714.12046
20. Kukulj S, Jaganjac M, Boranic M, Krizanac S, Santic Z, Poljak-Blazi M, et al. Altered iron metabolism, inflammation, transferrin receptors, and ferritin expression in non-small-cell lung cancer. Med Oncol (2010) 27:268–77. doi: 10.1007/s12032-009-9203-2
21. Sandoval-Acuña C, Torrealba N, Tomkova V, Jadhav SB, Blazkova K, Merta L, et al. Targeting mitochondrial iron metabolism suppresses tumor growth and metastasis by inducing mitochondrial dysfunction and mitophagy. Cancer Res (2021) 81:2289–303. doi: 10.1158/0008-5472.CAN-20-1628
22. Blanco R, Iwakawa R, Tang M, Kohno T, Angulo B, Pio R, et al. A gene-alteration profile of human lung cancer cell lines. Hum Mutat (2009) 30:1199–206. doi: 10.1002/humu.21028
23. Acunzo M, Romano G, Nigita G, Veneziano D, Fattore L, Laganà A, et al. Selective targeting of point-mutated KRAS through artificial microRNAs. Proc Natl Acad Sci U.S.A. (2017) 114:E4203–12. doi: 10.1073/pnas.1620562114
24. Matsumoto S, Iwakawa R, Takahashi K, Kohno T, Nakanishi Y, Matsuno Y, et al. Prevalence and specificity of LKB1 genetic alterations in lung cancers. Oncogene (2007) 26:5911–8. doi: 10.1038/sj.onc.1210418
25. Chen X, Su C, Ren S, Zhou C, Jiang T. Pan-cancer analysis of KEAP1 mutations as biomarkers for immunotherapy outcomes. Ann Trans Med (2019) 8(4):141. doi: 10.21037/atm.2019.11.52
26. Taguchi K, Yamamoto M. The KEAP1-NRF2 system in cancer. Front Oncol (2017) 7:85. doi: 10.3389/fonc.2017.00085
27. Malhotra J, Ryan B, Patel M, Chan N, Guo Y, Aisner J, et al. Clinical outcomes and immune phenotypes associated with STK11 co-occurring mutations in non-small cell lung cancer. J Thorac Dis (2022) 14(6):1772–83. doi: 10.21037/jtd-21-1377
28. Lushchak OV, Piroddi M, Galli F, Lushchak VI. Aconitase post-translational modification as a key in linkage between Krebs cycle, iron homeostasis, redox signaling, and metabolism of reactive oxygen species. null (2014) 19:8–15. doi: 10.1179/1351000213Y.0000000073
29. Volz K. The functional duality of iron regulatory protein 1. Curr Opin Struct Biol (2008) 18:106–11. doi: 10.1016/j.sbi.2007.12.010
30. Casey JL, Hentze MW, Koeller DM, Caughman SW, Rouault TA, Klausner RD, et al. Iron-responsive elements: regulatory RNA sequences that control mRNA levels and translation. Science (1988) 240:924–8. doi: 10.1126/science.2452485
31. Erlitzki R, Long JC, Theil EC. Multiple, conserved iron-responsive elements in the 3′-untranslated region of transferrin receptor mRNA enhance binding of iron regulatory protein 2 *. J Biol Chem (2002) 277:42579–87. doi: 10.1074/jbc.M207918200
32. Müllner EW. & kühn, l. c. a stem-loop in the 3′ untranslated region mediates iron-dependent regulation of transferrin receptor mRNA stability in the cytoplasm. Cell (1988) 53:815–25. doi: 10.1016/0092-8674(88)90098-0
33. Casey JL, Koeller DM, Ramin VC, Klausner RD, Harford JB. Iron regulation of transferrin receptor mRNA levels requires iron-responsive elements and a rapid turnover determinant in the 3′ untranslated region of the mRNA. EMBO J (1989) 8:3693–9. doi: 10.1002/j.1460-2075.1989.tb08544.x
34. Hentze Matthias W, Caughman SW, Rouault TA, Barriocanal JG, Dancis A, Harford JB, et al. Identification of the iron-responsive element for the translational regulation of human ferritin mRNA. Science (1987) 238:1570–3. doi: 10.1126/science.3685996
35. Leibold EA, Munro HN. Cytoplasmic protein binds in vitro to a highly conserved sequence in the 5’ untranslated region of ferritin heavy- and light-subunit mRNAs. Proc Natl Acad Sci USA (1988) 85:2171. doi: 10.1073/pnas.85.7.2171
36. Hentze MW, Muckenthaler MU, Andrews NC. Balancing acts: molecular control of mammalian iron metabolism. Cell (2004) 117:285–97. doi: 10.1016/S0092-8674(04)00343-5
37. Gammella E, Buratti P, Cairo G, Recalcati S. The transferrin receptor: the cellular iron gate. Metallomics (2017) 9:1367–75. doi: 10.1039/C7MT00143F
38. Harrison PM, Arosio P. The ferritins: molecular properties, iron storage function and cellular regulation. Biochim Biophys Acta (BBA) - Bioenergetics (1996) 1275:161–203. doi: 10.1016/0005-2728(96)00022-9
39. Akam EA, Gautam R, Tomat E. Metal-binding effects of sirtuin inhibitor sirtinol. Supramol Chem (2016) 28:108–16. doi: 10.1080/10610278.2015.1092537
40. Singh A, Misra V, Thimmulappa RK, Lee H, Ames S, Hoque MO, et al. Dysfunctional KEAP1–NRF2 interaction in non-Small-Cell lung cancer. PloS Med (2006) 3:e420. doi: 10.1371/journal.pmed.0030420
41. Taguchi K, Yamamoto M. The KEAP1–NRF2 system in cancer. Front Oncol (2017) 7. doi: 10.3389/fonc.2017.00085
42. Gong M, Li Y, Ye X, Zhang L, Wang Z, Xu X, et al. Loss-of-function mutations in KEAP1 drive lung cancer progression via KEAP1/NRF2 pathway activation. Cell Communication Signaling (2020) 18:98. doi: 10.1186/s12964-020-00568-z
43. Vollrath V, Wielandt AM, Iruretagoyena M, Chianale J. Role of Nrf2 in the regulation of the Mrp2 (ABCC2) gene. Biochem J (2006) 395:599–609. doi: 10.1042/BJ20051518
44. Saleh MM, Scheffler M, Merkelbach-Bruse S, Scheel AH, Ulmer B, Wolf J, et al. Comprehensive analysis of TP53 and KEAP1 mutations and their impact on survival in localized- and advanced-stage NSCLC. J Thorac Oncol (2022) 17:76–88. doi: 10.1016/j.jtho.2021.08.764
45. Case AJ, McGill JL, Tygrett LT, Shirasawa T, Spitz DR, Waldschmidt TJ, et al. Elevated mitochondrial superoxide disrupts normal T cell development, impairing adaptive immune responses to an influenza challenge. Free Radic Biol Med (2011) 50:448–58. doi: 10.1016/j.freeradbiomed.2010.11.025
Keywords: cancer therapy, iron metabolism, non-small cell lung cancer (NSCLC), iron, cancer biology
Citation: Petronek MS, Bayanbold K, Amegble K, Tomanek-Chalkley AM, Allen BG, Spitz DR and Bailey CK (2023) Evaluating the iron chelator function of sirtinol in non-small cell lung cancer. Front. Oncol. 13:1185715. doi: 10.3389/fonc.2023.1185715
Received: 13 March 2023; Accepted: 25 May 2023;
Published: 15 June 2023.
Edited by:
Pranav Kumar Prabhakar, Lovely Professional University, IndiaReviewed by:
Hiu Chuen Lok, The University of Sydney, AustraliaMiranda Lynch, Hauptman-Woodward Medical Research Institute, United States
Copyright © 2023 Petronek, Bayanbold, Amegble, Tomanek-Chalkley, Allen, Spitz and Bailey. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael S. Petronek, bWljaGFlbC1wZXRyb25la0B1aW93YS5lZHU=; Charvann K. Bailey, YmFpbGV5Y2hAZ3Jpbm5lbGwuZWR1