 Harjot Athwal
Harjot Athwal Arpitha Kochiyanil
Arpitha Kochiyanil Vasudeva Bhat1,3
Vasudeva Bhat1,3 Armen Parsyan
Armen Parsyan- 1Department of Anatomy and Cell Biology, Schulich School of Medicine and Dentistry, Western University, London, ON, Canada
- 2Faculty of Science, Schulich School of Medicine and Dentistry, Western University, London, ON, Canada
- 3London Regional Cancer Program, London Health Sciences Centre, Lawson Health Research Institute, London, ON, Canada
- 4Department of Oncology, Schulich School of Medicine and Dentistry, Western University, London, ON, Canada
- 5Division of General Surgery, Department of Surgery, Schulich School of Medicine and Dentistry, Western University, London, ON, Canada
- 6Department of Surgery, St. Joseph’s Health Care London and London Health Sciences Centre, London, ON, Canada
Breast cancer is the most prevalent malignancy among women worldwide. Despite significant advances in treatment, it remains one of the leading causes of female mortality. The inability to effectively treat advanced and/or treatment-resistant breast cancer demonstrates the need to develop novel treatment strategies and targeted therapies. Centrosomes and their associated proteins have been shown to play key roles in the pathogenesis of breast cancer and thus represent promising targets for drug and biomarker development. Centrosomes are fundamental cellular structures in the mammalian cell that are responsible for error-free execution of cell division. Centrosome amplification and aberrant expression of its associated proteins such as Polo-like kinases (PLKs), Aurora kinases (AURKs) and Cyclin-dependent kinases (CDKs) have been observed in various cancers, including breast cancer. These aberrations in breast cancer are thought to cause improper chromosomal segregation during mitosis, leading to chromosomal instability and uncontrolled cell division, allowing cancer cells to acquire new genetic changes that result in evasion of cell death and the promotion of tumor formation. Various chemical compounds developed against PLKs and AURKs have shown meaningful antitumorigenic effects in breast cancer cells in vitro and in vivo. The mechanism of action of these inhibitors is likely related to exacerbation of numerical genomic instability, such as aneuploidy or polyploidy. Furthermore, growing evidence demonstrates enhanced antitumorigenic effects when inhibitors specific to centrosome-associated proteins are used in combination with either radiation or chemotherapy drugs in breast cancer. This review focuses on the current knowledge regarding the roles of centrosome and centrosome-associated proteins in breast cancer pathogenesis and their utility as novel targets for breast cancer treatment.
Introduction
Breast cancer is a commonly diagnosed malignancy and one of the leading causes of cancer-related deaths among women worldwide (1). Current treatment strategies for breast cancer are based on the molecular subtype classification that takes into account cancer cell expression of hormone (estrogen and progesterone) receptors and the human epidermal growth factor 2 receptor (HER2) (2). Treatment modalities include surgery and/or radio-, chemo-, anti-endocrine, targeted and immune therapies. Despite advances in treatment options, the recurrence of breast cancer, chemoresistance, radioresistance and metastatic disease remain major areas of concern in achieving desired patient outcomes (3–5). Therefore, the development of novel drugs and therapies for breast cancer is of utmost clinical importance. Recent efforts in targeted drug development in oncology have been focused on inhibitors of cell cycle regulation. Aberrant regulation of cell cycle events is often observed in cancer cells (2, 6) and has been associated with breast cancer pathogenesis and the development of treatment resistance. This has led to the investigation of novel targets and the development of therapeutic agents targeting the cell cycle (2, 6–9). A large body of recent research indicates that centrosomes and their associated proteins play an important role in cell cycle progression and are thus highly promising targets for drug development in breast cancer (10–13). Here, we aim to review current knowledge on the role of centrosomes in breast cancer pathogenesis and highlight the development of novel drugs that target this critical hub of the cell cycle.
Centrosomes and their associated proteins during the cell cycle
The mammalian cell cycle is an intricately coordinated and controlled process that ensures the proper division of cells (6) resulting in efficient segregation of genetic information. It is regulated by the activation and deactivation of various proteins throughout the five different stages of the cell cycle, including G0 (gap 0), G1, S (synthesis), G2 and M (mitosis) (6). The first four stages are known as interphase, the process by which cells grow and duplicate their genetic material to prepare for cell division (14). In the G0 phase, cells are quiescent and remain in a state of rest (14). Upon external stimulation, cells can enter G1 and produce mRNA and proteins needed for DNA replication in the S phase (14). Cells in G2 then ensure DNA replication is accurate and synthesize enzymes required for cell division (10, 14). Cells then proceed to divide during the M phase, which progresses through five main stages: prophase, metaphase, anaphase, telophase and cytokinesis (14). In prophase, the chromosomes condense, and the nuclear membrane starts to break down (15). The latter is a central step for assembling the mitotic spindle, consisting of the centrosome and microtubules (15). The microtubules of the developing spindle then attach to the kinetochore of chromosomes and pull them in opposite directions, thereby having a bi-directional orientation by the end of prophase (15). In metaphase, chromosomes are aligned at the equatorial plate and ensure the correct attachment of microtubules to the kinetochore, commonly referred to as the spindle assembly checkpoint (10, 16). Cells can then proceed to anaphase where sister chromatids separate to the spindle poles (16), followed by telophase where chromosomes decondense and the nuclear membrane reforms (16). As the last and final stage, the cytoplasm and chromosomes are equally divided into two daughter cells during cytokinesis (15).
Centrosomes comprise a pair of centrioles surrounded by pericentriolar material and function as the major microtubule organizing centers in mammalian cells (17). They play an important role in regulating many cellular processes during interphase, such as cell morphology and polarization; as well as during mitosis, such as spindle formation, chromosome segregation and cytokinesis (18). These intracellular organelles are regulated by a large variety of centrosome-associated proteins throughout the cell cycle (19) (Figure 1, Table 1). The complex process of centriole duplication and maturation is orchestrated by a number of proteins during various stages of the cell cycle (Figures 1-4). During G1, centrioles prepare to divide with the help of Polo-like kinase 4 (PLK4) activity (29, 73) (Figure 2). Upon being recruited by centrosomal C-terminal encoded protein (CEP) 152 and CEP192 (53), PLK4 binds and phosphorylates SCL/TAL-interrupting locus protein (STIL) and associates with spindle assembly abnormal protein 6 (SAS6) to initiate centriole duplication (68, 74) (Figure 2). The phosphorylation of STIL promotes its binding to centrosomal P4.1-associated protein (CPAP; also known as centromere protein J [CENPJ]) (63), which interacts with CEP120 and is responsible for regulating centriole assembly, length and duplication during the S phase (46, 64) (Figure 2). During the G1 and S phases, Cyclin-dependent kinase (CDK) proteins also interact with their cyclin partners to facilitate centriole duplication (Figure 3). Centriole maturation then occurs in G2 and is primarily regulated by PLK1 and Aurora kinase A (AURKA) (Figure 4) (6). PLK1 regulates the localization of AURKA to the centrosome, which phosphorylates and activates PLK1 to promote bipolar spindle formation and mitotic entry (20, 31). PLK1 also regulates NIMA-related-kinase 2 (NEK2), which participates in bipolar spindle assembly to promote mitotic entry as well as mediates centrosome separation during mitosis by interacting with Centrosomal NEK2-Associated Protein 1 (C-NAP1; also known as CEP250) (21, 55, 66, 75, 76) (Figure 4). At this stage, the separating centrosomes form the poles of the mitotic spindle to facilitate chromosome segregation (Figure 1) (19).
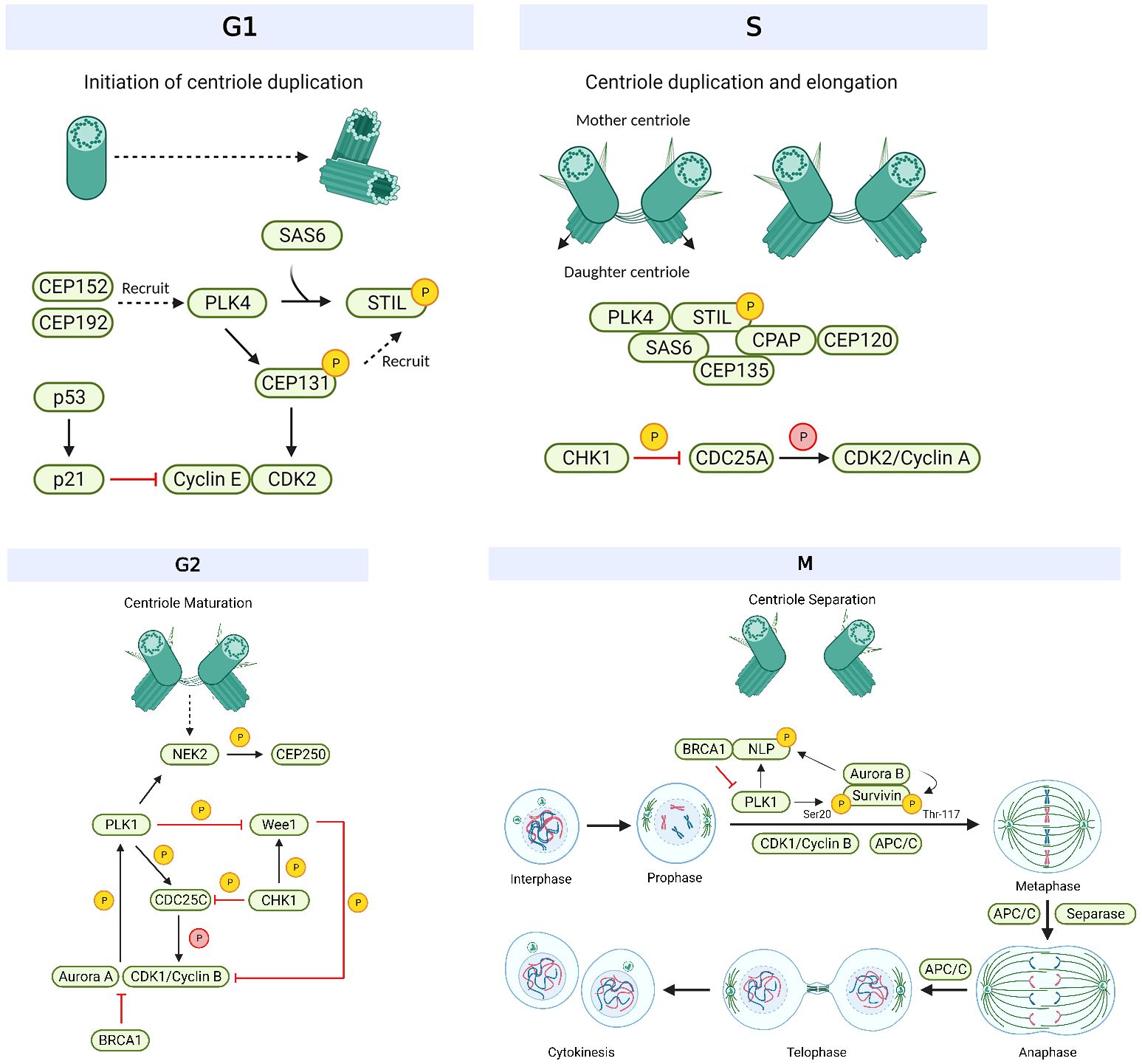
Figure 1 Overview of the mechanism of centrosomes during the cell cycle. Initiation of centriole duplication begins in G1. CEP152 and CEP192 recruit PLK4, which binds and phosphorylates SCL/TAL-interrupting locus protein (STIL) and associates with spindle assembly abnormal protein 6 (SAS6). STIL is recruited by CEP131, which gets phosphorylated by Polo-like kinase 4 (PLK4) and is responsible for regulating the centriole positioning of Cyclin-dependent kinase 2 (CDK2). CDK2 binds to its cyclin partner (cyclin E) but this interaction is inhibited by p21, the latter which gets activated by p53. Centriole duplication and elongation commences in S phase. The phosphorylation of STIL promotes its binding to centrosomal P4.1-associated protein (CPAP), which interacts with C-terminal encoded protein 120 (CEP120). CEP135 associates with SAS6 to link it to CPAP. Checkpoint kinase 1 (CHK1) phosphorylates and inhibits cell division cycle 25 (CDC25) A/C, which is responsible for dephosphorylating and activating CDK2/Cyclin A and CDK1/Cyclin B, respectively. CHK1 also phosphorylates and activates WEE1 during G2 (centriole maturation), which can phosphorylate and inhibit CDK1 activity. CDK1/Cyclin B interacts with Aurora kinase A to enhance its phosphorylation and activation of PLK1. Aurora kinase A activity is inhibited by BRCA1. PLK1 induces CDC25C activation and WEE1 degradation as well as regulates NIMA-related-kinase 2 (NEK2) activity. NEK2 phosphorylates CEP250 and mediates centrosome separation (M phase). Mitosis (M) is broken down into 6 steps: interphase, prophase, metaphase, anaphase, telophase, and cytokinesis. During prophase, PLK1 and Aurora kinase B phosphorylate Ninein-like protein (NLP) and survivin. The former interacts with BRCA1, and the latter activates Aurora kinase B activity. BRCA1 also inhibits PLK1 activity. CDK1/Cyclin B activates APC/C which regulates chromosome separation and degrades centrosomal components (such as SAS6, STIL, etc.) throughout mitosis. Separase regulates chromosome and centriole separation by cleaving sister chromatids during metaphase. Based on references 9, 13, 21 and 22. Created with BioRender.com.
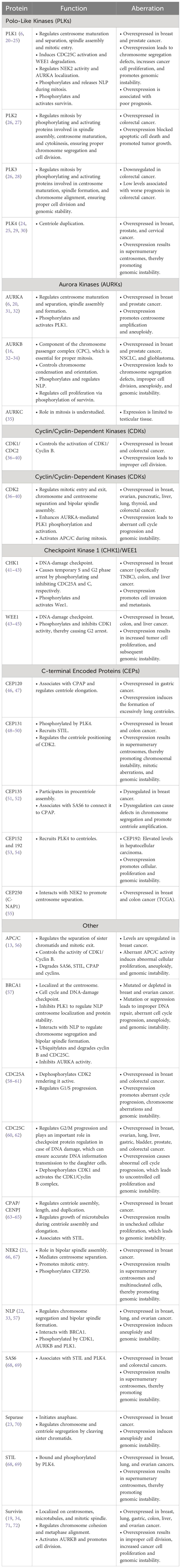
Table 1 Overview of the function and aberrations of centrosome-associated proteins.
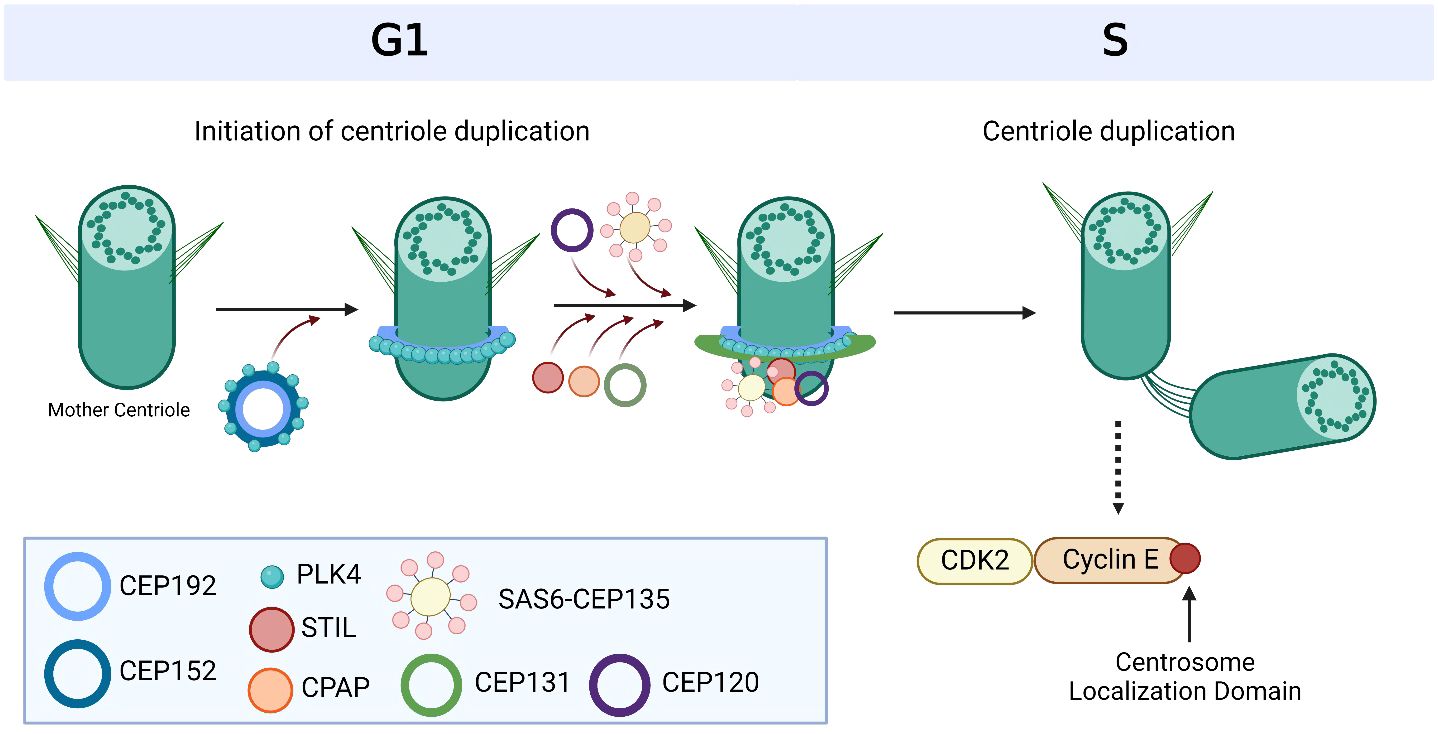
Figure 2 Overview of centriole duplication. To initiate centriole duplication, C-terminal encoded protein (CEP) 152 and CEP192 recruit Polo-like kinase 4 (PLK4) to the mother centriole, where it phosphorylates CEP131, which in turn recruits SCL/TAL-interrupting locus protein (STIL). PLK4 then binds and phosphorylates STIL as well as associates with spindle assembly abnormal protein 6 (SAS6) during late G1 and early S phase to initiate centriole duplication. CEP135 interacts with SAS6 to link it to centrosomal P4.1-associated protein (CPAP), which directly binds to CEP120 to induce centriole elongation of the nascent centriole. Cyclin E is recruited to the centrosome via its centrosome localization domain to promote entry into the S phase and facilitate centriole duplication. It is bound to both the centrosome and its Cyclin-dependent kinase (CDK) partner, CDK2, to facilitate centriole duplication and progression of the cell cycle. Based on references 24, 27, 150 and 212. Created with BioRender.com.
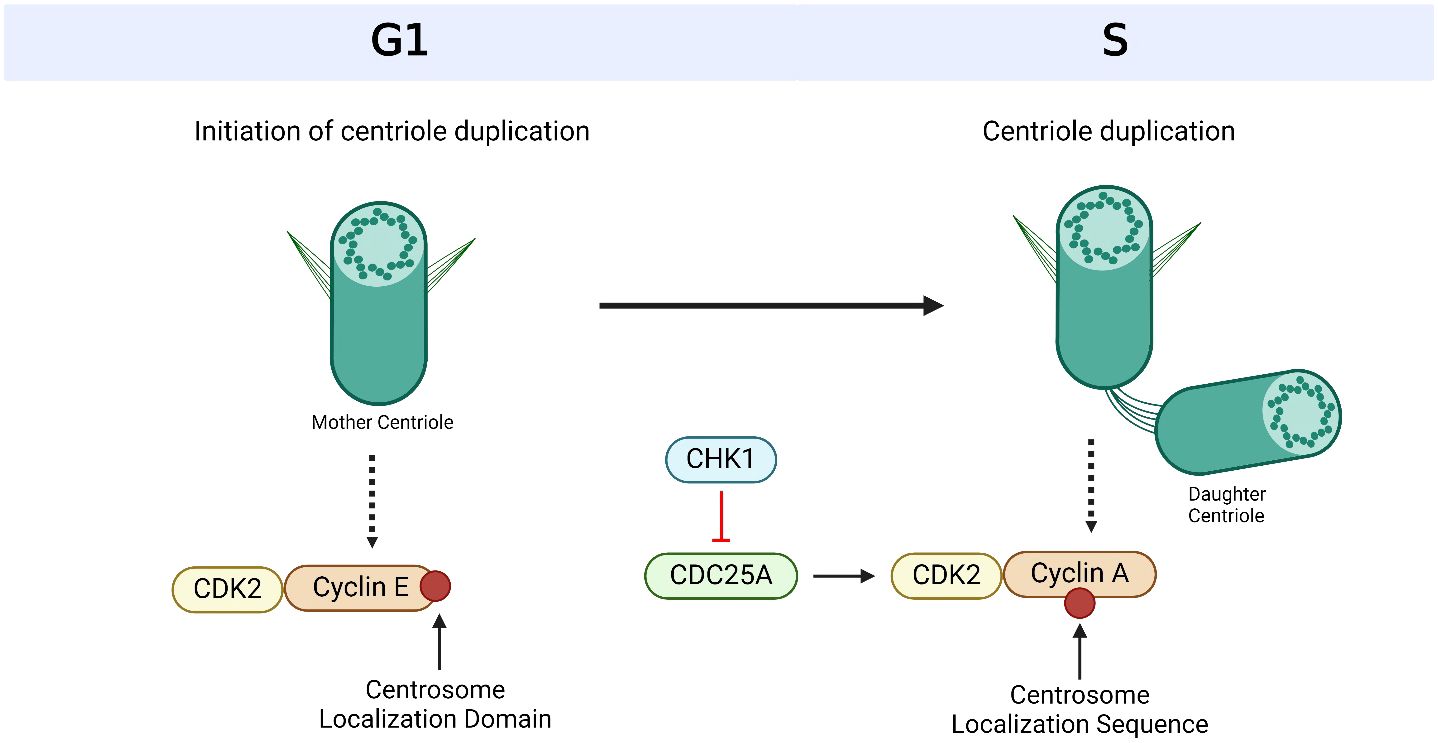
Figure 3 Cyclin-dependent kinases (CDKs) and their cyclin partners facilitate centriole duplication. Cyclin E is recruited to the centrosome via its centrosome localization domain and binds to the centrosome and its Cyclin-dependent kinase (CDK) partner, CDK2, to promote S phase entry and facilitate centriole duplication. Cyclin A has a centrosome localization sequence that allows it to bind to the centrosome as well as CDK2. The complex formation of cyclin A and CDK2 is promoted by cell division cycle 25A (CDC25A), which in turn is regulated by Checkpoint Kinase 1 (CHK1). Based on references 150, 151 and 238. Created with BioRender.com.
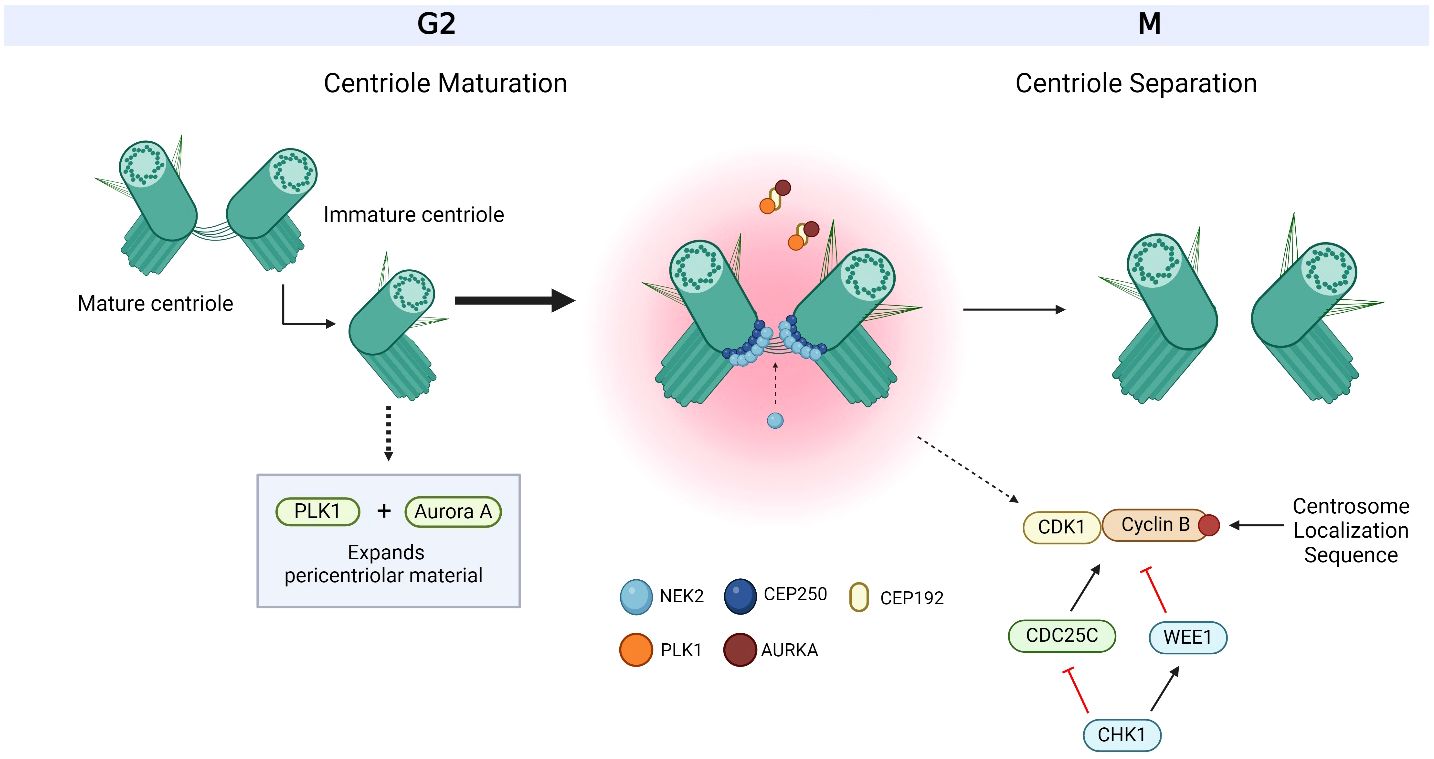
Figure 4 Overview of centriole maturation. Centriole maturation occurs during G2/M and is governed by both Polo-like kinase 1 (PLK1) and Aurora kinase A (AURKA). Both centrosome-associated proteins are bound and recruited to the centrosome by C-terminal encoded protein 192 (CEP192) and are responsible for expanding the pericentriolar material in order to prepare for mitotic spindle formation. CEP250 is one of the many factors that form the linkage between both centrioles and acts as a docking site for other proteins to bind to. CEP250 is bound and phosphorylated by NIMA-related-kinase 2 (NEK2) at the proximal ends of both centrioles. Together, PLK1, AURKA, CEP250 and NEK2 orchestrate centriole maturation and separation. Cyclin B is bound to the centrosome via a centrosome localization sequence at its C-terminus. The activation of cyclin B with its Cyclin-dependent kinase (CDK) partner, CDK1, occurs at the centrosome through the dephosphorylation by cell division cycle 25C (CDC25C), where its activity is regulated by Checkpoint Kinase 1 (CHK1). CHK1 also targets WEE1 for activation, which inactivates CDK1/Cyclin B. The centrosome-associated proteins, CHK1, WEE1, CDK1/Cyclin B and CDC25C also work together to facilitate centriole maturation and separation. Based on references 21, 36, 37, 152 and 234. Created with BioRender.com.
In the M phase, PLK1 also phosphorylates Ninein-like protein (NLP), which interacts with BRCA1 to mediate spindle assembly and centriole separation (22, 57). NLP is also phosphorylated and regulated by AURKB (33), a major component of the chromosome passenger complex (CPC), which is responsible for correcting improper spindle attachments to the kinetochores during metaphase in order to facilitate the proper execution of mitosis (16). AURKB also controls chromosome condensation and orientation by regulating survivin, another CPC subunit (34). Survivin, which localizes to the mitotic spindle, microtubules and centrosome (19) is phosphorylated by PLK1 and activates AURKB to promote mitotic exit and complete cell division (71). During mitotic exit, separase cleaves the sister chromatids and initiates anaphase (23). Anaphase-promoting complex/cyclosome (APC/C) then facilitates the separation of the sister chromatids, thereby promoting chromosome separation (13). Chromosomes are then evenly divided into two daughter cells during cytokinesis (19). As such, centrosomes and their associated players have a crucial role throughout the cell cycle (10) and centrosome aberrations, such as defects in the structure and function of centrosomes as well as the dysregulation of their associated proteins, could lead to known hallmarks of cancer such as mitotic catastrophe and aneuploidy (17, 19, 77) (Figure 1).
Centrosomes and their associated proteins in breast cancer
Centrosome aberrations are a result of the centrosome cycle being deregulated or a direct consequence of cytokinesis failure (78). This has been linked to chromosomal instability, which may support genomic changes and mutation accumulation that can encourage tumor development and resistance to therapy (79). Hence, it’s not surprising that centrosome aberrations have been implicated in various cancers and contribute to their development, growth and metastatic progression (80–83). A common centrosome aberration found in many cancers is centrosome amplification, which leads to alterations in the intricate process of cell division, thereby resulting in numerical chromosomal instability (i.e., aneuploidy or polyploidy) and the accumulation of genetic changes (84). This in turn can enhance tumorigenic properties of cancer cells. Numerical chromosomal instability induced by centrosome amplification can also accelerate the onset and spread of carcinomas via the accumulation of genetic mutations (19, 83, 85). Hence, the genomic instability that drives the development of cancer is exacerbated by these anomalies, which hinders the proper segregation of chromosomes during cell division.
Specific to this review, centrosome amplification has been strongly associated with the development and progression of breast cancer (10, 30) and numerical chromosomal instability is frequently observed in this disease (86, 87). It is a common feature of the most aggressive triple-negative breast cancer subtype, or TNBC, a highly genetically unstable disease (88). Breast tumors are fueled by this instability (89) and subsequently promote the acquisition of new mutations, which contributes to breast tumor heterogeneity (90). In vitro and in vivo studies in breast cancer have confirmed that centrosome amplification promotes tumorigenesis (10, 91, 92). However, the underlying causes of centrosome aberrations in cancer in general and in breast cancer in particular are not fully understood due to the complexities of the regulation of centrosomes and multiple proteins involved in regulating this key component of cell division. Here we highlight key proteins linked to the regulation of centrosomes and their utility as targets for therapeutic development.
Polo-like kinases
A family of serine/threonine protein kinases known as the PLKs are involved in the control of many different cellular functions, with the regulation of centrosomes being of particular importance (26). There are five members in this family (PLK1-5) and each member has a unique function and subcellular location (26). PLK2 and PLK3 aid in regulating mitosis through centriole duplication and DNA replication respectively (26), with studies reporting tumor-promoting properties in colorectal cancer (27, 28). Meanwhile, PLK1 and PLK4 appear to be essential for centrosome regulation (93), with PLK1 controlling centrosome maturation (94) and PLK4 controlling centriole duplication (63). Hence, aberrations of PLK1 and PLK4 can cause centrosome-related errors, which in turn impact genomic stability and cell division accuracy and thereby play a vital role in cancer, including breast cancer (24, 25) (Table 1).
PLK1
PLK1 is a key regulatory protein involved in cell division and preservation of genomic integrity (90). Specifically, PLK1 controls the process of centrosome maturation by phosphorylating pericentriolar material proteins to drive their expansion (Figure 4) (95). It was noted that in healthy cells, PLK1 expression is strictly regulated, ensuring that it is activated precisely when needed throughout G2 and mitosis (96). PLK1 has been found to be overexpressed in breast cancer (97), with up to 92% of TNBC patients showing overexpressed levels of this protein (98). Overexpression of PLK1 in breast cancer is thought to result in aberrant cell division and genomic instability (99) (Table 1). Higher PLK1 levels have also been linked to chemotherapy resistance (100) and aggressive tumor cell morphologies (101), making this protein an attractive biomarker candidate (102, 103) and a drug target. A number of PLK1 inhibitors, such as Rigosertib (ON-01910), Volasertib (BI 6726) and BI 2536 (104–106), have been developed (Table 2). These highly selective PLK1 inhibitors have been investigated in preclinical studies (104–106, 189) and have been reported to promote apoptosis, G2/M arrest and to induce anticancer effects in head and neck square cell carcinoma (Rigosertib) (105), non-small cell lung carcinoma and melanoma (Volasertib) (106), colorectal cancer (Volasertib and BI 2536) and pancreatic and cervical cancers (BI 2536) (104). BI 2536 has also been shown to reduce the invasion and metastasis of breast cancer (107). In response to BI 2536, tamoxifen-resistant MCF-7 breast cancer cells display antiproliferative effects in vitro and antitumor effects in vivo (107). Currently, there is a lack of knowledge of the efficacy of Rigosertib and Volasertib monotherapy in breast cancer, hence clinical trials are focused on other carcinomas (115, 116, 118) (squamous cell, pancreatic, acute myeloid leukemia) and disorders (117) (myelodysplastic syndrome). However, BI 2536 has entered phase II clinical trials for patients with recurrent or metastatic solid tumors, such as breast cancer (108).
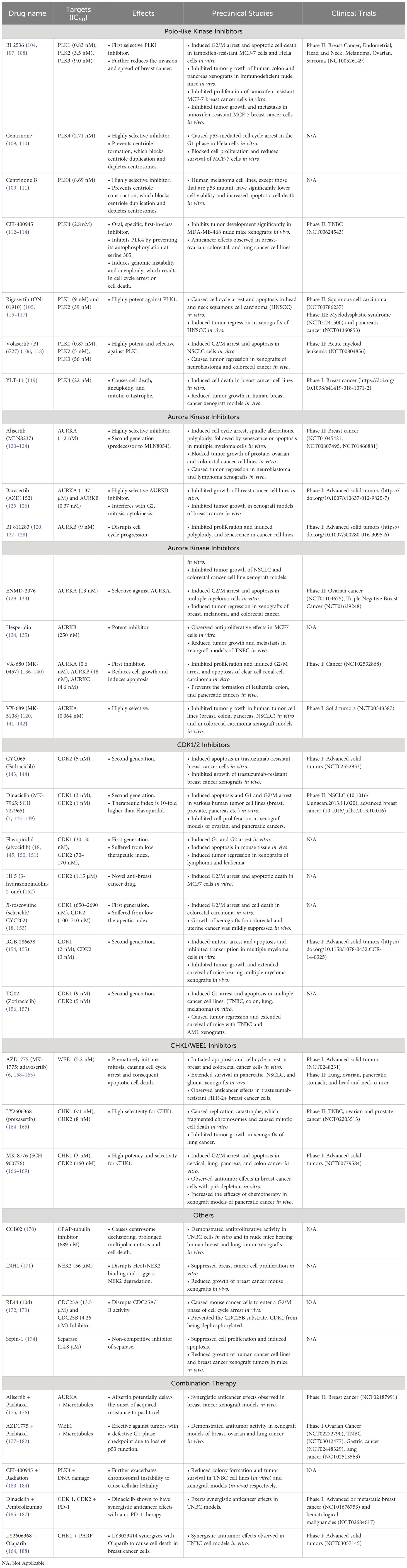
Table 2 Overview of drugs targeting centrosomes and associated proteins.
PLK4
PLK4 plays an important role in centrosome amplification in breast cancer, overseeing the essential process of centriole duplication (10, 30). By phosphorylating and activating STIL, a central part of the centriole, PLK4 allows its interaction with other centrosome-related proteins, such as SAS6 and CPAP (63) (Figure 2). The binding and recruitment of these other centrosome-associated proteins facilitate the process of centriole duplication (63). PLK4 is often aberrantly expressed in breast cancer, which can lead to abnormal centrosome duplication and aneuploidy, thereby inducing genomic instability and consequently contributing to breast cancer tumorigenesis and poor clinical prognosis (30). High levels of PLK4 are especially significant in TNBC, as overexpression of this gene has been observed in 48% of TNBC tumors, demonstrating the potential correlation between PLK4 and the development of TNBC (190). A more comprehensive investigation further demonstrates that PLK4 overexpression occurs in 26% of all breast cancer tumors, and this overexpression has been associated with a reduced survival rate among breast cancer patients (191). In addition to being correlated with centrosome amplification, the increased expression of PLK4 suggests that it may play a role in the development of more aggressive tumors that exhibit high-grade malignancy and a dedifferentiated cellular state (30). Understanding the relationship between centrosome amplification, PLK4 overexpression, and the poor prognosis linked to aggressive tumors, PLK4 becomes a compelling target for therapy in high-grade breast malignancies (192) (Table 1).
Various targeted therapeutic compounds against PLK4 have been developed, such as Centrinone, Centrinone B, CFI-400945 and YLT-11 (109, 112, 119) (Table 2). Centrinone and Centrinone B are highly selective PLK4 inhibitors (109). They both act by preventing centriole formation, which results in the depletion of centrosomes and subsequent cell cycle arrest (109). Centrinone has been shown to cause p53-mediated G1 arrest in cervical cancer (HeLa) cells (109), while Centrinone B induces apoptotic death in melanoma cells (111). In MCF-7 human breast cancer cells, Centrinone caused centrosome loss, which subsequently inhibited cellular proliferation and reduced survival in vitro (110). However, in some cancer cells (HeLa, NIH/3T3), Centrinone treatment blocked cellular proliferation independent of centrosome loss and its effect was suggested to be insufficient for use as a single agent for cancer therapy (189). YLT-11 is another novel PLK4 inhibitor which has shown substantial antiproliferative and antitumor effects in breast cancer cells in vitro and in vivo respectively (119). Moreover, this agent has demonstrated a good safety profile with no significant toxic effects (119).
CFI-400945 emerges as the most promising candidate drug for PLK4 inhibition (Table 2). It is the first orally available inhibitor that targets PLK4 (112) and can also inhibit AURKB activity (113). This drug inhibits PLK4 by preventing its autophosphorylation at serine 305 (113). Upon inhibition of PLK4, it induces genomic instability and aneuploidy, eventually leading to cell death or cell cycle arrest (113). Inhibition of PLK4 has also been shown to significantly reduce tumor growth in vivo (113). CFI-400945 has shown antitumor activity in preclinical studies, including TNBC, and has demonstrated to be a safe and well-tolerated drug in a phase 1 clinical trial (112, 113). It is currently being investigated in phase 2 trials (NCT03624543) in patients with TNBC (114). Although CFI-400945 is demonstrating encouraging clinical responses from monotherapy treatment, combination therapies with other DNA-damaging agents or radiotherapy which promote mitotic catastrophe may result in greater anticancer effects than each treatment alone, thereby proving to be an effective treatment strategy for breast cancer patients. Multimodality combination treatment has been investigated in TNBC models, where CFI-400945 with radiation (183, 184) has been shown to synergize anticancer effects (Table 2). Namely, compared to single agent treatment, the combination treatment significantly reduced colony formation in TNBC cell lines and patient-derived organoids (PDOs) in vitro and decreased tumor growth and improved humane endpoint survival in mouse xenografts in vivo (183). Both CFI-400945 (113) and radiotherapy induce genomic instability and genotoxic stress, thus acting in combination potentially further exacerbates chromosomal instability to eventually cause cell death (192, 193).
Aurora kinases
AURKs are a family of serine/threonine protein kinases and are vital for controlling several parts of the cell cycle, most notably mitosis (194, 195). Three members make up the AURK family (AURKA, AURKB, AURKC) (194). However, the role of AURKC in mitosis is understudied and its expression is limited to testicular tissue (35). Hence, knowledge of AURKC’s role in tumorigenesis is limited (35). Conversely, AURKA and AURKB have piqued the most interest among researchers due to their critical roles in centrosome regulation (194, 195). AURKA plays a key role in centrosome maturation and separation (16, 33, 196), specifically by phosphorylating several pericentriolar material proteins to organize the mitotic spindle (195) (Figure 4). While AURKB is essential for controlling chromosomal alignment and segregation during mitosis (78, 79, 197). This ensures the correct arrangement and operation of centrosomes during cell division. Overexpression of AURKA and AURKB disrupts G2 phase checkpoints (198), which results in improper chromosomal segregation during mitosis, chromosomal instability, and promotes uncontrolled cell division. These factors may contribute to tumor formation, as well as the development of more aggressive and genetically heterogeneous cancer cells (6, 199). AURKA/B have been observed to be overexpressed in various cancers, including breast (32, 200, 201). High expression of these kinases in breast cancer strongly correlates with poor survival outcomes (202). It was reported that inhibition of AURKA significantly decreased the survival of luminal and HER-2 cancer models (202). In light of this, the aberrant expression of AURKA/B in cancer emphasizes their potential value as therapeutic and drug-development targets (32, 203, 204) (Table 1).
Various inhibitors that target AURKA/B have been developed, such as VX-680 (MK-0457), VX-689 (MK-5108), Alisertib (MLN8237), ENMD-2076, Barasertib (AZD1152), Hesperidin, and BI 811283 (120, 134, 136) (Table 2). VX-680 is a first-in-class AURKA inhibitor (136). It has been shown to inhibit cellular proliferation and promote apoptotic death in various human cancers, such as ovarian and cervical cancers (136–138). It was shown to induce G2/M arrest and apoptosis in clear cell renal cell carcinoma in vitro (139) as well as tumor regression in colon and pancreatic tumors in vivo (140). Although initial results were encouraging, clinical trials using VX-680 were discontinued due to the concern regarding its toxicity (205). However, VX-689, a potent inhibitor of AURKA was well tolerated in preclinical models and human studies with no reports of toxic adverse effects (120). VX-689 monotherapy was shown to suppress cell proliferation in a wide range of tumors, such as breast, colon, pancreas, and NSCLC using in vitro models (141) and elicited anti-tumor effects in colorectal xenograft models in vivo (120). A phase I clinical trial of VX-689 monotherapy in advanced solid tumor patients showed that the drug is generally well-tolerated (142). Alisertib is another highly selective and second-generation AURKA inhibitor (120, 121). Upon inhibition of AURKA, it induces G2/M arrest and aneuploidy, which subsequently leads to senescence or apoptosis in multiple myeloma cells in vitro (122). Alisertib monotherapy has also been shown to inhibit tumor cell proliferation in prostate, ovarian and colorectal cancer cells in vitro (121). In vivo studies have shown reduced tumor growth in xenografts of neuroblastoma (123) and lymphoma (121). There are currently more than 30 clinical trials using Alisertib in a wide variety of cancers (breast, lung, ovarian, prostate) (6). In particular, there are ongoing phase II studies in breast cancer that show patient response rates of 18% when given alisertib monotherapy (124). Inhibition of AURKA by ENMD-2076 also promotes cell cycle arrest in the G2/M phase and cell death via apoptosis in multiple myeloma cells in vitro (129). ENMD-2076 treatment has also been shown to diminish the growth of breast, melanoma and colorectal tumor xenografts in vivo (130). Moreover, phase II trials of ENMD-2076 monotherapy in ovarian cancer have demonstrated a median overall survival of approximately 12 months, compared to 11 months without treatment (131, 132). Single-agent treatment with ENMD-2076 also resulted in clinical benefit or partial response in 16.7% of patients with TNBC in phase II trials (133), thereby highlighting its utility as a novel therapeutic for cancer patients.
AURKB inhibitors have shown mixed results in terms of their efficacy and clinical applicability (18) (Table 2). The selective AURKB inhibitor Barasertib showed promising results in preclinical studies in breast cancer (125) and was well tolerated in phase I trials in patients with advanced solid tumors (126). However, phase II trials were suspended after failing to show any significant clinical benefit in most patients with advanced solid tumors, including breast tumors (6). Other highly selective AURKB inhibitors include Hesperidin (134) and BI 811283 (120). Hesperidin has demonstrated antiproliferative activity in MCF7 breast cancer cells in vitro (134) and anticancer effects, such as reduced tumor growth and metastasis in mouse xenograft models of TNBC in vivo (135). Thus, further investigation regarding its efficacy and safety profile is warranted. BI 811283 has also shown efficacy in several solid tumors by inhibiting cell proliferation and inducing polyploidy and senescence in vitro (127) and reducing tumor growth in mouse xenograft models in vivo (127). Additionally, it has been demonstrated to be well-tolerated in a phase I clinical trial in patients with advanced solid tumors, including breast cancer (128).
Other novel therapeutic approaches have investigated the combination of Alisertib (175) or AZD1775 (177) with paclitaxel, a widely used anticancer agent that targets microtubules in a vast array of tumors (206) (Table 2). When treating breast cancer xenograft models with Alisertib and paclitaxel, synergistic or additive effects were observed (176). Moreover, in a phase II clinical study, the combination of Alisertib and paclitaxel significantly improved median progression-free survival (10.2 months) in metastatic breast cancer patients compared to either treatment alone (7.1 months) (175). This suggests that the addition of Alisertib could potentially delay the onset of acquired resistance to paclitaxel (175). Moreover, combining AZD1775 with paclitaxel in a mouse xenograft model of breast cancer resulted in the inhibition of tumor growth and extended animal survival (178). Currently, multiple clinical trials are studying this combination in various cancers harboring p53 mutations, including TNBC (179–182); the type of mutations in which AZD-1775 monotherapy has been proven to be most effective (177).
Cyclin-dependent kinases
Another group of regulatory proteins known as CDKs is involved in centrosome control and cell cycle progression (36). There are 20 known members of the CDK family in humans, and they all play different roles in regulating different facets of cell division and proliferation (37, 38). In particular, CDK1 and CDK2 are important cell cycle regulators that play a critical role in coordinating various phases of cell division, including DNA replication and segregation (39). The activity of CDK1/2 is closely regulated by the binding of cyclins A/B (37), ensuring proper progression through the cell cycle checkpoints (201). In particular, CDK1/2 are essential for centrosome duplication and segregation through their regulatory roles in the G1/S and G2/M phases of the cell cycle (37, 207–209) (Figures 2-4, Table 1). Specifically in G2, CDK1 promotes centrosome maturation and separation once activated by cyclin B (210) and CDK2 phosphorylates proteins required for centriole duplication and separation (211) (Figures 3, 4).
The dysregulation of CDKs can result in cell cycle aberrations and centrosome abnormalities, specifically affecting centrosome duplication and segregation, and ultimately promoting tumorigenesis (40) (Table 1). Studies in breast cancer have revealed elevated CDK1/2 levels in approximately 60% of tumor samples compared to normal breast tissue (32). Elevated levels of these kinases or their activating cyclins lead to disrupted cell cycle checkpoints and abnormal cell division (212). This genomic instability allows cell cycle progression even in the presence of DNA damage, enabling breast cancer cells to evade arrest and divide uncontrollably (213–215). Moreover, the overexpression of CDK1/2 interferes with DNA repair pathways, causing genomic instability and tumor growth progression (215). In HER2-positive breast tumors, overexpression of CDKs appears to promote unregulated cell cycle progression and uncontrolled tumor development (216). The control of the cell cycle in luminal breast tumors (217–219) may also be influenced by CDK overexpression, which can accelerate cell division. Consequently, targeting abnormal CDK1/2 expression and activity has emerged as a promising therapeutic strategy to suppress cancer cell proliferation and restore proper cell cycle regulation (213).
Various CDK1/2 inhibitors have been developed and investigated in preclinical and clinical studies, such as Flavopiridol (Alvocidib), R-roscovitine (Seliciclib or CYC202), Dinaciclib (MK-7965 or SCH 727965), TG02 (Zotiraciclib), CYC065 (Fadraciclib), RGB-286638 and HI-5 (18, 143, 145, 152, 154, 156) (Table 2). Flavopiridol and R-roscovitine are classified as first-generation pan-CDK inhibitors, which demonstrate limited specificity to various CDKs (18). Flavopiridol especially is the most extensively investigated CDK inhibitor, with over 60 clinical trials conducted between 1998 and 2014 (145). This compound effectively inhibits several CDKs by inducing G1 and G2 arrest; thereby blocking cell cycle progression and subsequently inhibiting cancer cell growth (150). These anticancer effects have been observed in breast and lung cancer cells in vitro (150). Flavopiridol also reduced tumor growth in xenograft models of leukemia and lymphoma in vivo (151). However, despite demonstrating strong antitumor activity in preclinical studies, phase II clinical trials with Flavopiridol alone failed to show efficacy against solid carcinomas (18). R-roscovitine also demonstrated antitumor activity by inducing cell death and G2/M arrest in colorectal carcinoma cells in vitro but only mildly suppressed tumor growth of colorectal and uterine carcinoma xenografts in vivo (153), thereby exhibiting a lack of clinical application as a therapeutic agent. Moreover, first-generation pan-CDK inhibitors have led to relatively high rates of adverse effects (220), causing toxicities at concentrations required to effectively inhibit their targets (18).
To overcome the aforementioned drawbacks observed with first-generation CDK inhibitors, second-generation pan-CDK inhibitors have been developed (Table 2) (145). Dinaciclib was one of the most extensively studied second-generation pan-CDK inhibitor (145). Compared to Flavopiridol, it has a ten-fold higher therapeutic index (146) and selectively targets CDK2 with an IC50 of 1 nM (145). In preclinical studies, this compound induced cell cycle arrest in G1 and G2/M as well as initiated apoptotic cell death in multiple human cancer cell lines in vitro, including breast cancer (146). Additionally, dinaciclib treatment suspended tumor cell proliferation in ovarian (146) and pancreatic (147) xenograft models in vivo. However, early clinical trials with Dinaciclib monotherapy showed limited efficacy towards a range of solid tumors, including breast cancer (7, 148, 149). However, combining Dinaciclib with anti-PD-1 therapy showed synergistic antitumor effects in colorectal cancer compared to single-agent treatments (185). Hence, the combination of dinaciclib with PD-1 inhibitors, such as pembrolizumab has been investigated (186, 187) (Table 2). Dinaciclib and pembrolizumab treatment caused synergistic anticancer effects in TNBC models via attenuation of metastasis and synthetic lethality (186). Encouraging antitumor activity in hematological malignancies was also observed upon combination treatment (187). Currently, phase I trials with this combination modality are being tested in TNBC patients as well as those with leukemia, multiple myeloma and lymphoma (186, 187).
Other second-generation pan-CDK inhibitors, such as TG02, promote antiproliferative effects in a broad range of cancer cell lines (TNBC, melanoma, lung, colon) by initiating G1 arrest and apoptosis in vitro (Table 2) (156, 157). TG02 also reduced tumor growth and survival in mouse xenograft models of acute myeloid leukemia (AML) (156) and TNBC (157) in vivo. These results support the development of trials focused on assessing the clinical efficacy of TG02. Furthermore, CYC065 has been reported to cause apoptotic death of trastuzumab-resistant breast cancer cells in vitro as well as induce tumor regression in trastuzumab-resistant breast cancer xenograft models in vivo (143). A phase I clinical trial in patients with advanced solid tumors also demonstrated manageable levels of toxicity upon CYC065 treatment, thereby supporting the potential of this agent as a clinical therapeutic (144). Lastly, RGB-286638 has been shown to block transcription and initiate cell cycle arrest and cell death via apoptosis in multiple myeloma cells in vitro (154). Additionally, it has inhibited tumor growth and survival of multiple melanoma xenograft models in vivo (154). The information regarding this compound’s efficacy in breast cancer is still lacking, although a phase I study of RGB-286638 monotherapy demonstrated to be safe and well tolerated in patients with advanced solid tumors (155). Recently, a novel and potent CDK2 inhibitor, 3-hydrazonoindolin-2-one scaffold, called HI 5 has emerged as a potential anti-breast cancer agent (152). It elicited encouraging preclinical results by demonstrating antiproliferative effects in MCF-7 breast cancer cells through the induction of growth arrest at the G2/M phase and apoptotic death in vitro (152). Further studies of H1 5 are warranted to advance its development to clinical trials.
Checkpoint Kinase 1 (CHK1)/WEE1
CHK1 and WEE1 are checkpoint kinases that control the cell cycle by slowing it down to ensure there is enough time for precise DNA replication and DNA damage repair (221). This helps preserve genomic stability and prevents the spread of potentially detrimental genetic changes (41, 221, 222). CHK1 mainly controls the G2/M checkpoint to monitor DNA damage and cell cycle progression (41), whereas WEE1 serves as a negative regulator (44) of the cell cycle by blocking CDKs (Table 1). In terms of their role in centrosome regulation, CHK1 localizes to interphase centrosomes and controls centrosome separation through CDK1 activity, whereas WEE1 inactivates the CDK1/Cyclin B complex through inhibitory phosphorylation to also participate in the control of centrosome separation (223) (Figure 4).
Overexpression of CHK1 and WEE1 has been reported in the setting of cancer (42, 43) and has been shown to aid in the growth and development of tumors (41). The overexpression of CHK1 and WEE1 in cancer cells provides them with a survival advantage over cells with normal expression levels. These proteins facilitate cancer cells to overcome genotoxic stress, promote extended cell cycle arrest, and actively assist DNA repair pathways. Thus, cancer cells are able to avoid dying and preserve the integrity of their DNA (41, 42). As a result, the normal checkpoints that prevent DNA replication errors, genomic instability, and the buildup of DNA damage are overridden by cancer cells (224). Various studies have reported overexpression of CHK1 and WEE1 in breast cancer patients (45, 225) as well as in colon and liver cancer (43) (Table 1). This overexpression negatively affects the sensitivity of these cells to chemicals that damage DNA in luminal breast tumors (226). Increased expression in CHK1 and WEE1 also appears to facilitate long-term survival of HER2-positive breast tumor cells (45) through the repair of DNA damage, dysregulation of the cell cycle and the evasion of apoptosis. On the other hand, inhibition of CHK1/WEE1 halts the progression of the cell cycle, causing DNA damage and premature entry of the cell into the M phase, which may trigger selective destruction of cancer cells (227).
CHK1/WEE1 are emerging as promising candidates for anticancer drug development (158, 164, 166). MK-8776, a selective and potent inhibitor of CHK1, has been shown to cause G2/M arrest and apoptotic death of cervical, lung, pancreas and colon cancer cells in vitro (166). There has been a lack of research regarding its role as a monotherapy in breast cancer and in vivo studies. However, it has shown promising results as a radiosensitizer in TNBC models in vitro by inhibiting autophagy (167), as well as a chemosensitizer in pancreatic cancer xenograft models in vivo (168). Phase I studies in patients with advanced solid tumor carcinomas (but not breast cancer patients) demonstrated that MK-8776 monotherapy was well tolerated (169) (Table 2).
LY2606368 (Prexasertib) has also demonstrated high selectivity for CHK1 (164). Treatment of cancer cells with LY2606368 triggers the activation of cell division cycle (CDC) 25A, resulting in increased levels of CDK2 (18). This drives S phase progression, which causes the accumulation of replication forks and subsequently the formation of DNA double-stranded breaks, commonly referred to as replication catastrophe (164). When studying the antitumor effects of LY2606368 in preclinical models, the compound caused the fragmentation of chromosomes and induced mitotic cell death in HeLa cells in vitro (164) as well as exhibited enhanced tumor regression in a lung cancer xenograft model in vivo (164). In TNBC cell lines in vitro, LY2606368 monotherapy caused the degradation of homologous recombination proteins (BRCA1 and RAD51) as well as induced cell cycle arrest in the S phase (228). These alterations in the homologous recombination machinery suggest the possibility of LY2606368’s utility as a combination modality with Olaparib, a well-known PARP inhibitor that targets homologous recombination deficiency in breast cancer (164). The combination of LY26006348 with Olaparib induced DNA damage and cell cycle arrest in TNBC cells in vitro, leading to mitotic catastrophe, genomic instability and cell death (164). A phase II clinical trial of LY26006348 monotherapy in patients with TNBC has shown limited activity (165), hence clinical trials are focused on its combination with other targeted agents, such as Olaparib (188). The combination modality of LY26006348 and Olaparib has been investigated in a phase I study in patients with metastatic solid tumors, including advanced-stage breast cancer (188) (Table 2).
AZD1775 (MK-1775; Adavosertib), a potent, selective and first-in-class WEE1 inhibitor, causes increased CDK1 and CDK2 levels, leading to the premature entry of cells into mitosis (158). This results in the induction of mitotic arrest and cell death via apoptosis (158). These effects were observed in colorectal cancer cells in vitro (158). AZD1775 monotherapy has also been reported to extend the survival of pancreatic (159), NSCLC (160) and glioma (161) mouse xenografts in vivo. In HER-2 positive breast cancer, AZD1775 has shown remarkable anticancer effects by overcoming anti-HER2 agent trastuzumab resistance through the initiation of apoptosis and G2/M arrest in breast cancer cells in vitro (162). These promising preclinical studies have led to numerous clinical trials of single-agent AZD1775 and its combination with chemotherapies in a variety of carcinomas (6). One, in particular, was a phase I trial of patients with advanced solid tumors such as ovarian cancer and TNBC where AZD1775 monotherapy was found to be well tolerated (163) (Table 2).
C-terminal encoded proteins
The CEP family of proteins is the active component within the centrosome and consists of 31 members (48). They play a key role in cell cycle control and centriole duplication (48). In particular, CEP120, CEP131, CEP135, CEP152, CEP192 and CEP250 hold potential as therapeutic targets (48) (Table 1). CEP120 is known to directly interact with CPAP to elongate centrioles (Figure 2), and its depletion has been shown to suppress CPAP-mediated centriole elongation (46). Its overexpression is known to produce overly long centrioles, which disrupts the process of centriole duplication and promotes atypical supernumerary centrioles (46). Elevated levels of CEP120 have been observed in gastric cancers with minimal information regarding its expression in breast cancer (47). CEP131 has a vital role in the maintenance of genomic instability during cell cycle progression by regulating centriole duplication (48, 49). It does so through the phosphorylation by PLK4, which facilitates the recruitment of STIL to the centriole (50) (Figure 2). Upon elevated levels of CEP131, STIL is excessively recruited, which promotes supernumerary centrosomes (50). This has been observed in breast cancer, where the deubiquitinating enzyme USP9X, an integral component of the centrosome and required for centriole duplication, causes excessive levels of CEP131, thereby contributing to the pathogenesis of breast cancer (49). These findings support the promise of CEP131 as a novel target for anti-cancer interventions, particularly for breast cancer. CEP135 also plays a key role in regulating centriole duplication by interacting with SAS6 to link it to CPAP (51) (Figure 2). Similar to CEP120, depletion of CEP135 is known to suppress CPAP-induced centriole elongation (51), and its overexpression induces centrosome amplification (52). This has been shown to cause chromosome segregation errors in breast cancer cells, thereby promoting its carcinogenic properties (52). CEP152 and CEP192 are both involved in the recruitment of PLK4 to the centriole in order to ensure proper centriole duplication (53) (Figure 2). However, little is known about its tumor-promoting properties in breast cancer. A study by Liu et al. did identify CEP192 as a novel prognostic marker in hepatocellular carcinoma based on increased expression with tumor stage and association with a high mortality and recurrence rate (54). Inhibition of CEP192 blocked the proliferation of hepatocellular carcinoma cell lines, suggesting its utility as a novel anti-cancer target (54). Given these findings, more studies are warranted to determine the role of CEP152 and CEP192 in breast cancer tumorigenesis as well as its potential as a therapeutic target. Lastly, CEP250 is phosphorylated by NEK2 in order to induce centrosome separation and bipolar spindle formation (55). This is important for proper progression of the cell cycle and maintenance of genomic instability. Limited knowledge is available about the aberrant expression of CEP250 in breast cancer and cancer in general. Instead, most research has focused on studying elevated levels and inhibition of NEK2 in breast cancer tumorigenesis (229–231) (see below). Overall, there is limited knowledge about the carcinogenic mechanism of CEPs in the progression of breast cancer and more research in this field is warranted.
Other proteins
The SAS6 and STIL proteins are vital for controlling cell division and forming the mitotic spindle, a structure that is necessary for appropriate chromosomal segregation during cell division (68). Along with PLK4, SAS6 and STIL are the core components of centriole duplication, and together they initiate the creation of a new centriole (68) (Figure 2). However, the development of cancer has been linked to the deregulation of these proteins (69) (Table 1). Changes in STIL and SAS6 levels have been linked to several subtypes of breast cancer, such as TNBC (232, 233), luminal breast cancer (234, 235), and HER2-positive breast cancer (236, 237). Tumor growth can be aided by the deregulation of these proteins, which can result in abnormal cell division and genetic instability (238, 239). By providing new paths for precision medicine in the treatment of luminal, HER2+, and TNBC subtypes, an understanding of the molecular processes involving STIL and SAS6 in breast cancer subtypes holds promise for the development of novel targeted therapeutics.
CPAP/CENPJ are essential proteins for regular cellular functions, especially those involving the control of centrosome activity and cell division (240). In order to maintain centrosome stability and appropriate chromosomal segregation, which guarantees accurate genetic material transfers to daughter cells, CPAP/CENPJ interacts with various centrosome-associated proteins (241) (Figure 2). These interactions allow CPAP to positively regulate centriole length and to aid in the duplication of centrosomes (63). Hence, the development of cancer may be facilitated by abnormal cell division and genomic instability brought on by disruptions in these proteins’ regular activity. A variety of cancers, including breast cancer, have been linked to overexpression in CPAP/CENPJ (65) (Table 1). The unchecked cell proliferation observed in luminal and HER2-positive breast cancer subtypes is thought to be associated with deregulation of centrosome activity mediated by CPAP/CENPJ (242). Moreover, abnormalities in these proteins may also affect the genomic stability of cells, which may contribute to the emergence and advancement of TNBC (243). Understanding the specific roles of CPAP/CENPJ in breast cancer subtypes provides valuable insights for the development of targeted therapeutic approaches in the future. Currently, drug treatments addressing CPAP/CENPJ are understudied regarding their use in the treatment of breast cancer. Bridging this research gap could result in new opportunities for targeted therapies and progress the field toward more tailored and successful treatments for breast cancer patients.
A family of dual-specific phosphatases known as CDC25 are essential to the control of the cell cycle (244, 245). CDC25 exists in three primary isoforms: CDC25A, CDC25B, and CDC25C (244, 245). The G1/S transition is mostly regulated by CDC25A, the G2/M transition is regulated by CDC25B and the G2/M checkpoint is dependent on CDC25C (58–60). A vital component in the proper progression of the cell cycle, including the control of centrosomes and chromosomes, is the cell cycle regulator CDC25A (61) (Table 1). CDC25A participates in centrosome regulation by dephosphorylating CDKs (61) (Figure 3). This promotes cell cycle advancement, as well as regulates centrosome duplication and maturation (61). Furthermore, CDC25A regulates apoptosis, highlighting the complexity of its role in cellular functioning (61). CDC25A dysregulation, however, has been linked to a number of malignancies, including breast cancer (58–60) (Table 1). CDC25A has been shown to be overexpressed in HER2-positive and TNBC subtypes (246–248). The overexpression of HER2 in HER2-positive breast tumors is correlated with overexpression of CDC25A, which may lead to unchecked cell proliferation (246). Moreover, abnormal cell cycle progression has been linked to CDC25A dysregulation in TNBC tumors (247, 248). Thus, CDC25A might serve as a potential therapeutic target in breast cancer (247). One of the compounds that has an inhibitory effect towards CDC25A/B is an o-hydroxybenzyl derivative RE44 (10d), which has shown promise in models of mouse tsFT210 breast cancer cell line in vivo (172, 173) (Table 2).
Another key protein involved in the control of the cell cycle is CDC25C (60, 62). It typically functions as a phosphatase, helping to promote the G2 phase transition to mitosis by removing inhibitory phosphate groups from CDKs (244, 245) (Figure 4). It participates in centrosome regulation through the co-localization with cyclin B at the centrosomes in G2 (249) and is responsible for dephosphorylating CDK1 to activate the CDK1/Cyclin B complex to regulate centrosome maturation and separation (249). Dysregulation of CDC25C has been linked to the uncontrolled proliferation of cells (62). The fast and uncontrollable cell division observed in TNBC can be attributed to CDC25C overexpression (245, 248). Acquiring insight into CDC25C’s function in these subtypes of breast cancer could assist with developing more focused treatment strategies (Table 1).
A major force in controlling the progression of the cell cycle is the kinase NEK2 (250). Its typical role in mitosis consists of the maintenance of correct chromosomal segregation by aiding in the precise arrangement of centrosomes and microtubules (67). It also assists in the regulation of centrosome separation by localizing to the centrosome and controlling bipolar spindle formation as well as facilitating correct spindle attachments (67, 230). However, a number of malignancies, particularly breast cancer, have been linked to the deregulation of NEK2 (67) (Table 1). NEK2 overexpression has been linked to TNBC (230), luminal breast cancer (230), and HER2-positive (251) breast cancer subtypes. Increased genomic instability, tumor development, and abnormal cell cycle progression are all influenced by elevated activity of NEK2 (252). Following the inhibition of NEK2, a subsequent step involves the inhibition of CDK4/6 (253). Notably, these two therapies used together significantly reduced the extent of breast tumor in mice without causing harm to the animals in vivo (253). The combination of these treatments changed the mitotic spindle genes’ mechanistic features in vivo, suggesting a higher degree of genomic instability (253). All these findings point to NEK2 as a potential therapeutic target for the treatment of aggressive breast malignancies like TNBC, when combined with FDA-approved CDK4/6 inhibitors. As the function of NEK2 varies in luminal, HER2-positive, and TNBC settings, knowing the precise impact of NEK2 dysregulation in distinct breast cancer subtypes can provide important insights for targeted therapy approaches. Moreover, small molecule inhibitors against NEK2, such as INH1, blocked the proliferation of several breast cancer cell lines in vitro as well as reduced the growth of tumor mouse xenografts of breast cancer in vivo (171) (Table 2). Therefore, these pathways also serve as potential therapeutic strategies for breast cancer.
A crucial component of cellular machinery, APC/C controls how the cell cycle progresses. In a typical cell cycle, APC/C coordinates the degradation of certain proteins at various phases to enable appropriate cell division (254). In relation to centrosome regulation, APC/C is known to localize to the centrosome during mitosis and interact with various centrosome-associated proteins (such as CEPs) to facilitate mitotic spindle assembly (255). Dysregulation of APC/C, however, has been linked to a number of cancers, including breast cancer (56) (Table 1). The onset and advancement of breast cancer have been linked to aberrant APC/C activity, which affects vital cellular functions like proliferation and genomic integrity (256). For example, dysregulation of APC/C can cause unchecked cell division and genomic instability, which can aid in the development and spread of breast cancer (256). Inhibition of APC/C (13) or interference with the CPAP-tubulin interaction via CCB02 (170) prevents extra centrosomes from aggregating during mitosis, commonly referred to as centrosome clustering (Table 2). This helps reduce the number of cancer cells with amplified centrosomes, thereby reducing tumor growth (13, 50). These effects have been observed in TNBC and colon cancer cells in vitro as well as mice bearing breast and lung cancer xenografts in vivo (13, 50) (Table 2).
Survivin, another centrosome-associated protein, is essential for controlling cell division and preventing apoptosis (257). In order to properly assemble the mitotic spindle and guarantee precise chromosomal segregation during cell division, survivin regulates centrioles and centrosomes (258). It also plays a critical function in the accurate coordination of cellular activities during mitosis by aiding in cytokinesis and stabilizing microtubules (258). Hence, abnormal survivin expression has been linked to a number of malignancies, including breast cancer (72) (Table 1). Survivin is frequently overexpressed in breast cancer (72) and linked to the pathogenesis of its various molecular subtypes, such as luminal (259), HER2-positive (260), and TNBC (261). Thus, there is a potential for using therapeutic targeting of survivin in breast cancer (262). However, preclinical studies of survivin inhibitors in breast cancer are limited.
Another protein that is implicated in cell division is separase, which is involved in sister chromatid separation during mitosis (263). Its regular function is to break the cohesin protein, which keeps chromatids joined together and enables precise gene transfer to daughter cells (264). During metaphase, it is known to cleave the centrosomal protein, pericentrin, allowing for centrioles to separate and be ready for the next round of centriole duplication (265). On the other hand, elevated levels of separase activity have been linked to a number of cancers, including breast cancer (70) (Table 1). Separase dysregulation has been connected to luminal (266), HER2-positive (267), and TNBC (268) breast cancer subtypes. The presence of hormone receptors in luminal breast tumors leads to separase hyperactivity, which can cause unchecked cell proliferation (269, 270). Separase dysregulation can render HER2+ breast tumors to be more aggressive by exacerbating the overexpression of HER2 (271). Thus, investigation of separase inhibition in breast cancer is a promising strategy. Sepin-1 has been shown to be a non-competitive inhibitor of separase, thereby preventing its enzymatic activity (174) (Table 2). This drug exhibits the ability to suppress cell proliferation and induce apoptosis in order to hinder the growth of human cancer cell lines and breast cancer xenograft tumors in mice (174) (Table 2).
Aspects of regulation of centrosomes and their associated proteins by tumor suppressors and oncogenes in the context of breast cancer
The vastness of proteins participating in centriole duplication and maturation and regulation of centrosomes, combined with dynamic changes that take place in these processes during various phases of the cell cycle requires complex regulatory mechanisms that are provided by various cell signaling pathways, involving tumor suppressors and oncogenes, which are outside of the scope of this review. However, several such molecules are worth noting in the context of breast cancer. A guardian of the genome and a well-known tumor suppressor protein, p53, is known to participate in breast cancer pathogenesis and is found mutated in 30% of all breast tumors (272). p53 targets p21, a gene that regulates centrosome stability by inhibiting CDK2/Cyclin E during early S phase (Figure 1) (273). Inactivation of p53 along with the involvement of p21 leads to the induction and increase of centrosome amplification and aneuploidy in breast cancer, which is known to be associated with its poor prognosis (273). There has been emerging evidence indicating a mutual crosstalk between PLKs and p53 in cancer cells (274). For instance, PLK1 is tightly regulated by p53 and the regulation of DNA-damage repair by PLK1 is mediated through p53 among other proteins (275). However, in p53-null breast cancer cells, the regulation of PLK1 becomes dysregulated, leading to abnormal levels and in turn contributes to the development of breast cancer (275). There have also been reports of the direct interaction of AURK proteins and p53 (276). For example, the overexpression of AURKA in breast cancer may lead to enhanced degradation of p53, which in turn facilitates the development and progression of breast tumors (277).
BRCA1/2 are tumor-suppressor proteins participating in DNA damage repair pathways whose hereditary mutations are linked to the increased risk of breast cancer development (278, 279). BRCA1 is known to localize to the interphase and mitotic centrosomes and binds specifically to the mother centrioles (280). It adjusts centrosome function by inhibiting AURKA and CDK1/Cyclin B (Figure 1) (281). It also inhibits PLK1 in order to regulate the centrosomal localization and stability of NLP (Figure 1) (22). It has been reported that BRCA1 inhibition leads to centrosome amplification and aneuploidy, which promotes breast cancer tumor progression (280). It has also been shown that 62% of breast cancers overexpress AURKA, which is responsible for blocking the function of BRCA1 during G2, the phase of the cell cycle when its activity is crucial for regulating the centrosome (282). This inhibition led to the induction of supernumerary centrosomes and a more aggressive breast cancer (282).
PIK3CA is a key oncogenic protein involved in cell growth, survival and migration, and is known to be mutated in 40% of hormone receptor and HER2-positive advanced breast tumors (283). Mutations in PIK3CA lead to sustained activation of the PI3K pathway, which has been shown to induce centrosome amplification through CDK2/Cyclin E pathways (283). It was found that increased levels of PIK3CA led to higher levels of cyclin E, which in turn caused enhanced activation of centrosome duplication (284). Thereby, contributing to the overamplification of centrosomes observed in breast tumors (284).
Discussion
Despite significant advances in diagnosis and treatment, breast cancer remains one of the leading causes of cancer-related deaths among women. It is essential to develop novel treatment approaches for the improvement of oncological outcomes in breast cancer patients. Due to abnormalities in cell division (a well-known characteristic of breast cancer cells and cancer cells in general), a focus of interest in the study of breast cancer treatment is the utilization of cell cycle proteins as therapeutic targets. In particular, targeting the centrosome and centrosome-associated proteins has piqued interest as a viable therapeutic approach due to their important role in cell cycle control.
Centrosomes are key players in the normal process of cellular division and are regulated by a large variety of centrosome-associated proteins (17, 19) (Figure 1, Table 1). The most notable proteins studied are PLKs, AURKs, CDKs, CHK1 and WEE1. These proteins have been shown to be linked to breast cancer by contributing to its development and progression, particularly through centrosome amplification, which is a common centrosome abnormality (10, 80, 84). Hence, targeting proteins involved in the regulation and function of centrosomes are attractive therapeutic targets for this aggressive disease. Currently, there are targeted therapeutics against centrosome-associated proteins in development with some in clinical trials (Table 2). These include the PLK4 inhibitor CFI-400945; PLK1 inhibitor BI 2536; AURKA inhibitors Alisertib and EMND-2076; AURKB inhibitor BI 811283; CHK1 inhibitor LY2606368; CDK2 inhibitor Dinaciclib and others. These agents as well as the majority of those described in this review have been shown to cause severe mitotic spindle abnormalities, such as the promotion of chromosome missegregation during mitosis in breast cancer cells. This leads to mitotic catastrophe, such as mitotic arrest and aneuploidy, which subsequently results in senescence or apoptotic cell death. Since inhibitors against these proteins have shown promising potential in preclinical models as well as clinical trials, they have strong utility to benefit breast cancer patients in the clinic.
This review synthesized a complex topic of centrosomes and their associated proteins in breast cancer, examining a vast amount of information that serves to emphasize their potential role in the future in terms of biomarker and drug discovery. While there is a number of proteins described here in the context of centrosomes and breast cancer, many of them have multiple roles that are not directly related to centrosome regulation. For example, proteins such as CHK1, WEE1 and CDKs, are known to participate in other mechanisms and pathways such as DNA-damage and DNA replication. On the other hand, some proteins have multiple roles in the complex process of centrosome regulation. For example, PLKs participate in both the maturation and duplication of centrosomes. Hence, it is difficult to discern the degree to which these proteins play a role in breast cancer through centrosome regulation and at which exact stages of this process their dysregulation participates in breast cancer pathogenesis. Furthermore, some of these proteins are not well studied in cancer, particularly in breast cancer, limiting the scope of this review. To address this, the implications of these proteins in other cancers were addressed. Lastly, the number of proteins discussed in this study are regulated by a plethora of regulatory pathways involving tumor suppressors and oncogenes, some of which have been linked to breast cancer pathogenesis. While here we address such key pathways, their detailed overview in the context of centrosomes and their regulation in breast cancer deserve a separate analysis of the literature.
Overall, this review highlights the importance of centrosomes and their associated proteins in breast cancer pathogenesis and as potential therapeutic targets for breast cancer treatment. Advances in research and drug development in this area provide encouraging results in terms of augmenting precision oncology-based treatment approaches in breast cancer required to improve outcomes in this prevalent and deadly disease. To our knowledge this is the first comprehensive review of centrosomes and their associated proteins in breast cancer, summarizing a large amount of information that could help set the stage for further scientific and clinical developments in this field.
Author contributions
AP: Conceptualization, Writing – review & editing. HA: Conceptualization, Methodology, Supervision, Writing – original draft, Writing – review & editing. AK: Conceptualization, Writing – original draft, Writing – review & editing. VB: Conceptualization, Writing – review & editing. AA: Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. AP lab research is supported by the Grant 942549 from the Cancer Research Society (CRS) and Canadian Institutes of Health Research (CIHR, Funding Reference Number 184668)/Institute of Cancer Research (ICR), Operating Grants 2022 Competition, Targeted Funding Opportunity, and by the Opportunities Fund of the Academic Health Sciences Centre Alternative Funding Plan of the Academic Medical Organization of Southwestern Ontario (AMOSO, Project #F20-007). AP is also supported by the Clinical Scientist Award, Department of Surgery, Western University. AA’s breast cancer research is supported by a Project Grant from the Canadian Institutes of Health Research (CIHR; # PJT-75100). AA, VB and HA are supported by Breast Cancer Canada.
Acknowledgments
We thank Ms. Anna Bonvissuto-Pin (from Dr. Parsyan’s laboratory) for help with the editing and formatting of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Arnold M, Morgan E, Rumgay H, Mafra A, Singh D, Laversanne M, et al. Current and future burden of breast cancer: Global statistics for 2020 and 2040. Breast Off J Eur Soc Mastology. (2022) 66:15–23. doi: 10.1016/j.breast.2022.08.010
2. Quereda V, Bayle S, Vena F, Frydman SM, Monastyrskyi A, Roush WR, et al. Therapeutic targeting of CDK12/CDK13 in triple-negative breast cancer. Cancer Cell. (2019) 36:545–558.e7. doi: 10.1016/j.ccell.2019.09.004
3. Kumar P, Aggarwal R. An overview of triple-negative breast cancer. Arch Gynecol. Obstet. (2016) 293:247–69. doi: 10.1007/s00404-015-3859-y
4. Nedeljković M, Damjanović A. Mechanisms of chemotherapy resistance in triple-negative breast cancer-how we can rise to the challenge. Cells. (2019) 8:957. doi: 10.3390/cells8090957
5. Kyndi M, Sørensen FB, Knudsen H, Overgaard M, Nielsen HM, Overgaard J, et al. Estrogen receptor, progesterone receptor, HER-2, and response to postmastectomy radiotherapy in high-risk breast cancer: the Danish Breast Cancer Cooperative Group. J Clin Oncol Off J Am Soc Clin Oncol. (2008) 26:1419–26. doi: 10.1200/JCO.2007.14.5565
6. Otto T, Sicinski P. Cell cycle proteins as promising targets in cancer therapy. Nat Rev Cancer. (2017) 17:93–115. doi: 10.1038/nrc.2016.138
7. Mita MM, Joy AA, Mita A, Sankhala K, Jou YM, Zhang D, et al. Randomized phase II trial of the cyclin-dependent kinase inhibitor dinaciclib (MK-7965) versus capecitabine in patients with advanced breast cancer. Clin Breast Cancer. (2014) 14:169–76. doi: 10.1016/j.clbc.2013.10.016
8. Turner NC, Ro J, André F, Loi S, Verma S, Iwata H, et al. Palbociclib in hormone-receptor–positive advanced breast cancer. N Engl J Med. (2015) 373:209–19. doi: 10.1056/NEJMoa1505270
9. Tolaney SM, Beeram M, Beck JT, Conlin AK, Dees EC, Dickler MN, et al. A phase Ib study of abemaciclib with therapies for metastatic breast cancer. J Clin Oncol. (2015) 33:522–2. doi: 10.1200/jco.2015.33.15_suppl.522
10. Zhang Y, Tian J, Qu C, Peng Y, Lei J, Sun L, et al. A look into the link between centrosome amplification and breast cancer. Biomed Pharmacother. (2020) 132:110924. doi: 10.1016/j.biopha.2020.110924
11. Sampson PB, Liu Y, Forrest B, Cumming G, Li SW, Patel NK, et al. The discovery of Polo-like kinase 4 inhibitors: identification of (1R,2S).2-(3-((E).4-(((cis).2,6-dimethylmorpholino)methyl)styryl). 1H.indazol-6-yl)-5'-methoxyspiro[cyclopropane-1,3'-indolin]-2'-one (CFI-400945) as a potent, orally active antitumor agent. J Med Chem. (2015) 58:147–69. doi: 10.1021/jm5005336
12. Cheung CHA, Sarvagalla S, Lee JY-C, Huang Y-C, Coumar MS. Aurora kinase inhibitor patents and agents in clinical testing: an update (2011 - 2013). Expert Opin Ther Pat. (2014) 24:1021–38. doi: 10.1517/13543776.2014.931374
13. Drosopoulos K, Tang C, Chao WCH, Linardopoulos S. APC/C is an essential regulator of centrosome clustering. Nat Commun. (2014) 5:3686. doi: 10.1038/ncomms4686
14. Dominguez-Brauer C, Thu KL, Mason JM, Blaser H, Bray MR, Mak TW. Targeting mitosis in cancer: emerging strategies. Mol Cell. (2015) 60:524–36. doi: 10.1016/j.molcel.2015.11.006
16. Lara-Gonzalez P, Westhorpe FG, Taylor SS. The spindle Assembly checkpoint. Curr Biol. (2012) 22:R966–80. doi: 10.1016/j.cub.2012.10.006
17. Gönczy P. Centrosomes and cancer: revisiting a long-standing relationship. Nat Rev Cancer. (2015) 15:639–52. doi: 10.1038/nrc3995
18. Nigg EA, Holland AJ. Once and only once: mechanisms of centriole duplication and their deregulation in disease. Nat Rev Mol Cell Biol. (2018) 19:297–312. doi: 10.1038/nrm.2017.127
19. Luan Y, Li M, Zhao Y, Li Q, Wen J, Gao S, et al. Centrosomal-associated Proteins: potential therapeutic targets for solid tumors? Biomed Pharmacother. (2021) 144:112292. doi: 10.1016/j.biopha.2021.112292
20. De Luca M, Lavia P, Guarguaglini G. A functional interplay between Aurora-A, Plk1 and TPX2 at spindle poles: Plk1 controls centrosomal localization of Aurora-A and TPX2 spindle association. Cell Cycle Georget. Tex. (2006) 5:296–303. doi: 10.4161/cc.5.3.2392
21. Mbom BC, Siemers KA, Ostrowski MA, Nelson WJ, Barth AIM. Nek2 phosphorylates and stabilizes β-catenin at mitotic centrosomes downstream of Plk1. Mol Biol Cell. (2014) 25:977–91. doi: 10.1091/mbc.e13-06-0349
22. Casenghi M, Barr FA, Nigg EA. Phosphorylation of Nlp by Plk1 negatively regulates its dynein-dynactin-dependent targeting to the centrosome. J Cell Sci. (2005) 118:5101–8. doi: 10.1242/jcs.02622
23. Moschou PN, Smertenko AP, Minina EA, Fukada K, Savenkov EI, Robert S, et al. The caspase-related protease separase (EXTRA SPINDLE POLES) regulates cell polarity and cytokinesis in arabidopsis. Plant Cell. (2013) 25:2171–86. doi: 10.1105/tpc.113.113043
24. Kahl I, Mense J, Finke C, Boller AL, Lorber C, Győrffy B, et al. The cell cycle-related genes RHAMM, AURKA, TPX2, PLK1, and PLK4 are associated with the poor prognosis of breast cancer patients. J Cell Biochem. (2022) 123:581–600. doi: 10.1002/jcb.30205
25. Jiawei W, Xiajun B, Tian S, Xuzheng G, Zhenwang Z. Comprehensive analysis of PLKs expression and prognosis in breast cancer. Cancer Genet. (2022) 268–9:83–92. doi: 10.1016/j.cancergen.2022.09.007
26. Lee S-Y, Jang C, Lee K-A. Polo-like kinases (Plks), a key regulator of cell cycle and new potential target for cancer therapy. Dev Reprod. (2014) 18:65–71. doi: 10.12717/DR.2014.18.1.065
27. Ou B, Zhao J, Guan S, Wangpu X, Zhu C, Zong Y, et al. Plk2 promotes tumor growth and inhibits apoptosis by targeting Fbxw7/Cyclin E in colorectal cancer. Cancer Lett. (2016) 380:457–66. doi: 10.1016/j.canlet.2016.07.004
28. Ou B, Sun H, Zhao J, Xu Z, Liu Y, Feng H, et al. Polo-like kinase 3 inhibits glucose metabolism in colorectal cancer by targeting HSP90/STAT3/HK2 signaling. J Exp Clin Cancer Res. (2019) 38:426. doi: 10.1186/s13046-019-1418-2
29. Xu X, Huang S, Zhang B, Huang F, Chi W, Fu J, et al. DNA replication licensing factor Cdc6 and Plk4 kinase antagonistically regulate centrosome duplication via Sas-6. Nat Commun. (2017) 8:15164. doi: 10.1038/ncomms15164
30. Denu RA, Zasadil LM, Kanugh C, Laffin J, Weaver BA, Burkard ME. Centrosome amplification induces high grade features and is prognostic of worse outcomes in breast cancer. BMC Cancer. (2016) 16:47. doi: 10.1186/s12885-016-2083-x
31. Seki A, Coppinger JA, Jang C-Y, Yates JR, Fang G. Bora and the kinase Aurora a cooperatively activate the kinase Plk1 and control mitotic entry. Science. (2008) 320:1655–8. doi: 10.1126/science.1157425
32. Miralaei N, Majd A, Ghaedi K, Peymani M, Safaei M. Integrated pan-cancer of AURKA expression and drug sensitivity analysis reveals increased expression of AURKA is responsible for drug resistance. Cancer Med. (2021) 10:6428–41. doi: 10.1002/cam4.4161
33. Schuyler SC, Pellman D. Search, capture and signal: games microtubules and centrosomes play. J Cell Sci. (2001) 114:247–55. doi: 10.1242/jcs.114.2.247
34. Kelly AE, Ghenoiu C, Xue JZ, Zierhut C, Kimura H, Funabiki H. Survivin reads phosphorylated histone H3 threonine 3 to activate the mitotic kinase Aurora B. Science. (2010) 330:235–9. doi: 10.1126/science.1189505
35. Boss DS, Beijnen JH, Schellens JHM. Clinical experience with aurora kinase inhibitors: A review. Oncologist. (2009) 14:780–93. doi: 10.1634/theoncologist.2009-0019
37. Ding L, Cao J, Lin W, Chen H, Xiong X, Ao H, et al. The roles of cyclin-dependent kinases in cell-cycle progression and therapeutic strategies in human breast cancer. Int J Mol Sci. (2020) 21:1960. doi: 10.3390/ijms21061960
38. Pharmacology. Tocris bioscience. Available online at: https://www.tocris.com/pharmacology/cyclin-dependent-protein-kinases.
39. Enserink JM, Kolodner RD. An overview of Cdk1-controlled targets and processes. Cell Div. (2010) 5:11. doi: 10.1186/1747-1028-5-11
40. Kargbo RB. Cyclin-dependent kinase degradation via E3 ligase binding for potential disease modulation in cancer treatment. ACS Med Chem Lett. (2023) 14:1035–7. doi: 10.1021/acsmedchemlett.3c00275
41. Patil M, Pabla N, Dong Z. Checkpoint kinase 1 in DNA damage response and cell cycle regulation. Cell Mol Life Sci CMLS. (2013) 70:4009–21. doi: 10.1007/s00018-013-1307-3
42. Zhang Y, Hunter T. Roles of chk1 in cell biology and cancer therapy. Int J Cancer J Int Cancer. (2014) 134:1013–23. doi: 10.1002/ijc.28226
43. Cicenas J, Zalyte E, Bairoch A, Gaudet P. Kinases and cancer. Cancers. (2018) 10:63. doi: 10.3390/cancers10030063
44. Tang Z, Coleman TR, Dunphy WG. Two distinct mechanisms for negative regulation of the Wee1 protein kinase. EMBO J. (1993) 12:3427–36. doi: 10.1002/embj.1993.12.issue-9
45. Fallah Y, Demas DM, Jin L, He W, Shajahan-Haq AN. Targeting WEE1 inhibits growth of breast cancer cells that are resistant to endocrine therapy and CDK4/6 inhibitors. Front Oncol. (2021) 11:681530. doi: 10.3389/fonc.2021.681530
46. Lin Y-N, Wu CT, Lin YC, Hsu WB, Tang CJC, Chang CW, et al. CEP120 interacts with CPAP and positively regulates centriole elongation. J Cell Biol. (2013) 202:211–9. doi: 10.1083/jcb.201212060
47. Zhang C, Ma X, Wei G, Zhu X, Hu P, Chen X, et al. Centrosomal protein 120 promotes centrosome amplification and gastric cancer progression via USP54-mediated deubiquitination of PLK4. iScience. (2023) 26:105745. doi: 10.1016/j.isci.2022.105745
48. Kumar A, Rajendran V, Sethumadhavan R, Purohit R. CEP proteins: the knights of centrosome dynasty. Protoplasma. (2013) 250:965–83. doi: 10.1007/s00709-013-0488-9
49. Li X, Song N, Liu L, Liu X, Ding X, Song X, et al. USP9X regulates centrosome duplication and promotes breast carcinogenesis. Nat Commun. (2017) 8:14866. doi: 10.1038/ncomms14866
50. Kim DH, Ahn JS, Han HJ, Kim HM, Hwang J, Lee KH, et al. Cep131 overexpression promotes centrosome amplification and colon cancer progression by regulating Plk4 stability. Cell Death Dis. (2019) 10:1–16. doi: 10.1038/s41419-019-1778-8
51. Lin Y-C, Chang CW, Hsu WB, Tang CJC, Lin YN, Chou EJ, et al. Human microcephaly protein CEP135 binds to hSAS-6 and CPAP, and is required for centriole assembly. EMBO J. (2013) 32:1141–54. doi: 10.1038/emboj.2013.56
52. Ohta T, Essner R, Ryu JH, Palazzo RE, Uetake Y, Kuriyama R. Characterization of Cep135, a novel coiled-coil centrosomal protein involved in microtubule organization in mammalian cells. J Cell Biol. (2002) 156:87–99. doi: 10.1083/jcb.200108088
53. Sonnen KF, Gabryjonczyk A-M, Anselm E, Stierhof Y-D, Nigg EA. Human Cep192 and Cep152 cooperate in Plk4 recruitment and centriole duplication. J Cell Sci. (2013) 126:3223–33. doi: 10.1242/jcs.129502
54. Liu Y, Liang W, Chang Y, He Z, Wu M, Zheng H, et al. CEP192 is a novel prognostic marker and correlates with the immune microenvironment in hepatocellular carcinoma. Front Immunol. (2022) 13:950884. doi: 10.3389/fimmu.2022.950884
55. Harrison LE, Bleiler M, Giardina C. A look into centrosome abnormalities in colon cancer cells, how they arise and how they might be targeted therapeutically. Biochem Pharmacol. (2018) 147:1–8. doi: 10.1016/j.bcp.2017.11.003
56. Schrock MS, Stromberg BR, Scarberry L, Summers MK. APC/C ubiquitin ligase: functions and mechanisms in tumorigenesis. Semin Cancer Biol. (2020) 67:80–91. doi: 10.1016/j.semcancer.2020.03.001
57. Jin S, Gao H, Mazzacurati L, Wang Y, Fan W, Chen Q, et al. BRCA1 interaction of centrosomal protein Nlp is required for successful mitotic progression. J Biol Chem. (2009) 284(34):22970–7. doi: 10.1074/jbc.M109.009134
58. Xu X, Yamamoto H, Sakon M, Yasui M, Ngan CY, Fukunaga H, et al. Overexpression of CDC25A phosphatase is associated with hypergrowth activity and poor prognosis of human hepatocellular carcinomas. Clin Cancer Res Off J Am Assoc Cancer Res. (2003) 9:1764–72.
59. Cangi MG, Cukor B, Soung P, Signoretti S, Moreira G, Ranashinge M, et al. Role of the Cdc25A phosphatase in human breast cancer. J Clin Invest. (2000) 106:753–61. doi: 10.1172/JCI9174
60. Boutros R, Lobjois V, Ducommun B. CDC25 phosphatases in cancer cells: key players? Good targets? Nat Rev Cancer. (2007) 7:495–507. doi: 10.1038/nrc2169
61. Shen T, Huang S. The role of Cdc25A in the regulation of cell proliferation and apoptosis. Anticancer Agents Med Chem. (2012) 12:631–9. doi: 10.2174/187152012800617678
62. Liu K, Zheng M, Lu R, Du J, Zhao Q, Li Z, et al. The role of CDC25C in cell cycle regulation and clinical cancer therapy: a systematic review. Cancer Cell Int. (2020) 20:213. doi: 10.1186/s12935-020-01304-w
63. Moyer TC, Holland AJ. PLK4 promotes centriole duplication by phosphorylating STIL to link the procentriole cartwheel to the microtubule wall. eLife. (2019) 8:e46054. doi: 10.7554/eLife.46054
64. Zheng X, Ramani A, Soni K, Gottardo M, Zheng S, Ming Gooi L, et al. Molecular basis for CPAP-tubulin interaction in controlling centriolar and ciliary length. Nat Commun. (2016) 7:11874. doi: 10.1038/ncomms11874
65. Li Y, Wang X, Vural S, Mishra NK, Cowan KH, Guda C. Exome analysis reveals differentially mutated gene signatures of stage, grade and subtype in breast cancers. PloS One. (2015) 10:e0119383. doi: 10.1371/journal.pone.0119383
66. O’Regan L, Fry AM. The nek6 and nek7 protein kinases are required for robust mitotic spindle formation and cytokinesis. Mol Cell Biol. (2009) 29:3975–90. doi: 10.1128/MCB.01867-08
67. Fang Y, Zhang X. Targeting NEK2 as a promising therapeutic approach for cancer treatment. Cell Cycle. (2016) 15:895. doi: 10.1080/15384101.2016.1152430
68. Arquint C, Nigg EA. The PLK4-STIL-SAS-6 module at the core of centriole duplication. Biochem Soc Trans. (2016) 44:1253–63. doi: 10.1042/BST20160116
69. Martin TA, Ye L, Sanders AJ, Lane J, Jiang WG. Cancer invasion and metastasis: molecular and cellular perspective. In: Madame curie bioscience database. Metastatic Cancer: Clinical and Biological Perspectives (2013).
70. Zhang N, Ge G, Meyer R, Sethi S, Basu D, Pradhan S, et al. Overexpression of Separase induces aneuploidy and mammary tumorigenesis. Proc Natl Acad Sci. (2008) 105:13033–8. doi: 10.1073/pnas.0801610105
71. Garlapati C, Joshi S, Bhattarai S, Krishnamurthy J, Turaga RC, Nguyen T, et al. PLK1 and AURKB phosphorylate survivin differentially to affect proliferation in racially distinct triple-negative breast cancer. Cell Death Dis. (2023) 14:1–13. doi: 10.1038/s41419-022-05539-5
72. Martínez-Sifuentes MA, Bassol-Mayagoitia S, Nava-Hernández MP, Ruiz-Flores P, Ramos-Treviño J, Haro-Santa cruz J, et al. Survivin in breast cancer: A review. Genet Test Mol biomark. (2022) 26:411–21. doi: 10.1089/gtmb.2021.0286
73. Tang TK. Centriole biogenesis in multiciliated cells. Nat Cell Biol. (2013) 15:1400–2. doi: 10.1038/ncb2892
74. Ohta M, Ashikawa T, Nozaki Y, Kozuka-Hata H, Goto H, Inagaki M, et al. Direct interaction of Plk4 with STIL ensures formation of a single procentriole per parental centriole. Nat Commun. (2014) 5:5267. doi: 10.1038/ncomms6267
75. Wang G, Jiang Q, Zhang C. The role of mitotic kinases in coupling the centrosome cycle with the assembly of the mitotic spindle. J Cell Sci. (2014) 127:4111–22. doi: 10.1242/jcs.151753
76. Hames RS, Crookes RE, Straatman KR, Merdes A, Hayes MJ, Faragher AJ, et al. Dynamic recruitment of nek2 kinase to the centrosome involves microtubules, PCM-1, and localized proteasomal degradation. Mol Biol Cell. (2005) 16:1711–24. doi: 10.1091/mbc.e04-08-0688
77. Vitre B, Holland AJ, Kulukian A, Shoshani O, Hirai M, Wang Y, et al. Chronic centrosome amplification without tumorigenesis. Proc Natl Acad Sci U. S. A. (2015) 112:E6321–6330. doi: 10.1073/pnas.1519388112
78. Jusino S, Fernández-Padín FM, Saavedra HI. Centrosome aberrations and chromosome instability contribute to tumorigenesis and intra-tumor heterogeneity. J Cancer Metastasis Treat. (2018) 4:43. doi: 10.20517/2394-4722.2018.24
79. Roylance R, Endesfelder D, Gorman P, Burrell RA, Sander J, Tomlinson I, et al. Relationship of extreme chromosomal instability with long-term survival in a retrospective analysis of primary breast cancer. Cancer Epidemiol. biomark Prev Publ. Am Assoc Cancer Res Cosponsored Am Soc Prev Oncol. (2011) 20:2183–94. doi: 10.1158/1055-9965.EPI-11-0343
80. Pihan GA, Purohit A, Wallace J, Knecht H, Woda B, Quesenberry P, et al. Centrosome defects and genetic instability in Malignant tumors. Cancer Res. (1998) 58:3974–85.
81. Chan JY. A clinical overview of centrosome amplification in human cancers. Int J Biol Sci. (2011) 7:1122–44. doi: 10.7150/ijbs.7.1122
82. Rivera-Rivera Y, Saavedra HI. Centrosome – a promising anti-cancer target. Biol Targets Ther. (2016) 10:167–76. doi: 10.2147/BTT
83. LoMastro GM, Holland AJ. The emerging link between centrosome aberrations and metastasis. Dev Cell. (2019) 49:325–31. doi: 10.1016/j.devcel.2019.04.002
84. Jin L, Han B, Siegel E, Cui Y, Giuliano A, Cui X. Breast cancer lung metastasis: Molecular biology and therapeutic implications. Cancer Biol Ther. (2018) 19:858–68. doi: 10.1080/15384047.2018.1456599
85. Song S, Jung S, Kwon M. Expanding roles of centrosome abnormalities in cancers. BMB Rep. (2023) 56:216–24. doi: 10.5483/BMBRep.2023-0025
86. Sansregret L, Swanton C. The role of aneuploidy in cancer evolution. Cold Spring Harb. Perspect Med 7 a028373. (2017). doi: 10.1101/cshperspect.a028373
87. Singh A, Denu RA, Wolfe SK, Sperger JM, Schehr J, Witkowsky T, et al. Centrosome amplification is a frequent event in circulating tumor cells from subjects with metastatic breast cancer. Mol Oncol. (2020) 14:1898–909. doi: 10.1002/1878-0261.12687
88. Fallahpour S, Navaneelan T, De P, Borgo A. Breast cancer survival by molecular subtype: a population-based analysis of cancer registry data. CMAJ Open. (2017) 5:E734–9. doi: 10.9778/cmajo.20170030
89. Lingle WL, Lutz WH, Ingle JN, Maihle NJ, Salisbury JL. Centrosome hypertrophy in human breast tumors: implications for genomic stability and cell polarity. Proc Natl Acad Sci U. S. A. (1998) 95:2950–5. doi: 10.1073/pnas.95.6.2950
90. Weichert W, Kristiansen G, Winzer KJ, Schmidt M, Gekeler V, Noske A, et al. Polo-like kinase isoforms in breast cancer: expression patterns and prognostic implications. Virchows Arch Int J Pathol. (2005) 446:442–50. doi: 10.1007/s00428-005-1212-8
91. Ogden A, Rida PCG, Aneja R. Centrosome amplification: A suspect in breast cancer and racial disparities. Endocr. Relat Cancer. (2017) 24:T47–64. doi: 10.1530/ERC-17-0072
92. Yang J, Ikezoe T, Nishioka C, Tasaka T, Taniguchi A, Kuwayama Y, et al. AZD1152, a novel and selective aurora B kinase inhibitor, induces growth arrest, apoptosis, and sensitization for tubulin depolymerizing agent or topoisomerase II inhibitor in human acute leukemia cells. Vitro vivo. Blood. (2007) 110:2034–40. doi: 10.1182/blood-2007-02-073700
93. Wong S-S, Wilmott ZM, Saurya S, Alvarez-Rodrigo I, Zhou FY, Chau KY, et al. Centrioles generate a local pulse of Polo/PLK1 activity to initiate mitotic centrosome assembly. EMBO J. (2022) 41:e110891. doi: 10.15252/embj.2022110891
94. Lee K, Rhee K. PLK1 phosphorylation of pericentrin initiates centrosome maturation at the onset of mitosis. J Cell Biol. (2011) 195:1093–101. doi: 10.1083/jcb.201106093
95. Ohta M, Zhao Z, Wu D, Wang S, Harrison JL, Gómez-Cavazos JS, et al. Polo-like kinase 1 independently controls microtubule-nucleating capacity and size of the centrosome. J Cell Biol. (2021) 220:e202009083. doi: 10.1083/jcb.202009083
96. van Vugt MATM, Brás A, Medema RH. Polo-like kinase-1 controls recovery from a G2 DNA damage-induced arrest in mammalian cells. Mol Cell. (2004) 15:799–811. doi: 10.1016/j.molcel.2004.07.015
97. Liu Z, Sun Q, Wang X. PLK1, A potential target for cancer therapy. Transl Oncol. (2016) 10:22–32. doi: 10.1016/j.tranon.2016.10.003
98. Salama ME, Dina A DA. Polo-like kinase 1(PLK1) immunohistochemical expression in triple negative breast carcinoma: A probable therapeutic target. Asian Pac. J Cancer Prev APJCP. (2021) 22:3921–5. doi: 10.31557/APJCP.2021.22.12.3921
99. Ueda A, Oikawa K, Fujita K, Ishikawa A, Sato E, Ishikawa T, et al. Therapeutic potential of PLK1 inhibition in triple-negative breast cancer. Lab Investig J Tech. Methods Pathol. (2019) 99:1275–86. doi: 10.1038/s41374-019-0247-4
100. Su S, Chhabra G, Singh CK, Ndiaye MA, Ahmad N. PLK1 inhibition-based combination therapies for cancer management. Transl Oncol. (2021) 16:101332. doi: 10.1016/j.tranon.2021.101332
101. de Cárcer G, Venkateswaran SV, Salgueiro L, El Bakkali A, Somogyi K, Rowald K, et al. Plk1 overexpression induces chromosomal instability and suppresses tumor development. Nat Commun. (2018) 9:3012. doi: 10.1038/s41467-018-05429-5
102. de Cárcer G. The mitotic cancer target polo-like kinase 1: oncogene or tumor suppressor? Genes. (2019) 10:208. doi: 10.3390/genes10030208
103. Liu B, Meng LB, Su JZ, Fan B, Zhao SB, Wang HY, et al. PLK1 as one novel target for the poor prognosis of bladder cancer: An observational study. Med (Baltimore). (2022) 101:e30723. doi: 10.1097/MD.0000000000030723
104. Steegmaier M, Hoffmann M, Baum A, Lénárt P, Petronczki M, Krssák M, et al. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth. vivo. Curr Biol CB. (2007) 17:316–22. doi: 10.1016/j.cub.2006.12.037
105. Anderson RT, Keysar SB, Bowles DW, Glogowska MJ, Astling DP, Morton JJ, et al. The dual pathway inhibitor rigosertib is effective in direct patient tumor xenografts of head and neck squamous cell carcinomas. Mol Cancer Ther. (2013) 12:1994–2005. doi: 10.1158/1535-7163.MCT-13-0206
106. Gorlick R, Kolb EA, Keir ST, Maris JM, Reynolds CP, Kang MH, et al. Initial testing (Stage 1) of the polo-like kinase inhibitor volasertib (BI 6727), by the pediatric preclinical testing program. Pediatr Blood Cancer. (2014) 61:158–64. doi: 10.1002/pbc.24616
107. Jeong SB, Im JH, Yoon JH, Bui QT, Lim SC, Song JM, et al. Essential role of polo-like kinase 1 (Plk1) oncogene in tumor growth and metastasis of tamoxifen-resistant breast cancer. Mol Cancer Ther. (2018) 17:825–37. doi: 10.1158/1535-7163.MCT-17-0545
108. Schöffski P, Blay JY, De Greve J, Brain E, Machiels JP, Soria JC, et al. Multicentric parallel phase II trial of the polo-like kinase 1 inhibitor BI 2536 in patients with advanced head and neck cancer, breast cancer, ovarian cancer, soft tissue sarcoma and melanoma. The first protocol of the European Organization for Research and Treatment of Cancer (EORTC) Network Of Core Institutes (NOCI). Eur J Cancer. (2010) 46:2206–15. Available at: https://clinicaltrials.gov/study/NCT00526149.
109. Zhao Y, Wang X. PLK4: a promising target for cancer therapy. J Cancer Res Clin Oncol. (2019) 145:2413–22. doi: 10.1007/s00432-019-02994-0
110. Yeow ZY, Lambrus BG, Marlow R, Zhan KH, Durin MA, Evans LT, et al. Exploiting TRIM37-driven centrosome dysfunction to kill breast cancer cells. Nature. (2020) 585:447–52. doi: 10.1038/s41586-020-2690-1
111. Denu RA, Shabbir M, Nihal M, Singh CK, Longley BJ, Burkard ME, et al. Centriole overduplication is the predominant mechanism leading to centrosome amplification in melanoma. Mol Cancer Res MCR. (2018) 16:517–27. doi: 10.1158/1541-7786.MCR-17-0197
112. Veitch ZW, Cescon DW, Denny T, Yonemoto LM, Fletcher G, Brokx R, et al. Safety and tolerability of CFI-400945, a first-in-class, selective PLK4 inhibitor in advanced solid tumours: a phase 1 dose-escalation trial. Br J Cancer. (2019) 121:318–24. doi: 10.1038/s41416-019-0517-3
113. Mason JM, Lin DCC, Wei X, Che Y, Yao Y, Kiarash R, et al. Functional characterization of CFI-400945, a polo-like kinase 4 inhibitor, as a potential anticancer agent. Cancer Cell. (2014) 26:163–76. doi: 10.1016/j.ccr.2014.05.006
114. Canadian Cancer Trials Group. A phase II study of CFI-400945 in patients with advanced/metastatic breast cancer. (2023). Available online at: https://clinicaltrials.gov/study/NCT03624543.
115. Bauer PJ, Phase II. Open study to assess efficacy and safety of rigosertib in patients with recessive dystrophic epidermolysis bullosa associated locally advanced/metastatic squamous cell carcinoma. https://clinicaltrials.gov/study/NCT03786237. (2022).
116. Scott AJ, O’Neil BH, Ma WW, Cohen SJ, Aisner D, Menter AR, et al. A phase II/III randomized study to compare the efficacy and safety of rigosertib plus gemcitabine versus gemcitabine alone in patients with previously untreated metastatic pancreatic cancer. JCO (2015) 33:4117.
117. Onconova Therapeutics, IncPhase III multiCenter randomized controlled study to assess efficacy and safety of ON 01910.Na 72-hr continuous IV infusion in MDS patients with excess blasts relapsing after or refractory to or intolerant to azacitidine or decitabine. (2020). Available online at: https://clinicaltrials.gov/study/NCT01241500.
118. Boehringer Ingelheim. An open phase I/IIa trial to investigate the maximum tolerated dose, safety, pharmacokinetics, and efficacy of intravenous BI 6727 as monotherapy or in combination with subcutaneous cytarabine in patients with acute myeloid leukaemia. (2022). Available online at: https://clinicaltrials.gov/study/NCT00804856.
119. Lei Q, Xiong L, Xia Y, Feng Z, Gao T, Wei W, et al. YLT-11, a novel PLK4 inhibitor, inhibits human breast cancer growth via inducing maladjusted centriole duplication and mitotic defect. Cell Death Dis. (2018) 9:1066. doi: 10.1038/s41419-018-1071-2
120. Kollareddy M, Zheleva D, Dzubak P, Brahmkshatriya PS, Lepsik M, Hajduch M. Aurora kinase inhibitors: Progress towards the clinic. Invest. New Drugs. (2012) 30:2411–32. doi: 10.1007/s10637-012-9798-6
121. Manfredi MG, Ecsedy JA, Chakravarty A, Silverman L, Zhang M, Hoar KM, et al. Characterization of Alisertib (MLN8237), an investigational small-molecule inhibitor of aurora A kinase using novel in vivo pharmacodynamic assays. Clin Cancer Res Off J Am Assoc Cancer Res. (2011) 17:7614–24. doi: 10.1158/1078-0432.CCR-11-1536
122. Görgün G, Calabrese E, Hideshima T, Ecsedy J, Perrone G, Mani M, et al. A novel Aurora-A kinase inhibitor MLN8237 induces cytotoxicity and cell-cycle arrest in multiple myeloma. Blood. (2010) 115:5202–13. doi: 10.1182/blood-2009-12-259523
123. Maris JM, Morton CL, Gorlick R, Kolb EA, Lock R, Carol H, et al. Initial testing of the aurora kinase A inhibitor MLN8237 by the pediatric preclinical testing program (PPTP). Pediatr Blood Cancer. (2010) 55:26–34. doi: 10.1002/pbc.22430
124. Melichar B, Adenis A, Lockhart AC, Bennouna J, Dees EC, Kayaleh O, et al. Safety and activity of alisertib, an investigational aurora kinase A inhibitor, in patients with breast cancer, small-cell lung cancer, non-small-cell lung cancer, head and neck squamous-cell carcinoma, and gastro-oesophageal adenocarcinoma: a five-arm phase 2 study. Lancet Oncol. (2015) 16:395–405. doi: 10.1016/S1470-2045(15)70051-3
125. Gully CP, Zhang F, Chen J, Yeung JA, Velazquez-Torres G, Wang E, et al. Antineoplastic effects of an Aurora B kinase inhibitor in breast cancer. Mol Cancer. (2010) 9:42. doi: 10.1186/1476-4598-9-42
126. Schwartz GK, Carvajal RD, Midgley R, Rodig SJ, Stockman PK, Ataman O, et al. Phase I study of barasertib (AZD1152), a selective inhibitor of Aurora B kinase, in patients with advanced solid tumors. Invest. New Drugs. (2013) 31:370–80. doi: 10.1007/s10637-012-9825-7
127. Gürtler U, Tontsch-Grunt U, Jarvis M, Zahn SK, Boehmelt G, Quant J, et al. Effect of BI 811283, a novel inhibitor of Aurora B kinase, on tumor senescence and apoptosis. J Clin Oncol. (2010) 28:e13632–2. doi: 10.1200/jco.2010.28.15_suppl.e13632
128. Mross K, Richly H, Frost A, Scharr D, Nokay B, Graeser R, et al. A phase I study of BI 811283, an Aurora B kinase inhibitor, in patients with advanced solid tumors. Cancer Chemother Pharmacol. (2016) 78:405–17. doi: 10.1007/s00280-016-3095-6
129. Wang X, Sinn AL, Pollok K, Sandusky G, Zhang S, Chen L, et al. Preclinical activity of a novel multiple tyrosine kinase and aurora kinase inhibitor, ENMD-2076, against multiple myeloma. Br J Haematol. (2010) 150:313–25. doi: 10.1111/j.1365-2141.2010.08248.x
130. Fletcher GC, Brokx RD, Denny TA, Hembrough TA, Plum SM, Fogler WE, et al. ENMD-2076 is an orally active kinase inhibitor with antiangiogenic and antiproliferative mechanisms of action. Mol Cancer Ther. (2011) 10:126–37. doi: 10.1158/1535-7163.MCT-10-0574
131. Matulonis UA, Lee J, Lasonde B, Tew WP, Yehwalashet A, Matei D, et al. ENMD-2076, an oral inhibitor of angiogenic and proliferation kinases, has activity in recurrent, platinum resistant ovarian cancer. Eur J Cancer Oxf. Engl. (1990) 49:121–31. doi: 10.1016/j.ejca.2012.07.020
132. Roncolato F, O’Connell R, Buizen L, Joly F, Lanceley A, Hilpert F, et al. Baseline quality of life (QOL) as a predictor of stopping chemotherapy early, and of overall survival, in platinum-resistant/refractory ovarian cancer (PRROC): The GCIG symptom benefit study (SBS). JCO. (2016) 34, 5508.
133. Diamond JR, Eckhardt SG, Pitts TM, van Bokhoven A, Aisner D, Gustafson DL, et al. A phase II clinical trial of the Aurora and angiogenic kinase inhibitor ENMD-2076 for previously treated, advanced, or metastatic triple-negative breast cancer. Breast Cancer Res BCR. (2018) 20:82. doi: 10.1186/s13058-018-1014-y
134. Al-Rikabi R, Al-Shmgani H, Dewir YH, El-Hendawy S. In vivo and in vitro evaluation of the protective effects of hesperidin in lipopolysaccharide-induced inflammation and cytotoxicity of cell. Molecules. (2020) 25:478. doi: 10.3390/molecules25030478
135. Pang S, Jia M, Gao J, Liu X, Guo W, Zhang H. Effects of dietary patterns combined with dietary phytochemicals on breast cancer metastasis. Life Sci. (2021) 264:118720. doi: 10.1016/j.lfs.2020.118720
136. Wang W, Feng X, Liu H-X, Chen S-W, Hui L. Synthesis and biological evaluation of 2,4-disubstituted phthalazinones as Aurora kinase inhibitors. Bioorg. Med Chem. (2018) 26:3217–26. doi: 10.1016/j.bmc.2018.04.048
137. Lin YG, Immaneni A, Merritt WM, Mangala LS, Kim SW, Shahzad MMK, et al. Targeting aurora kinase with MK-0457 inhibits ovarian cancer growth. Clin Cancer Res Off J Am Assoc Cancer Res. (2008) 14:5437–46. doi: 10.1158/1078-0432.CCR-07-4922
138. Jin X, Mo Q, Zhang Y, Gao Y, Wu Y, Li J, et al. The p38 MAPK inhibitor BIRB796 enhances the antitumor effects of VX680 in cervical cancer. Cancer Biol Ther. (2016) 17:566–76. doi: 10.1080/15384047.2016.1177676
139. Li Y, Zhang ZF, Chen J, Huang D, Ding Y, Tan MH, et al. VX680/MK-0457, a potent and selective Aurora kinase inhibitor, targets both tumor and endothelial cells in clear cell renal cell carcinoma. Am J Transl Res. (2010) 2:296–308.
140. de Groot CO, Hsia JE, Anzola JV, Motamedi A, Yoon M, Wong YL, et al. A cell biologist’s field guide to aurora kinase inhibitors. Front Oncol. (2015) 5:285. doi: 10.3389/fonc.2015.00285
141. Shimomura T, Hasako S, Nakatsuru Y, Mita T, Ichikawa K, Kodera T, et al. MK-5108, a highly selective Aurora-A kinase inhibitor, shows antitumor activity alone and in combination with docetaxel. Mol Cancer Ther. (2010) 9:157–66. doi: 10.1158/1535-7163.MCT-09-0609
142. Amin M, Minton SE, LoRusso PM, Krishnamurthi SS, Pickett CA, Lunceford J, et al. A phase I study of MK-5108, an oral aurora a kinase inhibitor, administered both as monotherapy and in combination with docetaxel, in patients with advanced or refractory solid tumors. Invest. New Drugs. (2016) 34:84–95. doi: 10.1007/s10637-015-0306-7
143. Scaltriti M, Eichhorn PJ, Cortés J, Prudkin L, Aura C, Jiménez J, et al. Cyclin E amplification/overexpression is a mechanism of trastuzumab resistance in HER2+ breast cancer patients. Proc Natl Acad Sci U. S. A. (2011) 108:3761–6. doi: 10.1073/pnas.1014835108
144. Chen R, Chen Y, Xiong P, Zheleva D, Blake D, Keating MJ, et al. Cyclin-dependent kinase inhibitor fadraciclib (CYC065) depletes anti-apoptotic protein and synergizes with venetoclax in primary chronic lymphocytic leukemia cells. Leukemia. (2022) 36:1596–608. doi: 10.1038/s41375-022-01553-w
145. Ettl T, Schulz D, Bauer RJ. The renaissance of cyclin dependent kinase inhibitors. Cancers. (2022) 14:293. doi: 10.3390/cancers14020293
146. Parry D, Guzi T, Shanahan F, Davis N, Prabhavalkar D, Wiswell D, et al. Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol Cancer Ther. (2010) 9:2344–53. doi: 10.1158/1535-7163.MCT-10-0324
147. Feldmann G, Mishra A, Bisht S, Karikari C, Garrido-Laguna I, Rasheed Z, et al. Cyclin-dependent kinase inhibitor Dinaciclib (SCH727965) inhibits pancreatic cancer growth and progression in murine xenograft models. Cancer Biol Ther. (2011) 12:598–609. doi: 10.4161/cbt.12.7.16475
148. Stephenson JJ, Nemunaitis J, Joy AA, Martin JC, Jou YM, Zhang D, et al. Randomized phase 2 study of the cyclin-dependent kinase inhibitor dinaciclib (MK-7965) versus erlotinib in patients with non-small cell lung cancer. Lung Cancer Amst. Neth. (2014) 83:219–23. doi: 10.1016/j.lungcan.2013.11.020
149. Gojo I, Sadowska M, Walker A, Feldman EJ, Iyer SP, Baer MR, et al. Clinical and laboratory studies of the novel cyclin-dependent kinase inhibitor dinaciclib (SCH 727965) in acute leukemias. Cancer Chemother Pharmacol. (2013) 72:897. doi: 10.1007/s00280-013-2249-z
150. Kaur G, Stetler-Stevenson M, Sebers S, Worland P, Sedlacek H, Myers C, et al. Growth inhibition with reversible cell cycle arrest of carcinoma cells by flavone L86-8275. J Natl Cancer Inst. (1992) 84:1736–40. doi: 10.1093/jnci/84.22.1736
151. Arguello F, Alexander M, Sterry JA, Tudor G, Smith EM, Kalavar NT, et al. Flavopiridol induces apoptosis of normal lymphoid cells, causes immunosuppression, and has potent antitumor activity In vivo against human leukemia and lymphoma xenografts. Blood. (1998) 91:2482–90. doi: 10.1182/blood.V91.7.2482
152. Al-Sanea MM, Obaidullah AJ, Shaker ME, Chilingaryan G, Alanazi MM, Alsaif NA, et al. A new CDK2 inhibitor with 3-hydrazonoindolin-2-one scaffold endowed with anti-breast cancer activity: design, synthesis, biological evaluation, and in silico insights. Molecules. (2021) 26:412. doi: 10.3390/molecules26020412
153. McClue SJ, Blake D, Clarke R, Cowan A, Cummings L, Fischer PM, et al. In vitro and in vivo antitumor properties of the cyclin dependent kinase inhibitor CYC202 (R-roscovitine). Int J Cancer. (2002) 102:463–8. doi: 10.1002/ijc.10738
154. Cirstea D, Hideshima T, Santo L, Eda H, Mishima Y, Nemani N, et al. Small-molecule multi-targeted kinase inhibitor RGB-286638 triggers P53-dependent and -independent anti-multiple myeloma activity through inhibition of transcriptional CDKs. Leukemia. (2013) 27:2366–75. doi: 10.1038/leu.2013.194
155. van der Biessen DAJ, Burger H, de Bruijn P, Lamers CHJ, Naus N, Loferer H, et al. Phase I study of RGB-286638, A novel, multitargeted cyclin-dependent kinase inhibitor in patients with solid tumors. Clin Cancer Res. (2014) 20:4776–83. doi: 10.1158/1078-0432.CCR-14-0325
156. Goh KC, Novotny-Diermayr V, Hart S, Ong LC, Loh YK, Cheong A, et al. TG02, a novel oral multi-kinase inhibitor of CDKs, JAK2 and FLT3 with potent anti-leukemic properties. Leukemia. (2012) 26:236–43. doi: 10.1038/leu.2011.218
157. Ortiz-Ruiz MJ, Álvarez-Fernández S, Parrott T, Zaknoen S, Burrows FJ, Ocaña A, et al. Therapeutic potential of ERK5 targeting in triple negative breast cancer. Oncotarget. (2014) 5:11308–18. doi: 10.18632/oncotarget.v5i22
158. Hirai H, Iwasawa Y, Okada M, Arai T, Nishibata T, Kobayashi M, et al. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol Cancer Ther. (2009) 8:2992–3000. doi: 10.1158/1535-7163.MCT-09-0463
159. Rajeshkumar NV, De Oliveira E, Ottenhof N, Watters J, Brooks D, Demuth T, et al. MK-1775, a potent Wee1 inhibitor, synergizes with gemcitabine to achieve tumor regressions, selectively in p53-deficient pancreatic cancer xenografts. Clin Cancer Res Off J Am Assoc Cancer Res. (2011) 17:2799–806. doi: 10.1158/1078-0432.CCR-10-2580
160. Guertin AD, Li J, Liu Y, Hurd MS, Schuller AG, Long B, et al. Preclinical evaluation of the WEE1 inhibitor MK-1775 as single-agent anticancer therapy. Mol Cancer Ther. (2013) 12:1442–52. doi: 10.1158/1535-7163.MCT-13-0025
161. Mueller S, Hashizume R, Yang X, Kolkowitz I, Olow AK, Phillips J, et al. Targeting Wee1 for the treatment of pediatric high-grade gliomas. Neuro-Oncol. (2014) 16:352–60. doi: 10.1093/neuonc/not220
162. Sand A, Piacsek M, Donohoe DL, Duffin AT, Riddell GT, Sun C, et al. WEE1 inhibitor, AZD1775, overcomes trastuzumab resistance by targeting cancer stem-like properties in HER2-positive breast cancer. Cancer Lett. (2020) 472:119–31. doi: 10.1016/j.canlet.2019.12.023
163. AstraZeneca. A phase ib, open-label, multi-centre study to assess the safety, tolerability, pharmacokinetics, and anti-tumour activity of AZD1775 monotherapy in patients with advanced solid tumours. (2023). Available online at: https://clinicaltrials.gov/study/NCT02482311.
164. King C, Diaz HB, McNeely S, Barnard D, Dempsey J, Blosser W, et al. LY2606368 causes replication catastrophe and antitumor effects through CHK1-dependent mechanisms. Mol Cancer Ther. (2015) 14:2004–13. doi: 10.1158/1535-7163.MCT-14-1037
165. Gatti-Mays ME, Karzai FH, Soltani SN, Zimmer A, Green JE, Lee M, et al. A phase II single arm pilot study of the CHK1 inhibitor prexasertib (LY2606368) in BRCA wild-type, advanced triple-negative breast cancer. Oncologist. (2020) 25:1013–e1824. doi: 10.1634/theoncologist.2020-0491
166. Guzi TJ, Paruch K, Dwyer MP, Labroli M, Shanahan F, Davis N, et al. Targeting the replication checkpoint using SCH 900776, a potent and functionally selective CHK1 inhibitor identified via high content screening. Mol Cancer Ther. (2011) 10:591–602. doi: 10.1158/1535-7163.MCT-10-0928
167. Zhou Z, Yang Z, Wang S, Zhang L, Luo J, Feng Y, et al. The Chk1 inhibitor MK-8776 increases the radiosensitivity of human triple-negative breast cancer by inhibiting autophagy. Acta Pharmacol Sin. (2017) 38:513–23. doi: 10.1038/aps.2016.136
168. Li J, Song C, Rong Y, Kuang T, Wang D, Xu X, et al. Chk1 inhibitor SCH 900776 enhances the antitumor activity of MLN4924 on pancreatic cancer. Cell Cycle. (2018) 17:191–9. doi: 10.1080/15384101.2017.1405194
169. Daud AI, Ashworth MT, Strosberg J, Goldman JW, Mendelson D, Springett G, et al. Phase I dose-escalation trial of checkpoint kinase 1 inhibitor MK-8776 as monotherapy and in combination with gemcitabine in patients with advanced solid tumors. J Clin Oncol Off J Am Soc Clin Oncol. (2015) 33:1060–6. doi: 10.1200/JCO.2014.57.5027
170. Mariappan A, Soni K, Schorpp K, Zhao F, Minakar A, Zheng X, et al. Inhibition of CPAP–tubulin interaction prevents proliferation of centrosome-amplified cancer cells. EMBO J. (2019) 38:e99876. doi: 10.15252/embj.201899876
171. Wu G, Qiu XL, Zhou L, Zhu J, Chamberlin R, Lau J, et al. Small molecule targeting the Hec1/Nek2 mitotic pathway suppresses tumor cell growth in culture and in animal. Cancer Res. (2008) 68:8393–9. doi: 10.1158/0008-5472.CAN-08-1915
172. Tsuchiya A, Hirai G, Koyama Y, Oonuma K, Otani Y, Osada H, et al. Dual-specificity phosphatase CDC25A/B inhibitor identified from a focused library with nonelectrophilic core structure. ACS Med Chem Lett. (2012) 3:294–8. doi: 10.1021/ml2002778
173. Zhang S, Jia Q, Gao Q, Fan X, Weng Y, Su Z. Dual-specificity phosphatase CDC25B was inhibited by natural product HB-21 through covalently binding to the active site. Front Chem. (2018) 6:531. doi: 10.3389/fchem.2018.00531
174. Zhang N, Scorsone K, Ge G, Kaffes CC, Dobrolecki LE, Mukherjee M, et al. Identification and characterization of separase inhibitors (Sepins) for cancer therapy. SLAS Discovery. (2014) 19:878–89. doi: 10.1177/1087057114520972
175. O’Shaughnessy J, McIntyre K, Wilks S, Ma L, Block M, Andorsky D, et al. Efficacy and safety of weekly paclitaxel with or without oral alisertib in patients with metastatic breast cancer: A randomized clinical trial. JAMA Netw Open. (2021) 4:e214103. doi: 10.1001/jamanetworkopen.2021.4103
176. Huck JJ, Zhang M, Mettetal J, Chakravarty A, Venkatakrishnan K, Zhou X, et al. Translational exposure-efficacy modeling to optimize the dose and schedule of taxanes combined with the investigational Aurora A kinase inhibitor MLN8237 (alisertib). Mol Cancer Ther. (2014) 13:2170–83. doi: 10.1158/1535-7163.MCT-14-0027
177. Oza AM, Weberpals JI, Provencher DM, Grischke EM, Hall M, Uyar D, et al. An international, biomarker-directed, randomized, phase II trial of AZD1775 plus paclitaxel and carboplatin (P/C) for the treatment of women with platinum-sensitive, TP53-mutant ovarian cancer. J Clin Oncol. (2015) 33:5506–6. doi: 10.1200/jco.2015.33.15_suppl.5506
178. Pitts TM, Simmons DM, Bagby SM, Hartman SJ, Yacob BW, Gittleman B, et al. Wee1 inhibition enhances the anti-tumor effects of capecitabine in preclinical models of triple-negative breast cancer. Cancers. (2020) 12:719. doi: 10.3390/cancers12030719
179. AstraZeneca. A multicentre phase II study of adavosertib plus chemotherapy in patients with platinum-resistant epithelial ovarian, fallopian tube, or primary peritoneal cancer. (2023). Available online at: https://clinicaltrials.gov/study/NCT02272790.
180. Tolaney S. A Phase II Study of Cisplatin + AZD1775 in Metastatic Triple-Negative Breast Cancer and Evaluation of pCDC2 as a Biomarker of Target Response (2021). Available online at: https://clinicaltrials.gov/study/NCT03012477.
181. H. Lee Moffitt Cancer Center and Research Institute. A phase II trial of AZD1775 plus carboplatin-paclitaxel in squamous cell lung cancer. (2023). Available online at: https://clinicaltrials.gov/study/NCT02513563.
182. Ahn YJ, Lee J. Study of AZD1775 in combination with paclitaxel, in advanced gastric adenocarcinoma patients harboring TP53 mutation as a second-line chemotherapy. In: ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine. Available at: https://clinicaltrials.gov/show/NCT02448329.
183. Parsyan A, Cruickshank J, Hodgson K, Wakeham D, Pellizzari S, Bhat V, et al. Anticancer effects of radiation therapy combined with Polo-Like Kinase 4 (PLK4) inhibitor CFI-400945 in triple negative breast cancer. Breast Off J Eur Soc Mastology. (2021) 58:6–9. doi: 10.1016/j.breast.2021.03.011
184. Pellizzari S, Bhat V, Athwal H, Cescon DW, Allan AL, Parsyan A. PLK4 as a potential target to enhance radiosensitivity in triple-negative breast cancer. Radiat. Oncol. (2024) 19:24. doi: 10.1186/s13014-024-02410-z
185. Hossain DMS, Javaid S, Cai M, Zhang C, Sawant A, Hinton M, et al. Dinaciclib induces immunogenic cell death and enhances anti-PD1-mediated tumor suppression. J Clin Invest. (2018) 128:644–54. doi: 10.1172/JCI94586
186. Chien AJ, Gliwa AS, Rahmaputri S, Dittrich HF, Majure MC, Rugo HS, et al. A phase Ib trial of the cyclin-dependent kinase inhibitor dinaciclib (dina) in combination with pembrolizumab (P) in patients with advanced triple-negative breast cancer (TNBC) and response correlation with MYC-overexpression. J Clin Oncol. (2020) 38:1076–6. doi: 10.1200/JCO.2020.38.15_suppl.1076
187. Gregory GP, Kumar S, Wang D, Mahadevan D, Walker P, Wagner-Johnston N, et al. Pembrolizumab plus dinaciclib in patients with hematologic Malignancies: the phase 1b KEYNOTE-155 study. Blood Adv. (2022) 6:1232–42. doi: 10.1182/bloodadvances.2021005872
188. Shapiro G. Phase 1 combination study of prexasertib (LY2606368), CHK1 inhibitor, and olaparib, PARP inhibitor, in patients with advanced solid tumors (2021). Available online at: https://clinicaltrials.gov/study/NCT03057145.
189. Zhang X, Wei C, Liang H, Han L. Polo-like kinase 4’s critical role in cancer development and strategies for plk4-targeted therapy. Front Oncol. (2021) 11:587554. doi: 10.3389/fonc.2021.587554
190. Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, et al. The clonal and mutational evolution spectrum of primary triple negative breast cancers. Nature. (2012) 486:395–9. doi: 10.1038/nature10933
191. Li Z, Dai K, Wang C, Song Y, Gu F, Liu F, et al. Expression of polo-like kinase 4(PLK4) in breast cancer and its response to taxane-based neoadjuvant chemotherapy. J Cancer. (2016) 7:1125–32. doi: 10.7150/jca.14307
192. Bakhoum SF, Kabeche L, Wood MD, Laucius CD, Qu D, Laughney AM, et al. Numerical chromosomal instability mediates susceptibility to radiation treatment. Nat Commun. (2015) 6:5990. doi: 10.1038/ncomms6990
193. Zhao Y, Yang J, Liu J, Cai Y, Han Y, Hu S, et al. Inhibition of Polo-like kinase 4 induces mitotic defects and DNA damage in diffuse large B-cell lymphoma. Cell Death Dis. (2021) 12:1–10. doi: 10.1038/s41419-021-03919-x
194. Tang A, Gao K, Chu L, Zhang R, Yang J, Zheng J. Aurora kinases: novel therapy targets in cancers. Oncotarget. (2017) 8:23937–54. doi: 10.18632/oncotarget.v8i14
195. Willems E, Dedobbeleer M, Digregorio M, Lombard A, Lumapat PN, Rogister B. The functional diversity of Aurora kinases: a comprehensive review. Cell Div. (2018) 13:7. doi: 10.1186/s13008-018-0040-6
196. Salisbury JL, D’Assoro AB, Lingle WL. Centrosome amplification and the origin of chromosomal instability in breast cancer. J Mammary Gland Biol Neoplasia. (2004) 9:275–83. doi: 10.1023/B:JOMG.0000048774.27697.30
197. Dodson H, Bourke E, Jeffers LJ, Vagnarelli P, Sonoda E, Takeda S, et al. Centrosome amplification induced by DNA damage occurs during a prolonged G2 phase and involves ATM. EMBO J. (2004) 23:3864–73. doi: 10.1038/sj.emboj.7600393
198. Goldenson B, Crispino JD. The aurora kinases in cell cycle and leukemia. Oncogene. (2015) 34:537–45. doi: 10.1038/onc.2014.14
199. Chieffi P. Aurora B: A new promising therapeutic target in cancer. Intractable Rare Dis Res. (2018) 7:141–4. doi: 10.5582/irdr.2018.01018
200. Sakakura C, Hagiwara A, Yasuoka R, Fujita Y, Nakanishi M, Masuda K, et al. Tumour-amplified kinase BTAK is amplified and overexpressed in gastric cancers with possible involvement in aneuploid formation. Br J Cancer. (2001) 84:824–31. doi: 10.1054/bjoc.2000.1684
201. Fernando M, Duijf PHG, Proctor M, Stevenson AJ, Ehmann A, Vora S, et al. Dysregulated G2 phase checkpoint recovery pathway reduces DNA repair efficiency and increases chromosomal instability in a wide range of tumours. Oncogenesis. (2021) 10:1–11. doi: 10.1038/s41389-021-00329-8
202. Korobeynikov V, Borakove M, Feng Y, Wuest WM, Koval AB, Nikonova AS, et al. Combined inhibition of Aurora A and p21-activated kinase 1 as a new treatment strategy in breast cancer. Breast Cancer Res Treat. (2019) 177:369–82. doi: 10.1007/s10549-019-05329-2
203. Du R, Huang C, Liu K, Li X, Dong Z. Targeting AURKA in Cancer: molecular mechanisms and opportunities for Cancer therapy. Mol Cancer. (2021) 20:15. doi: 10.1186/s12943-020-01305-3
204. Borah NA, Reddy MM. Aurora kinase B inhibition: A potential therapeutic strategy for cancer. Molecules. (2021) 26:1981. doi: 10.3390/molecules26071981
205. Seymour JF, Kim DW, Rubin E, Haregewoin A, Clark J, Watson P, et al. A phase 2 study of MK-0457 in patients with BCR-ABL T315I mutant chronic myelogenous leukemia and philadelphia chromosome-positive acute lymphoblastic leukemia. Blood Cancer J. (2014) 4:e238. doi: 10.1038/bcj.2014.60
206. Abu Samaan TM, Samec M, Liskova A, Kubatka P, Büsselberg D. Paclitaxel’s mechanistic and clinical effects on breast cancer. Biomolecules. (2019) 9:789. doi: 10.3390/biom9120789
207. Doxsey S, Zimmerman W, Mikule K. Centrosome control of the cell cycle. Trends Cell Biol. (2005) 15:303–11. doi: 10.1016/j.tcb.2005.04.008
208. Ferguson RL, Pascreau G, Maller JL. The cyclin A centrosomal localization sequence recruits MCM5 and Orc1 to regulate centrosome reduplication. J Cell Sci. (2010) 123:2743–9. doi: 10.1242/jcs.073098
209. Bentley AM, Normand G, Hoyt J, King RW. Distinct sequence elements of cyclin B1 promote localization to chromatin, centrosomes, and kinetochores during mitosis. Mol Biol Cell. (2007) 18:4847–58. doi: 10.1091/mbc.e06-06-0539
210. Crasta K, Huang P, Morgan G, Winey M, Surana U. Cdk1 regulates centrosome separation by restraining proteolysis of microtubule-associated proteins. EMBO J. (2006) 25:2551–63. doi: 10.1038/sj.emboj.7601136
211. Tokuyama Y, Horn HF, Kawamura K, Tarapore P, Fukasawa K. Specific phosphorylation of nucleophosmin on Thr(199) by cyclin-dependent kinase 2-cyclin E and its role in centrosome duplication. J Biol Chem. (2001) 276:21529–37. doi: 10.1074/jbc.M100014200
212. Wang Q, Bode AM, Zhang T. Targeting CDK1 in cancer: mechanisms and implications. NPJ Precis. Oncol. (2023) 7:1–14. doi: 10.1038/s41698-023-00407-7
213. Enomoto M, Goto H, Tomono Y, Kasahara K, Tsujimura K, Kiyono T, et al. Novel positive feedback loop between Cdk1 and Chk1 in the nucleus during G2/M transition. J Biol Chem. (2009) 284:34223–30. doi: 10.1074/jbc.C109.051540
214. Royou A, McCusker D, Kellogg DR, Sullivan W. Grapes(Chk1) prevents nuclear CDK1 activation by delaying cyclin B nuclear accumulation. J Cell Biol. (2008) 183:63–75. doi: 10.1083/jcb.200801153
215. Liao H, Ji F, Geng X, Xing M, Li W, Chen Z, et al. CDK1 promotes nascent DNA synthesis and induces resistance of cancer cells to DNA-damaging therapeutic agents. Oncotarget. (2017) 8:90662–73. doi: 10.18632/oncotarget.v8i53
216. O’Sullivan CC, Suman VJ, Goetz MP. The emerging role of CDK4/6i in HER2-positive breast cancer. Ther Adv Med Oncol. (2019) 11:1758835919887665. doi: 10.1177/1758835919887665
217. Gomes I, Gallego-Paez LM, Jiménez M, Santamaria PG, Mansinho A, Sousa R, et al. Co-targeting RANK pathway treats and prevents acquired resistance to CDK4/6 inhibitors in luminal breast cancer. Cell Rep Med. (2023) 4:101120. doi: 10.1016/j.xcrm.2023.101120
218. Mavratzas A, Marmé F. Treatment of luminal metastatic breast cancer beyond CDK4/6 inhibition: is there a standard of care in clinical practice? Breast Care. (2021) 16:115–28. doi: 10.1159/000514561
219. Li Z, Zou W, Zhang J, Zhang Y, Xu Q, Li S, et al. Mechanisms of CDK4/6 inhibitor resistance in luminal breast cancer. Front Pharmacol. (2020) 11:580251. doi: 10.3389/fphar.2020.580251
220. Cicenas J, Kalyan K, Sorokinas A, Jatulyte A, Valiunas D, Kaupinis A, et al. Highlights of the latest advances in research on CDK inhibitors. Cancers. (2014) 6:2224–42. doi: 10.3390/cancers6042224
221. Guertin AD, Martin MM, Roberts B, Hurd M, Qu X, Miselis NR, et al. Unique functions of CHK1 and WEE1 underlie synergistic anti-tumor activity upon pharmacologic inhibition. Cancer Cell Int. (2012) 12:45. doi: 10.1186/1475-2867-12-45
222. Gorecki L, Andrs M, Korabecny J. Clinical candidates targeting the ATR–CHK1–WEE1 axis in cancer. Cancers. (2021) 13:795. doi: 10.3390/cancers13040795
223. Krämer A, Mailand N, Lukas C, Syljuåsen RG, Wilkinson CJ, Nigg EA, et al. Centrosome-associated Chk1 prevents premature activation of cyclin-B–Cdk1 kinase. Nat Cell Biol. (2004) 6:884–91. doi: 10.1038/ncb1165
224. Oakes V, Wang W, Harrington B, Lee WJ, Beamish H, Chia KM, et al. Cyclin A/Cdk2 regulates Cdh1 and claspin during late S/G2 phase of the cell cycle. Cell Cycle. (2014) 13:3302–11. doi: 10.4161/15384101.2014.949111
225. Russell MR, Levin K, Rader J, Belcastro L, Li Y, Martinez D, et al. Combination therapy targeting the chk1 and wee1 kinases demonstrates therapeutic efficacy in neuroblastoma. Cancer Res. (2013) 73:776–84. doi: 10.1158/0008-5472.CAN-12-2669
226. Bukhari AB, Lewis CW, Pearce JJ, Luong D, Chan GK, Gamper AM. Inhibiting Wee1 and ATR kinases produces tumor-selective synthetic lethality and suppresses metastasis. J Clin Invest. (2019) 129:1329–44. doi: 10.1172/JCI122622
227. Hauge S, Naucke C, Hasvold G, Joel M, Rødland GE, Juzenas P, et al. Combined inhibition of Wee1 and Chk1 gives synergistic DNA damage in S-phase due to distinct regulation of CDK activity and CDC45 loading. Oncotarget. (2016) 8:10966–79. doi: 10.18632/oncotarget.v8i7
228. Mani C, Jonnalagadda S, Lingareddy J, Awasthi S, Gmeiner WH, Palle K. Prexasertib treatment induces homologous recombination deficiency and synergizes with olaparib in triple-negative breast cancer cells. Breast Cancer Res BCR. (2019) 21:104. doi: 10.1186/s13058-019-1192-2
229. Marina M, Saavedra HI. Nek2 and Plk4: prognostic markers, drivers of breast tumorigenesis and drug resistance. Front Biosci Landmark Ed. (2014) 19:352–65. doi: 10.2741/4212
230. Rivera-Rivera Y, Marina M, Jusino S, Lee M, Velázquez JV, Chardón-Colón C, et al. The Nek2 centrosome-mitotic kinase contributes to the mesenchymal state, cell invasion, and migration of triple-negative breast cancer cells. Sci Rep. (2021) 11:9016. doi: 10.1038/s41598-021-88512-0
231. Kokuryo T, Yokoyama Y, Yamaguchi J, Tsunoda N, Ebata T, Nagino M. NEK2 is an effective target for cancer therapy with potential to induce regression of multiple human Malignancies. Anticancer Res. (2019) 39:2251–8. doi: 10.21873/anticanres.13341
232. Abuhadra N, Sun R, Litton JK, Rauch GM, Yam C, Chang JT, et al. Prognostic impact of high baseline stromal tumor-infiltrating lymphocytes in the absence of pathologic complete response in early-stage triple-negative breast cancer. Cancers. (2022) 14:1323. doi: 10.3390/cancers14051323
233. Du L, Jing J, Wang Y, Xu X, Sun T, Shi Y, et al. Knockdown of SASS6 reduces growth of MDA−MB−231 triple−negative breast cancer cells through arrest of the cell cycle at the G2/M phase. Oncol Rep. (2021) 45:1–10. doi: 10.3892/or
234. García-Torralba E, Ivars-Rubio A, Perez-Ramos M, Navarro Manzano E, Blaya-Boluda N, de la Morena Barrio P, et al. Clinical meaning of stromal tumor infiltrating lymphocytes (sTIL) in luminal early breast cancer. J Clin Oncol. (2023) 41:572–2. doi: 10.1200/JCO.2023.41.16_suppl.572
235. Fong CS, Kim M, Yang TT, Liao J-C, Tsou M-FB. SAS-6 assembly templated by the lumen of cartwheel-less centrioles precedes centriole duplication. Dev Cell. (2014) 30:238–45. doi: 10.1016/j.devcel.2014.05.008
236. Lee M-Y, Marina M, King JL, Saavedra HI. Differential expression of centrosome regulators in Her2+ breast cancer cells versus non-tumorigenic MCF10A cells. Cell Div. (2014) 9:3. doi: 10.1186/1747-1028-9-3
237. Kolberg-Liedtke C, Feuerhake F, Garke M, Christgen M, Kates R, Grischke EM, et al. Impact of stromal tumor-infiltrating lymphocytes (sTILs) on response to neoadjuvant chemotherapy in triple-negative early breast cancer in the WSG-ADAPT TN trial. Breast Cancer Res BCR. (2022) 24:58. doi: 10.1186/s13058-022-01552-w
238. Patwardhan D, Mani S, Passemard S, Gressens P, El Ghouzzi V. STIL balancing primary microcephaly and cancer. Cell Death Dis. (2018) 9:65. doi: 10.1038/s41419-017-0101-9
239. Bolgioni AF, Ganem NJ. The interplay between centrosomes and the hippo tumor suppressor pathway. Chromosome Res Int J Mol Supramol. Evol Asp. Chromosome Biol. (2016) 24:93–104. doi: 10.1007/s10577-015-9502-8
240. Tang C-JC, Fu R-H, Wu K-S, Hsu W-B, Tang TK. CPAP is a cell-cycle regulated protein that controls centriole length. Nat Cell Biol. (2009) 11:825–31. doi: 10.1038/ncb1889
241. CENPJ centromere protein J [Homo sapiens (human)] - Gene - NCBI . Available online at: https://www.ncbi.nlm.nih.gov/gene/55835.
242. Rasool S, Baig JM, Moawia A, Ahmad I, Iqbal M, Waseem SS, et al. and CEP135 underlying autosomal recessive primary microcephaly in 32 consanguineous families from Pakistan. Mol Genet Genomic Med. (2020) 8:e1408. doi: 10.1002/mgg3.1408
243. Garvey DR, Chhabra G, Ndiaye MA, Ahmad N. Role of polo-like kinase 4 (PLK4) in epithelial cancers and recent progress in its small molecule targeting for cancer management. Mol Cancer Ther. (2021) 20:632–40. doi: 10.1158/1535-7163.MCT-20-0741
244. Sur S, Agrawal DK. Phosphatases and kinases regulating CDC25 activity in the cell cycle: clinical implications of CDC25 overexpression and potential treatment strategies. Mol Cell Biochem. (2016) 416:33–46. doi: 10.1007/s11010-016-2693-2
245. Cho Y-C, Park JE, Park BC, Kim JH, Jeong DG, Park SG, et al. Cell cycle-dependent Cdc25C phosphatase determines cell survival by regulating apoptosis signal-regulating kinase 1. Cell Death Differ. (2015) 22:1605–17. doi: 10.1038/cdd.2015.2
246. Brunetto E, Ferrara AM, Rampoldi F, Talarico A, Cin ED, Grassini G, et al. CDC25A protein stability represents a previously unrecognized target of HER2 signaling in human breast cancer: implication for a potential clinical relevance in trastuzumab treatment. Neoplasia N Y. N. (2013) 15:579–90. doi: 10.1593/neo.122054
247. Zacksenhaus E, Liu JC, Granieri L, Vorobieva I, Wang DY, Ghanbari-Azarnier R, et al. CDC25 as a common therapeutic target for triple-negative breast cancer - the challenges ahead. Mol Cell Oncol. (2018) 5:e1481814. doi: 10.1080/23723556.2018.1481814
248. Liu JC, Granieri L, Shrestha M, Wang DY, Vorobieva I, Rubie EA, et al. Identification of CDC25 as a common therapeutic target for triple-negative breast cancer. Cell Rep. (2018) 23:112–26. doi: 10.1016/j.celrep.2018.03.039
249. Bonnet J, Coopman P, Morris MC. Characterization of centrosomal localization and dynamics of Cdc25C phosphatase in mitosis. Cell Cycle Georget. Tex. (2008) 7:1991–8. doi: 10.4161/cc.7.13.6095
250. Helps NR, Luo X, Barker HM, Cohen PT. NIMA-related kinase 2 (Nek2), a cell-cycle-regulated protein kinase localized to centrosomes, is complexed to protein phosphatase 1. Biochem J. (2000) 349:509–18. doi: 10.1042/bj3490509
251. Harrison Pitner MK, Saavedra HI. Cdk4 and nek2 signal binucleation and centrosome amplification in a her2+ breast cancer model. PloS One. (2013) 8:e65971. doi: 10.1371/journal.pone.0065971
252. Hayward DG, Clarke RB, Faragher AJ, Pillai MR, Hagan IM, Fry AM. The centrosomal kinase Nek2 displays elevated levels of protein expression in human breast cancer. Cancer Res. (2004) 64:7370–6. doi: 10.1158/0008-5472.CAN-04-0960
253. Bobbitt JR, Cuellar-Vite L, Bryson B, Weber-Bonk K, Keri R. Abstract 1433: Targeting the mitotic kinase NEK2 potentiates CDK4/6 inhibitor efficacy in breast cancer. Cancer Res. (2023) 83:1433. doi: 10.1158/1538-7445.AM2023-1433
254. Barford D. Structural insights into anaphase-promoting complex function and mechanism. Philos Trans R Soc B Biol Sci. (2011) 366:3605–24. doi: 10.1098/rstb.2011.0069
255. Tischer T, Yang J, Barford D. The APC/C targets the Cep152–Cep63 complex at the centrosome to regulate mitotic spindle assembly. J Cell Sci. (2022) 135:jcs259273. doi: 10.1242/jcs.259273
256. VanGenderen C, Harkness TAA, Arnason TG. The role of Anaphase Promoting Complex activation, inhibition and substrates in cancer development and progression. Aging. (2020) 12:15818–55. doi: 10.18632/aging.v12i15
257. Wheatley SP, Altieri DC. Survivin at a glance. J Cell Sci. (2019) 132:jcs223826. doi: 10.1242/jcs.223826
258. Vong QP, Cao K, Li HY, Iglesias PA, Zheng Y. Chromosome alignment and segregation regulated by ubiquitination of survivin. Science. (2005) 310:1499–504. doi: 10.1126/science.1120160
259. Wright S, Burkholtz SR, Zelinsky C, Wittman C, Carback RT, Harris PE, et al. Survivin expression in luminal breast cancer and adjacent normal tissue for immuno-oncology applications. Int J Mol Sci. (2023) 24:11827. doi: 10.3390/ijms241411827
260. Kim J-S, Kim HA, Seong MK, Seol H, Oh JS, Kim EK, et al. STAT3-survivin signaling mediates a poor response to radiotherapy in HER2-positive breast cancers. Oncotarget. (2016) 7:7055–65. doi: 10.18632/oncotarget.v7i6
261. Zhang M, Zhang X, Zhao S, Wang Y, Di W, Zhao G, et al. Prognostic value of survivin and EGFR protein expression in triple-negative breast cancer (TNBC) patients. Targeting Oncol. (2014) 9:349–57. doi: 10.1007/s11523-013-0300-y
262. Li F, Aljahdali I, Ling X. Cancer therapeutics using survivin BIRC5 as a target: what can we do after over two decades of study? J Exp Clin Cancer Res. (2019) 38:368. doi: 10.1186/s13046-019-1362-1
263. Luo S, Tong L. Structure and function of the separase-securin complex. Subcell. Biochem. (2021) 96:217–32. doi: 10.1007/978-3-030-58971-4_4
264. Revenkova E, Jessberger R. Keeping sister chromatids together: cohesins in meiosis. Reprod Camb. Engl. (2005) 130:783–90. doi: 10.1530/rep.1.00864
265. Matsuo K, Ohsumi K, Iwabuchi M, Kawamata T, Ono Y, Kendrin TM. Kendrin is a novel substrate for separase involved in the licensing of centriole duplication. Curr Biol. (2012) 22:915–21. doi: 10.1016/j.cub.2012.03.048
266. Finetti P, Guille A, Adelaide J, Birnbaum D, Chaffanet M, Bertucci F. ESPL1 is a candidate oncogene of luminal B breast cancers. Breast Cancer Res Treat. (2014) 147:51–9. doi: 10.1007/s10549-014-3070-z
267. Gurvits N, Repo H, Löyttyniemi E, Nykänen M, Anttinen J, Kuopio T, et al. Prognostic implications of securin expression and sub-cellular localization in human breast cancer. Cell Oncol. (2016) 39:319–31. doi: 10.1007/s13402-016-0277-5
268. Gurvits N, Löyttyniemi E, Nykänen M, Kuopio T, Kronqvist P, Talvinen K. Separase is a marker for prognosis and mitotic activity in breast cancer. Br J Cancer. (2017) 117:1383–91. doi: 10.1038/bjc.2017.301
269. Yersal O, Barutca S. Biological subtypes of breast cancer: Prognostic and therapeutic implications. World J Clin Oncol. (2014) 5:412–24. doi: 10.5306/wjco.v5.i3.412
270. Staaf J, Ringnér M, Vallon-Christersson J, Jönsson G, Bendahl PO, Holm K, et al. Identification of subtypes in human epidermal growth factor receptor 2–positive breast cancer reveals a gene signature prognostic of outcome. J Clin Oncol Off J Am Soc Clin Oncol. (2010) 28:1813–20. doi: 10.1200/JCO.2009.22.8775
271. Chia S, Norris B, Speers C, Cheang M, Gilks B, Gown AM, et al. Human epidermal growth factor receptor 2 overexpression as a prognostic factor in a large tissue microarray series of node-negative breast cancers. J Clin Oncol Off J Am Soc Clin Oncol. (2008) 26:5697–704. doi: 10.1200/JCO.2007.15.8659
272. Marvalim C, Datta A, Lee SC. Role of p53 in breast cancer progression: An insight into p53 targeted therapy. Theranostics. (2023) 13:1421–42. doi: 10.7150/thno.81847
273. Tarapore P, Horn HF, Tokuyama Y, Fukasawa K. Direct regulation of the centrosome duplication cycle by the p53-p21Waf1/Cip1 pathway. Oncogene. (2011) 20:3173–84. doi: 10.1038/sj.onc.1204424
274. Strebhardt K. Multifaceted polo-like kinases: drug targets and antitargets for cancer therapy. Nat Rev Drug Discovery. (2010) 9:643–60. doi: 10.1038/nrd3184
275. King SI, Purdie CA, Bray SE, Quinlan PR, Jordan LB, Thompson AM, et al. Immunohistochemical detection of Polo-like kinase-1 (PLK1) in primary breast cancer is associated with TP53 mutation and poor clinical outcome. Breast Cancer Res BCR. (2012) 14:R40. doi: 10.1186/bcr3136
276. Tentler JJ, Ionkina A, Tan AC, Newton TP, Pitts TM, Glogowska MJ, et al. p53 family members regulate phenotypic response to aurora kinase A inhibition in triple-negative breast cancer. Mol Cancer Ther. (2015) 14:1117–29. doi: 10.1158/1535-7163.MCT-14-0538-T
277. Katayama H, Sasai K, Kawai H, Yuan ZM, Bondaruk J, Suzuki F, et al. Phosphorylation by aurora kinase A induces Mdm2-mediated destabilization and inhibition of p53. Nat Genet. (2004) 36:55–62. doi: 10.1038/ng1279
278. Rosen EM, Fan S, Pestell RG, Goldberg ID. BRCA1 gene in breast cancer. J Cell Physiol. (2003) 196:19–41. doi: 10.1002/jcp.10257
279. Brekelmans CTM, Tilanus-Linthorst MMA, Seynaeve C, vd Ouweland A, Menke-Pluymers MBE, Bartels CCM, et al. Tumour characteristics, survival and prognostic factors of hereditary breast cancer from BRCA2-, BRCA1- and non-BRCA1/2 families as compared to sporadic breast cancer cases. Eur J Cancer Oxf. Engl 1990. (2007) 43:867–76. doi: 10.1016/j.ejca.2006.12.009
280. Tarapore P, Hanashiro K, Fukasawa K. Analysis of centrosome localization of BRCA1 and its activity in suppressing centrosomal aster formation. Cell Cycle. (2012) 11:2931–46. doi: 10.4161/cc.21396
281. Zhan Q, Antinore MJ, Wang XW, Carrier F, Smith ML, Harris CC, et al. Association with Cdc2 and inhibition of Cdc2/Cyclin B1 kinase activity by the p53-regulated protein Gadd45. Oncogene. (1999) 18:2892–900. doi: 10.1038/sj.onc.1202667
282. Kais Z, Parvin JD. Regulation of centrosomes by the BRCA1-dependent ubiquitin ligase. Cancer Biol Ther. (2008) 7:1540–3. doi: 10.4161/cbt.7.10.7053
283. Vasan N, Razavi P, Johnson JL, Shao H, Shah H, Antoine A, et al. Double PIK3CA mutations in cis increase oncogenicity and sensitivity to PI3Kα inhibitors. Science. (2019) 366:714–23. doi: 10.1126/science.aaw9032
Keywords: breast cancer, centrosome, centriole, PLK, AURK, CDK, CHK1/WEE1, CEP
Citation: Athwal H, Kochiyanil A, Bhat V, Allan AL and Parsyan A (2024) Centrosomes and associated proteins in pathogenesis and treatment of breast cancer. Front. Oncol. 14:1370565. doi: 10.3389/fonc.2024.1370565
Received: 14 January 2024; Accepted: 04 March 2024;
Published: 28 March 2024.
Edited by:
Charles Theillet, Institut du Cancer de Montpellier (ICM), FranceReviewed by:
Muhammad Jameel, Washington University, United StatesMasanori Kawakami, University of Texas MD Anderson Cancer Center, United States
Copyright © 2024 Athwal, Kochiyanil, Bhat, Allan and Parsyan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Armen Parsyan, YXBhcnN5YW5AdXdvLmNh