 Katherina Bernadette Sreter
Katherina Bernadette Sreter Maria Joana Catarata2,3
Maria Joana Catarata2,3 Armin Frille
Armin Frille- 1Department of Pulmonology, University Hospital Centre “Sestre Milosrdnice”, Zagreb, Croatia
- 2Pulmonology Department, Hospital de Braga, Braga, Portugal
- 3Tumour & Microenvironment Interactions Group, I3S-Institute for Health Research & Innovation, University of Porto, Porto, Portugal
- 4Institute of Pathology, Leipzig University, Leipzig, Germany
- 5Department of Respiratory Medicine, Leipzig University, Leipzig, Germany
Lung cancer remains the leading cause of cancer death globally. More than 50% of new cases are diagnosed in an advanced or metastatic stage, thus contributing to the poor survival of such patients. Mutations in the KRAS (Kirsten rat sarcoma virus) gene occur in nearly a third of lung adenocarcinoma and have for decades been deemed an ‘undruggable’ target. Yet, in recent years, a growing number of small molecules, such as the GTPase inhibitors, has been investigated in clinical trials of lung cancer patients harboring KRAS mutations, yielding promising results with improved outcomes. Currently, there are only two approved targeted therapies (adagrasib and sotorasib) for advanced or metastatic KRAS-mutated NSCLC from the second-line setting onwards. In this narrative review, we will focus on KRAS, its molecular basis, the role of its co-mutations, clinical evidence for its inhibition, putative mutation to resistance, and future strategies to overcome resistance to KRAS inhibition.
1 Introduction
Lung cancer is the leading cause of cancer-related mortality worldwide (1). The poor survival rate of lung cancer patients is mainly due to the late stage of disease found in over half of them at the time of diagnosis (2). Therapeutic progress has been achieved in non-small cell lung cancer (NSCLC) through the introduction of immune checkpoint inhibitors (ICI) (3) and personalized treatment strategies against driver mutations within the tumor, including targeted therapy (4). These driver or oncogenic mutations are localized within kinase domains of receptor tyrosine kinases (RTKs) (5) and are not equally distributed among histologic subtypes of NSCLC (6). Most notably, lung adenocarcinoma (LUAD) harbors those driver mutations and rearrangements that can be therapeutically addressed, such as EGFR, BRAF, ALK, ROS1, RET, NTRK, and also KRAS (6, 7). Mutations in the KRAS (Kirsten rat sarcoma virus) gene occur in approximately 29–32% of LUAD and, until recently, have been considered to be ‘undruggable’ for the past several decades (8–10).
In the last few years, an increasing number of small-molecule anti-cancer drugs, the so-called GTPase inhibitors as well as others, has been tested in clinical trials, generating encouraging results with improved efficacy of lung cancer treatment for KRAS-mutated NSCLC. Presently, sotorasib and adagrasib are the only approved targeted therapies in locally advanced or metastatic KRAS-mutated NSCLC patients, but just in those who have received at least one prior systemic therapy. In this narrative review, we will focus on KRAS, its molecular basis, the role of its co-mutations, clinical evidence for its inhibition, putative mutation to resistance, and future strategies to overcome resistance to KRAS inhibition.
2 Molecular basis of KRAS as an oncogenic driver in lung cancer
The RAS proto-oncogenes encode intracellular guanine nucleotide binding proteins that belong to the GTPase family harboring a catalytic domain and a hypervariable region (11). The former binds guanine nucleotides and activates signaling while the latter determines how RAS proteins are localized on the cell membrane (11). RAS GTPases control downstream signaling by switching between the active nucleotide guanosine triphosphate (GTP)-bound and inactive nucleotide guanosine diphosphate (GDP)-bound states in response to extracellular signals (11). RAS-GTP commonly activates multiple signaling cascades including the canonical RAS-RAF-MEK-ERK (= mitogen-activated protein kinase, [MAPK]), PI3K-AKT-mTOR, and RAS-like (RAL and tumor invasion and metastasis-inducing protein 1 [TIAM1-RAC1]) pathways (11, 12). The first two signaling pathways are most relevant to tumor biology since they play an essential role in cell cycle regulation, thus cell proliferation, and tumor cell survival (Figure 1).
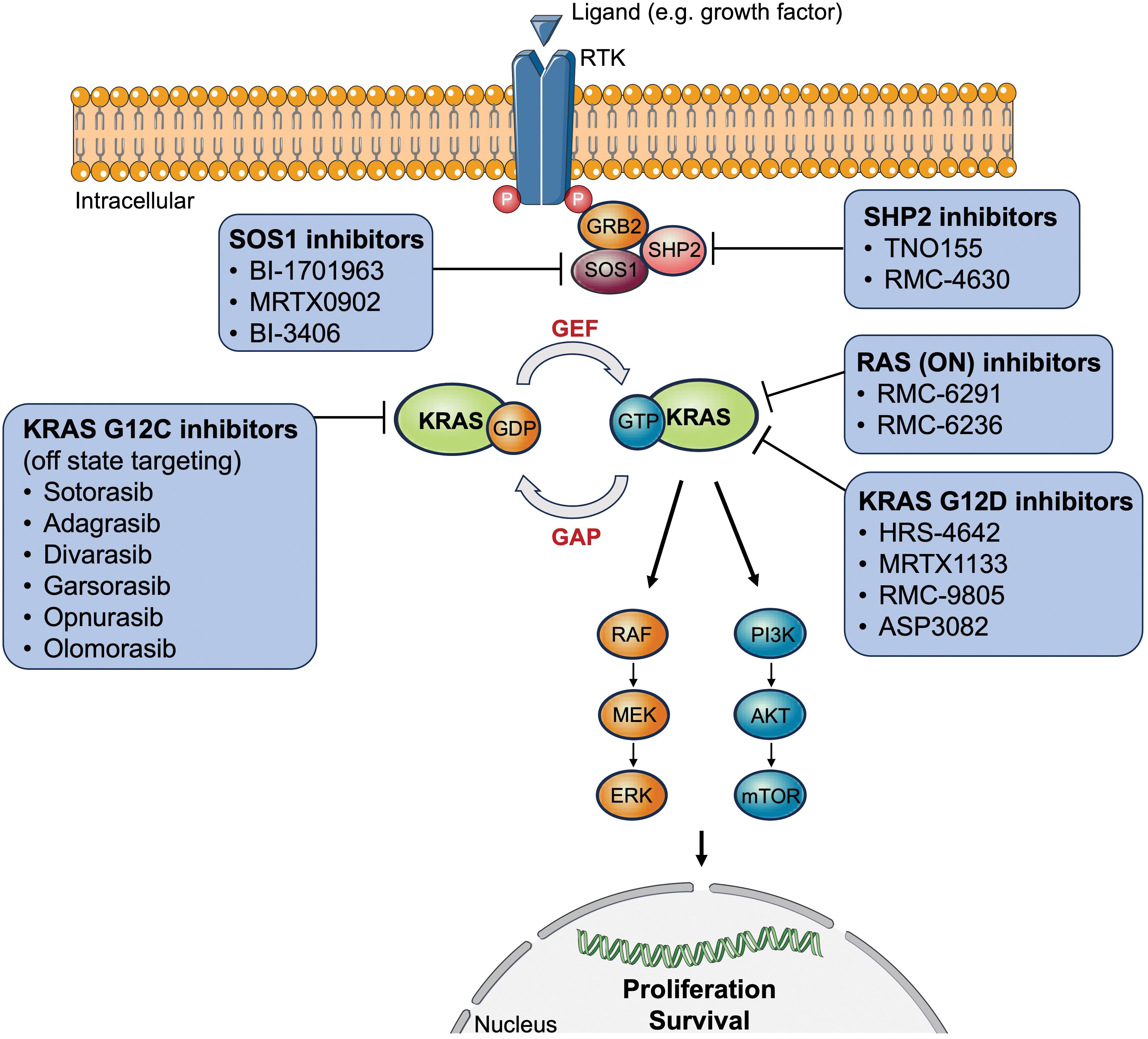
Figure 1 Overview of approved or clinically tested (direct/indirect) KRAS-targeted therapy inhibitors. AKT, protein kinase B; ERK, extracellular signal-regulated kinase; GAP, GTPase activating proteins; GDP, guanosine diphosphate; GEF, guanine nucleotide exchange factor; GRB2, growth factor receptor-bound protein 2; GTP, guanosine triphosphate; KRAS, Kirsten rat sarcoma virus; MEK, mitogen-activated protein kinase kinase; mTOR, mechanistic target of rapamycin; P, phosphorylated tyrosine residues; PI3K, phosphoinositide 3-kinases; RAF, rapidly accelerated fibrosarcoma; RTK, receptor tyrosine kinase; SOS1, son of sevenless 1; SHP2, Src homology region 2 domain-containing phosphatase-2.
In contrast to colorectal cancer and pancreatic adenocarcinoma, the point mutation G12C is the most prevalent genetic alteration in the KRAS gene of LUAD, occurring in 39% of cases, followed by the point mutations G12V (18.1%), G12D (13.8%), and G12A (7.2%) (13). However, to date, KRAS G12C is the only molecular target for which the two therapeutic agents, sotorasib and adagrasib, have been approved in NSCLC. Conversely, 61% of all KRAS point mutations in LUAD are still ineligible for targeted therapy.
3 Role of co-mutations with KRAS
It is well known that KRAS altered NSCLC frequently shows co-occurring mutations with other genes, including tumor protein 53 (TP53), serine/threonine kinase 11 (STK11), and Kelch-like ECH-associated protein 1 (KEAP1), also known as liver kinase B1 (LKB1), as well as concurrent amplifications in the MET and erb-b2 RTK 2 (ERBB2) genes (9, 14–18). Both the triple (KRAS + KEAP1 + STK11) and quadruple (KRAS + KEAP1 + STK11 + TP53) co-mutations have been shown to serve as a negative prognostic and predictive factor compared to the single KRAS mutational status (15, 18).
Chapter 9 will further elaborate on co-mutations in the context of mechanisms of resistance to KRAS inhibition.
4 Clinical evidence for KRAS inhibition in KRAS-mutant NSCLC
Historically considered undruggable, KRAS-mutant NSCLC now has two approved targeted therapies as well as other potential therapeutic agents that are still under clinical development (10, 13, 19–22). This recent milestone in modern medicine was achieved thanks to the discovery of the allosteric regulatory site of KRAS G12C, thereby leading to the design of irreversible covalent inhibitors (23). Such small compounds bind to the switch-II binding pocket of KRAS G12C (24). Previous crystallography studies were paramount in finding molecules capable of interacting with the unique conformation of the KRAS protein (25). A major scientific breakthrough was made in 2013 with the identification of the switch-II pocket of KRAS by the Shokat Lab, resulting in the structure-based validation of direct targeting of the compound binding region of KRAS in a “mutant-specific” and selective manner (26). The stage was set for the optimization of compounds, leading to the creation of the current KRAS G12C inhibitors available for clinical use today (27). By binding specifically to the inactive GDP-bound form of the KRAS oncoprotein in its switch-II pocket, a covalent bond is created with the mutant cysteine residue of KRAS G12C, blocking the reactivation of KRAS by nucleotide exchange (from GDP to GTP) (27, 28). Hence, KRAS G12C inhibitors essentially trap KRAS G12C in an inactive KRAS-GDP state (off state), hindering a switch to the active KRAS-GTP state (on state), and, thereby, impeding oncogenic activity. This has led to improved drug efficacy and selectivity (29). Currently, sotorasib and adagrasib are recommended by the National Comprehensive Cancer Network (NCCN) guidelines as a subsequent treatment option for patients with KRAS G12C-mutant NSCLC in second-line or beyond, if no previous KRAS G12C-targeted therapy was given (30). Given their similar mechanism of action, it is not recommended to switch between these two therapeutic agents at the time of progression (30). Table 1 summarizes the efficacy data of KRAS G12C inhibitors from published clinical trials.
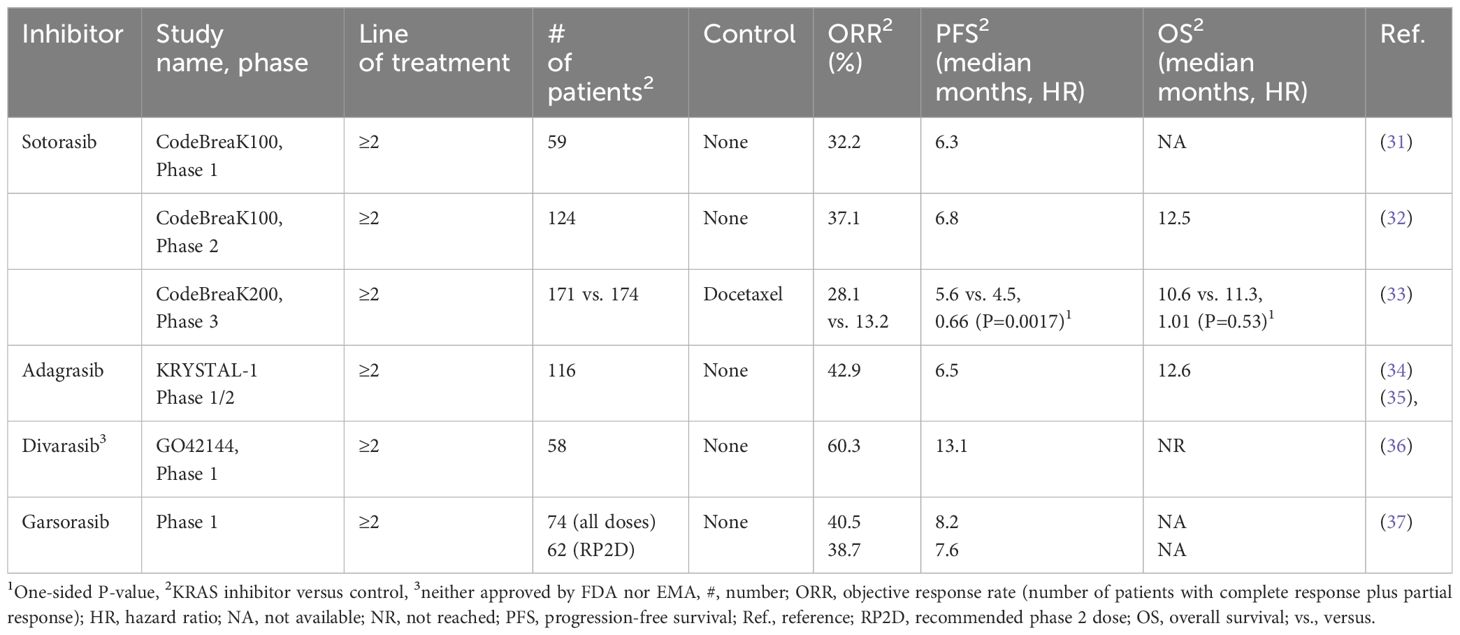
Table 1 Published clinical trials for KRAS G12C inhibitors.
The following chapters will give an overview of direct inhibitors of KRAS G12C in NSCLC.
5 Sotorasib (AMG 510)
The first drug to enter clinical trials geared toward targeting mutant KRAS, sotorasib (previously known as AMG 510), was granted accelerated approval by the U.S. Food and Drug Administration on May 28, 2021, for adult patients with previously treated (immunotherapy and/or chemotherapy) locally advanced or metastatic NSCLC harboring the KRAS p.G12C mutation (38, 39). In turn, Health Canada approved this KRAS G12C inhibitor in September 2021 (23), while the European Medicine Agency followed suit in January 2022 (Amgen, 2022). These approvals were based on the results of phase 2 of the CodeBreaK 100 trial (32). Preclinical analyses of sotorasib were very promising, showing inhibition of tumor cell growth in both in vitro and murine models (40). Sotorasib first entered clinical trial in 2018, and the results of the phase 1 CodeBreaK 100 trial demonstrated encouraging anticancer activity of sotorasib monotherapy in the NSCLC subgroup as follows: 32.2% had an objective response (complete or partial) rate (ORR), 88.1% had disease control (objective response or stable disease), and the median progression-free survival (PFS) was 6.3 months (31). A durable clinical benefit of monotherapy with daily sotorasib (administered orally at a dose of 960 mg) was confirmed in the phase 2 CodeBreaK 100 trial, showing a 37.1% ORR, median PFS of 6.8 months, and median overall survival (OS) of 12.5 months in KRAS p.G12C-mutant advanced NSCLC patients previously treated with standard therapies (Table 1) (32). The two-year pooled analysis of the CodeBreaK 100 phase 1/2 clinical trial showed that almost 25% of these previously treated advanced stage KRAS G12C-mutant NSCLC patients derived long-term benefit from additional sotorasib treatment, with few late-onset treatment-related toxicities (41). These results support the continuing clinical use of sotorasib both in the current therapeutic setting and in studies (ongoing and future) examining its potential role in earlier lines of therapy (41).
In the CodeBreaK 200 study, a randomized, open-label, phase 3 trial (June 2020 to April 2021) of sotorasib (n=171) versus docetaxel (n=174) in the second-line setting and beyond of advanced NSCLC patients with KRAS G12C mutation, sotorasib significantly increased PFS (i.e., median PFS 5.6 months [95% CI, 4.3–7.8] vs. 4.5 months [3.0–5.7]; hazard ratio 0.66 [0.51–0.86]; p=0.0017) and exhibited a better safety profile (33). In addition, sotorasib elicited a more rapid (1.4 months vs. 2.8 months) and longer response (8.36 months vs. 6.8 months) compared with docetaxel (33). Unfortunately, although PFS, ORR, and disease control rate (DCR) were improved in the sotorasib group, these results were disappointing when compared to the phase 1 and 2 CodeBreaK 100 trials that showed a longer PFS (6.3 and 6.8 months, respectively) and had a similar ORR and DCR (42).
In addition to sotorasib monotherapy, ongoing clinical studies are also investigating sotorasib-based combinations for the possible treatment of pretreated KRAS G12C-mutant NSCLC (20). The single-arm, phase-2 SCARLET study enrolled 30 patients with chemotherapy-naïve, advanced non-squamous, KRAS G12C-mutant NSCLC between October 2021 and July 2022 (43). Results from this clinical trial were recently presented in June 2023 at the American Society for Clinical Oncology (ASCO) Annual Meeting, and showed a favorable ORR (88.9%; 80% CI, 78.5–94.8%) (n=27) and tolerability (n=29) for sotorasib plus platinum-doublet chemotherapy (carboplatin/pemetrexed). The PFS and OS rates at 6 months were 61.2% and 87.0%, respectively; median PFS was not reached given the shorter follow-up period (median 4.2 months).
Most recently, exciting positive data from the study arm of sotorasib in combination with carboplatin and pemetrexed for KRAS G12C-mutant advanced NSCLC in the ongoing, phase 1b, CodeBreaK 101, global clinical trial have further endorsed the approach to repositioning sotorasib with novel therapeutic combinations into earlier lines of therapy within the treatment paradigm (44). These highly anticipated results, based on a median follow-up of 3 months, were presented at the 2023 International Association for the Study of Lung Cancer (IASLC) World Conference on Lung Cancer (2023) on September 10th, 2023, in Singapore. Patients (n=20) treated in the frontline (i.e., first-line) setting experienced a better ORR and DCR than their counterparts (n=13) treated in the second-line setting (ORR 65% vs. 54%, respectively; and DCR 100%; 95% CI: 83.2, 100, vs. 85%; 95% CI: 54.6, 98.1, respectively). Similar ORRs were reported among patients with programmed cell death ligand-1 (PD-L1) expression less than 1% (i.e., 62% vs. 50% in the frontline vs. second-line setting, respectively). Mature PFS and OS data were unavailable. Due to the very promising results from the global CodeBreaK 101 trial, a new multicenter, randomized, open-label, phase 3 study (CodeBreaK 202) of sotorasib plus carboplatin and pemetrexed as frontline therapy of PD-L1 negative, KRAS G12C-mutant advanced NSCLC has been recently initiated by Amgen and is currently recruiting patients (enrollment start date: November 26, 2023; estimated study completion date: March 1, 2031) (45).
6 Adagrasib (MRTX849)
Adagrasib is the second approved, orally administered, potent, covalent KRAS G12C inhibitor that selectively and irreversibly binds the switch-II pocket of KRAS G12C (46). Adagrasib was granted accelerated approval by the FDA in December 2022 as a targeted treatment option for locally advanced or metastatic NSCLC with a KRAS G12C mutation (47). This decision was based on the results of the ongoing phase 1/2 KRYSTAL-1 clinical trial (Table 1) (34, 35). This multicenter single-arm study included patients with histologically confirmed unresectable or metastatic KRAS G12C-mutant NSCLC whose disease progressed with frontline chemotherapy and/or immunotherapy. With respect to efficacy outcome measures, 42.9% (95% confidence interval [CI], 33.5 to 52.6) of the 112 patients with measurable disease at baseline had a confirmed objective response. The median duration of response (DOR) was 8.5 months (95% CI, 6.2 to 13.8) and the median PFS was 6.5 months. Confirmed ORRs were similar across PD-L1 expression subgroups (41.7 to 46.8%). The ORRs in patients with co-mutations in STK11, KEAP1, TP53, and CDKN2A ranged from 28.6% (KEAP1) to 58.3% (CDKN2A). As of January 15, 2022 (median follow-up, 15.6 months), the median OS was 12.6 months (95% CI, 9.2 to 19.2).
Updated, longer follow-up data from the KRYSTAL-1 trial, recently presented on September 10, 2023, at the World Congress on Lung Cancer 2023 (WCLC 2023) in Singapore, confirmed durable clinical activity and benefit of adagrasib in advanced KRAS G12C-mutant NSCLC across patient groups, including those with CNS metastases and co-mutations (48). Gadgeel and colleagues presented favorable safety and efficacy data (ORR, DOR, PFS, and OS) from a two-year follow-up pooled analysis of the Phase 1/1b Cohort and Phase 2 Cohort A of KRYSTAL-1. As of January 1, 2023, 132 patients received adagrasib, and showed an ORR of 43.0%, with a median DOR of 12.4 months. The median PFS was 6.9 months (95% CI 5.4–8.7), and the median OS was 14.1 months (95% CI 9.2–18.7). Approximately one in three patients (31.3%) remained alive at two years. Exploratory analyses suggested heterogeneity of clinical benefit based on the presence of co-mutations, requiring further evaluation. The safety profile was consistent with previous reports. A confirmatory, multi-center, randomized Phase 3 study, KRYSTAL-12, evaluating adagrasib monotherapy versus docetaxel in patients with previously treated advanced KRAS G12C-mutant NSCLC, is ongoing (Table 2) (51).
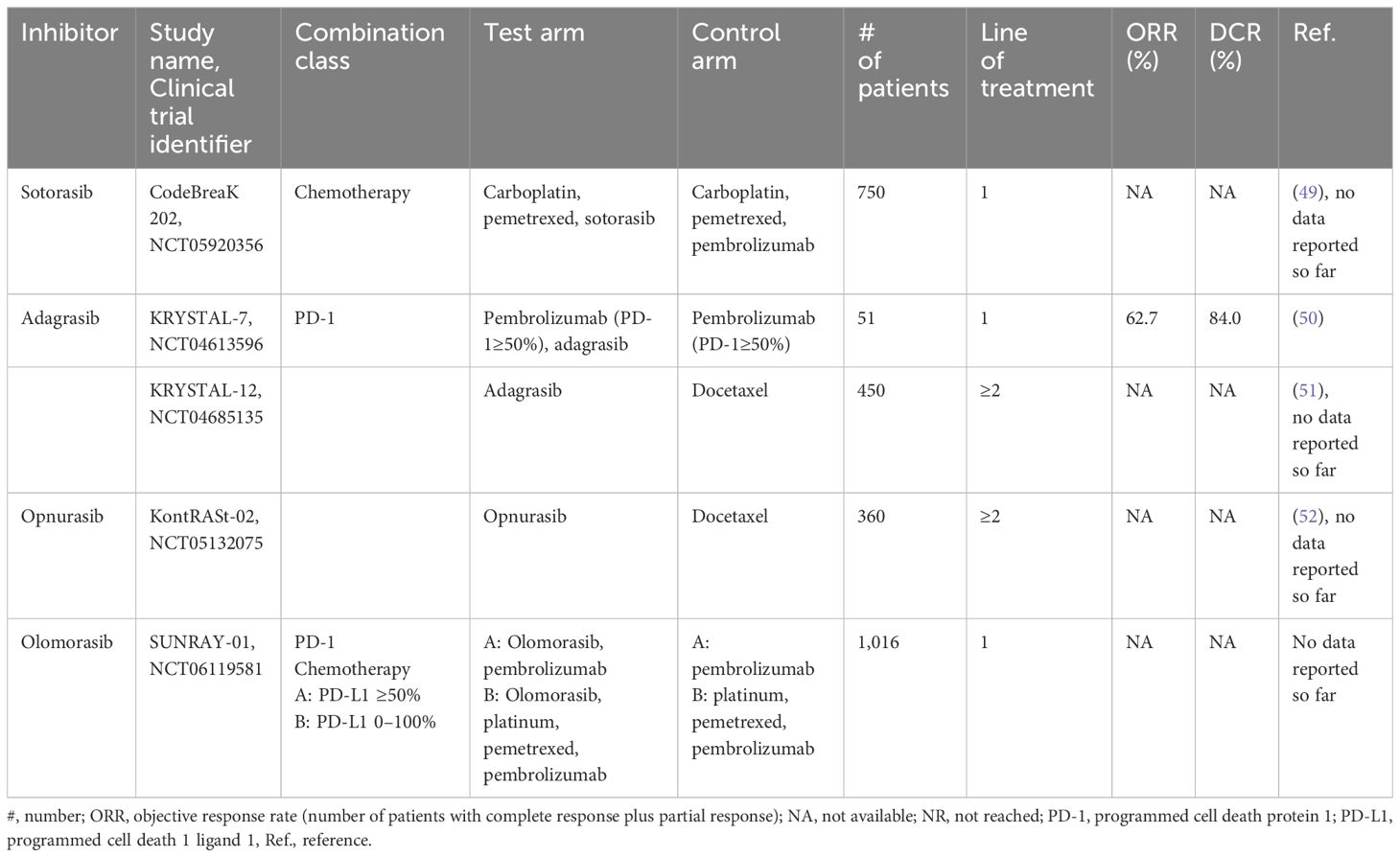
Table 2 Ongoing phase 3 trials targeting KRAS G12C.
It is important to mention that preliminary pharmacodynamics and mechanistic biomarker analysis on pre- and post-treatment tumor NSCLC biopsies of patients (n=3) treated with adagrasib (phase 1/1b and 2) demonstrated down-regulation of KRAS/MAPK pathway genes, including DUSP6 and SPRY4 (53). Patients with STK11-co-mutations had an impressive ORR of 64%. This was a surprising finding given that STK11 mutations typically portend a poor response and survival to immune checkpoint inhibitors in metastatic NSCLC (54). However, Riely et al. (2021) showed that treatment with adagrasib increased the expression of immune transcripts (e.g., CD4 and CD8) that are minimal at baseline, suggesting a potential immune response to therapy (53).
As noted by Cheema and colleagues (2022), data from preclinical and clinical studies have revealed that drug resistance to single-agent KRAS G12C-targeted therapy occurs quite early after treatment initiation (often within a few months) (23). This suggests that the use of KRAS G12C-targeted therapies in combination with other treatments may help overcome drug resistance observed with anti-G12C monotherapies. Updated, late-breaking data (safety and efficacy results) from the phase 2 KRYSTAL-7 study were recently presented at the European Society of Medical Oncology (ESMO) Congress 2023 in Madrid, Spain (October 20–24, 2023) (55). The results of the KRYSTAL-7 trial, with three patient cohorts stratified according to PD-L1 tumor proportion score (TPS), found that concurrent adagrasib and pembrolizumab in patients with treatment-naïve, advanced, unresectable, or metastatic NSCLC harboring KRAS G12C mutation demonstrated encouraging preliminary efficacy with clinically meaningful antitumor activity, especially in patients with high PD-L1 expression (TPS ≥ 50%), and a manageable safety profile (Table 2). The patients in this cohort (PD-L1 TPS ≥ 50%) had an ORR of 63% (32/51; 95% CI, 48–76) and a DCR of 84% (43/51; 95% CI, 12.6-not evaluable [NE]). This ORR for the adagrasib-pembrolizumab combination compares favorably with the ORR of pembrolizumab as a single agent (range: 39% to 45%). The median follow-up was longer for patients with PDL-1 TPS ≥ 50% versus all patients (10.1 months vs. 8.7 months, respectively). The median time to response was 1.4 months, and the median PFS was not reached (95% CI, 8.2-NE).
7 Intracranial responses with the selective KRAS-G12C inhibitors sotorasib and adagrasib
Patients with KRAS G12C-mutant NSCLC are prone to developing brain metastases (BMs) (56, 57). At diagnosis, BMs were detected in 27% to 42% of patients (56, 58–61). KRAS-mutant NSCLC patients with untreated central nervous system (CNS) metastases have poorer clinical outcomes (i.e., worse prognosis and higher CNS failure) compared to those without KRAS mutations (62–64). For this very important reason, the efficacy of selective G12C inhibitors in the CNS and untreated intracranial lesions remains the subject of intense active research (65). It should be noted that the initial KRYSTAL-1 and CodeBreak100 trials excluded patients with active, untreated BMs (66).
Despite their similarities as allele-specific inhibitors and covalent drugs, sotorasib and adagrasib are indeed different in many ways, reflecting the speed of drug development and their intrinsic properties (67). Notably, with respect to BMs in KRAS G12C-mutant NSCLC patients, efficacy data for adagrasib have become available earlier than for sotorasib. Preclinically, adagrasib has shown CNS penetration and its efficacy on KRAS G12C-BM in a LU99Luc mouse model showed CNS tumor regression with dose-dependent effects (56). Clinically, it has demonstrated cerebrospinal fluid penetration and BM regression in preliminary findings from the phase 1b portion of the KRYSTAL-1 trial; a retrospective database analysis was initially performed to better understand the clinicopathological features of KRAS G12C-mutant NSCLC patients with BM (56). The registrational phase 2 cohort of the KRYSTAL-1 reported findings consistent with the earlier preclinical models of tumor shrinkage, demonstrating an intracranial ORR of 33.3% (11/33 patients) with one intracranial complete response and a median duration of intracranial response of 11.2 months (35). Furthermore, Negrao and colleagues (2023) recently published the first prospective data for the KRAS G12C inhibitor adagrasib in patients with NSCLC and radiologically evaluable, active, and untreated CNS metastases (57). The results of this phase 1b limited BM expansion cohort of the KRYSTAL-1 trial provided proof-of-concept for adagrasib’s ability to penetrate the CNS and achieve promising intracranial activity, with a high concordance rate between intracranial and systemic activity (79%) and a low rate of CNS failure (37%). In early 2024, a case series taken from the KRYSTAL-1 CNS metastases cohort showed that most patients did not discontinue adagrasib because of CNS progression, which was consistent with the overall KRYSTAL-1 CNS metastases cohort and indicated that adagrasib may delay development of additional CNS metastases (68).
Until very recently, published CNS activity data for sotorasib remained relatively scant in comparison to adagrasib (65). Thus far, three case reports describe a remarkable intracranial response of previously untreated, active BMs (69–71). Both Koster et al. (2022) and Yeh et al. (2022) documented a rapid intracranial response in less than two months for their patients treated with sotorasib monotherapy following stereotactic body radiotherapy (SBRT) alone vs. postoperative stereotactic radiosurgery to the cranial resection cavity, respectively, and first-line systemic treatment (i.e., immunotherapy with pembrolizumab) (69, 70). Inno et al. (2023) reported the case of a long duration of intracranial response to sotorasib in the second-line setting lasting 16 months in a patient with both pretreated and untreated symptomatic BMs from KRAS G12C mutant NSCLC (71). The importance of exploring dose-dependent CNS response, control, and penetration of the selective inhibitor is emphasized by Lu & Husain (2023) in their case report (65). The patient showed intracranial stability for 5 months on the standard dose of second-line sotorasib monotherapy (960 mg daily), but following a reduction of the sotorasib to 480 mg daily as a result of seizures and vasogenic edema (without new BMs) developed new BMs 5 months later (65).
Clearly, further prospective clinical studies are required to fully characterize the intracranial efficacy of both sotorasib and adagrasib as currently approved therapies as well as other selective G12C inhibitors still in development, including divarasib (GDC-6036) and opnurasib (JDQ-443), among others (66).
8 Novel direct KRAS G12C inhibitors
In addition to sotorasib and adagrasib, several other direct KRAS G12C inhibitors, such as divarasib (GDC-6063), opnurasib (JDQ-443), garsorasib (D-1553), olomorasib (LY3537982), MK-1084, and JAB-21822 are now in clinical development as monotherapy or in combination with other treatments, as discussed in several recently published reviews (Tables 2, 3, Figure 1) (10, 13, 20–22, 82–84). A very recent review touches quite comprehensively and thoughtfully on the manifold combinatorial therapeutic strategies in RAS-driven cancers (84).
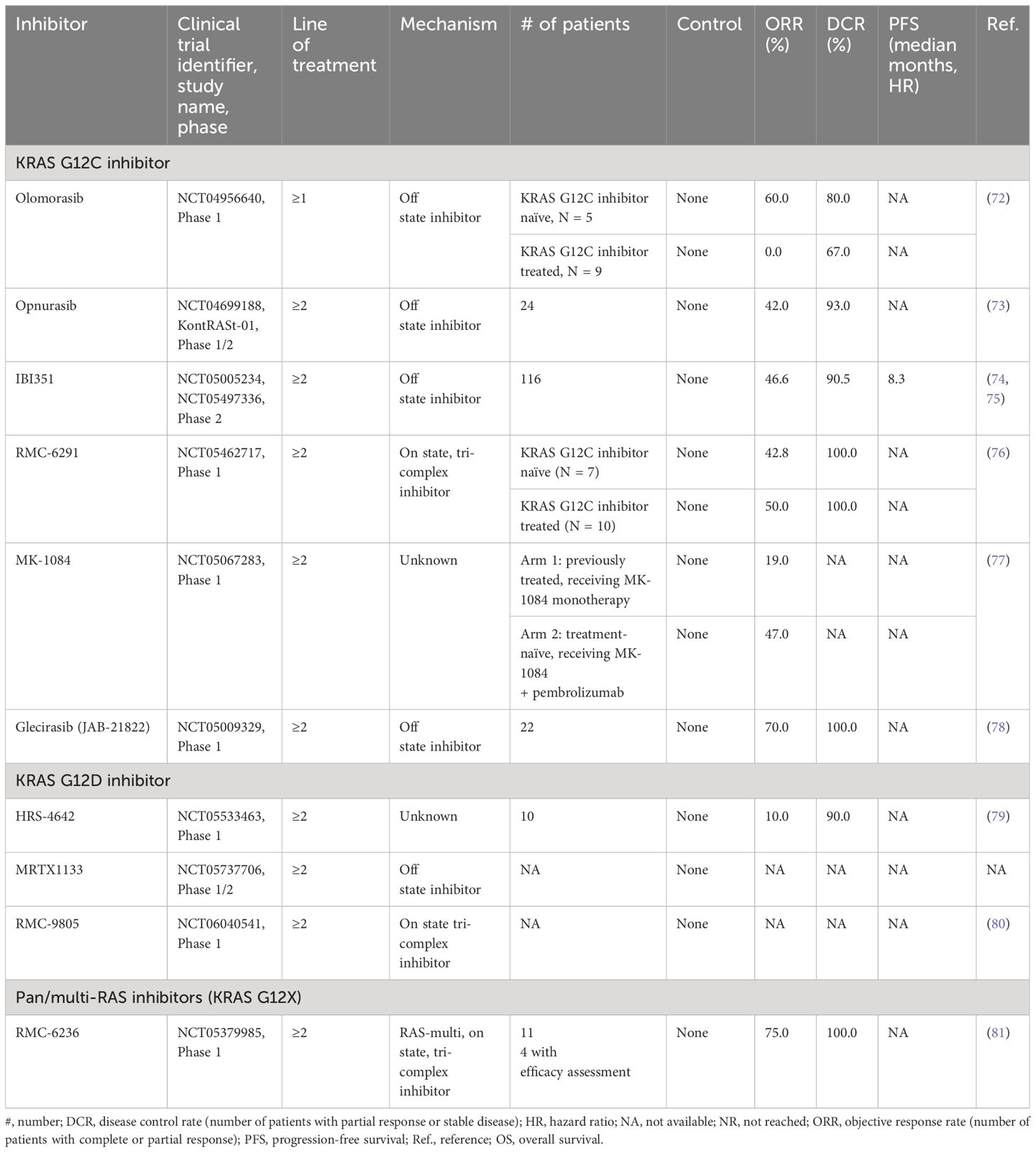
Table 3 Novel agents for KRAS inhibition.
Two formerly promising, orally available, investigational, small molecules, LY3499446 and JNJ-74699157 (ARS-3248), were abruptly removed from the G12C inhibitor landscape (82, 83). The discontinuation of the initial phase 1 trial of LY3499446 was due to unexpected toxicity (20, 27). Likewise, JNJ-74699157 (ARS-3248) was investigated in a phase 1 study of patients with advanced solid tumors, including NSCLC (n=5), but enrolment was terminated at just 10 patients due to dose-limiting skeletal muscle toxicities and the lack of efficacy at the lowest administered dose (100 mg) (83, 85).
Data from preclinical and in vitro studies have suggested that divarasib (GDC-6063) is more potent and selective than sotorasib or adagrasib (86). In a phase 1 clinical trial, among the 60 NSCLC patients who received divarasib, a confirmed response was observed in 53.4% of patients (95% confidence interval [CI], 39.9 to 66.7), and the median PFS was 13.1 months (95% CI, 8.8 to NE), with an acceptable safety profile (mainly low-grade adverse events) (36).
Opnurasib (JDQ-443), structurally unique and currently in clinical development, has been optimized by design to overcome resistance mechanisms through novel interactions with the binding pocket (83, 87–89). A stable atropisomer with PK/PD activity in vivo and dose-dependent antitumor activity in mouse xenograft models, opnurasib has performed in an encouraging manner as evidenced by the early phase data reported from an ongoing Phase 1b/2 clinical trial, with a confirmed ORR of 41.7% (83, 88, 89). As a promising therapy, opnurasib is being investigated in the combination arms of the ongoing, phase 1b/2, multicenter, KontRaSt-01 study, with either TNO155 (SHP2 inhibitor) or tislelizumab (anti-PD-1 monoclonal antibody), as well as in a phase 3 trial of opnurasib monotherapy versus docetaxel (Table 2) (73, 83, 90). An update of the KontRaSt-01 was recently presented at the ASCO 2023 Congress, demonstrating promising efficacy and well-tolerated safety data (73).
Garsorasib (D-1553), a novel small molecule inhibitor that selectively targets KRAS G12C, is currently in phase 2 clinical trials (91). Preclinical data have already demonstrated antitumor activity of garsorasib. In the phase 1, garsorasib dose-escalation study in KRAS G12C-mutant NSCLC patients (n=62), partial response occurred in 24 patients (ORR, 38.7%) and stable disease in 32 patients (DCR, 90.3%) (37).
Olomorasib (LY3537982) monotherapy was tested in a phase-1 clinical trial, in which 5 treatment-naïve and 9 previously treated patients with KRAS G12C mutational status showed an ORR of 60% or 0%, respectively, and a DCR of 80% or 67%, respectively (72). The phase-3 SUNRAY-01 trial (NCT06119581) will assess the efficacy of olomorasib in combination with pembrolizumab or pembrolizumab with chemotherapy in 1,016 patients with locally advanced or metastatic NSCLC.
MK-1084 is being tested for KRAS G12C mutations as monotherapy in pretreated patients with advanced solid tumors (arm 1) and in combination with pembrolizumab in previously untreated metastatic NSCLC with PD-L1 TPS≥1% in an ongoing, phase 1, global, dose-escalation trial (arm 2) (23). The preliminary results, presented at the ESMO Congress 2023 in October 2023, showed manageable safety and preliminary antitumor activity in both arms (ORR 19% and 47% in arm 1 and 2, respectively) (77).
JAB-21822, now designated glecirasib, was tested in a first-in-human clinical trial comprising 22 patients with advanced NSCLC. The results proved quite promising showing that ORR and DCR were 70% and 100%, respectively (78). Results from future clinical trials are awaited.
9 Mechanisms of resistance to KRAS inhibition
The vast majority of advanced NSCLC will progress due to treatment resistance. Tumor cell intrinsic mechanisms are the primary drivers of resistance to radiation, cytotoxic agents, and targeted therapies (6).
Resistance mechanisms to KRAS G12C inhibition cover primary resistance and acquired resistance (92, 93).
Primary resistance or early disease progression (PFS < 3 months) to KRAS G12C inhibitors occurs in about 36% of patients who received sotorasib therapy, as shown in recently published data from the 2-year analysis of the CodeBreaK100 study in NSCLC (41). In NSCLC, co-mutations with genetic alterations in KEAP1, SMARCA4 (SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily A, member 4), and CDKN2A (cyclin dependent kinase inhibitor 2A) are associated with inferior clinical outcomes to sotorasib therapy (94). Some studies have demonstrated that co-mutations in STK11, KEAP1, and TP53 could modulate the responsiveness of patients with KRAS alterations to either KRAS G12C inhibitors or to immunotherapy (14–16, 18, 95). Proulx-Rocray and colleagues (2021) showed that the presence of STK11 and/or KEAP1 mutations was associated with a negative impact on survival when compared with wild-type NSCLC patients treated with immune check point inhibitors (96). These authors also reported that in patients harboring KRAS mutation, improved prognosis was observed in STK11+KEAP1 wild-type tumors but not in STK11+/-KEAP1 mutant tumors. Interestingly, the presence of KRAS G12D is associated with diminished infiltration of CD8+ T cells in NSCLC (97). Patients harboring KRAS G12D mutations had worse clinical outcomes to PD-(L)1 inhibition compared to wild-type (97). The biological mechanism of resistance mediated by these mutations has yet to be explored. Co-occurring mutations that predict response to treatment might serve as markers for patient stratification and therapy intensification in randomized clinical trials (10).
In terms of allele amplification, high-level amplifications of the KRAS G12C allele were observed in some patients undergoing sotorasib treatment (98, 99).
Acquired resistance inevitably occurs and is responsible for disease progression after an initial benefit from targeted therapies. Principly, acquired resistance to KRAS G12C inhibitors are functionally divided into off-target and on-target mechanisms.
On-target resistance mechanisms include alterations that concern the molecular target, against which the inhibitor is directed, such as KRAS. These mechanisms comprise (92, 98, 100):
● Novel KRAS mutations in the switch II pocket (e.g. sotorasib: Y96c/d/s, R68S, adagrasib: H95D/Q/R);
● Acquired KRAS activating mutation (e.g. G12D on trans and G12W on cis, preventing inhibitor to bind);
● New production of KRAS G12C, and
● KRAS G12C gene amplification.
On-target resistance mechanisms were described in a recent in vitro study showing that secondary KRAS mutations (Y96D, A59T, A59S, R68M, R68M, M721, V8E, G13D, Q61L, Q99L, and H358) conferred resistance to the KRAS (G12C) inhibitors. Moreover, Y96D and Y96S secondary mutations caused resistance to both sotorasib and adagrasib, while the KRAS mutations G13D, R68M, A59S, and A59T were highly resistant only to sotorasib and Q99L was resistant to adagrasib but sensitive to sotorasib (101). These acquired mutations were also observed in a clinical study that included KRAS G12C-mutant cancer patients treated with adagrasib in monotherapy, of whom 71% were NSCLC patients (98). Furthermore, cell lines with co-mutations of KRAS G12C and G12V were described as acquired mechanisms of resistance to KRAS G12C inhibition in vitro (102). Similarly, a preclinical and clinical study from Tanaka and colleagues described two KRAS activating mutations (G12D, G12V) and a Y96D mutation affecting the cryptic Switch II pocket as mechanisms of resistance during adagrasib treatment (103). Interestingly, G12D-mutant cell lines are reported to have high levels of phosphorylated AKT, leading to the activation of the PI3K-AKT-mTOR pathway (102).
Off-target resistance mechanisms include alterations that comprise upstream and downstream signaling pathways of KRAS as well as histological transformation. These mechanisms comprise (92, 98, 100):
● Activating wild-type isoforms of RAS-proteins, such as NRAS and HRAS;
● Gain of function in oncogenes (e.g. downstream as in the MAPK pathway: NRAS, BRAF, MEK1, RET etc.);
● Loss of function in tumor suppressor genes (e.g. cell-cycle transition: CDKN2A);
● Gene amplifications, such as in cMET;
● Fusion of gene, such as ALK, RET, RAF1, BRAF, FGFR3, appear to be more common in colo-rectal cancer;
● Histological transformation (e.g. LUAD to squamous cell carcinoma).
A recent in vitro and in vivo study demonstrated that MET amplification in KRAS G12C was associated with resistance to sotorasib in vitro and the introduction of a MET inhibitor restored sensitivity by eliminating RAS–MEK–ERK and AKT signaling (104). Furthermore, MET copy level gain was an off-target mechanism of resistance to sotorasib in a patient with KRAS G12C-mutant LUAD (105). Activating mutations in NRAS, BRAF, MAP2K1, and RET; oncogenic fusions involving ALK, RET, BRAF, RAF1, and FGFR3; and loss-of-function mutations in tumor suppressor genes, such as PTEN and NF1, were described as acquired off-target resistance mechanisms of KRAS G12C inhibitors (19, 92, 101, 106).
Table 3 and Figure 1 give an overview of three potential agents targeting KRAS G12D mutations: HRS-4642, MRTX1133, and RMC-9805. Moreover, G12V mutations are shown to preferentially activate RAL signaling (102).
10 Future strategies to overcome resistance to KRAS inhibition
For NSCLC harboring a KRAS G12D mutation, there are several specific inhibitors undergoing testing in clinical and preclinical studies (Table 3, Figure 1). MRTX1133 is a non-covalent KRAS G12D inhibitor that showed significant preclinical antitumor activity in KRAS G12D-bearing tumor cells, especially pancreatic ductal adenocarcinoma (107). This compound might be a potential treatment in combination with KRAS G12C inhibitors for patients harboring co-mutations (KRAS G12C, G12D). Further studies are needed to clarify the role of adaptive resistance mechanisms in acquiring resistance to KRAS inhibitors.
RM-018, a tricomplex KRAS G12C active-state inhibitor, retains the ability to inhibit KRAS (G12C, Y96D) (103), thus being a promising therapy to address acquired resistance. Adaptive resistance mechanisms due to reactivation of MAPK pathway and upregulation of PI3K-AKT pathway were identified as likely resistance mechanisms and, according to in vitro and in vivo models, combination with PI3K inhibitors could overcome this resistance (108).
Several studies have uncovered the mechanisms underlying resistance to KRAS G12C inhibition and there have been pioneering efforts to overcome drug resistance using combinatorial treatments (108–111).
One approach is to target upstream effector proteins of the KRAS protein itself. For instance, the phosphatase son of sevenless homolog 1 (SOS1) is a RAS guanine nucleotide exchange factor (RasGEF), which is activated by SHP2 promoting RAS activation through GTP binding (Figure 1) (112). The combination of a novel SOS1 inhibitor (BI-3406) and trametinib exhibited potent activity against Y96D and Y96S (113). In addition, other SOS inhibitors, such as BI-1701963 and MRTX0902, are currently being tested in clinical trials (10).
SHP2 is another upstream adapter protein that is phosphorylated upon activation of RTK. Two SHP2 inhibitors are currently under clinical investigation: TNO155 and RMC-4630 (10, 13). KRAS G12C inhibitors in combination with SHP2 inhibition led to sustained RAS pathway suppression and improved efficacy in vitro and in vivo (111).
Recently, a phase 3 clinical trial showed that sotorasib in combination with panitumumab (EGFR inhibitor) resulted in longer PFS than standard treatment in metastatic colon cancer patients (114). Further studies are needed to test whether this combination could improve the outcome in lung cancer. Promising evidence has demonstrated that adagrasib plus pembrolizumab improves overall response rate in patients with newly diagnosed NSCLC harboring a KRAS G12C mutation, particularly in those with higher levels of PD-L1 (115).
As such, specific therapeutic combinations may help in cases of either intrinsic resistance or acquired resistance.
11 Conclusion
The KRAS mutation plays a major role in the development of tumor progression and resistance to treatment. Despite this, G12C point mutation (making up only 39% of all KRAS alterations) remains the only molecular target for which the two therapeutic agents, sotorasib and adagrasib, have been approved so far. The advent of novel inhibitors against KRAS mutations will further improve survival of lung cancer patients. Nevertheless, the co-occurrence of add-on mutations (co-mutations) and by-pass track pathways will remain challenging obstacles to overcome since they reduce treatment success. Future research efforts must be directed toward comprehensive molecular testing of lung cancer, allowing for the development of multimodal treatment strategies including immune checkpoint inhibitors, tyrosine kinase inhibitors, KRAS upstream inhibitors, and multi-kinase inhibitors against co-mutations.
Author contributions
KS: Writing – original draft, Writing – review & editing. MC: Writing – original draft, Writing – review & editing. MV: Writing – review & editing. AF: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This publication was funded by the Open Access Publishing Fund of Leipzig University, supported by the German Research Foundation (DFG). The funder was not involved in the study design; collection, analysis, and interpretation of data; writing of this article; or the decision to submit the article for publication.
Acknowledgments
Figure 1 contains modified graphic content provided by Servier Medical Art by Servier (https://smart.servier.com), licensed under a Creative Commons Attribution 4.0 unported license (CC BY 4.0). AF was supported by the postdoctoral fellowship “MetaRot program” from the Federal Ministry of Education and Research (BMBF), Germany (FKZ 01EO1501, IFB Adiposity Diseases), a research grant from the “Mitteldeutsche Gesellschaft für Pneumologie (MDGP) e.V.” (2018-MDGP-PA-002), a junior research grant from the Medical Faculty, University of Leipzig (934100–012), a graduate fellowship of the “Novartis Foundation”, and the “LuCaPET” project (ERAPerMed_324), which was funded with tax funds on the basis of the budget passed by the Saxon State Parliament (Germany) under the framework of ERA PerMed (Horizon 2020).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2021) 71:209–49. doi: 10.3322/caac.21660
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. (2020) 70:7–30. doi: 10.3322/caac.21590
3. Addeo A, Banna GL, Metro G, Di Maio M. Chemotherapy in combination with immune checkpoint inhibitors for the first-line treatment of patients with advanced non-small cell lung cancer: A systematic review and literature-based meta-analysis. Front Oncol. (2019) 9:264. doi: 10.3389/fonc.2019.00264
4. Reck M, Heigener DF, Mok T, Soria JC, Rabe KF. Management of non-small-cell lung cancer: recent developments. Lancet. (2013) 382:709–19. doi: 10.1016/S0140–6736(13)61502–0
5. Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. (2010) 141:1117–34. doi: 10.1016/j.cell.2010.06.011
6. Wang M, Herbst RS, Boshoff C. Toward personalized treatment approaches for non-small-cell lung cancer. Nat Med. (2021) 27:1345–56. doi: 10.1038/s41591–021-01450–2
7. Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature. (2014) 511:543–50. doi: 10.1038/nature13385
8. Riely GJ, Marks J, Pao W. KRAS mutations in non-small cell lung cancer. Proc Am Thorac Soc. (2009) 6:201–5. doi: 10.1513/pats.200809-107LC
9. Skoulidis F, Heymach JV. Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat Rev Cancer. (2019) 19:495–509. doi: 10.1038/s41568–019-0179–8
10. Singhal A, Li BT, O’Reilly EM. Targeting KRAS in cancer. Nat Med. (2024) 30:969–83. doi: 10.1038/s41591–024-02903–0
11. Friedlaender A, Drilon A, Weiss GJ, Banna GL, Addeo A. KRAS as a druggable target in NSCLC: Rising like a phoenix after decades of development failures. Cancer Treat Rev. (2020) 85:101978. doi: 10.1016/j.ctrv.2020.101978
12. Seger R, Krebs EG. The MAPK signaling cascade. FASEB J. (1995) 9:726–35. doi: 10.1096/fasebj.9.9.7601337
13. Chen Y, Liu QP, Xie H, Ding J. From bench to bedside: current development and emerging trend of KRAS-targeted therapy. Acta Pharmacol Sin. (2024) 45:686–703. doi: 10.1038/s41401–023-01194–4
14. Arbour KC, Jordan E, Kim HR, Dienstag J, Yu HA, Sanchez-Vega F, et al. Effects of co-occurring genomic alterations on outcomes in patients with KRAS-mutant non-small cell lung cancer. Clin Cancer Res. (2018) 24:334–40. doi: 10.1158/1078–0432.CCR-17–1841
15. Boeschen M, Kuhn CK, Wirtz H, Seyfarth HJ, Frille A, Lordick F, et al. Comparative bioinformatic analysis of KRAS, STK11 and KEAP1 (co-)mutations in non-small cell lung cancer with a special focus on KRAS G12C. Lung Cancer. (2023) 184:107361. doi: 10.1016/j.lungcan.2023.107361
16. Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, et al. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discovery. (2018) 8:822–35. doi: 10.1158/2159–8290.CD-18–0099
17. Scheffler M, Ihle MA, Hein R, Merkelbach-Bruse S, Scheel AH, Siemanowski J, et al. K-ras mutation subtypes in NSCLC and associated co-occuring mutations in other oncogenic pathways. J Thorac Oncol. (2019) 14:606–16. doi: 10.1016/j.jtho.2018.12.013
18. Frille A, Boeschen M, Wirtz H, Stiller M, Bläker H, von Laffert M. TP53 co-mutations in advanced lung adenocarcinoma: comparative bioinformatic analyses suggest ambivalent character on overall survival alongside KRAS, STK11 and KEAP1 mutations. Front Oncol. (2024) 14:1357583. doi: 10.3389/fonc.2024.1357583
19. Huang L, Guo Z, Wang F, Fu L. KRAS mutation: from undruggable to druggable in cancer. Signal Transduct Target Ther. (2021) 6:386. doi: 10.1038/s41392–021-00780–4
20. O’Sullivan E, Keogh A, Henderson B, Finn SP, Gray SG, Gately K. Treatment strategies for KRAS-mutated non-small-cell lung cancer. Cancers (Basel). (2023) 15:1635. doi: 10.3390/cancers15061635
21. Salem ME, El-Refai SM, Sha W, Puccini A, Grothey A, George TJ, et al. Landscape of KRAS(G12C), associated genomic alterations, and interrelation with immuno-oncology biomarkers in KRAS-mutated cancers. JCO Precis Oncol. (2022) 6:e2100245. doi: 10.1200/PO.21.00245
22. Molina-Arcas M, Downward J. Exploiting the therapeutic implications of KRAS inhibition on tumor immunity. Cancer Cell. (2024) 42:338–57. doi: 10.1016/j.ccell.2024.02.012
23. Cheema PK, Banerji SO, Blais N, Chu QS, Juergens RA, Leighl NB, et al. Canadian consensus recommendations on the management of KRAS G12C-mutated NSCLC. Curr Oncol. (2023) 30:6473–96. doi: 10.3390/curroncol30070476
24. Ceddia S, Landi L, Cappuzzo F. KRAS-mutant non-small-cell lung cancer: from past efforts to future challenges. Int J Mol Sci. (2022) 23:9391. doi: 10.3390/ijms23169391
25. Maurer T, Garrenton LS, Oh A, Pitts K, Anderson DJ, Skelton NJ, et al. Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proc Natl Acad Sci U.S.A. (2012) 109:5299–304. doi: 10.1073/pnas.1116510109
26. Ostrem JM, Peters U, Sos ML, Wells JA. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. (2013) 503:548–51. doi: 10.1038/nature12796
27. Kwan AK, Piazza GA, Keeton AB, Leite CA. The path to the clinic: a comprehensive review on direct KRAS(G12C) inhibitors. J Exp Clin Cancer Res. (2022) 41:27. doi: 10.1186/s13046-021-02225-w
28. Lito P, Solomon M, Li LS, Hansen R, Rosen N. Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science. (2016) 351:604–8. doi: 10.1126/science.aad6204
29. Li HY, Qi WL, Wang YX, Meng LH. Covalent inhibitor targets KRas(G12C): A new paradigm for drugging the undruggable and challenges ahead. Genes Dis. (2023) 10:403–14. doi: 10.1016/j.gendis.2021.08.011
30. NCCN Clinical Practice Guidelines in Oncology: Non-Small Cell Lung Cancer. Version 5.2023 (2023). Available online at: https://www.nccn.org/professionals/physician_gls/pdf/nscl.pdf (Accessed November 11, 2023).
31. Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. KRAS(G12C) inhibition with sotorasib in advanced solid tumors. N Engl J Med. (2020) 383:1207–17. doi: 10.1056/NEJMoa1917239
32. Skoulidis F, Li BT, Dy GK, Price TJ, Falchook GS, Wolf J, et al. Sotorasib for lung cancers with KRAS p.G12C mutation. N Engl J Med. (2021) 384:2371–81. doi: 10.1056/NEJMoa2103695
33. de Langen AJ, Johnson ML, Mazieres J, Dingemans AC, Mountzios G, Pless M, et al. Sotorasib versus docetaxel for previously treated non-small-cell lung cancer with KRAS(G12C) mutation: a randomised, open-label, phase 3 trial. Lancet. (2023) 401:733–46. doi: 10.1016/S0140–6736(23)00221–0
34. Ou SI, Janne PA, Leal TA, Rybkin II, Sabari JK, Barve MA, et al. First-in-human phase I/IB dose-finding study of adagrasib (MRTX849) in patients with advanced KRAS(G12C) solid tumors (KRYSTAL-1). J Clin Oncol. (2022) 40:2530–8. doi: 10.1200/JCO.21.02752
35. Janne PA, Riely GJ, Gadgeel SM, Heist RS, Ou SI, Pacheco JM, et al. Adagrasib in non-small-cell lung cancer harboring a KRAS(G12C) mutation. N Engl J Med. (2022) 387:120–31. doi: 10.1056/NEJMoa2204619
36. Sacher A, LoRusso P, Patel MR, Miller WH Jr., Garralda E, Forster MD, et al. Single-agent divarasib (GDC-6036) in solid tumors with a KRAS G12C mutation. N Engl J Med. (2023) 389:710–21. doi: 10.1056/NEJMoa2303810
37. Li Z, Song Z, Zhao Y, Wang P, Jiang L, Gong Y, et al. D-1553 (Garsorasib), a potent and selective inhibitor of KRAS(G12C) in patients with NSCLC: phase 1 study results. J Thorac Oncol. (2023) 18:940–51. doi: 10.1016/j.jtho.2023.03.015
38. Nakajima EC, Drezner N, Li X, Mishra-Kalyani PS, Liu Y, Zhao H, et al. FDA approval summary: sotorasib for KRAS G12C-mutated metastatic NSCLC. Clin Cancer Res. (2022) 28:1482–6. doi: 10.1158/1078–0432.CCR-21–3074
39. U.S. Food & Drug Administration. FDA grants accelerated approval to sotorasib for KRAS G12C mutated NSCLC (2021). Available online at: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-sotorasib-kras-g12c-mutated-nsclc.
40. Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. (2019) 575:217–23. doi: 10.1038/s41586–019-1694–1
41. Dy GK, Govindan R, Velcheti V, Falchook GS, Italiano A, Wolf J, et al. Long-term outcomes and molecular correlates of sotorasib efficacy in patients with pretreated KRAS G12C-mutated non-small-cell lung cancer: 2-year analysis of codeBreaK 100. J Clin Oncol. (2023) 41:3311–7. doi: 10.1200/JCO.22.02524
42. Zhang SS, Lee A, Nagasaka M. CodeBreak 200: sotorasib has not broken the KRAS(G12C) enigma code. Lung Cancer (Auckl). (2023) 14:27–30. doi: 10.2147/LCTT.S403461
43. Sakata S, Akamatsu H, Azuma K, Uemura T, Tsuchiya-Kawano Y, Yoshioka H, et al. The primary endpoint analysis of SCARLET study: A single-arm, phase II study of sotorasib plus carboplatin-pemetrexed in patients with advanced non-squamous, non-small cell lung cancer with KRAS G12C mutation (WJOG14821L). J Clin Oncol. (2023) 41:9006–6. doi: 10.1200/JCO.2023.41.16_suppl.9006
44. Clarke JM, Felip E, Li BT, Ruffinelli JC, Garrido P, Zugazagoitia J, et al. MA06.05 codeBreaK 101: safety and efficacy of sotorasib with carboplatin and pemetrexed in KRAS G12C-mutated advanced NSCLC. J Thorac Oncol. (2023) 18:S118–9. doi: 10.1016/j.jtho.2023.09.153
45. Amgen presents new Lumakras® (sotorasib) plus chemotherapy data in first-line KRAS G12C NSCLC at WCLC (2023). Available online at: https://www.amgen.com/newsroom/press-releases/2023/09/amgen-presents-new-lumakras-sotorasib-plus-chemotherapy-data-in-firstline-kras-g12c-nsclc-at-wclc (Accessed November 5, 2023).
46. Fell JB, Fischer JP, Baer BR, Blake JF, Bouhana K, Briere DM, et al. Identification of the clinical development candidate MRTX849, a covalent KRAS(G12C) inhibitor for the treatment of cancer. J Med Chem. (2020) 63:6679–93. doi: 10.1021/acs.jmedchem.9b02052
47. U.S. Food & Drug Administration. FDA grants accelerated approval to adagrasib for KRAS G12C-mutated NSCLC (2022). Available online at: www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-adagrasib-kras-g12cmutated-nsclc.
48. Gadgeel S, Jänne PA, Spira AI, Ou SHI, Heist RS, Pacheco JM, et al. MA06.04 KRYSTAL-1: two-year follow-up of adagrasib (MRTX849) monotherapy in patients with advanced/metastatic KRASG12C-mutated NSCLC. J Thorac Oncol. (2023) 18:S118. doi: 10.1016/j.jtho.2023.09.152
49. Barlesi F, Felip E, Popat S, Solomon B, Wolf J, Li BT, et al. 103TiP Sotorasib versus pembrolizumab in combination with platinum doublet chemotherapy as first-line treatment for metastatic or locally advanced, PD-L1 negative, KRAS G12C-mutated NSCLC (CodeBreaK 202). ESMO Open. (2024) 9:102682. doi: 10.1016/j.esmoop.2024.102682
50. Garassino MC, Theelen WSME, Jotte R, Laskin J, de Marinis F, Aguado C, et al. LBA65 KRYSTAL-7: Efficacy and safety of adagrasib with pembrolizumab in patients with treatment-naive, advanced non-small cell lung cancer (NSCLC) harboring a KRASG12C mutation. Ann Oncol. (2023) 34:S1309–10. doi: 10.1016/j.annonc.2023.10.066
51. Mok TSK, Lawler WE, Shum MK, Dakhil SR, Spira AI, Barlesi F, et al. KRYSTAL-12: A randomized phase 3 study of adagrasib (MRTX849) versus docetaxel in patients (pts) with previously treated non-small-cell lung cancer (NSCLC) with KRAS mutation. J Clin Oncol. (2021) 39:TPS9129–9. doi: 10.1200/JCO.2021.39.15_suppl.TPS9129
52. Cappuzzo F, Castro G, Kang J-H, Wu Y-L, Brustugun OT, Cheema PK, et al. KontRASt-02: A phase III trial investigating the efficacy and safety of the KRASG12C inhibitor JDQ443 vs docetaxel in patients with previously treated, locally advanced or metastatic, KRAS G12C-mutated NSCLC. J Clin Oncol. (2023) 41:TPS9144. doi: 10.1200/JCO.2023.41.16_suppl.TPS9144
53. Riely GJ, Ou SHI, Rybkin I, Spira A, Papadopoulos K, Sabari JK, et al. KRYSTAL-1: Activity and preliminary pharmacodynamic (PD) analysis of adagrasib (MRTX849) in patients (Pts) with advanced non-small cell lung cancer (NSCLC) harboring KRASG12C mutation. J Thorac Oncol. (2021) 16:S751–2. doi: 10.1016/S1556-0864(21)01941-9
54. Cordeiro de Lima VC, Corassa M, Saldanha E, Freitas H, Arrieta O, Raez L, et al. STK11 and KEAP1 mutations in non-small cell lung cancer patients: Descriptive analysis and prognostic value among Hispanics (STRIKE registry-CLICaP). Lung Cancer. (2022) 170:114–21. doi: 10.1016/j.lungcan.2022.06.010
55. Garassino MC, Theelen WSME, Jotte R, Laskin J, de Marinis F, Aguado C, et al. KRYSTAL-7: Efficacy and safety of adagrasib with pembrolizumab in patients with treatment-naive, advanced non-small cell lung cancer (NSCLC) harboring a KRASG12C mutation. Ann Oncol. (2023) 34:S1309–10. doi: 10.1016/j.annonc.2023.10.066
56. Sabari JK, Velcheti V, Shimizu K, Strickland MR, Heist RS, Singh M, et al. Activity of adagrasib (MRTX849) in brain metastases: preclinical models and clinical data from patients with KRASG12C-mutant non-small cell lung cancer. Clin Cancer Res. (2022) 28:3318–28. doi: 10.1158/1078–0432.CCR-22–0383
57. Negrao MV, Spira AI, Heist RS, Jaenne PA, Pacheco JM, Weiss J, et al. Intracranial efficacy of adagrasib in patients from the KRYSTAL-1 trial with-mutated non-small-cell lung cancer who have untreated CNS metastases. J Clin Oncol. (2023) 41:4472–7. doi: 10.1200/Jco.23.00046
58. Cui W, Franchini F, Alexander M, Officer A, Wong HL, IJ M, et al. Real world outcomes in KRAS G12C mutation positive non-small cell lung cancer. Lung Cancer. (2020) 146:310–7. doi: 10.1016/j.lungcan.2020.06.030
59. Sebastian M, Eberhardt WEE, Hoffknecht P, Metzenmacher M, Wehler T, Kokowski K, et al. KRAS G12C-mutated advanced non-small cell lung cancer: A real-world cohort from the German prospective, observational, nation-wide CRISP Registry (AIO-TRK-0315). Lung Cancer. (2021) 154:51–61. doi: 10.1016/j.lungcan.2021.02.005
60. Wu MY, Zhang EW, Strickland MR, Mendoza DP, Lipkin L, Lennerz JK, et al. Clinical and imaging features of non-small cell lung cancer with G12C KRAS mutation. Cancers (Basel). (2021) 13:3572. doi: 10.3390/cancers13143572
61. Vassella E, Kashani E, Zens P, Kundig A, Fung C, Scherz A, et al. Mutational profiles of primary pulmonary adenocarcinoma and paired brain metastases disclose the importance of KRAS mutations. Eur J Cancer. (2021) 159:227–36. doi: 10.1016/j.ejca.2021.10.006
62. Tomasini P, Serdjebi C, Khobta N, Metellus P, Ouafik L, Nanni I, et al. EGFR and KRAS mutations predict the incidence and outcome of brain metastases in non-small cell lung cancer. Int J Mol Sci. (2016) 17:2132. doi: 10.3390/ijms17122132
63. Parikh NR, Likhacheva A, Pinnix C, Allen PK, Prabhu SS, Guha-Thakurta N, et al. Prognostic significance of EGFR and KRAS mutations in NSCLC patients with brain metastases treated with radiosurgery(dagger). J Radiosurg SBRT. (2015) 3:171–8.
64. Lauko A, Kotecha R, Barnett A, Li H, Tatineni V, Ali A, et al. Impact of KRAS mutation status on the efficacy of immunotherapy in lung cancer brain metastases. Sci Rep. (2021) 11:18174. doi: 10.1038/s41598-021-97566-z
65. Lu K, Husain H. Intracranial responses with selective KRAS-G12C inhibitors in non-small cell lung cancer. Transl Lung Cancer Res. (2023) 12:1335–7. doi: 10.21037/tlcr-23–26
66. Palma G, Khurshid F, Lu K, Woodward B, Husain H. Selective KRAS G12C inhibitors in non-small cell lung cancer: chemistry, concurrent pathway alterations, and clinical outcomes. NPJ Precis Oncol. (2021) 5:98. doi: 10.1038/s41698–021-00237–5
67. Herzberg BO, Manji GA. KRAS: druggable at last. Oncologist. (2023) 28:283–6. doi: 10.1093/oncolo/oyad014
68. Bernstein E, Luo J, Wang K, Negrao MV, Janne PA, Sabari JK. Safety and intracranial activity of adagrasib in patients with KRAS(G12C)-mutated non-small-cell lung cancer and untreated CNS metastases in the KRYSTAL-1 trial: A case series. JCO Precis Oncol. (2024) 8:e2300447. doi: 10.1200/PO.23.00447
69. Koster KL, Appenzeller C, Lauber A, Fruh M, Schmid S. Sotorasib shows intracranial activity in patients with KRAS G12C-mutated adenocarcinoma of the lung and untreated active brain metastases. Case Rep Oncol. (2022) 15:720–5. doi: 10.1159/000525341
70. Yeh J, Marks JA, Alzeer AH, Sloan EA, Varghese R, Paudel N, et al. Remarkable intracranial response to sotorasib in a patient with KRAS (G12C)-mutated lung adenocarcinoma and untreated brain metastases: A case report. JTO Clin Res Rep. (2022) 3:100428. doi: 10.1016/j.jtocrr.2022.100428
71. Inno A, Marchetti F, Valerio M, Giaj Levra N, Alongi F, Foti G, et al. Activity of sotorasib against brain metastases from NSCLC harboring KRAS p.G12C mutation: a case report. Drug Target Insights. (2023) 17:90–1. doi: 10.33393/dti.2023.2593
72. Murciano-Goroff YR, Heist RS, Kuboki Y, Koyama T, Ammakkanavar NR, Hollebecque A, et al. Abstract CT028: A first-in-human phase 1 study of LY3537982, a highly selective and potent KRAS G12C inhibitor in patients with KRAS G12C-mutant advanced solid tumors. Cancer Res. (2023) 83:CT028–8. doi: 10.1158/1538–7445.Am2023-ct028
73. Cassier PA, Dooms CA, Gazzah A, Felip E, Steeghs N, Rohrberg KS, et al. KontRASt-01 update: Safety and efficacy of JDQ443 in KRAS G12C-mutated solid tumors including non-small cell lung cancer (NSCLC). J Clin Oncol. (2023) 41:9007–7. doi: 10.1200/JCO.2023.41.16_suppl.9007
74. Yuan Y, Deng Y, Jin Y, Pan Y, Wang C, Wang Z, et al. 106P Efficacy and safety of IBI351 (GFH925) monotherapy in metastatic colorectal cancer harboring KRASG12C mutation: Updated results from a pooled analysis of two phase I studies. Ann Oncol. (2023) 34:S1512. doi: 10.1016/j.annonc.2023.10.241
75. Zhou Q, Meng X, Sun L, Huang D, Yang N, Yu Y, et al. LBA12 Efficacy and safety of IBI351 (GFH925), a selective KRASG12C inhibitor, monotherapy in patients (pts) with advanced non-small cell lung cancer (NSCLC): Initial results from a registrational phase II study. Ann Oncol. (2023) 34:S1662. doi: 10.1016/j.annonc.2023.10.584
76. Jänne PA, Bigot F, Papadopoulos K, Eberst L, Sommerhalder D, Lebellec L, et al. Abstract PR014: Preliminary safety and anti-tumor activity of RMC-6291, a first-in-class, tri-complex KRASG12C(ON) inhibitor, in patients with or without prior KRASG12C(OFF) inhibitor treatment. Mol Cancer Ther. (2023) 22:PR014. doi: 10.1158/1535–7163.Targ-23-pr014
77. Rojas C, Lugowska I, Juergens R, Sacher A, Weindler S, Sendur MAN, et al. Safety and preliminary efficacy of the KRAS G12C Inhibitor MK-1084 in solid tumors and in combination with pembrolizumab in NSCLC. Ann Oncol. (2023) 34:S466–7. doi: 10.1016/j.annonc.2023.09.1849
78. Li J, Zhao J, Cao B, Fang J, Li X, Wang M, et al. A phase I/II study of first-in-human trial of JAB-21822 (KRAS G12C inhibitor) in advanced solid tumors. J Clin Oncol. (2022) 40:3089–9. doi: 10.1200/JCO.2022.40.16_suppl.3089
79. Zhou C, Li W, Song Z, Zhang Y, Zhang Y, Huang D, et al. LBA33 A first-in-human phase I study of a novel KRAS G12D inhibitor HRS-4642 in patients with advanced solid tumors harboring KRAS G12D mutation. Ann Oncol. (2023) 34:S1273. doi: 10.1016/j.annonc.2023.10.025
80. Knox JE, Burnett GL, Weller C, Jiang L, Zhang D, Vita N, et al. Abstract ND03: Discovery of RMC-9805, an oral, covalent tri-complex KRASG12D(ON) inhibitor. Cancer Res. (2024) 84:ND03. doi: 10.1158/1538–7445.Am2024-nd03
81. Arbour KC, Punekar S, Garrido-Laguna I, Hong DS, Wolpin B, Pelster MS, et al. 652O Preliminary clinical activity of RMC-6236, a first-in-class, RAS-selective, tri-complex RAS-MULTI(ON) inhibitor in patients with KRAS mutant pancreatic ductal adenocarcinoma (PDAC) and non-small cell lung cancer (NSCLC). Ann Oncol. (2023) 34:S458. doi: 10.1016/j.annonc.2023.09.1838
82. Corral de la Fuente E, Olmedo Garcia ME, Gomez Rueda A, Lage Y, Garrido P. Targeting KRAS in non-small cell lung cancer. Front Oncol. (2021) 11:792635. doi: 10.3389/fonc.2021.792635
83. Batrash F, Kutmah M, Zhang J. The current landscape of using direct inhibitors to target KRAS(G12C)-mutated NSCLC. Exp Hematol Oncol. (2023) 12:93. doi: 10.1186/s40164–023-00453–8
84. Perurena N, Situ L, Cichowski K. Combinatorial strategies to target RAS-driven cancers. Nat Rev Cancer. (2024) 24:316–37. doi: 10.1038/s41568–024-00679–6
85. Wang J, Martin-Romano P, Cassier P, Johnson M, Haura E, Lenox L, et al. Phase I study of JNJ-74699157 in patients with advanced solid tumors harboring the KRAS G12C mutation. Oncologist. (2022) 27:536–e553. doi: 10.1093/oncolo/oyab080
86. Purkey H. Discovery of GDC-6036, a clinical stage treatment for KRAS G12C-positive cancers. Cancer Res. (2022) 82:ND11–1. doi: 10.1158/1538-7445.AM2022-ND11
87. Brachmann SM, Weiss A, Guthy DA, Beyer K, Voshol J, Maira M, et al. JDQ443, a covalent irreversible inhibitor of KRAS G12C, exhibits a novel binding mode and demonstrates potent anti-tumor activity and favorable pharmacokinetic properties in preclinical models. Mol Cancer Ther. (2021) 20:P124–4. doi: 10.1158/1535–7163.Targ-21-P124
88. Lorthiois E, Gerspacher M, Beyer KS, Vaupel A, Leblanc C, Stringer R, et al. JDQ443, a structurally novel, pyrazole-based, covalent inhibitor of KRAS(G12C) for the treatment of solid tumors. J Med Chem. (2022) 65:16173–203. doi: 10.1021/acs.jmedchem.2c01438
89. Weiss A, Lorthiois E, Barys L, Beyer KS, Bomio-Confaglia C, Burks H, et al. Discovery, preclinical characterization, and early clinical activity of JDQ443, a structurally novel, potent, and selective covalent oral inhibitor of KRASG12C. Cancer Discov. (2022) 12:1500–17. doi: 10.1158/2159–8290.CD-22–0158
90. Tan DS, Shimizu T, Solomon B, Heist RS, Schuler M, Luken MJDM, et al. Abstract CT033: KontRASt-01: A phase Ib/II, dose-escalation study of JDQ443 in patients (pts) with advanced, KRAS G12C-mutated solid tumors. Cancer Res. (2022) 82:CT033–3. doi: 10.1158/1538–7445.Am2022-ct033
91. Shi Z, Weng J, Niu H, Yang H, Liu R, Weng Y, et al. D-1553: A novel KRAS(G12C) inhibitor with potent and selective cellular and in vivo antitumor activity. Cancer Sci. (2023) 114:2951–60. doi: 10.1111/cas.15829
92. Reita D, Pabst L, Pencreach E, Guerin E, Dano L, Rimelen V, et al. Direct targeting KRAS mutation in non-small cell lung cancer: focus on resistance. Cancers (Basel). (2022) 14:1321. doi: 10.3390/cancers14051321
93. Camidge DR, Pao W, Sequist LV. Acquired resistance to TKIs in solid tumours: learning from lung cancer. Nat Rev Clin Oncol. (2014) 11:473–81. doi: 10.1038/nrclinonc.2014.104
94. Negrao MV, Araujo HA, Lamberti G, Cooper AJ, Akhave NS, Zhou T, et al. Comutations and KRASG12C inhibitor efficacy in advanced NSCLC. Cancer Discov. (2023) 13:1556–71. doi: 10.1158/2159–8290.CD-22–1420
95. Lietman CD, Johnson ML, McCormick F, Lindsay CR. More to the RAS story: KRAS(G12C) inhibition, resistance mechanisms, and moving beyond KRAS(G12C). Am Soc Clin Oncol Educ Book. (2022) 42:1–13. doi: 10.1200/EDBK_351333
96. Proulx-Rocray F, Routy B, Nassabein RM, El Ouarzadi O, Belkaid W, Tran-Thanh D, et al. The prognostic impact of KRAS, TP53, STK11 and KEAP1 mutations and the influence of the NLR in NSCLC patients treated with immunotherapy. J Clin Oncol. (2021) 39:100767. doi: 10.1200/JCO.2021.39.15_suppl.e21010
97. Ricciuti B, Alessi JV, Elkrief A, Wang X, Cortellini A, Li YY, et al. Dissecting the clinicopathologic, genomic, and immunophenotypic correlates of KRAS(G12D)-mutated non-small-cell lung cancer. Ann Oncol. (2022) 33:1029–40. doi: 10.1016/j.annonc.2022.07.005
98. Awad MM, Liu S, Rybkin II, Arbour KC, Dilly J, Zhu VW, et al. Acquired resistance to KRAS(G12C) inhibition in cancer. N Engl J Med. (2021) 384:2382–93. doi: 10.1056/NEJMoa2105281
99. Priest K, Le A, Aisner D, Gebregzabheir A, Nijmeh H, Mandell M, et al. Evolution of therapy resistance through acquired KRAS amplification in ROS1 fusion KRAS G12C double positive NSCLC. Cancer Res. (2022) 82:9. doi: 10.1158/1538-7445.AM2022-5233
100. Di Federico A, Ricciotti I, Favorito V, Michelina SV, Scaparone P, Metro G, et al. Resistance to KRAS G12C inhibition in non-small cell lung cancer. Curr Oncol Rep. (2023) 25:1017–29. doi: 10.1007/s11912-023-01436-y
101. Koga T, Suda K, Fujino T, Ohara S, Hamada A, Nishino M, et al. KRAS secondary mutations that confer acquired resistance to KRAS G12C inhibitors, sotorasib and adagrasib, and overcoming strategies: insights from in vitro experiments. J Thorac Oncol. (2021) 16:1321–32. doi: 10.1016/j.jtho.2021.04.015
102. Parikh K, Banna G, Liu SV, Friedlaender A, Desai A, Subbiah V, et al. Drugging KRAS: current perspectives and state-of-art review. J Hematol Oncol. (2022) 15:152. doi: 10.1186/s13045–022-01375–4
103. Tanaka N, Lin JJ, Li C, Ryan MB, Zhang J, Kiedrowski LA, et al. Clinical acquired resistance to KRAS(G12C) inhibition through a novel KRAS switch-II pocket mutation and polyclonal alterations converging on RAS-MAPK reactivation. Cancer Discov. (2021) 11:1913–22. doi: 10.1158/2159–8290.CD-21–0365
104. Suzuki S, Yonesaka K, Teramura T, Takehara T, Kato R, Sakai H, et al. KRAS inhibitor resistance in MET-amplified KRAS (G12C) non-small cell lung cancer induced by RAS- and non-RAS-mediated cell signaling mechanisms. Clin Cancer Res. (2021) 27:5697–707. doi: 10.1158/1078–0432.CCR-21–0856
105. Qin K, Hong L, Zhang J, Le X. MET amplification as a resistance driver to TKI therapies in lung cancer: clinical challenges and opportunities. Cancers (Basel). (2023) 15:612. doi: 10.3390/cancers15030612
106. Liu J, Kang R, Tang D. The KRAS-G12C inhibitor: activity and resistance. Cancer Gene Ther. (2022) 29:875–8. doi: 10.1038/s41417–021-00383–9
107. Tang D, Kang R. Glimmers of hope for targeting oncogenic KRAS-G12D. Cancer Gene Ther. (2023) 30:391–3. doi: 10.1038/s41417–022-00561–3
108. Misale S, Fatherree JP, Cortez E, Li C, Bilton S, Timonina D, et al. KRAS G12C NSCLC models are sensitive to direct targeting of KRAS in combination with PI3K inhibition. Clin Cancer Res. (2019) 25:796–807. doi: 10.1158/1078–0432.CCR-18–0368
109. Jiao D, Yang S. Overcoming resistance to drugs targeting KRAS(G12C) mutation. Innovation (Camb). (2020) 1:100035. doi: 10.1016/j.xinn.2020.100035
110. Xue JY, Zhao Y, Aronowitz J, Mai TT, Vides A, Qeriqi B, et al. Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition. Nature. (2020) 577:421–5. doi: 10.1038/s41586-019-1884-x
111. Ryan MB, Fece de la Cruz F, Phat S, Myers DT, Wong E, Shahzade HA, et al. Vertical pathway inhibition overcomes adaptive feedback resistance to KRAS(G12C) inhibition. Clin Cancer Res. (2020) 26:1633–43. doi: 10.1158/1078–0432.CCR-19–3523
112. Rosen JC, Sacher A, Tsao MS. Direct GDP-KRAS(G12C) inhibitors and mechanisms of resistance: the tip of the iceberg. Ther Adv Med Oncol. (2023) 15:1–23. doi: 10.1177/17588359231160141
113. Nagasaka M, Potugari B, Nguyen A, Sukari A, Azmi AS, Ou SI. KRAS Inhibitors- yes but what next? Direct targeting of KRAS- vaccines, adoptive T cell therapy and beyond. Cancer Treat Rev. (2021) 101:102309. doi: 10.1016/j.ctrv.2021.102309
114. Fakih MG, Salvatore L, Esaki T, Modest DP, Lopez-Bravo DP, Taieb J, et al. Sotorasib plus panitumumab in refractory colorectal cancer with mutated KRAS G12C. N Engl J Med. (2023) 389:2125–39. doi: 10.1056/NEJMoa2308795
Keywords: non-small cell lung cancer, lung adenocarcinoma, KRAS, co-mutations, resistance to therapy
Citation: Sreter KB, Catarata MJ, von Laffert M and Frille A (2024) Resistance to KRAS inhibition in advanced non-small cell lung cancer. Front. Oncol. 14:1357898. doi: 10.3389/fonc.2024.1357898
Received: 18 December 2023; Accepted: 06 May 2024;
Published: 23 May 2024.
Edited by:
Wouter H. Van Geffen, Medical Center Leeuwarden, NetherlandsReviewed by:
Oke Dimas Asmara, University of Groningen, NetherlandsIlona Tietzova, Charles University, Czechia
Copyright © 2024 Sreter, Catarata, von Laffert and Frille. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Armin Frille, YXJtaW4uZnJpbGxlQG1lZGl6aW4udW5pLWxlaXB6aWcuZGU=