 Christopher J. Ferreri
Christopher J. Ferreri Manisha Bhutani*
Manisha Bhutani*- Department of Hematologic Oncology and Blood Disorders, Levine Cancer Institute, Atrium Health Wake Forest University School of Medicine, Charlotte, NC, United States
Chimeric antigen receptor (CAR) T cell therapies have dramatically improved treatment outcomes for patients with relapsed or refractory B-cell acute lymphoblastic leukemia, large B-cell lymphoma, follicular lymphoma, mantle cell lymphoma, and multiple myeloma. Despite unprecedented efficacy, treatment with CAR T cell therapies can cause a multitude of adverse effects which require monitoring and management at specialized centers and contribute to morbidity and non-relapse mortality. Such toxicities include cytokine release syndrome, immune effector cell-associated neurotoxicity syndrome, neurotoxicity distinct from ICANS, immune effector cell-associated hemophagocytic lymphohistiocytosis-like syndrome, and immune effector cell-associated hematotoxicity that can lead to prolonged cytopenias and infectious complications. This review will discuss the current understanding of the underlying pathophysiologic mechanisms and provide guidelines for the grading and management of such toxicities.
Introduction
Chimeric antigen receptors (CAR) are engineered recombinant receptors consisting of an extracellular target antigen-binding domain, hinge region, transmembrane domain, and intracellular signaling domain that can be transduced into immune effector cells such as T cells. This allows the engineered T cells to bind the target antigen in a major histocompatibility complex (MHC)-independent fashion, followed by robust activation and expansion of the CAR T cell population which promotes tumor elimination. (1) The anti-CD19 CAR T cell therapies tisagenlecleucel (tisa-cel) and axicabtagene autoleucel (axi-cel) were the first to receive regulatory approval in 2017 after demonstrating unprecedented efficacy for the treatment of relapsed/refractory pediatric B-cell acute lymphoblastic leukemia (B-ALL) and large B-cell lymphoma (LBCL). (2–4) Subsequently, lisocabtagene maraleucel (liso-cel) received approval for relapsed/refractory LBCL, followed by both liso-cel and axi-cel becoming indicated for second-line use in LBCL based on positive phase 3 trial data. (5–7) Anti-CD19 CAR-T has additionally received regulatory approval for relapsed/refractory follicular lymphoma, adult B-ALL, and relapsed/refractory mantle cell lymphoma. (8–10) Idecabtagene vicleucel (ide-cel) and ciltacabtagene autoleucel (cilta-cel) are both B-cell maturation antigen (BCMA)-targeted CAR T cell therapies approved for the treatment of relapsed/refractory multiple myeloma after four or more prior lines of therapy (11, 12).
Despite remarkable efficacy noted with these therapies, supraphysiologic T cell activation, expansion, and systemic hyperinflammatory response mediated by cytokine production can result in potentially severe and life-threatening complications. Prototypical toxicities include cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS). Beyond CRS and ICANS, the systemic inflammatory response triggered by CAR-T can culminate in less frequent but potentially fatal immune effector cell-associated hemophagocytic lymphohistiocytosis-like syndrome (IEC-HS). Cytopenias after CAR T cell therapy, recently termed immune effector cell-associated hematotoxicity (ICAHT), can lead to significant transfusion dependence and predispose patients to bleeding and infectious sequelae. Neurologic complications distinct from ICANS such as delayed movement and neurocognitive treatment–emergent adverse events (MNTs) are also associated with cellular therapy. (13) Post-infusion monitoring for these toxicities at specialized centers and the interventions required for toxicity management contribute significantly to the total cost of care. (14, 15) With the expected expanded indications for CAR T cell therapy in hematologic malignancies, as well as potential applications in solid tumors and autoimmune disease, optimizing prevention and treatment of the associated toxicities will become increasingly important to establish frameworks for institutions and health care providers to ultimately improve patient outcomes (16, 17).
Cytokine release syndrome (CRS): clinical manifestations and mechanisms
The American Society for Transplantation and Cellular Therapy (ASTCT) consensus grading criteria defines cytokine release syndrome as “a supraphysiologic response following any immune therapy that results in the activation or engagement of endogenous or infused T cells and/or other immune effector cells. Symptoms can be progressive, must include fever at the onset, and may include hypotension, capillary leak (hypoxia) and end organ dysfunction.” (18)While fever is the first symptom of CRS and typically occurs within the first 14 days after infusion, clinical manifestations can range across a spectrum of severity. In less severe forms, self-limited cases may consist of fever, myalgias, fatigue, and headache. With escalating severity, CRS can lead to increased vascular permeability and capillary leak causing hypotension, pulmonary edema, or acute lung injury, requiring ICU level care. (13, 19–21) Sustained inflammation with CRS has been associated with cardiovascular toxicities of elevated serum troponin reflective of myocardial injury, decreased left ventricular ejection fraction, and cardiac arrhythmias. (22) Electrolyte derangements, liver function test abnormalities, and renal insufficiency potentially requiring temporary hemodialysis support have also been associated with CRS. (13) Table 1 includes a summary of the incidence rate and median time to onset of CRS observed in the respective registrational clinical trial for each of the commercially available anti-CD19 and anti-BCMA CAR T cell therapies.
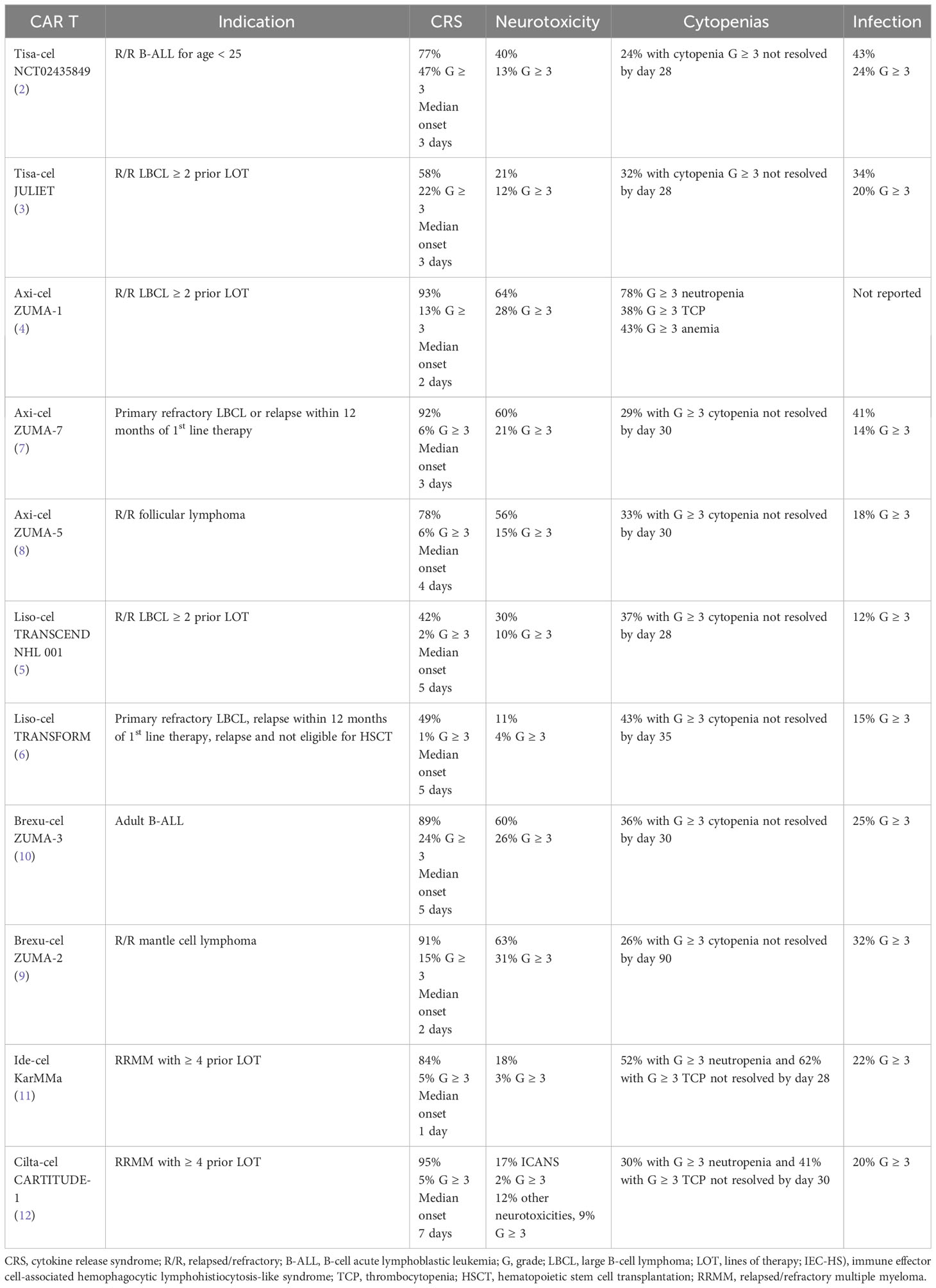
Table 1 Incidence of immune effector cell-associated toxicities seen on registrational trials for approved CAR T cell therapies.
Inflammatory markers and cytokines that have been implicated in CRS include tumor necrosis factor-alpha (TNF-α), interferon-gamma (IFN-ɣ), interleukin-1 (IL-1), IL-2, soluble IL2Rα, IL-4, IL-6, IL-8, IL-10, ferritin, C-reactive protein (CRP), granulocyte/macrophage colony stimulating factor (GM-CSF), macrophage inflammatory protein-1α (MIP-1α), monocyte chemoattractant protein-1 (MCP-1), granzyme B, and soluble gp130. (23) The term “cytokine release syndrome” was first used in the early 1990s after systemic reactions associated with elevations in TNF-α and IFN-ɣ were noted after immunosuppressive treatment with the anti-T-cell antibody muromonab-CD3 (OKT3), which was tempered with glucocorticoids. Subsequently, CRS has been associated with various immunotherapies including monoclonal antibodies, antibody-drug conjugates, immune checkpoint inhibitors, bispecific T cell redirecting antibodies, and CAR T cell therapies. (24) Similar elevations in serum cytokines were observed in the first clinical studies of CAR T cell therapies for chronic lymphocytic leukemia (CLL) and B-ALL (19, 25, 26).
Activation of the vascular endothelium has also been implicated in the pathophysiology of CRS. RNA in-situ hybridization studies from a patient who died due to CRS after treatment with anti-CD19 CAR T cell therapy noted that both interstitial cells and vascular endothelium lining cells expressed IL-6, which was markedly elevated. (27) Patients with grade ≥2 CRS after anti CD19 CAR T infusion had increased prothrombin time (PT), activated partial thromboplastin time (aPTT), fibrinogen, D-dimer, factor VIII, von Willebrand factor (vWF), and decreased platelet count and antithrombin levels compared to those with lower grade or no CRS, suggesting that endothelial activation was predictive for greater CRS severity and development of disseminated intravascular coagulopathy (DIC). (28) A multivariable analysis of 133 adult patients treated with anti-CD19 CAR T noted that biomarkers of endothelial activation including angiopoietin-2 and vWF were increased during severe CRS, and also relatively increased prior to lymphodepletion chemotherapy in patients who subsequently developed CRS. Severe thrombocytopenia prior to lymphodepletion was predictive for development of CRS, and it was postulated that these patients were more susceptible to endothelial activation due to platelets being a primary source of the endothelial stabilizing cytokine angiopoietin-1 (29).
While retrospective analysis of serum cytokines in relation to CRS severity have been informative in understanding the underlying pathophysiology, the development of animal models of cytokine release syndrome has also been critical to further elucidating these mechanisms. Experiments from a murine model of B-ALL treated with anti-CD19 CAR T demonstrated that CAR T cells produced negligible levels of IL-6, but instead bystander proinflammatory monocytes and macrophages were responsible for IL-1 and IL-6 production. It was noted that IL-1 secretion preceded IL-6 production by several hours, and treatment with the IL-1 receptor inhibitor anakinra was equally effective as the IL-6 receptor inhibitor tocilizumab in treating CRS in this model. (30) Another murine model of CRS developing after anti-CD19 CAR T cell therapy for B-ALL also demonstrated that CRS severity was mediated by IL-6, IL-1, and nitric oxide production from recipient macrophages rather than directly from the CAR T cells themselves (31).
Though IL-1 and IL-6 secretion primarily occurs from bystander monocytes and macrophages in CRS, upstream IFN-ɣ secretion occurs from activated CAR T cells and hence IFN-ɣ production is used as a potency assay for CAR T cell effector function. IFN-ɣ activates innate immune cells such as macrophages to upregulate antigen-presentation pathways, which also leads to immune checkpoint expression. In vitro experiments utilizing xenograft models of hematologic malignancy and serum samples obtained from patients who developed clinically significant CRS demonstrated that IFN-ɣ inhibition and deletion resulted in decreased macrophage activation and proinflammatory cytokine production, as well as reduced immune checkpoint expression. Compared to inhibition of IL-1 and IL-6, IFN-ɣ blockade dampened downstream proinflammatory cytokine production to a greater extent. (32) In a simplified humanized murine model of CRS/neurotoxicity after treatment with anti-CD19 CAR T, treatment with the IFN-ɣ inhibitor emapalumab led to decreased inflammatory signaling and toxicity mitigation without impacting CAR T efficacy. (33) However, studies from a fully immunocompetent mouse model demonstrated that treatment with IFN-ɣ knockout CAR T cells resulted in similar levels of proinflammatory cytokine elevation and occurrence of clinical CRS in the treated mice compared to those treated with the wild-type CAR T. While possible that IFN-ɣ signaling plays a more significant role in humans compared to mice, these results suggest that in vivo IFN-ɣ signaling may be redundant or not directly causal for CRS development (34).
Lastly, an in vitro model of the macrophage-endothelial interface suggested that endothelial inflammation occurs after CAR T cell therapy independent of signaling from macrophages, which in turn amplifies macrophage-mediated inflammatory signaling. The transcription factor STAT3 was implicated in hyperinflammatory signaling driven by the activated endothelium, and inhibition of the JAK-STAT pathway with ruxolitinib in combination with dexamethasone resulted in the greatest reduction of pro-inflammatory cytokine secretion. (35) Further refinement of in vivo animal models will be pivotal for discovering the more precise molecular underpinnings of CRS development and severity.
Factors associated with CRS risk
Identifying factors associated with an increased risk for the development of severe CRS can help inform decisions about ensuing monitoring and management. Such factors may be intrinsic to the CAR product or related to patient and disease specific factors. The dose of infused CAR T cells has been shown to be one such intrinsic factor, with several studies noting that CRS was observed more commonly in patients treated with a higher cell dose. (29, 36). Analysis of clinical trials and real-world experience studies have suggested that treatment with CARs incorporating a CD28 costimulatory motif may pose a greater risk for incidence and severity of CRS compared to those with a 4–1BB costimulatory domain. (21, 36, 37) Several studies have suggested that CD4-postiive CAR T cells contribute to CRS development to a greater degree than CD8-positive CAR T cells, indicating that the CD4/CD8 ratio of the CAR product may be potentially be predictive for CRS (34, 38).
Regarding disease and patient specific factors, a higher disease burden prior to CAR T cell infusion (e.g., degree of bone marrow involvement prior to lymphodepletion) has generally been associated with increased severity of CRS. (21, 39–42) The underlying disease biology also impacts CRS risk, with patients having a more proliferative malignancy such as B-ALL or LBCL seemingly at risk for higher grade CRS compared to those with more indolent disease such as follicular lymphoma or multiple myeloma. (8, 11, 12, 43–45) The modified Endothelial Activation and Stress Index (m-EASIX) score consists of laboratory parameters that are readily available (lactate dehydrogenase [LDH in U/L] x C-reactive protein [CRP in mg/dL]/platelets [PLTs in 109 cells/L]). In a retrospective analysis of 118 patients receiving anti-CD19 CAR T cell therapy for the treatment of either B-ALL or LBCL, the m-EASIX score prior to infusion was associated with onset of any grade and severe CRS, and m-EASIX score on days +1 and +3 post-infusion were associated with onset of severe CRS. Furthermore, when analyzing individual variables (CRP, LDH, PLTs, creatinine), lower platelet counts and higher CRP were independently associated with the onset of severe CRS. Thus, the m-EASIX score can be used as a predictor of endothelial activation which may indicate increased risk for more severe CRS (46).
Grading and management of CRS
There were initially multiple heterogeneous definition and grading systems for CRS when CAR T cell therapies were first being studied in humans, such as the Common Terminology Criteria for Adverse Events (CTCAE), University of Pennsylvania, Memorial Sloan Kettering Cancer Center (MSKCC), and CAR-T cell therapy-associated toxicity (CARTOX) criteria. (41, 47–49) A multi-institutional effort to refine CRS grading, known as the Lee criteria, defined fever as a hallmark of CRS and included hypotension responsive to a low dose of one vasopressor as grade 2 CRS. Furthermore, the Lee criteria incorporated a treatment algorithm that corresponded with each grade of toxicity. (50) Given the differences among the published grading systems for CAR T-associated toxicity, an expert panel was convened to draft the ASTCT consensus grading criteria for CRS and neurologic toxicity associated with immune effector cells in 2018. (18) The ASTCT consensus criteria have since been applied as a uniform grading system for subsequent clinical trials and real-world analyses.
Given that symptoms of CRS are non-specific, the symptoms defining CRS must be attributed specifically to treatment with the immune effector cell therapy. In addition to having a reasonable temporal relation, typically with onset occurring within 14 days of therapy, other causes of fever, hypotension, or hypoxia such as sepsis should be excluded. While there is overlap between CRS and other immune effector cell-associated toxicities (ICANS, IEC-HS), these toxicities are excluded from the definition of CRS and are graded separately. Laboratory parameters of inflammation such as CRP and ferritin are often trended for patients receiving cell therapy; however, specific laboratory parameters are excluded from the definition and grading of CRS due to the non-specificity of these markers of inflammation, and because cytokine panels are not readily available at most institutions. For the purposes of defining CRS, fever is defined by the CTCAE version 5.0 criteria as a temperature ≥ 38.0 °C. The severity of CRS is defined by the degree of hypotension and hypoxia, as other specific organ toxicities generally occur in the setting of hypotension and hypoxia and do not influence treatment decisions with glucocorticoids or anti-cytokine therapy. While fever may resolve quickly after the initiation of anti-cytokine therapies, CRS is not considered resolved until all signs and symptoms leading to the original diagnosis of CRS have resolved unless they can be clearly attributed to an alternative etiology. The ASTCT CRS consensus grading system is summarized in Table 2 (18).

Table 2 The ASTCT consensus grading criteria for cytokine release syndrome (CRS).
Acknowledging the relatively high incidence of CRS with most approved CAR T cell therapies, patients require close monitoring initially with frequent vital sign assessment, laboratory studies (complete blood count, complete metabolic panel, coagulation studies, inflammatory markers), and neurologic assessment. (13) Outpatient administration has been successfully demonstrated in both clinical trial and real-world settings; however, the majority of CAR T cell therapies administered to date have occurred inpatient with a defined period of monitoring for the development of the associated toxicities. (51–53) Management of CRS involves evaluating for infectious etiologies of fever, supportive care, and varying degrees of immunosuppressive therapy based on CRS severity. (13) Tocilizumab is a monoclonal antibody antagonist of the IL-6 receptor that has received regulatory approval for the treatment of CRS. (54) Each approved CAR T cell therapy is administered through a Risk Evaluation and Mitigation Strategy (REMS) program which mandates tocilizumab availability at the site of treatment in case it is needed for the management of CRS. Furthermore, the prescribing information for each CAR product includes management guidance for each grade of CRS (55–60).
As fever without hypotension or hypoxia defines grade 1 CRS, initial management consists of excluding infectious etiology with blood cultures, urine culture, and chest radiography. The underlying hematologic malignancy and lymphodepletion chemotherapy administered prior to CAR T often result in neutropenia concomitant with onset of fever, and thus neutropenic fever should be managed with broad spectrum antibiotics and granulocyte stimulating factor (G-CSF) per institutional guidelines. Grade 1 CRS can often be managed with supportive care alone, such as antipyretics, intravenous fluids, and symptomatic management; however, patients with recurrent or persistent fever may be managed with tocilizumab as per grade ≥ 2 CRS (61).
Management of grade 2 CRS includes intravenous fluids for hypotension and/or supplemental oxygenation as needed. Patients with grade ≥2 CRS should undergo more intensive monitoring with continuous pulse oximetry and cardiac telemetry. All patients with grade 2 CRS should receive tocilizumab 8 mg/kg IV (dose capped at 800 mg/dose). Tocilizumab may be given every eight hours for a maximum of four total doses. For patients with persistent borderline hypotension after reasonable intravenous fluid bolus administration and one to two doses of tocilizumab, the addition of glucocorticoid therapy with dexamethasone 10 mg IV (or equivalent) every 12 hours should be considered. If brisk improvement is not observed, vasopressor therapy should be initiated for grade 3 CRS and level of care should be escalated to an intensive care unit. The management of grade 3 CRS includes supportive care for hypotension and hypoxia with vasopressors and escalating supplemental oxygenation as necessary, and echocardiography should be performed to assess cardiac function. Tocilizumab and glucocorticoid should be administered concurrently, with tocilizumab dosing as per labeled use above and with glucocorticoid dose increased to dexamethasone 10 mg IV every 6 hours (or equivalent) followed by a rapid taper once symptoms are noted to improve. Refractory grade 3 or escalation to grade 4 CRS may necessitate further titration of glucocorticoid dose up to a maximum methylprednisolone dose of 1,000 mg IV every 12 hours plus consideration of alternative anti-cytokine or immunosuppressive therapies after tocilizumab has been administered for the maximum of four doses (61).
The management of CRS refractory to standard anti-inflammatory therapy with tocilizumab and glucocorticoids can pose significant challenges, with data often limited to case reports or retrospective series. It is first prudent to exclude active infection or progression of malignancy as a contributing factor. (62) As murine models of CAR T-associated toxicities have implied IL-1 as an important pathophysiologic mediator of both CRS and ICANS, the IL-1 receptor antagonist anakinra, which is approved for the treatment of rheumatoid arthritis and neonatal-onset multisystem inflammatory disease, has become increasingly used for the treatment of refractory toxicity. (30, 31, 62) While anakinra has demonstrated greater efficacy in the treatment of refractory ICANS, it has been shown to facilitate resolution of refractory CRS in case reports and retrospective studies (63, 64).
Siltuximab is a monoclonal antibody that binds directly to IL-6 preventing its interaction with IL-6 receptors, whereas tocilizumab is an inhibitor of the IL-6 receptor. Siltuximab has been demonstrated to be efficacious in the treatment of CRS both alone and in combination with tocilizumab. (5, 65, 66) The TNF-α receptor inhibitor etanercept, approved for various rheumatologic indications, has been shown to abate CRS in case reports of both anti-CD19 and anti-BCMA CAR T cell therapies. (26, 67) Emapalumab is a monoclonal antibody inhibitor of IFN-ɣ approved for the treatment of primary hemophagocytic lymphohistiocytosis (HLH). Preclinical studies have supported a potential role for its use in CAR T-associated toxicities, and emapalumab was given with success for a case of refractory grade 4 CRS with secondary HLH. (33, 68) Furthermore, in a case series of pediatric patients with B-ALL who had refractory CRS despite IL-6 inhibition, glucocorticoids, and IL-1 blockade, a single dose of emapalumab 1 mg/kg resulted in a profound decrease in inflammatory markers and significant clinical improvement in five of six patients treated (69).
The tyrosine kinase inhibitor dasatinib has been demonstrated to inhibit the lymphocyte-specific protein tyrosine kinase, which impairs downstream signaling in various CAR constructs leading to a cessation in cytokine production, cytolytic activity, and in decreased CAR T cell proliferation in both in vitro and in vivo models. In a murine CRS model, dasatinib was able to mitigate the effects of CRS and CAR-T cells were able to regain antitumor cytolytic activity after dasatinib discontinuation. (70, 71) In one published case of grade 3 CRS and grade 4 ICANS observed after anti-CD19 CAR T cell therapy for LBCL despite treatment with four doses of tocilizumab and high-dose glucocorticoids, treatment with dasatinib 100 mg daily for seven days resulted in substantial clinical improvement. While CAR T expansion was noted to decline markedly for this patient after dasatinib initiation without evidence of re-expansion after discontinuation, it did not appear to hamper efficacy as the patient was in an ongoing complete remission over two years from the time of CAR T cell infusion. (72)
Inflammatory cytokines such as IFN-ɣ, IL-6, IL-12, and TNF-α signal through Janus kinase 1 (JAK1), with activation of JAK leading to phosphorylation of signal transducer and activator of transcription proteins (STATs) which subsequently modulates gene expression. The selective JAK1 inhibitor itacitinib was able to effectively reduce CRS-related cytokines produced by anti-CD19 CAR T cells in a dose-dependent fashion in both in vitro and in vivo models without impacting CAR T proliferation or cytolytic function. (73) This preclinical rationale for JAK-STAT inhibition has been translated to the clinical setting with several case reports noting resolution of refractory CRS after treatment with the JAK-inhibitor ruxolitinib. (74–77) Lastly, a variety of different inducible safety switches to either reversibly or irreversibly impair CAR T cell function have been designed as a mechanism for potentially managing severe toxicities; however, they have yet to become significant in clinical practice. (78) Pharmacotherapy for the management of CRS is summarized in Table 3.
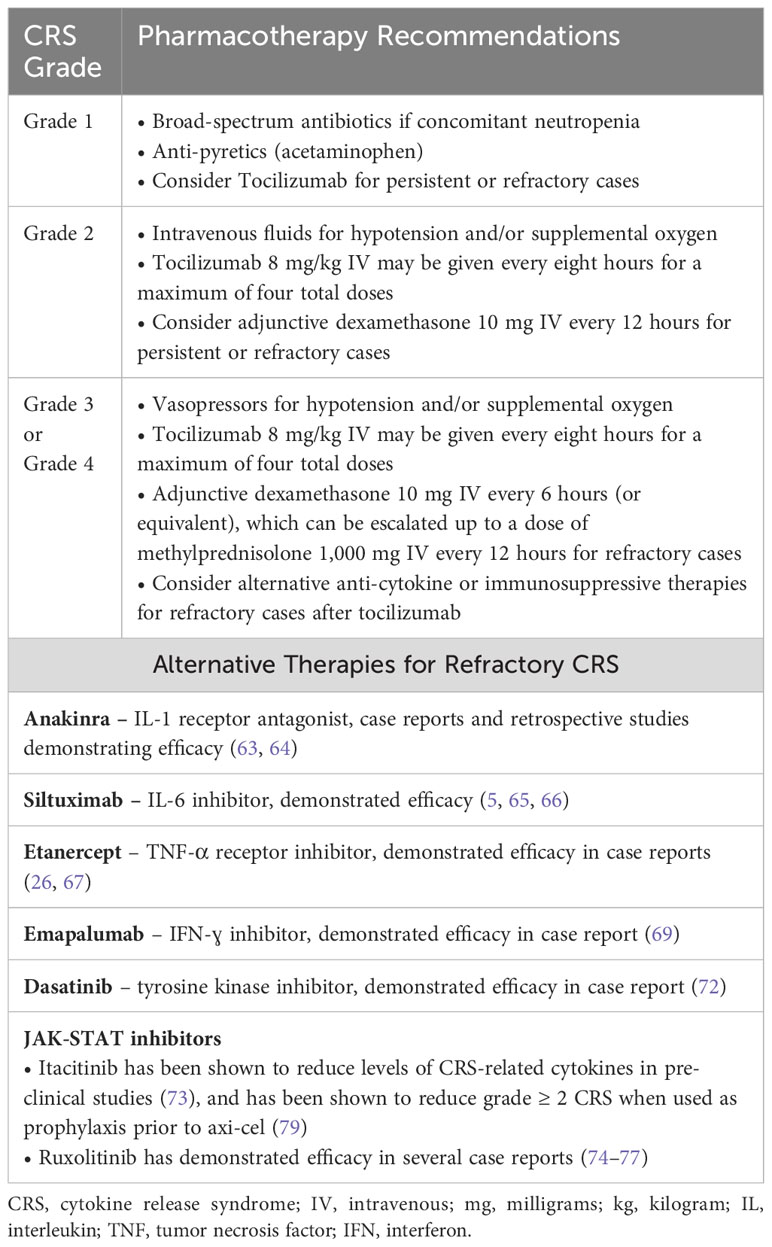
Table 3 Pharmacotherapy for the treatment of cytokine release syndrome.
Because the management of CRS often requires pharmacologic intervention with anti-cytokine therapy or glucocorticoids, there has been theoretical concern that such anti-inflammatory therapy may hamper the efficacy of CAR T cell therapy. Preclinical studies did not suggest hindrance of antitumor activity with IL-1, IL-6 and IFN-ɣ inhibition respectively. (30, 33) A retrospective analysis regarding the timing of tocilizumab administration for patients treated with anti-BCMA CAR T cell therapy demonstrated that patients receiving tocilizumab earlier (<12 hours from CRS onset) experienced a shorter median duration of CRS without negative effect on response or median progression-free survival outcomes. (80) There have been conflicting reports regarding corticosteroid therapy, with one retrospective study demonstrating no impact of corticosteroid therapy on response rate and CAR T expansion/persistence in B-ALL patients, whereas another retrospective analysis in LBCL patients suggested that a higher cumulative dose and prolonged use of corticosteroids had a negative impact on progression-free survival. (81, 82) Ultimately, randomized studies with long-term follow up will be required to discern whether certain toxicity management strategies have a positive or negative effect on CAR T cell therapy efficacy.
There has also been investigation into whether prophylactic or preemptive approaches to toxicity management can decrease the incidence and severity of both CRS and ICANS. Such attempts include early intervention with tocilizumab or corticosteroids for lower grade toxicity, as well as prophylaxis strategies with corticosteroids, tocilizumab, anakinra, or the JAK1 inhibitor itacitinib. (79, 83–90) Results from studies investigating prophylactic interventions are summarized in Table 4.
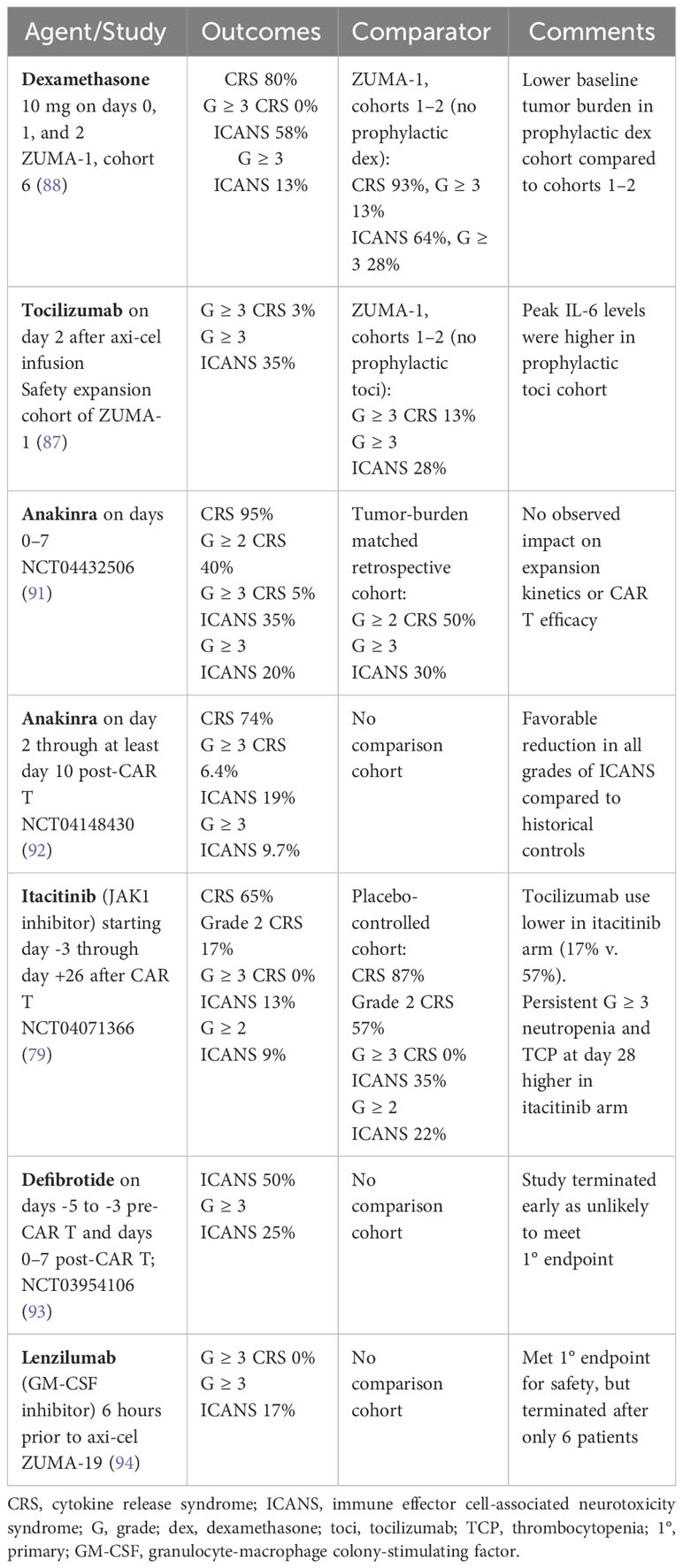
Table 4 Prophylactic approaches for mitigation of CRS/ICANS in patients treated with anti-CD19 CAR T.
Immune effector cell-associated neurotoxicity syndrome (ICANS): clinical manifestations and mechanisms
The ASTCT consensus grading criteria defines ICANS as “a disorder characterized by a pathologic process involving the central nervous system following any immune therapy that results in the activation or engagement of endogenous or infused T cells and/or other immune effector cells. Symptoms or signs can be progressive and may include aphasia, altered level of consciousness, impairment of cognitive skills, motor weakness, seizures, and cerebral edema.” Other forms of neurotoxicity potentially attributable to CAR T cell therapies such as headache, tremor, myoclonus, and asterixis are not considered as ICANS-defining, are graded separately by NCI-CTCAE criteria, and are managed symptomatically. (18) ICANS has been noted to occur concurrently with CRS, shortly after CRS subsides, in the absence of CRS, and in some instances with delayed onset up to one month after infusion. It often first manifests with tremor, mild expressive aphasia, impaired attention, dysgraphia, apraxia, and mild lethargy. Expressive aphasia appears to be the most common and specific symptom of ICANS. At higher grades of severity, ICANS may result in refractory seizures, necessitate intubation for airway protection, and can rarely result in fatal cerebral edema (18, 61).
Observations from imaging of the central nervous system in patients with ICANS has generally observed that findings are symmetric and have a propensity to affect uniquely susceptible brain regions. Involvement of the thalami and deep gray matter indicate that ICANS is likely prompted by a systemic process such as the inflammation mediated by inflammatory cytokines. Specific imaging findings such as leptomeningeal enhancement and T2 hyperintensity of the cerebral sulci, T2 hyperintensity and swelling of the bilateral thalami, and T2 hyperintensities in the supratentorial white matter can bear resemblance to injury patterns seen in other infectious or inflammatory central nervous system (CNS) conditions. Elevation of cerebrospinal fluid (CSF) protein on lumbar puncture samples has been commonly observed in patients with ICANS, and elevated cytokine levels in the CSF typically mirror cytokine elevations observed in serum samples. (95) Electroencephalogram (EEG) findings in patients with ICANS have most commonly been notable for generalized periodic discharges generated from both cortical and subcortical regions which may be induced by proinflammatory cytokines or directly mediated by T cells (96).
Elevated serum and CSF cytokines have been implicated in the pathophysiology of ICANS. Despite the strong association with CRS and inflammatory cytokines, the pathophysiology remains poorly understood in that ICANS can occur without antecedent CRS, and patients with severe CRS do not always develop ICANS. Based on serum and CSF cytokine analyses pooled from multiple human studies, IFN-ɣ, IL-15, IL-6, IL-10, GM-CSF, and IL-1RA appear to be most strongly associated with ICANS. Additionally, IL-2, IL-2Rα, CXCL10, and Granzyme b seem to be possibly associated with ICANS. (95) In a retrospective analysis of 53 patients treated with anti-CD19 CAR T for B-ALL, higher serum elevations of the proinflammatory cytokines IL1α, IL2, IL3, IL5, IL6, IL10, IL15, INF-γ, interferon-gamma inducible protein-10 (IP10), G-CSF, GM-CSF, and MCP1 by day three after CAR T was associated with severe neurotoxicity. Those with severe neurotoxicity were observed to have disproportionately high CSF levels of IL6, IL8, MCP1, and IP10, potentially indicating localized CNS production rather than just mirroring serum cytokine elevations (97).
Studies from animal models have also supported cytokine elevations in association with neurotoxicity. In the murine model of CRS and ICANS which demonstrated that IL-1 and IL-6 were primarily derived from activated monocytes and macrophages, treatment with tocilizumab did not abate symptoms of neurotoxicity when given at the onset of fever, whereas anakinra was able to improve neurotoxicity severity and survival. Furthermore, prophylactic tocilizumab did not protect the mice from delayed lethal neurotoxicity, whereas prophylactic anakinra prevented both CRS and neurotoxicity development. These murine studies suggested that IL-1 may be more integral to the pathophysiology of ICANS and further supported investigation of IL-1 inhibition for ICANS treatment and prevention in subsequent human studies. (30) In a rhesus macaque model of neurotoxicity after adoptive transfer of anti-CD20 CAR T cells, neurotoxicity was associated with serum elevations in IL-8, IL-8, IL-1RA, CXCL9, and CXCL11, and with disproportionately elevated CSF levels of IL-6, IL-2, GM-CSF, and VEGF levels. Pan-T cell encephalitis was observed in the brain parenchyma of the non-human primates experiencing neurotoxicity as evidenced by the accumulation of both CAR T and non-CAR T cells (98).
As seen with CRS, evidence from multiple studies have demonstrated endothelial activation and disruption as a mechanism underlying the pathophysiology of ICANS. Endothelial activation resulting from systemic cytokine release after CAR T cell therapy may predispose to the CNS microvascular dysfunction noted in ICANS. Perturbations in the angiopoietin (Ang)-Tie 2 axis have been associated with ICANS, as Ang-2 is released from endothelial cells upon activation from proinflammatory cytokines. Ang-1 binding to the Tie-2 receptor typically facilitates vascular quiescence and integrity; however, Ang-2 displaces Ang-1 from its receptor and induces vascular permeability. (95) In two separate retrospective analyses of patients treated with CAR T for B-ALL, patients who developed severe neurotoxicity were more likely to have laboratory findings consistent with DIC and capillary leak, lowers levels of Ang-1, higher levels of Ang-2, and a higher Ang-2:Ang-1 ratio suggestive of endothelial activation. (97, 99) Another retrospective study found that ICANS occurrence was associated with an increased PT, D-dimer, Factor VII level, and vWF level, and with decreased serum levels of fibrinogen and platelet count. (28) CSF samples from patients experiencing ICANS compared to baseline were notable for elevated protein concentrations, leukocyte counts (including CAR T cells), and serum cytokines reflective of increased blood-brain barrier permeability. When human brain vascular pericytes were exposed in vitro to IFN-ɣ and TNF-α, pericyte secretion of IL-6 and VEGF was observed which further increased blood-brain barrier disruption. An autopsy specimen from a patient who developed fatal neurotoxicity after anti-CD19 CAR T was notable for platelet aggregation, vWF binding in small capillaries, and multifocal vascular disruption with multifocal hemorrhages indicative of endothelial activation (99).
A subset of patients with ICANS develop excitatory neurotoxicity manifested by seizures. Glutamate and quinolinic acid are endogenous excitatory agonists of the N-methyl-D-aspartate (NMDA) receptor. Analysis of CSF from 13 patients with pre-treatment samples who then developed excitatory neurotoxicity revealed that both glutamate and quinolinic acid were significantly elevated during the neurotoxicity period compared to baseline. On the contrary, these endogenous NMDA agonists were not elevated in a patient without ICANS who underwent pre-treatment and day 14 lumbar punctures. These findings indicate that endothelial activation and increased blood-brain barrier permeability contributing to CSF cytokine elevation may be linked to elevations of these excitatory agonists in the CNS. (97) It has been proposed that neurotoxicity may reflect an on-target, off-tumor effect of anti-CD19 CAR T cell therapy given that CD19 expression was observed by single-cell RNA sequencing analysis of human brain mural cells, which support the vasculature. However, this appears to be an unlikely mechanism given that ICANS has subsequently been observed with CAR T therapies targeting alternative antigens such as BCMA and GPRC5D (11, 12, 100).
Factors associated with ICANS risk
Risk factors for ICANS intrinsic to the CAR T product identified from several patient cohorts include higher CAR T dose, higher peak CAR T expansion, and early/higher elevations of serum proinflammatory cytokines likely related to CAR T expansion kinetics. (97, 99) Additionally, the incorporation of a CD28 costimulatory domain as in axi-cel and brexu-cel appears to be associated with higher incidence and severity of ICANS compared to the other approved products that incorporate a 4–1BB costimulatory domain (Table 1) (4, 7, 9, 10).
In terms of patient and disease-specific factors, a high burden of disease prior to lymphodepletion has been associated with increased ICANS risk. While ICANS can occur in the absence of CRS, developing early and severe CRS after CAR T infusion has been associated with increased incidence and severity of ICANS. (97, 99) One analysis identified pre-existing neurologic comorbidities as a risk factor for subsequent ICANS development. (99) Another study observed that thrombocytopenia was significantly associated with an increased risk for grade ≥ 3 neurotoxicity. (101) The modified EASIX score at day three after CAR T infusion was found to be associated with onset of severe ICANS, though the pre-infusion m-EASIX score was not associated with the development of ICANS. Analyzing the individual components of the m-EASIX score, higher CRP and lower platelet count were both associated with ensuing onset of severe ICANS (46).
Grading and management of ICANS
While ICANS has become the preferred term for this specific manifestation of neurotoxicity due to immune effector cell therapies, the registrational trials for the approved CAR products were done at a time when the NCI-CTCAE criteria for neurotoxicity were being utilized and thus there is more heterogeneity in the way neurotoxicity has been defined, graded, and reported in the literature. (18, 55–60) The CARTOX criteria provided the first step toward streamlining the objective grading of neurotoxicity with the incorporation of a screening tool called the CARTOX-10, which included a ten point scale to assess the patient for alterations in speech, orientation, concentration, and handwriting. The complete CARTOX neurotoxicity grading system involved evaluation of level of consciousness, motor symptoms, seizures, and for signs of elevated intracranial pressure (ICP) with CSF opening pressure measurement and papilledema grading, which can be challenging to obtain and objectively quantify. (49) The ASTCT consensus criteria incorporates a modified version of the CARTOX-10 screening tool called the Immune Effector Cell-Associated Encephalopathy (ICE) score (Table 5), and simplifies assessment of the other domains to facilitate more objective ICANS grading (Table 6) (18).

Table 5 The immune effector cell-associated encephalopathy (ICE) score.
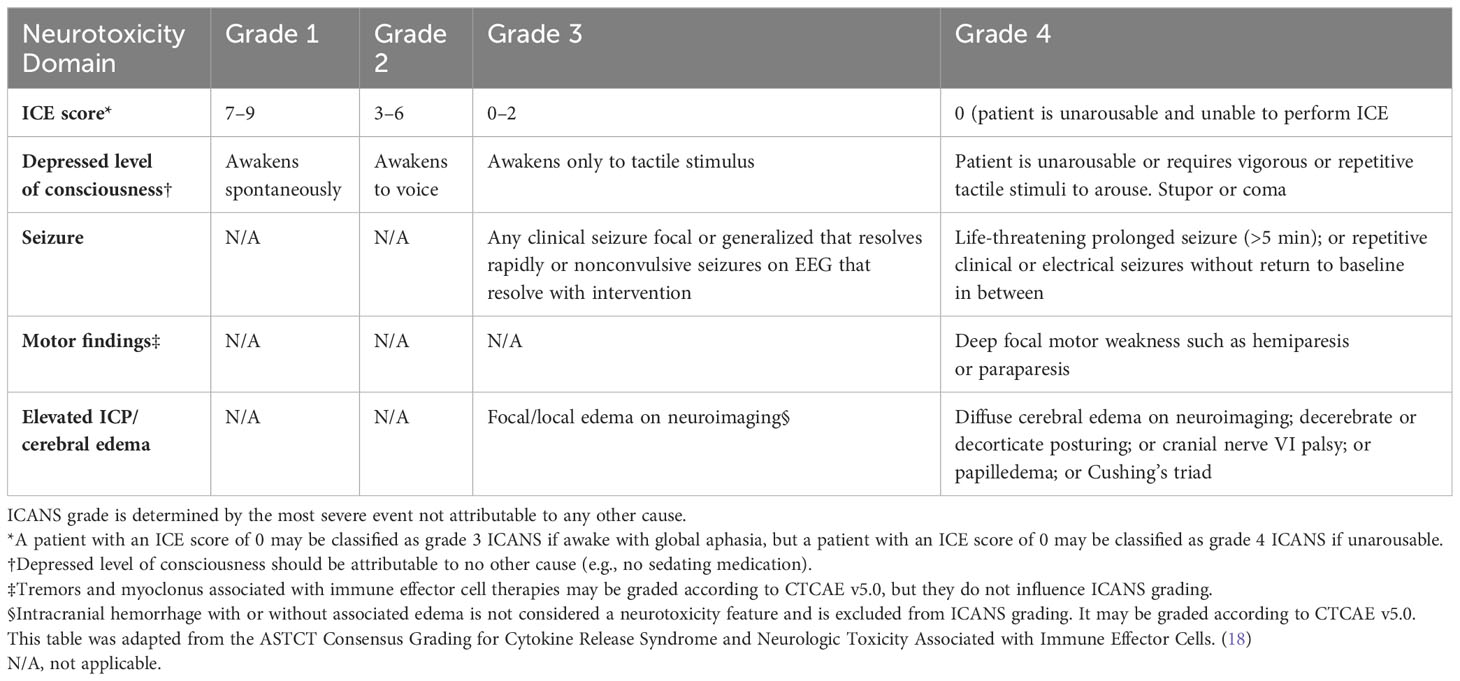
Table 6 ASTCT consensus grading for ICANS in adult patients.
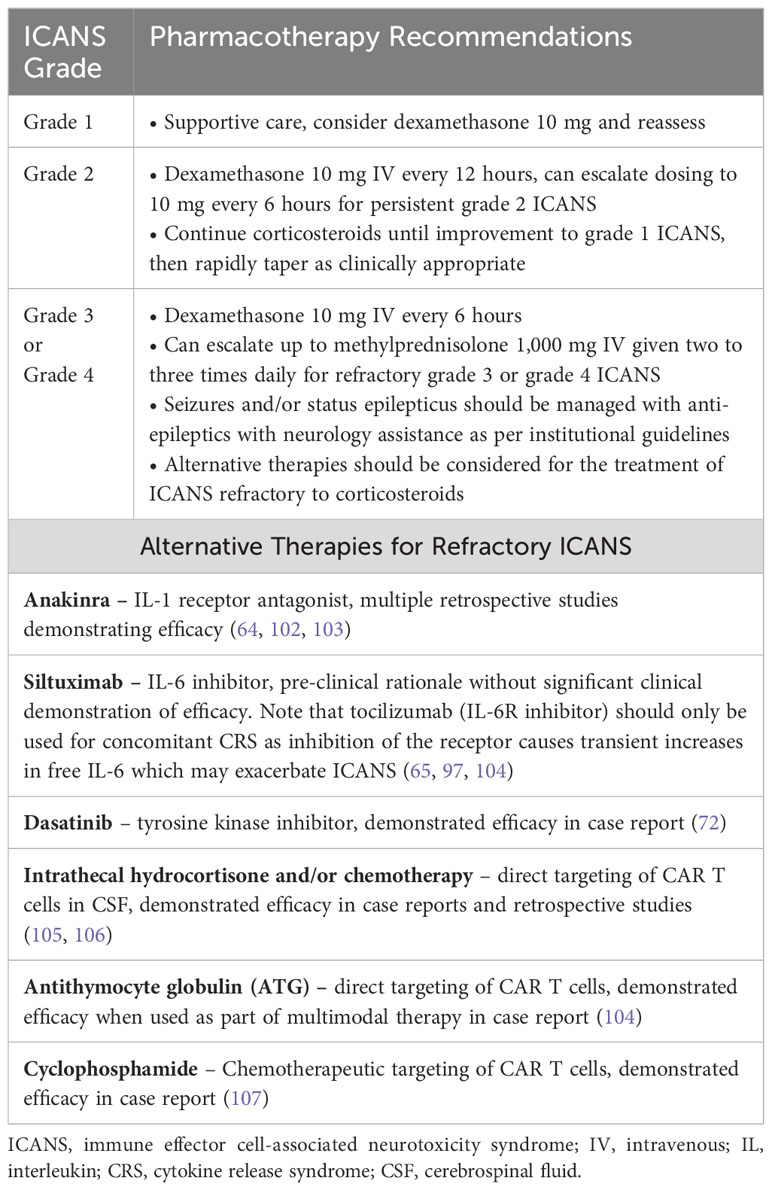
Table 7 Pharmacotherapy for the treatment of ICANS.
The incidence of neurotoxicity observed with each approved CAR T product in their respective registrational trials is summarized in Table 1; however, it is important to note that the ASTCT consensus criteria were not yet implemented for these trials. The ASTCT consensus grading criteria for ICANS is outlined in Table 6. The overall ICANS grade is dictated by the most severe grade observed across the five neurotoxicity domains (ICE score, depressed level of consciousness, seizure, motor findings, and elevated ICP/cerebral edema). Grade 1 ICANS is defined by an ICE score in the range of 7–9 and the patient must be able to awaken spontaneously. An ICE score of 3–6 constitutes grade 2 ICANS, with the patient being able to awaken to voice. Any clinical or electrical seizure that resolves rapidly with intervention or focal areas of edema on neuroimaging qualify as grade 3 ICANS, as does an ICE score ranging 0–2 if patient awakens only to tactile stimulus. A patient with an ICE score of 0 who is unarousable and unable to perform the ICE questionnaire is defined as having grade 4 ICANS. Other qualifiers for grade 4 ICANS include life-threatening prolonged seizures or repetitive clinical or electrical without interim return to baseline, deep focal motor weakness, diffuse cerebral edema on neuroimaging, decerebrate or decorticate posturing, cranial nerve VI palsy, papilledema, and Cushing’s triad. Patients with grade 4 ICANS often require intubation and mechanical ventilation for airway protection and seizure management, but this should not also be classified as grade 4 CRS if the need for mechanical ventilation is not secondary to refractory hypoxia (18).
Pharmacotherapy for the management of ICANS is summarized in Table 7. The prescribing information for each approved CAR T includes guidance for the management of neurologic toxicities. (55–60) After CAR T infusion, patients should be monitored closely with documentation of the ICE score and assessment of motor strength at least twice daily. Patients should have serial laboratory monitoring (CBC, CMP, coagulation studies, inflammatory markers) and severe hyponatremia should be corrected. Initiating anti-epileptic therapy for seizure prophylaxis is common at most institutions. Neurology consultation should be sought after noted onset of ICANS, and grade ≥ 2 neurotoxicity should prompt further evaluation with neuroimaging and EEG. Lumbar puncture for CSF analysis and opening pressure can be considered for patients with grade 2 ICANS and strongly encouraged for those with grade ≥ 3 ICANS. Tocilizumab may be administered for situations where CRS is concurrent with ICANS, but in the absence of CRS the mainstay of ICANS treatment is supportive care and corticosteroid therapy (61).
Patients with grade 1 ICANS can often be monitored and managed with supportive care alone, but it is reasonable to treat with dexamethasone 10 mg and reassess. Increasingly severe ICANS is generally managed with increased corticosteroid dosing, such as dexamethasone 10 mg every 12 hours for grade 2 ICANS, and escalation to 10 mg every 6 hours for persistent grade 2 or development of grade 3 ICANS. Corticosteroids should be continued until improvement to grade 1 ICANS and then rapidly tapered as deemed clinically appropriate. Patients with grade ≥ 3 ICANS should be monitored in an ICU setting. Seizures and status epilepticus should be managed with neurology expertise per institutional guidelines. With persistent grade 3 ICANS or emergence of grade 4 toxicity, corticosteroid dose can be further increased to 1,000 mg methylprednisolone given two to three times daily. Alternative therapies should be considered for management of ICANS refractory to corticosteroids, which may include anakinra, ruxolitinib, siltuximab, cyclophosphamide, antithymocyte globulin, or intrathecal hydrocortisone with or without chemotherapy. (61) As with CRS, attempts to prevent or mitigate the severity of ICANS prior to its onset are being investigated. Table 4 summarizes recent investigations into such prophylactic approaches.
There is a lack of high-quality evidence to inform the management of ICANS refractory to corticosteroid therapy, and it can be challenging to isolate the effect of a particular intervention in the setting of patients receiving simultaneous or overlapping therapies. With preclinical studies suggesting a fundamental role for IL-1 in the pathogenesis of ICANS, anakinra has become a preferred agent for the management of refractory ICANS. (30) Clinical responses with ICANS improvement or resolution have been reported in retrospective analyses from multiple institutions, though timing of initiation, dosing, and concurrent therapies have been heterogeneous. (64, 102, 103) A recent retrospective analysis across nine institutions identified 40 patients treated with anakinra for grade ≥ 2 ICANS without improvement on corticosteroid therapy, which was demonstrated to be well tolerated. Treatment with a high-dose anakinra regimen (defined as >200 mg/day IV) was associated with improved clinical outcomes compared to a lower dose (100–200 mg/day), as evidenced by 0% treatment-related mortality in the high-dose group compared to 47% in the low-dose group, and a median cumulative incidence of CRS/ICANS resolution from the time of anakinra initiation of seven days in the high-dose group compared to median time to resolution not being reached in the low-dose due to the higher rate of treatment-related mortality (64).
The IL-6 receptor inhibitor tocilizumab has not been shown to be effective in the treatment of ICANS. Due to its inhibition of IL-6R rather than IL-6 itself, transient increases in serum and CSF IL-6 have been documented after tocilizumab initiation and it has been hypothesized that this may exacerbate ICANS as tocilizumab is not thought to penetrate the blood-brain barrier well. For example, in 16 patients with severe neurotoxicity who received tocilizumab, 56% had their peak neurotoxicity grade occur after the first dose of tocilizumab administration. (97) Whereas siltuximab is an antagonist of IL-6, there is some rationale that treatment with this agent would be advantageous for ICANS outcomes compared to tocilizumab. (65) However, clinical experience with siltuximab specifically for the treatment of refractory ICANS has not been well documented. (65, 104) The tyrosine kinase inhibitor dasatinib has demonstrated an ability to impair CAR T proliferation and downstream cytokine secretion in both in vitro and in vivo models, supporting a rationale for clinical use. (70, 71) In one patient who received dasatinib for refractory grade 4 ICANS with concurrent grade 3 CRS, treatment with dasatinib led to a rapid improvement in neurologic function and allowed for extubation one day later (72).
In addition to interfering with cytokine signaling, direct targeting of CAR T cells has been explored for the management of refractory ICANS. A case report of two patients treated with intrathecal hydrocortisone plus chemotherapy for corticosteroid-refractory grade 3 and 4 ICANS respectively led to substantial clinical improvement. (105) A retrospective analysis noted that for seven patients who received early intrathecal hydrocortisone with or without intrathecal chemotherapy within five days of developing high-grade ICANS, all seven patients had subsequent resolution of ICANS. Of the four patients with refractory high-grade ICANS who did not receive intrathecal therapy or received it late (>5 days after onset), only two patients recovered from ICANS. Additionally, the median duration of steroid use and cumulative steroid dose were lower for the patients receiving early intrathecal therapy. (106) In one case of refractory grade 4 ICANS with cerebral edema, three doses of rabbit antithymocyte globulin (ATG) were given as part of the multimodal therapy with a temporal suggestion of benefit as the patient was extubated five days after starting ATG and ICANS had resolved completely within two weeks. (104) Another case described a patient who developed refractory rebound neurotoxicity characterized by the development of brain MRI findings consistent with posterior reversible encephalopathy syndrome (PRES) which coincided with a continued rise to peak circulating CAR T cell levels in the peripheral blood. The patient was treated with cyclophosphamide 1.5 g/m2 to target the CAR T cells and significant clinical improvement was noted over the subsequent days and with resolution of MRI abnormalities four weeks later (107).
Other neurotoxicities distinct from ICANS
Neurotoxicity distinct from ICANS has also been observed after CAR T cell therapies. Of particular concern are the described movement and neurocognitive treatment-emergent adverse events (MNTs) that have been described after administration of anti-BCMA CAR T. These defined MNTs consist of a potential range of symptoms falling into one of three categories: movement disorder (e.g., micrographia, resting tremor), cognitive impairment (e.g., memory loss, disturbance in attention), or personality changes (e.g., reduced facial expression, tremors). For patients treated with cilta-cel on the CARTITUDE-1 study, patients were defined as having MNTs if they reported symptoms in at least two of these categories. Symptoms must have occurred after recovery from CRS and/or ICANS and felt to be attributable to the CAR T cell therapy. Using these criteria, five patients (5%) developed MNTs after cilta-cel with median onset of 27 days post-infusion, and a median onset of 17 days (range, 3–94) after resolution from CRS/ICANS. These toxicities were generally not responsive to corticosteroids or other supportive measures, which included anakinra, siltuximab, systemic and intrathecal chemotherapy, dasatinib, and carbidopa/levodopa for parkinsonism. One patient’s death was attributed to such neurotoxicity, and only one patient’s MNTs were resolving at data cutoff. Patients who experienced MNTs were retrospectively determined to have at least two of the following features: high tumor burden, grade ≥ 2 CRS or any grade ICANS, and high CAR T expansion/persistence in peripheral blood. After mitigation strategies were implemented, which included more aggressive bridging therapy to reduce disease burden prior to cilta-cel infusion, early intervention for CRS/ICANS, handwriting assessments for early detection, and an extended monitoring period for neurotoxicity, incidence of MNTs has been noted to be < 1% for patients treated with cilta-cel on subsequent trials. (108)
While the clinical manifestations of MNTs seen after anti-BCMA CAR T can have a presentation similar to Parkinson disease, autopsy results from two of the patients who developed such toxicity after cilta-cel suggest an alternative pathophysiology. Both patients were noted to have normal pigmentation in the substantia nigra and symptoms did not improve with a trial of carbidopa/levodopa. There was notable focal gliosis and T cell infiltrate (CD8 > CD4) in the basal ganglia. It was not determined whether these were CAR-positive T cells, however these patients did have persistent CAR T elevations in the blood and CSF. (108, 109)One patient was noted to have detectable BCMA expression on neurons and astrocytes in the caudate nucleus of the basal ganglia, as well as in neurons of the adjacent frontal cortex. (109) This autopsy finding coupled with RNA sequencing analysis of healthy donors observing low levels of BCMA RNA expression in the basal ganglia suggest a possible on-target, off-tumor mechanism for this form of neurotoxicity. (110) While more commonly observed after cilta-cel, MNTs manifesting as parkinsonism have also been observed after treatment with ide-cel. (59) Clinical management of MNTs remains challenging, as they have generally not been responsive to corticosteroids or anti-inflammatory therapies. One case report demonstrated that treatment with cyclophosphamide 2 grams/m2 aimed at significantly reducing the persistent CAR T cell population resulted in resolution of refractory parkinsonism symptoms. (111)
In addition to ICANS and MNTs, other neurotoxicities observed after anti-BCMA CAR T cell therapy include peripheral sensory and motor neuropathies, ataxia, nystagmus, facial nerve paralysis and other cranial nerve palsies, diplopia, and concentration impairment. (108) CAR T cell therapy targeting the G protein-coupled receptor, class C, group 5, member D (GPRC5D) are under clinical investigation for the treatment of multiple myeloma with promising preliminary results. At the highest dose-level evaluated, two patients developed dose-limiting toxicities of cerebellar toxicity characterized by wide-based gait, saccadic eye movements, appendicular and truncal ataxia, and dysarthria. Treatment with steroids and intravenous immune globulin (IVIG) resulted in stabilization of symptoms but without resolution. Given that microarray data from six healthy donors noted that GPRC5D expression was most enriched in the olivary nucleus, it is possible that this represents on-target, off-tumor toxicity. (100) Other rare distinct neurotoxicities observed after CAR T cell therapy include transverse myelitis/myelopathy, human herpesvirus 6-associated myelitis, acute leukoencephalopathy, central diabetes insipidus, Guillain-Barre-like syndrome, persistent postural tremors resembling essential tremor, new onset migraine headaches, and various peripheral neuropathies (112–117).
Immune effector cell-associated hemophagocytic lymphohistiocytosis-like syndrome (IEC-HS): clinical manifestations, risk factors, and mechanisms
Secondary hemophagocytic lymphohistiocytosis (HLH) can occur after CAR T due to overactivation of the immune system. Typically characterized by hyperferritinemia, coagulopathy, hepatic dysfunction and cytopenias among other HLH-like manifestations, this is a lesser known but potentially fatal complication of CAR T that has become increasingly recognized. Consequently, US Food and Drug Administration package inserts incorporate the black box warning risk of HLH for both commercially approved BCMA -targeted CAR T cell therapies. (59, 60) The chronology and symptomatology of HLH-like toxicities can be difficult to distinguish from severe CRS. The earliest of the reports described laboratory features mimicking HLH/MAS superimposed on severe CRS in a 7-year-old girl with recurrent B-ALL who was treated with CD19 CAR T cell therapy. (26) Subsequent reports have demonstrated that HLH-like syndrome can occur as a late manifestation when signs of conventional CRS appear to be resolving. (118, 119) Recognizing the need to better delineate HLH-like toxicities following CAR T and to provide a framework for cross-trial comparisons, an ASTCT expert panel recently named this complication as IEC-HS, which is defined as “the development of a pathological and biochemical hyperinflammatory syndrome independent from CRS and ICANS that (1) manifests with features of MAS/HLH (2), is attributable to immune effector cell therapy, and (3) is associated with progression or new onset of cytopenias, hyperferritinemia, coagulopathy with hypofibrinogenemia, and/or transaminitis.” (120)
Clinical suspicion and early recognition of IEC-HS is critical. CRS in moderate to severe forms may manifest with overlapping features of IEC-HS (e.g., elevated ferritin, elevated transaminases, hemophagocytosis, coagulopathy, cytopenia, and multi-organ dysfunction) with indistinguishable cytokine and proteomic profiles. (18, 118, 121) Given significant overlap, it is critically important to distinguish between IEC-HS and prolonged severe or recurrent CRS as treatment implications differ between the two. Timing of IEC-HS in relation to antecedent CRS can vary, but it is a distinct manifestation independent from CRS that occurs once CRS is resolved/resolving. While CRS typically develops and resolves within 2 weeks of CAR-T administration, IEC-HS arises after CRS and may persist for weeks. The consensus panel strongly cautions against using IEC-HS nomenclature to describe patients with severe CRS involving multiorgan dysfunction. Malignancy progression, underlying rheumatologic/metabolic disorders, severe infections, or other etiologies that can trigger secondary HLH should be excluded (120).
The HLH-2004 criteria and the H score, the common scoring systems used for diagnosis of secondary HLH, have low validity and applicability in identifying IEC-HS as most patients treated with CAR T have alternative explanations for traditional criteria and thresholds used for diagnosing HLH (e.g., elevated ferritin, fever and cytopenias). (122–124) In 2017, a CARTOX Working Group was formed that proposed a diagnosis of HLH should be made if the ferritin levels peak at >10,000 ng/mL within the CRS window of 5 days after CAR T-cell therapy, and if the patient develops any two of the following: grade 3 or greater organ toxicities involving the liver, kidneys, or lungs, or hemophagocytosis in the bone marrow or other organs (49). Considering vast heterogeneity in baseline ferritin values among patients undergoing CAR T, waiting to reach an arbitrarily set threshold for ferritin elevation could cause unnecessary delays in diagnosis of IEC-HS, therefore the ASTCT working group has not incorporated specific ferritin values in their proposed diagnostic criteria of IEC-HS. Although there is no specific cutoff, a substantial elevation (defined as >2 times the upper limit of normal or baseline at time of infusion) or rapidly rising serum ferritin is a prerequisite for the diagnosis of IEC-HS. Acknowledging that not all patients with antecedent or ongoing CRS will progress to IEC-HS, the ASTCT working group considers the timing as a key factor in diagnosing IEC-HS. While CRS develops within 14 days post-infusion, IEC-HS is often of delayed onset and is associated with progression or new onset laboratory abnormalities or clinical deterioration as CRS is resolved/resolving. Common manifestations of IEC-HS include hepatic transaminase elevation, hypofibrinogenemia, other coagulation abnormalities, hemophagocytes in bone marrow or other tissue, cytopenias (new onset, worsening, or refractory), elevated LDH, direct hyperbilirubinemia, and new onset splenomegaly. Other manifestations that may be present include renal insufficiency, pulmonary complications, neurotoxicity, fever, and hypertriglyceridemia (120).
Risk factors that have been implicated in the development of IEC-HS include baseline disease burden, CAR T proliferation dynamics, immune resistance mechanisms, target antigen, CAR T cell construct, costimulatory domain, T cell selection, cell dose and patient characteristics (baseline inflammation, immune suppression, cytopenias, genetic predisposition). (45, 118, 119, 125, 126). While the underlying pathophysiology of IEC-HS remains to be fully elucidated, similarities to CRS have been observed. The sustained T cell activation via engineered CAR recognition of tumor antigen results in a hyperinflammatory response, T cell activation and proliferation, cytokine release, and resultant macrophage activation and release of soluble factors creating a positive feedback loop. (120) Like primary and secondary HLH, where defects in cytolytic function of NK cells and CTLs can lead to pathologic T-cell expansion, it has been postulated that the profound and persistent NK cell lymphopenia in concert with heightened CAR T cell expansion and delayed T cell contraction can predispose select patients with CRS to develop a secondary hyperinflammatory response in form of IEC-HS. (118) In the context of 59 patients infused with CD22 CAR T cells where a substantial proportion developed IEC-HS, disproportionately low absolute number of NK cells relative to CD8 T cells in peripheral blood pre-infusion, and higher bone marrow T cell to NK cell ratio was noted in patients who later developed IEC-HS. (118) In line with this observation, a recent murine model of HLH demonstrated that both CTL and cytotoxic NK cells contribute to the development of HLH-like syndrome in mice after infection with lymphocytic choriomeningitis virus (127).
Certain cytokines (e.g., IFN-γ, IL-6, IL-10, IL-18, sIL-2R, CXCL9), and proteomic profiles overlap between HLH and severe CRS. (118, 121). Unlike in CRS, IL-1 appears to be a central player in IEC-HS. IL-1β levels were particularly high among those patients with IEC-HS, which supports the use of anakinra, an IL-1 receptor antagonist, for management of this syndrome. (118) IFN-ɣ, a mediator of systemic inflammation and macrophage activation, is thought to play a key role in development of IEC-HS, which is supported by the high serum level observed in a patient who developed IEC-HS post tisa-cel and was treated with emapalumab with rapid defervescence and improvement in clinical and laboratory parameters (128).
Grading and management of IEC-HS
The ASTCT expert panel developed a grading schema for IEC-HS formed based on the NCI-CTCAE category “Immune system disorder, other.” Grades of IEC-HS range from 1–5 with grade 1 toxicity representative of asymptomatic or mild symptoms not requiring intervention, and grade 5 reflecting death due to IEC-HS. Toxicity warranting intervention is labeled as grade 2, with grade 3 toxicity being severe but not immediately life-threatening, and grade 4 IEC-HS is considered life-threatening (e.g., life-threatening bleeding or hypotension, respiratory distress requiring intubation, dialysis indicated for acute kidney injury) (120).
The ASTCT expert panel has proposed a stepwise approach to the treatment of IEC-HS. Therapy should be initiated prior to the development of life-threatening complications. The use of tocilizumab or siltuximab in patients with IEC-HS without CRS is discouraged. First line therapy with the IL-1 receptor antagonist anakinra, with or without corticosteroids, is recommended given its potential to disrupt secondary HLH/MAS associated with rheumatologic and other disorders. Anakinra has a short half-life allowing rapid titration to effect, a well-established side effect profile, and pharmacokinetic data to support both intravenous and subcutaneous use. (120) Anakinra has been observed to be efficacious in IEC-HS in patients treated with CD19 CAR T cell therapy for pediatric B-ALL, DLBCL, and mantle cell lymphoma. (129, 130) Anakinra can be given at a dose of 100–200 mg every 6–12 hours in adults, or up to 10 mg/kg IV daily. If there is no improvement within 48 hours despite escalation of anakinra to target dose with concurrent corticosteroids, initiation of the JAK1/2 inhibitor ruxolitinib as second line therapy is recommended. Ruxolitinib, via inhibition of JAK/STAT signaling pathways, can reduce secretion of the pro-inflammatory cytokines implicated in the development of IEC-HS such as IL-2, IL-6, and IFN-γ. (120) Case reports and retrospective studies have demonstrated its clinical efficacy for primary and secondary HLH (131, 132).
For severe IEC-HS refractory to anakinra and ruxolitinib, additional agents like the anti-IFN-γ monoclonal antibody emapalumab or low-dose etoposide chemotherapy can be considered. Treatment with emapalumab may be more appropriate in the context of significantly elevated serum IFN-γ, whereas etoposide to deplete T cells may be more applicable for a documented persistent elevation of CAR T cells. (120) Emapalumab binds both free and receptor-bound IFN-γ, and elevated levels of each have been documented in both primary HLH and CAR-T associated toxicities. (133, 134) It is FDA-approved for primary HLH in children and adults in the setting of refractory, recurrent or progressive disease or intolerance to other standard treatments. The initial dosing is a twice-weekly intravenous infusion of 1 mg/kg. Evidence pertaining to its efficacy in IEC-HS is limited to case reports describing the clinical recovery of patients with severe IEC-HS and CRS refractory to corticosteroids and multiple anti-cytokine treatments who received emapalumab as salvage therapy. (68, 128) The topoisomerase inhibitor etoposide is cytotoxic to T cells and has been shown to have efficacy in primary and secondary HLH in protocols such as HLH-94. (135, 136) The suggested dosing in adults is 50–100 mg/m2 once to twice weekly. (137) Antithymocyte globulin (ATG) and alemtuzumab have been proposed as salvage therapy options in primary pediatric HLH, but their use for IEC-HS is not well established (120).
Patients with IEC-HS require frequent clinical assessment, daily laboratory studies (CBC, CMP, coagulation studies, inflammatory markers), and diligent supportive care. Such supportive care includes transfusion support for cytopenias, cryoprecipitate for hypofibrinogenemia, vitamin K and/or fresh frozen plasma for severe coagulopathy, and infectious disease consultation for both diagnostic purposes and infection prophylaxis recommendations given prolonged immunosuppression. As clinical and laboratory parameters stabilize, tapering immunosuppression is encouraged while monitoring for recrudescence of symptoms. Further retrospective and prospective studies are necessary to characterize and differentiate severe CRS complicated by HLH-like manifestations versus IEC-HS, and to inform optimal monitoring and therapeutic strategies (120).
Immune effector cell-associated hematotoxicity (ICAHT): clinical manifestations, risk factors, and mechanisms
Anemia, thrombocytopenia, and leukopenia with neutropenia are among the most common toxicities observed after treatment with CAR T cell therapy. The incidence of cytopenias observed with each approved CAR T cell therapy on the respective registrational study is summarized in Table 1, noting the limitations of comparisons among trials. In a pooled real-world analysis of 235 patients treated with anti-CD19 CAR T, incidence of grade ≥3 neutropenia, anemia, and thrombocytopenia were 91%, 69%, and 62% respectively. While early cytopenias can be attributed to the effects of lymphodepletion chemotherapy prior to CAR T, persistent or late recurrence of cytopenias is likely multifactorial in nature and can result in significant transfusion dependence, bleeding complications, and infectious sequelae (138–140).
Three distinct clinical phenotypes of neutrophil recovery have been observed after CAR T. The most common recovery pattern observed in a multicenter retrospective analysis was that of intermittent recovery in 52%, consisting of initial recovery with G-CSF stimulation for the neutropenia following lymphodepletion followed by a biphasic second reduction in absolute neutrophil count (ANC) in month two post-CAR T, and then with another recovery in the subsequent weeks. A subset of 25% demonstrated quick recovery from the initial neutropenia, whereas 23% of patients displayed an aplastic phenotype characterized by a protracted course of neutropenia despite G-CSF use (139).
A multitude of risk factors have been associated with the development of cytopenias after CAR T cell therapy. Such risk factors include malignancy-related features (e.g., underlying disease and disease burden), number and type of prior therapies, baseline bone marrow characteristics (e.g., pre-existing cytopenias, clonal hematopoiesis of indeterminate potential [CHIP]), inflammatory status prior to CAR T, and factors intrinsic to the CAR T product. Additionally, post-infusion toxicities such as CRS, IEC-HS, or infection influence the risk of developing prolonged cytopenias. (140) Calculated prior to lymphodepletion (day −5), the CAR-HEMATOTOX score incorporates both factors that relate to the baseline inflammatory state (e.g., CRP, ferritin) and the patient’s hematopoietic reserve (e.g., hemoglobin, ANC, platelet count). This model was first developed to predict severe hematotoxicity in relapsed/refractory LBCL and was then subsequently extended to patients receiving CAR-T therapy for mantle cell lymphoma and multiple myeloma. High-risk patients with score ≥2 had a longer duration of neutropenia, higher incidence of severe thrombocytopenia and anemia, increased rates of severe infection, higher non-relapse mortality, and inferior treatment outcomes compared to their low-risk counterparts (score 0–1) (139–142).
There are several factors to consider regarding the pathophysiology of ICAHT. Baseline cytopenias prior to CAR T treatment likely reflect underlying impairment of the hematopoietic stem and progenitor cell compartment secondary to prior cytotoxic therapies. The presence of preexisting CHIP may contribute to an underlying inflammatory state and contribute to the subsequent development of prolonged cytopenias. (143, 144) Patients with cytopenia prior to CAR T are more likely to have increased bone marrow disease burden, which in turn is correlated with a longer time to hematologic recovery after CAR T. Patients with delayed or prolonged cytopenias after anti-BCMA CAR T were shown to have a higher percentage of persistent CAR T cells in the bone marrow aspirate two months after infusion, suggesting that CAR T-mediated inflammation may contribute to these cytopenias. (145) This is further supported by the observation that patients developing hyperinflammatory complications such as CRS and IEC-HS are more likely to develop prolonged cytopenias. (146, 147) In patients with an aplastic phenotype, single cell sequencing studies have shown an inflammatory micromilieu in the form of oligoclonal CAR-T-cell expansion, T-cell receptor restriction, clonally expanded CXCR1hi, and IFN-γ expressing cytotoxic T cells (148, 149) Additionally, reactivation of certain viruses (human herpesvirus 6, cytomegalovirus, Epstein-Barr virus, adenovirus etc.) in the setting of immune suppression can directly suppress hematopoietic cell recovery (140).
Grading and management of ICAHT
The NCI-CTCAE criteria have been used for grading the hematologic adverse events in the respective registrational CAR T studies. With the goal of more accurately representing the neutropenia observed after T-cell-based immunotherapies, the European Hematology Association (EHA) and European Society for Blood and Marrow Transplantation (EBMT) formed an expert panel that established the terminology and grading system for ICAHT. Early ICAHT is defined as occurring within the first 30 days after CAR T, and late ICAHT refers to cytopenia observed after day 30. The grading system (grade 1–4) incorporates both depth and duration of neutropenia for early ICAHT, whereas late ICAHT grading is reflective of the depth of neutropenia (140).
The EHA/EBMT expert panel recommends the use of the CAR-HEMATOTOX score to identify patients at elevated risk for developing severe neutropenia and continued use of current grading systems for anemia and thrombocytopenia. A tiered approach to diagnostic evaluation has been proposed for persistent cytopenia. Initial workup includes ruling out common infectious etiologies, vitamin deficiencies, contribution from medications, and persistent inflammatory etiologies (CRS/IEC-HS). For those with sustained ICAHT, additional evaluation to rule out less common infectious etiologies and bone marrow aspiration and biopsy to investigate underlying bone marrow disease is warranted (140).
Management of ICAHT consists primarily of transfusion support, growth factor administration, and infectious prophylaxis. Administration of G-CSF has been demonstrated to shorten the duration of severe neutropenia and reduce infectious complications. (140) Retrospective analyses from real-word data sets have demonstrated faster neutrophil recovery and an acceptable safety profile with early G-CSF without increases in the rate of high-grade CRS or ICANS. (150–152) Notably, G-CSF did not impact CAR-T expansion or efficacy. (153) Most patients will adequately respond to growth factor support with count recovery. (146) While data for patients treated with CAR T remains limited, thrombopoietin (TPO) agonists have been given successfully to patients with prolonged or late thrombocytopenia, and have also been noted to improve anemia in some instances. (140) B-cell aplasia and hypogammaglobulinemia associated with CAR T can compound the risk of infections, therefore IgG replacement should be considered for those with high infection risk or experiencing recurrent infections with a recommended goal IgG trough > 400 mg/dL. (154) For patients with persistent ICAHT refractory to the aforementioned interventions, hematopoietic stem cell boost with cryopreserved autologous CD34+ stem cells has been observed to result in improvement or resolution of neutropenia in a majority of cases (155–157).
Secondary malignancies observed after CAR T cell therapy
The US Food and Drug Administration (FDA) released a statement on November 28, 2023, regarding the potential risk of secondary T cell malignancies in patients treated with CAR T cell therapy, which include CAR-positive T cell lymphoma. There have been 20 cases of T cell malignancies reported after CAR T cell infusion in approximately 8,000 cases conveyed to the FDA Adverse Events Reporting System. With an estimated total number of commercial CAR T infusions greater than 30,000, the suggested incidence of secondary T cell malignancies is quite low, but further studies are needed to better define the true incidence and to further elucidate the pathogenesis. Furthermore, the Centers for International Blood and Marrow Transplant Research (CIBMTR) database has documented 565 cases of secondary or subsequent malignancies in 485 individual patients out of a total of 8,060 enrolled in post-authorization safety studies. Such malignancies are most commonly non-melanomatous skin cancers and therapy-related myelodysplastic syndrome (MDS) or acute myeloid leukemia (AML). (158) For example, 10 of the 97 patients who received cilta-cel on the CARTITUDE-1 study have since been noted to have secondary myeloid malignancies. (60, 159) As such, the FDA has stated that patients who have received CAR T cell therapy should be monitored indefinitely for the development of subsequent malignancies but that benefits of CAR T in treating the malignancy at hand continues to outweigh such risks. (158) It is particularly challenging to determine the causal association of CAR T with the development of secondary malignancy given the existing confounders related to prior systemic therapies received (including high-dose alkylating chemotherapy for autologous stem cell transplant), patient age, and immortal time bias. Long-term follow up of patients enrolled to studies with CAR T in earlier line treatment settings may help to elucidate this further (159).
Conclusion
In conclusion, CAR T cell therapy has revolutionized the treatment of hematologic malignancies and efforts are ongoing to extend the benefits of cellular therapies to patients with solid tumors and rheumatologic disease. Despite remarkable efficacy, the development of potentially severe immune effector cell-associated toxicities require that patients receive close monitoring at centers with expertise in the diagnosis and management of these adverse effects. Significant progress has been made in the understanding of the underlying pathophysiology of such toxicities by means of retrospective analyses and animal models, which have informed therapeutic approaches to toxicity management. The development of consensus guidelines for the definition and grading of these distinct toxicities will further facilitate future cross-trial comparisons and prospective studies, which will be critical for optimizing risk-stratification and management approaches. For the future, active areas of investigation include the optimization of preclinical models to better elucidate the mechanisms of CAR T toxicities, identification of predictive biomarkers, prospective trials of prophylactic or preemptive toxicity mitigation, and creative CAR designs to further ameliorate the development of such toxicities.
Author contributions
CF: Conceptualization, Writing – original draft, Writing – review & editing. MB: Conceptualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
The authors express their deep appreciation to the Leon Levine Foundation and the Kerry and Simone Vickar Family Foundation for their support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. (2021) 11:69. doi: 10.1038/s41408–021-00459–7
2. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. New Engl J Med. (2018) 378:439–48. doi: 10.1056/NEJMoa1709866
3. Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. New Engl J Med. (2019) 380:45–56. doi: 10.1056/NEJMoa1804980
4. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. New Engl J Med. (2017) 377:2531–44. doi: 10.1056/NEJMoa1707447
5. Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang M, Arnason J, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet. (2020) 396:839–52. doi: 10.1016/S0140–6736(20)31366–0
6. Abramson JS, Solomon SR, Arnason J, Johnston PB, Glass B, Bachanova V, et al. Lisocabtagene maraleucel as second-line therapy for large B-cell lymphoma: primary analysis of the phase 3 TRANSFORM study. Blood. (2023) 141:1675–84. doi: 10.1182/blood.2022018730
7. Locke FL, Miklos DB, Jacobson CA, Perales MA, Kersten MJ, Oluwole OO, et al. Axicabtagene ciloleucel as second-line therapy for large B-cell lymphoma. New Engl J Med. (2022) 386:640–54. doi: 10.1056/NEJMoa2116133
8. Jacobson CA, Chavez JC, Sehgal AR, William BM, Munoz J, Salles G, et al. Axicabtagene ciloleucel in relapsed or refractory indolent non-Hodgkin lymphoma (ZUMA-5): a single-arm, multicentre, phase 2 trial. Lancet Oncol. (2022) 23:91–103. doi: 10.1016/S1470-2045(21)00591-X
9. Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, et al. KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. New Engl J Med. (2020) 382:1331–42. doi: 10.1056/NEJMoa1914347
10. Shah BD, Ghobadi A, Oluwole OO, Logan AC, Boissel N, Cassaday RD, et al. KTE-X19 for relapsed or refractory adult B-cell acute lymphoblastic leukaemia: phase 2 results of the single-arm, open-label, multicentre ZUMA-3 study. Lancet. (2021) 398:491–502. doi: 10.1016/S0140–6736(21)01222–8
11. Munshi NC, Anderson LD, Shah N, Madduri D, Berdeja J, Lonial S, et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N Engl J Med. (2021) 384:705–16. doi: 10.1056/NEJMoa2024850
12. Berdeja JG, Madduri D, Usmani SZ, Jakubowiak A, Agha M, Cohen AD, et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): a phase 1b/2 open-label study. Lancet. (2021) 398:314–24. doi: 10.1016/S0140–6736(21)00933–8
13. Brudno JN, Kochenderfer JN. Recent advances in CAR T-cell toxicity: Mechanisms, manifestations and management. Blood Rev. (2019) 34:45–55. doi: 10.1016/j.blre.2018.11.002
14. Zhu F, Wei G, Zhang M, Zhao H, Wu W, Yang L, et al. Factors associated with costs in chimeric antigen receptor T-cell therapy for patients with relapsed/refractory B-cell Malignancies. Cell Transpl. (2020) 29:963689720919434. doi: 10.1177/0963689720919434
15. Palomba ML, Jun MP, Lymp J, Nguyen A, McGarvey N, Gitlin M, et al. Postinfusion monitoring costs by site of care for patients with relapsed/refractory large B-cell lymphoma receiving third- or later-line treatment with lisocabtagene maraleucel in the TRANSCEND NHL 001 and OUTREACH trials. Leuk Lymphoma. (2021) 62:2169–76. doi: 10.1080/10428194.2021.1910686
16. Maalej KM, Merhi M, Inchakalody VP, Mestiri S, Alam M, Maccalli C, et al. CAR-cell therapy in the era of solid tumor treatment: current challenges and emerging therapeutic advances. Mol Cancer. (2023) 22:20. doi: 10.1186/s12943-023-01723-z
17. Schett G, Mackensen A, Mougiakakos D. CAR T-cell therapy in autoimmune diseases. Lancet. (2023) 402:2034–44. doi: 10.1016/S0140–6736(23)01126–1
18. Lee DW, Santomasso BD, Locke FL, Ghobadi A, Turtle CJ, Brudno JN, et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transpl. (2019) 25:625–38. doi: 10.1016/j.bbmt.2018.12.758
19. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. New Engl J Med. (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
20. Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. (2014) 6:224ra25. doi: 10.1126/scitranslmed.3008226
21. Brudno JN, Maric I, Hartman SD, Rose JJ, Wang M, Lam N, et al. T cells genetically modified to express an anti–B-Cell maturation antigen chimeric antigen receptor cause remissions of poor-prognosis relapsed multiple myeloma. J Clin Oncol. (2018) 36:2267–80. doi: 10.1200/JCO.2018.77.8084
22. Alvi RM, Frigault MJ, Fradley MG, Jain MD, Mahmood SS, Awadalla M, et al. Cardiovascular events among adults treated with chimeric antigen receptor T-cells (CAR-T). J Am Coll Cardiol. (2019) 74:3099–108. doi: 10.1016/j.jacc.2019.10.038
23. Wang Z, Han W. Biomarkers of cytokine release syndrome and neurotoxicity related to CAR-T cell therapy. biomark Res. (2018) 6:4. doi: 10.1186/s40364–018-0116–0
24. Shimabukuro-Vornhagen A, Gödel P, Subklewe M, Stemmler HJ, Schlößer HA, Schlaak M, et al. Cytokine release syndrome. J Immunother Cancer. (2018) 6:56. doi: 10.1186/s40425–018-0343–9
25. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor–modified T cells in chronic lymphoid leukemia. New Engl J Med. (2011) 365:725–33. doi: 10.1056/NEJMoa1103849
26. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor–modified T cells for acute lymphoid leukemia. New Engl J Med. (2013) 368:1509–18. doi: 10.1056/NEJMoa1215134
27. Obstfeld AE, Frey NV, Mansfield K, Lacey SF, June CH, Porter DL, et al. Cytokine release syndrome associated with chimeric-antigen receptor T-cell therapy: clinicopathological insights. Blood. (2017) 130:2569–72. doi: 10.1182/blood-2017–08-802413
28. Galli E, Sorà F, Hohaus S, Fresa A, Pansini I, Autore F, et al. Endothelial activation predicts disseminated intravascular coagulopathy, cytokine release syndrome and prognosis in patients treated with anti-CD19 CAR-T cells. Br J Haematol. (2023) 201:86–94. doi: 10.1111/bjh.18596
29. Hay KA, Hanafi LA, Li D, Gust J, Liles WC, Wurfel MM, et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T-cell therapy Key Points. Blood. (2017) 130:2295–306. doi: 10.1182/blood-2017–06-793141
30. Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med. (2018) 24:739–48. doi: 10.1038/s41591–018-0036–4
31. Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, Sadelain M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade letter. Nat Med. (2018) 24:731–8. doi: 10.1038/s41591–018-0041–7
32. Bailey SR, Vatsa S, Larson RC, Bouffard AA, Scarfò I, Kann MC, et al. Blockade or deletion of IFNγ reduces macrophage activation without compromising CAR T-cell function in hematologic Malignancies. Blood Cancer Discovery. (2022) 3:136–53. doi: 10.1158/2643–3230.BCD-21–0181
33. Manni S, Del Bufalo F, Merli P, Silvestris DA, Guercio M, Caruso S, et al. Neutralizing IFNγ improves safety without compromising efficacy of CAR-T cell therapy in B-cell Malignancies. Nat Commun. (2023) 14:3423. doi: 10.1038/s41467-023-38723-y
34. Boulch M, Cazaux M, Cuffel A, Ruggiu M, Allain V, Corre B, et al. A major role for CD4+ T cells in driving cytokine release syndrome during CAR T cell therapy. Cell Rep Med. (2023) 4:101161. doi: 10.1016/j.xcrm.2023.101161
35. Rosen RS, Yang JH, Peña JS, Schloss R, Yarmush ML. An in vitro model of the macrophage-endothelial interface to characterize CAR T-cell induced cytokine storm. Sci Rep. (2023) 13:18835. doi: 10.1038/s41598-023-46114-y
36. Raje N, Berdeja J, Lin Y, Siegel D, Jagannath S, Madduri D, et al. Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. New Engl J Med. (2019) 380:1726–37. doi: 10.1056/NEJMoa1817226
37. Bachy E, Le Gouill S, Di Blasi R, Sesques P, Manson G, Cartron G, et al. A real-world comparison of tisagenlecleucel and axicabtagene ciloleucel CAR T cells in relapsed or refractory diffuse large B cell lymphoma. Nat Med. (2022) 28:2145–54. doi: 10.1038/s41591-022-01969-y
38. Bove C, Arcangeli S, Falcone L, Camisa B, El Khoury R, Greco B, et al. CD4 CAR-T cells targeting CD19 play a key role in exacerbating cytokine release syndrome, while maintaining long-term responses. J Immunother Cancer. (2023) 11:e005878. doi: 10.1136/jitc-2022–005878
39. Perez A, Johnson G, Patel K, Arciola B, Wood A, Bachmeier CA, et al. Primary progression during frontline CIT associates with decreased efficacy of subsequent CD19 CAR T-cell therapy in LBCL. Blood Adv. (2022) 6:3970–3. doi: 10.1182/bloodadvances.2022007006
40. Dean EA, Mhaskar RS, Lu H, Mousa MS, Krivenko GS, Lazaryan A, et al. High metabolic tumor volume is associated with decreased efficacy of axicabtagene ciloleucel in large B-cell lymphoma. Blood Adv. (2020) 4:3268–76. doi: 10.1182/bloodadvances.2020001900
41. Park JH, Rivière I, Gonen M, Wang X, Sénéchal B, Curran KJ, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. New Engl J Med. (2018) 378:449–59. doi: 10.1056/NEJMoa1709919
42. Locke FL, Rossi JM, Neelapu SS, Jacobson CA, Miklos DB, Ghobadi A, et al. Tumor burden, inflammation, and product attributes determine outcomes of axicabtagene ciloleucel in large B-cell lymphoma. Blood Adv. (2020) 4:4898–911. doi: 10.1182/bloodadvances.2020002394
43. Lemoine J, Vic S, Houot R. Disease-specific outcomes after chimeric antigen receptor T-cell therapy. Eur J Cancer. (2022) 160:235–42. doi: 10.1016/j.ejca.2021.10.022
44. Scholler N, Perbost R, Locke FL, Jain MD, Turcan S, Danan C, et al. Tumor immune contexture is a determinant of anti-CD19 CAR T cell efficacy in large B cell lymphoma. Nat Med. (2022) 28:1872–82. doi: 10.1038/s41591-022-01916-x
45. Faramand R, Jain M, Staedtke V, Kotani H, Bai R, Reid K, et al. Tumor microenvironment composition and severe cytokine release syndrome (CRS) influence toxicity in patients with large B-cell lymphoma treated with axicabtagene ciloleucel. Clin Cancer Res. (2020) 26:4823–31. doi: 10.1158/1078–0432.CCR-20–1434
46. Pennisi M, Sanchez-Escamilla M, Flynn JR, Shouval R, Tomas AA, Silverberg ML, et al. Modified EASIX predicts severe cytokine release syndrome and neurotoxicity after chimeric antigen receptor T cells. Blood Adv. (2021) 5:3397–406. doi: 10.1182/bloodadvances.2020003885
47. National Cancer Institute. Common terminology criteria for adverse events (CTCAE). Version 5.0 . Available online at: https://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03/CTCAE_4.03_2010–06-14_QuickReference_8.5x11.pdf.
48. Porter D, Frey N, Wood PA, Weng Y, Grupp SA. Grading of cytokine release syndrome associated with CAR T cell therapy tisagenlecleucel. J Hematol Oncol. (2018) 11:35. doi: 10.1186/s13045-018-0571-y
49. Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, et al. Chimeric antigen receptor T-cell therapy - assessment and management of toxicities. Nat Rev Clin Oncol. (2018) 15:47–62. doi: 10.1038/nrclinonc.2017.148
50. Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. (2014) 124:188–95. doi: 10.1182/blood-2014–05-552729
51. Myers GD, Verneris MR, Goy A, Maziarz RT. Perspectives on outpatient administration of CAR-T cell therapy in aggressive B-cell lymphoma and acute lymphoblastic leukemia. J Immunother Cancer. (2021) 9:e002056. doi: 10.1136/jitc-2020–002056
52. Nasta SD, Hughes ME, Namoglu EC, Garfall A, DiFilippo H, Ballard HJ, et al. Outcomes of tisagenlecleucel in lymphoma patients with predominant management in an ambulatory setting. Clin Lymphoma Myeloma Leuk. (2022) 22:e730–7. doi: 10.1016/j.clml.2022.04.012
53. Borogovac A, Keruakous A, Bycko M, Holter Chakrabarty J, Ibrahimi S, Khawandanah M, et al. Safety and feasibility of outpatient chimeric antigen receptor (CAR) T-cell therapy: experience from a tertiary care center. Bone Marrow Transpl. (2022) 57:1025–7. doi: 10.1038/s41409-022-01664-z
54. Le RQ, Li L, Yuan W, Shord SS, Nie L, Habtemariam BA, et al. FDA approval summary: tocilizumab for treatment of chimeric antigen receptor T cell-induced severe or life-threatening cytokine release syndrome. Oncologist. (2018) 23:943–7. doi: 10.1634/theoncologist.2018–0028
55. US Food and Drug Administration. KYMRIAH® (tisagenlecleucel) suspension for intravenous infusion. Initial U.S. Approval (2017).
56. US Food and Drug Administration. YESCARTA® (axicabtagene ciloleucel) suspension for intravenous infusion. Initial U.S. Approval (2017).
57. US Food and Drug Administration. TECARTUS® (brexucabtagene autoleucel) suspension for intravenous infusion. Initial U.S. Approval (2020).
58. US Food and Drug Administration. BREYANZI® (lisocabtagene maraleucel) suspension for intravenous infusion. Initial U.S. Approval (2021).
59. US Food and Drug Administration. ABECMA® (idecabtagene vicleucel) suspension for intravenous infusion. Initial U.S. Approval (2021).
60. US Food and Drug Administration. CARVYKTI® (ciltacabtagene autoleucel) suspension for intravenous infusion. Initial U.S. Approval (2022).
61. Santomasso BD, Nastoupil LJ, Adkins S, Lacchetti C, Schneider BJ, Anadkat M, et al. Management of immune-related adverse events in patients treated with chimeric antigen receptor T-cell therapy: ASCO Guideline. J Clin Oncol. (2021) 39:3978–92. doi: 10.1200/JCO.21.01992
62. Jain MD, Smith M, Shah NN. How I treat refractory CRS and ICANS after CAR T-cell therapy. Blood. (2023) 141:2430–42. doi: 10.1182/blood.2022017414
63. Jatiani SS, Aleman A, Madduri D, Chari A, Cho HJ, Richard S, et al. Myeloma CAR-T CRS management with IL-1R antagonist anakinra. Clin Lymphoma Myeloma Leuk. (2020) 20:632–636.e1. doi: 10.1016/j.clml.2020.04.020
64. Gazeau N, Liang EC, Wu Q“Vicky,”, Voutsinas JM, Barba P, Iacoboni G, et al. Anakinra for refractory cytokine release syndrome or immune effector cell-associated neurotoxicity syndrome after chimeric antigen receptor T cell therapy. Transplant Cell Ther. (2023) 29:430–7. doi: 10.1016/j.jtct.2023.04.001
65. Chen F, Teachey DT, Pequignot E, Frey N, Porter D, Maude SL, et al. Measuring IL-6 and sIL-6R in serum from patients treated with tocilizumab and/or siltuximab following CAR T cell therapy. J Immunol Methods. (2016) 434:1–8. doi: 10.1016/j.jim.2016.03.005
66. Lipe BC, Renaud T. Siltuximab as a primary treatment for cytokine release syndrome in a patient receiving a bispecific antibody in a clinical trial setting. J Oncol Pharm Pract. (2023) 29:1006–10. doi: 10.1177/10781552221140320
67. Zhang L, Wang S, Xu J, Zhang R, Zhu H, Wu Y, et al. Etanercept as a new therapeutic option for cytokine release syndrome following chimeric antigen receptor T cell therapy. Exp Hematol Oncol. (2021) 10:16. doi: 10.1186/s40164–021-00209–2
68. McNerney KO, DiNofia AM, Teachey DT, Grupp SA, Maude SL. Potential role of IFNγ inhibition in refractory cytokine release syndrome associated with CAR T-cell therapy. Blood Cancer Discovery. (2022) 3:90–4. doi: 10.1158/2643–3230.BCD-21–0203
69. Schuelke MR, Bassiri H, Behrens EM, Canna S, Croy C, DiNofia A, et al. Emapalumab for the treatment of refractory cytokine release syndrome in pediatric patients. Blood Adv. (2023) 7:5603–7. doi: 10.1182/bloodadvances.2023010712
70. Mestermann K, Giavridis T, Weber J, Rydzek J, Frenz S, Nerreter T, et al. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Sci Transl Med. (2019) 11:eaau5907. doi: 10.1126/scitranslmed.aau5907
71. Weber EW, Lynn RC, Sotillo E, Lattin J, Xu P, Mackall CL. Pharmacologic control of CAR-T cell function using dasatinib. Blood Adv. (2019) 3:711–7. doi: 10.1182/bloodadvances.2018028720
72. Baur K, Heim D, Beerlage A, Poerings AS, Kopp B, Medinger M, et al. Dasatinib for treatment of CAR T-cell therapy-related complications. J Immunother Cancer. (2022) 10:e005956. doi: 10.1136/jitc-2022–005956
73. Huarte E, O’Connor RS, Peel MT, Nunez-Cruz S, Leferovich J, Juvekar A, et al. Itacitinib (INCB039110), a JAK1 inhibitor, reduces cytokines associated with cytokine release syndrome induced by CAR T-cell therapy. Clin Cancer Res. (2020) 26:6299–309. doi: 10.1158/1078–0432.CCR-20–1739
74. Pan J, Deng B, Ling Z, Song W, Xu J, Duan J, et al. Ruxolitinib mitigates steroid-refractory CRS during CAR T therapy. J Cell Mol Med. (2021) 25:1089–99. doi: 10.1111/jcmm.16176
75. Zi FM, Ye LL, Zheng JF, Cheng J, Wang QM. Using JAK inhibitor to treat cytokine release syndrome developed after chimeric antigen receptor T cell therapy for patients with refractory acute lymphoblastic leukemia: A case report. Med (Baltimore). (2021) 100:e25786. doi: 10.1097/MD.0000000000025786
76. Wei S, Gu R, Xu Y, Liu X, Xing Y, Gong X, et al. Adjuvant ruxolitinib therapy relieves steroid-refractory cytokine-release syndrome without impairing chimeric antigen receptor-modified T-cell function. Immunotherapy. (2020) 12:1047–52. doi: 10.2217/imt-2020–0116
77. Gu C, Wu Q, Zhang J, Kang L, Yu L, Qiu H, et al. Successful treatment of severe cytokine release syndrome after CAR-T therapy by ruxolitinib without compromising CAR-T efficacy. Leuk Lymphoma. (2023) 64:495–8. doi: 10.1080/10428194.2022.2148209
78. Sahillioglu AC, Schumacher TN. Safety switches for adoptive cell therapy. Curr Opin Immunol. (2022) 74:190–8. doi: 10.1016/j.coi.2021.07.002
79. Frigault MJ, Maziarz RT, Park JH, Lazaryan A, Shah NN, Svoboda J, et al. Itacitinib for the prevention of immune effector cell therapy-associated cytokine release syndrome: results from the phase 2 Incb 39110–211 placebo-controlled randomized cohort. Blood. (2023) 142:356. doi: 10.1182/blood-2023–180205
80. Banerjee R, Marsal J, Huang CY, Lo M, Kambhampati S, Kennedy VE, et al. Early time-to-tocilizumab after B cell maturation antigen-directed chimeric antigen receptor T cell therapy in myeloma. Transplant Cell Ther. (2021) 27:477.e1–7. doi: 10.1016/j.jtct.2021.03.004
81. Liu S, Deng B, Yin Z, Pan J, Lin Y, Ling Z, et al. Corticosteroids do not influence the efficacy and kinetics of CAR-T cells for B-cell acute lymphoblastic leukemia. Blood Cancer J. (2020) 10:15. doi: 10.1038/s41408-020-0280-y
82. Strati P, Ahmed S, Furqan F, Fayad LE, Lee HJ, Iyer SP, et al. Prognostic impact of corticosteroids on efficacy of chimeric antigen receptor T-cell therapy in large B-cell lymphoma. Blood. (2021) 137:3272–6. doi: 10.1182/blood.2020008865
83. Kadauke S, Myers RM, Li Y, Aplenc R, Baniewicz D, Barrett DM, et al. Risk-adapted preemptive tocilizumab to prevent severe cytokine release syndrome after CTL019 for pediatric B-cell acute lymphoblastic leukemia: a prospective clinical trial. J Clin Oncol. (2021) 39:920–30. doi: 10.1200/JCO.20.02477
84. Gardner RA, Ceppi F, Rivers J, Annesley C, Summers C, Taraseviciute A, et al. Preemptive mitigation of CD19 CAR T-cell cytokine release syndrome without attenuation of antileukemic efficacy. Blood. (2019) 134:2149–58. doi: 10.1182/blood.2019001463
85. Topp MS, van Meerten T, Houot R, Minnema MC, Bouabdallah K, Lugtenburg PJ, et al. Earlier corticosteroid use for adverse event management in patients receiving axicabtagene ciloleucel for large B-cell lymphoma. Br J Haematol. (2021) 195:388–98. doi: 10.1111/bjh.17673
86. Caimi PF, Pacheco Sanchez G, Sharma A, Otegbeye F, Ahmed N, Rojas P, et al. Prophylactic tocilizumab prior to anti-CD19 CAR-T cell therapy for non-Hodgkin lymphoma. Front Immunol. (2021) 12:745320. doi: 10.3389/fimmu.2021.745320
87. Locke FL, Neelapu SS, Bartlett NL, Lakakis LJ, Jacobson CA, Braunschweig I, et al. Preliminary results of prophylactic tocilizumab after axicabtagene ciloleucel (axi-cel; KTE-C19) treatment for patients with refractory, aggressive non-Hodgkin lymphoma (NHL). Blood. (2017) 130:1547. doi: 10.1182/blood.V130.Suppl_1.1547.1547
88. Oluwole OO, Bouabdallah K, Muñoz J, De Guibert S, Vose JM, Bartlett NL, et al. Prophylactic corticosteroid use in patients receiving axicabtagene ciloleucel for large B-cell lymphoma. Br J Haematol. (2021) 194:690–700. doi: 10.1111/bjh.17527
89. Strati P, Li X, Deng Q, Feng L, Sun R, Jallouk A, et al. Primary analysis of a pilot study of prophylactic anakinra to mitigate CAR T cell-associated toxicity in patients with relapsed or refractory large B-cell lymphoma. Blood. (2022) 140:7493–5. doi: 10.1182/blood-2022–157448
90. Park JH, Sauter CS, Palomba ML, Shah GL, Dahi PB, Lin RJ, et al. A phase II study of prophylactic anakinra to prevent CRS and neurotoxicity in patients receiving CD19 CAR T cell therapy for relapsed or refractory lymphoma. Blood. (2021) 138:96. doi: 10.1182/blood-2021–150431
91. Strati P, Jallouk A, Deng Q, Li X, Feng L, Sun R, et al. A phase 1 study of prophylactic anakinra to mitigate ICANS in patients with large B-cell lymphoma. Blood Adv. (2023) 7:6785–9. doi: 10.1182/bloodadvances.2023010653
92. Park JH, Nath K, Devlin SM, Sauter CS, Palomba ML, Shah G, et al. CD19 CAR T-cell therapy and prophylactic anakinra in relapsed or refractory lymphoma: phase 2 trial interim results. Nat Med. (2023) 29:1710–7. doi: 10.1038/s41591–023-02404–6
93. Jacobson CA, Rosenthal AC, Arnason J, Agarwal S, Zhang P, Wu W, et al. A phase 2 trial of defibrotide for the prevention of chimeric antigen receptor T-cell–associated neurotoxicity syndrome. Blood Adv. (2023) 7:6790–9. doi: 10.1182/bloodadvances.2023009961
94. Oluwole OO, Kenderian SS, Shiraz P, Karmali R, Reshef R, McCarthy PL, et al. ZUMA-19: A phase 1/2 study of axicabtagene ciloleucel plus lenzilumab in patients with relapsed or refractory large B-cell lymphoma. Blood. (2022) 140:10318–20. doi: 10.1182/blood-2022–167688
95. Gust J, Ponce R, Liles WC, Garden GA, Turtle CJ. Cytokines in CAR T cell–associated neurotoxicity. Front Immunol. (2020) 11:577027. doi: 10.3389/fimmu.2020.577027
96. Herlopian A, Dietrich J, Abramson JS, Cole AJ, Westover MB. EEG findings in CAR T-cell therapy-related encephalopathy. Neurology. (2018) 91:227–9. doi: 10.1212/WNL.0000000000005910
97. Santomasso BD, Park JH, Salloum D, Riviere I, Flynn J, Mead E, et al. Clinical and biological correlates of neurotoxicity associated with car t-cell therapy in patients with B-cell acute lymphoblastic leukemia. Cancer Discovery. (2018) 8:958–71. doi: 10.1158/2159–8290.CD-17–1319
98. Taraseviciute A, Tkachev V, Ponce R, Turtle CJ, Snyder JM, Liggitt HD, et al. Chimeric antigen receptor T cell–mediated neurotoxicity in nonhuman primates. Cancer Discovery. (2018) 8:750–63. doi: 10.1158/2159–8290.CD-17–1368
99. Gust J, Hay KA, Hanafi LA, Li D, Myerson D, Gonzalez-Cuyar LF, et al. Endothelial activation and blood–brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T cells. Cancer Discovery. (2017) 7:1404–19. doi: 10.1158/2159–8290.CD-17–0698
100. Mailankody S, Devlin SM, Landa J, Nath K, Diamonte C, Carstens EJ, et al. GPRC5D-targeted CAR T cells for myeloma. New Engl J Med. (2022) 387:1196–206. doi: 10.1056/NEJMoa2209900
101. Karschnia P, Jordan JT, Forst DA, Arrillaga-Romany IC, Batchelor TT, Baehring JM, et al. Clinical presentation, management, and biomarkers of neurotoxicity after adoptive immunotherapy with CAR T cells. Blood. (2019) 133:2212–21. doi: 10.1182/blood-2018–12-893396
102. Strati P, Ahmed S, Kebriaei P, Nastoupil LJ, Claussen CM, Watson G, et al. Clinical efficacy of anakinra to mitigate CAR T-cell therapy-associated toxicity in large B-cell lymphoma. Blood Adv. (2020) 4:3123–7. doi: 10.1182/bloodadvances.2020002328
103. Wehrli M, Gallagher K, Chen YB, Leick MB, McAfee SL, El-Jawahri AR, et al. Single-center experience using anakinra for steroid-refractory immune effector cell-Associated neurotoxicity syndrome (ICANS). J Immunother Cancer. (2022) 10:e003847. doi: 10.1136/jitc-2021–003847
104. Wang M, Jain P, Chi TL, Chen SE, Heimberger A, Weathers SP, et al. Management of a patient with mantle cell lymphoma who developed severe neurotoxicity after chimeric antigen receptor T-cell therapy in ZUMA-2. J Immunother Cancer. (2020) 8:e001114. doi: 10.1136/jitc-2020–001114
105. Shah NN, Johnson BD, Fenske TS, Raj RV, Hari P. Intrathecal chemotherapy for management of steroid-refractory CAR T-cell-associated neurotoxicity syndrome. Blood Adv. (2020) 4:2119–22. doi: 10.1182/bloodadvances.2020001626
106. Zurko JC, Johnson BD, Aschenbrenner E, Fenske TS, Hamadani M, Hari P, et al. Use of early intrathecal therapy to manage high-grade immune effector cell-associated neurotoxicity syndrome. JAMA Oncol. (2022) 8:773–5. doi: 10.1001/jamaoncol.2022.0070
107. Garfall AL, Lancaster E, Stadtmauer EA, Lacey SF, Dengel K, Ambrose DE, et al. Posterior reversible encephalopathy syndrome (PRES) after infusion of anti-BCMA CAR T cells (CART-BCMA) for multiple myeloma: successful treatment with cyclophosphamide. Blood. (2016) 128:5702. doi: 10.1182/blood.V128.22.5702.5702
108. Cohen AD, Parekh S, Santomasso BD, Pérez-Larraya JG, van de Donk NWCJ, Arnulf B, et al. Incidence and management of CAR-T neurotoxicity in patients with multiple myeloma treated with ciltacabtagene autoleucel in CARTITUDE studies. Blood Cancer J. (2022) 12:32. doi: 10.1038/s41408–022-00629–1
109. Van Oekelen O, Aleman A, Upadhyaya B, Schnakenberg S, Madduri D, Gavane S, et al. Neurocognitive and hypokinetic movement disorder with features of parkinsonism after BCMA-targeting CAR-T cell therapy. Nat Med. (2021) 27:2099–103. doi: 10.1038/s41591–021-01564–7
110. Marella M, Yao X, Carreira V, Bustamante MF, Clark HB, Jackson CC, et al. Comprehensive BCMA expression profiling in adult normal human brain suggests a low risk of on-target neurotoxicity in BCMA-targeting multiple myeloma therapy. J Histochem Cytochem. (2022) 70:273–87. doi: 10.1369/00221554221079579
111. Graham CE, Lee W, Wiggin HR, Supper VM, Leick MB, Birocchi F, et al. Chemotherapy-induced reversal of ciltacabtagene autoleucel-associated movement and neurocognitive toxicity. Blood. (2023) 142:1248–52. doi: 10.1182/blood.2023021429
112. Sheikh S, Mokhtari S, Silverman JA, Reid K, Faramand R, Davila ML, et al. Transverse myelitis after anti-CD19 directed CAR T cell therapy for relapsed large B cell lymphoma. EJHaem. (2022) 3:223–7. doi: 10.1002/jha2.286
113. Aghajan Y, Yu A, Jacobson CA, Kim AI, Kean L, Robertson M, et al. Myelopathy because of CAR-T-related neurotoxicity treated with siltuximab. Neurol Clin Pract. (2021) 11:e944–6. doi: 10.1212/CPJ.0000000000001078
114. Handley G, Khawaja F, Kondapi DS, Lee HJ, Kaufman GP, Neelapu SS, et al. Human herpesvirus 6 myelitis after chimeric antigen receptor T-cell therapy. Int J Infect Dis. (2021) 112:327–9. doi: 10.1016/j.ijid.2021.09.061
115. Nair R, Drillet G, Lhomme F, Le Bras A, Michel L, Rossi J, et al. Acute leucoencephalomyelopathy and quadriparesis after CAR T-cell therapy. Haematologica. (2021) 106:1504–6. doi: 10.3324/haematol.2020.259952
116. Koch C, Fleischer J, Popov T, Frontzek K, Schreiner B, Roth P, et al. Diabetes insipidus and Guillain-Barré-like syndrome following CAR-T cell therapy: a case report. J Immunother Cancer. (2023) 11:e006059. doi: 10.1136/jitc-2022–006059
117. Santomasso BD, Gust J, Perna F. How I treat unique and difficult-to-manage cases of CAR T-cell therapy-associated neurotoxicity. Blood. (2023) 141:2443–51. doi: 10.1182/blood.2022017604
118. Lichtenstein DA, Schischlik F, Shao L, Steinberg SM, Yates B, Wang HW, et al. Characterization of HLH-like manifestations as a CRS variant in patients receiving CD22 CAR T cells. Blood. (2021) 138:2469–84. doi: 10.1182/blood.2021011898
119. Hines MR, Keenan C, Maron Alfaro G, Cheng C, Zhou Y, Sharma A, et al. Hemophagocytic lymphohistiocytosis-like toxicity (carHLH) after CD19-specific CAR T-cell therapy. Br J Haematol. (2021) 194:701–7. doi: 10.1111/bjh.17662
120. Hines MR, Knight TE, McNerney KO, Leick MB, Jain T, Ahmed S, et al. Immune effector cell-associated hemophagocytic lymphohistiocytosis-like syndrome. Transplant Cell Ther. (2023) 29:438.e1–438.e16. doi: 10.1016/j.jtct.2023.03.006
121. Diorio C, Shraim R, Myers R, Behrens EM, Canna S, Bassiri H, et al. Comprehensive serum proteome profiling of cytokine release syndrome and immune effector cell–associated neurotoxicity syndrome patients with B-cell ALL receiving CAR T19. Clin Cancer Res. (2022) 28:3804–13. doi: 10.1158/1078–0432.CCR-22–0822
122. Henter JI, Horne AC, Aricó M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. (2007) 48:124–31. doi: 10.1002/pbc.21039
123. Fardet L, Galicier L, Lambotte O, Marzac C, Aumont C, Chahwan D, et al. Development and validation of the hscore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. (2014) 66:2613–20. doi: 10.1002/art.38690
124. Kim DW, Bukhari A, Lutfi F, Zafforoni F, Merechi F, Mustafa Ali MK, et al. Low utility of the H-Score and HLH-2004 criteria to identify patients with secondary hemophagocytic lymphohistiocytosis after CAR-T cell therapy for relapsed/refractory diffuse large B-Cell lymphoma. Leuk Lymphoma. (2022) 63:1339–47. doi: 10.1080/10428194.2021.2024817
125. Shah NN, Highfill SL, Shalabi H, Yates B, Jin J, Wolters PL, et al. CD4/CD8 T-cell selection affects chimeric antigen receptor (CAR) T-cell potency and toxicity: updated results from a phase I anti-CD22 CAR T-cell trial. J Clin Oncol. (2020) 38:1938–50. doi: 10.1200/JCO.19.03279
126. McNerney KO, Si Lim SJ, Ishikawa K, Dreyzin A, Vatsayan A, Chen JJ, et al. HLH-like toxicities predict poor survival after the use of tisagenlecleucel in children and young adults with B-ALL. Blood Adv. (2023) 7:2758–71. doi: 10.1182/bloodadvances.2022008893
127. Sepulveda FE, Maschalidi S, Vosshenrich CAJ, Garrigue A, Kurowska M, Enasche GM, et al. A novel immunoregulatory role for NK-cell cytotoxicity in protection from HLH-like immunopathology in mice. Blood. (2015) 125:1427–34. doi: 10.1182/blood-2014–09-602946
128. Rainone M, Ngo D, Baird JH, Budde LE, Htut M, Aldoss I, et al. Interferon-γ blockade in CAR T-cell therapy-associated macrophage activation syndrome/hemophagocytic lymphohistiocytosis. Blood Adv. (2023) 7:533–6. doi: 10.1182/bloodadvances.2022008256
129. Porter TJ, Lazarevic A, Ziggas JE, Fuchs E, Kim K, Byrnes H, et al. Hyperinflammatory syndrome resembling haemophagocytic lymphohistiocytosis following axicabtagene ciloleucel and brexucabtagene autoleucel. Br J Haematol. (2022) 199:720–7. doi: 10.1111/bjh.18454
130. Major A, Collins J, Craney C, Heitman AK, Bauer E, Zerante E, et al. Management of hemophagocytic lymphohistiocytosis (HLH) associated with chimeric antigen receptor T-cell (CAR-T) therapy using anti-cytokine therapy: an illustrative case and review of the literature. Leuk Lymphoma. (2021) 62:1765–9. doi: 10.1080/10428194.2021.1881507
131. Zhang Q, Zhao YZ, Ma HH, Wang D, Cui L, Li WJ, et al. A study of ruxolitinib response-based stratified treatment for pediatric hemophagocytic lymphohistiocytosis. Blood. (2022) 139:3493–504. doi: 10.1182/blood.2021014860
132. Wang J, Wang Y, Wu L, Wang X, Jin Z, Gao Z, et al. Ruxolitinib for refractory/relapsed hemophagocytic lymphohistiocytosis. Haematologica. (2020) 105:e210–2. doi: 10.3324/haematol.2019.222471
133. Larson RC, Kann MC, Bailey SR, Haradhvala NJ, Llopis PM, Bouffard AA, et al. CAR T cell killing requires the IFNγR pathway in solid but not liquid tumours. Nature. (2022) 604:563–70. doi: 10.1038/s41586–022-04585–5
134. Bracaglia C, De Graaf K, Marafon DP, Guilhot F, Ferlin W, Prencipe G, et al. Elevated circulating levels of interferon-γ and interferon-γ-induced chemokines characterize patients with macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. Ann Rheum Dis. (2017) 76:166–72. doi: 10.1136/annrheumdis-2015–209020
135. Johnson TS, Terrell CE, Millen SH, Katz JD, Hildeman DA, Jordan MB. Etoposide selectively ablates activated T cells to control the immunoregulatory disorder hemophagocytic lymphohistiocytosis. J Immunol. (2014) 192:84–91. doi: 10.4049/jimmunol.1302282
136. Trottestam H, Horne AC, Aricò M, Egeler RM, Filipovich AH, Gadner H, et al. Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: long-term results of the HLH-94 treatment protocol. Blood. (2011) 118:4577–84. doi: 10.1182/blood-2011–06-356261
137. Rosée P, Rosée R, Horne A, Hines M, Von Bahr Greenwood T, Machowicz R, et al. Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood. (2019) 133:2465–77. doi: 10.1182/blood.2018894618
138. Cordeiro A, Bezerra ED, Hirayama AV, Hill JA, Wu QV, Voutsinas J, et al. Late events after treatment with CD19-targeted chimeric antigen receptor modified T cells. Biol Blood Marrow Transpl. (2020) 26:26–33. doi: 10.1016/j.bbmt.2019.08.003
139. Rejeski K, Perez A, Sesques P, Hoster E, Berger C, Jentzsch L, et al. CAR-HEMATOTOX: a model for CAR T-cell-related hematologic toxicity in relapsed/refractory large B-cell lymphoma. Blood. (2021) 138:2499–513. doi: 10.1182/blood.2020010543
140. Rejeski K, Subklewe M, Aljurf M, Bachy E, Balduzzi A, Barba P, et al. Immune effector cell-associated hematotoxicity: EHA/EBMT consensus grading and best practice recommendations. Blood. (2023) 142:865–77. doi: 10.1182/blood.2023020578
141. Rejeski K, Wang Y, Albanyan O, Munoz J, Sesques P, Iacoboni G, et al. The CAR-HEMATOTOX score identifies patients at high risk for hematological toxicity, infectious complications, and poor treatment outcomes following brexucabtagene autoleucel for relapsed or refractory MCL. Am J Hematol. (2023) 98:1699–710. doi: 10.1002/ajh.27056
142. Rejeski K, Hansen DK, Bansal R, Sesques P, Ailawadhi S, Logue JM, et al. The CAR-HEMATOTOX score as a prognostic model of toxicity and response in patients receiving BCMA-directed CAR-T for relapsed/refractory multiple myeloma. J Hematol Oncol. (2023) 16:88. doi: 10.1186/s13045-023-01465-x
143. Miller PG, Sperling AS, Brea EJ, Leick MB, Fell GG, Jan M, et al. Clonal hematopoiesis in patients receiving chimeric antigen receptor T-cell therapy. Blood Adv. (2021) 5:2982–6. doi: 10.1182/bloodadvances.2021004554
144. Saini NY, Swoboda DM, Greenbaum U, Ma J, Patel RD, Devashish K, et al. Clonal hematopoiesis is associated with increased risk of severe neurotoxicity in axicabtagene ciloleucel therapy of large B-cell lymphoma. Blood Cancer Discovery. (2022) 3:385–93. doi: 10.1158/2643–3230.BCD-21–0177
145. Brudno JN, Natrakul D, Lam N, Dulau-Florea A, Yuan CM, Kochenderfer JN. Acute and delayed cytopenias following CAR T-cell therapy: an investigation of risk factors and mechanisms. Leuk Lymphoma. (2022) 63:1849–60. doi: 10.1080/10428194.2022.2056172
146. Jain T, Knezevic A, Pennisi M, Chen Y, Ruiz JD, Purdon TJ, et al. Hematopoietic recovery in patients receiving chimeric antigen receptor T-cell therapy for hematologic Malignancies. Blood Adv. (2020) 4:3776–87. doi: 10.1182/bloodadvances.2020002509
147. Juluri KR, Wu QV, Voutsinas J, Hou J, Hirayama AV, Mullane E, et al. Severe cytokine release syndrome is associated with hematologic toxicity following CD19 CAR T-cell therapy. Blood Adv. (2022) 6:2055–68. doi: 10.1182/bloodadvances.2020004142
148. Rejeski K, Wu Z, Blumenberg V, Kunz WG, Müller S, Kajigaya S, et al. Oligoclonal T-cell expansion in a patient with bone marrow failure after CD19 CAR-T therapy for Richter-transformed DLBCL. Blood. (2022) 140:2175–9. doi: 10.1182/blood.2022017015
149. Strati P, Li X, Deng Q, Marques-Piubelli ML, Henderson J, Watson G, et al. Prolonged cytopenia following CD19 CAR T cell therapy is linked with bone marrow infiltration of clonally expanded IFNγ-expressing CD8 T cells. Cell Rep Med. (2023) 4:101158. doi: 10.1016/j.xcrm.2023.101158
150. Barreto JN, Bansal R, Hathcock MA, Doleski CJ, Hayne JR, Truong TA, et al. The impact of granulocyte colony stimulating factor on patients receiving chimeric antigen receptor T-cell therapy. Am J Hematol. (2021) 96:E399–402. doi: 10.1002/ajh.26313
151. Galli E, Allain V, Di Blasi R, Bernard S, Vercellino L, Morin F, et al. G-CSF does not worsen toxicities and efficacy of CAR-T cells in refractory/relapsed B-cell lymphoma. Bone Marrow Transpl. (2020) 55:2347–9. doi: 10.1038/s41409-020-01006-x
152. Liévin R, Di Blasi R, Morin F, Galli E, Allain V, De Jorna R, et al. Effect of early granulocyte-colony-stimulating factor administration in the prevention of febrile neutropenia and impact on toxicity and efficacy of anti-CD19 CAR-T in patients with relapsed/refractory B-cell lymphoma. Bone Marrow Transpl. (2022) 57:431–9. doi: 10.1038/s41409–021-01526–0
153. Miller KC, Johnson PC, Abramson JS, Soumerai JD, Yee AJ, Branagan AR, et al. Effect of granulocyte colony-stimulating factor on toxicities after CAR T cell therapy for lymphoma and myeloma. Blood Cancer J. (2022) 12:146. doi: 10.1038/s41408–022-00741–2
154. Wat J, Barmettler S. Hypogammaglobulinemia after chimeric antigen receptor (CAR) T-cell therapy: characteristics, management, and future directions. J Allergy Clin Immunol Pract. (2022) 10:460–6. doi: 10.1016/j.jaip.2021.10.037
155. Rejeski K, Burchert A, Iacoboni G, Sesques P, Fransecky L, Bücklein V, et al. Safety and feasibility of stem cell boost as a salvage therapy for severe hematotoxicity after CD19 CAR T-cell therapy. Blood Adv. (2022) 6:4719–25. doi: 10.1182/bloodadvances.2022007776
156. Gagelmann N, Fischer M, Penack O, Holtick U, Thomson J, Dreger P, et al. Hematopoietic stem cell boost for persistent neutropenia after CAR-T cell therapy: a GLA/DRST study. Blood Adv. (2023) 7:555–9. doi: 10.1182/bloodadvances.2022008042
157. Davis JA, Sborov DW, Wesson W, Julian K, Abdallah AO, McGuirk JP, et al. Efficacy and safety of CD34+ stem cell boost for delayed hematopoietic recovery after BCMA directed CAR T-cell therapy. Transplant Cell Ther. (2023) 29:567–71. doi: 10.1016/j.jtct.2023.05.012
158. Levine BL, Pasquini MC, Connolly JE, Porter DL, Gustafson MP, Boelens JJ, et al. Unanswered questions following reports of secondary Malignancies after CAR-T cell therapy. Nat Med. (2024) 30(2):338–41. doi: 10.1038/s41591-023-02767-w
Keywords: chimeric antigen receptor (CAR T), cytokine release syndrome (CRS), ICANS - immune effector cell-associated neurotoxicity syndrome, HLH - hemophagocytic lymphohistiocytosis, tocilizumab (IL-6 inhibitor), large B cell lymphoma, multiple myeloma
Citation: Ferreri CJ and Bhutani M (2024) Mechanisms and management of CAR T toxicity. Front. Oncol. 14:1396490. doi: 10.3389/fonc.2024.1396490
Received: 05 March 2024; Accepted: 07 May 2024;
Published: 21 May 2024.
Edited by:
Alice Di Rocco, Sapienza University of Rome, ItalyReviewed by:
Jacopo Mariotti, Humanitas Research Hospital, ItalyCopyright © 2024 Ferreri and Bhutani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manisha Bhutani, bWFuaXNoYS5iaHV0YW5pQGF0cml1bWhlYWx0aC5vcmc=