 Gizem Yayli
Gizem Yayli Alexa Tokofsky
Alexa Tokofsky Utthara Nayar
Utthara Nayar- 1Department of Biochemistry and Molecular Biology, Bloomberg School of Public Health, Johns Hopkins University, Baltimore, MD, United States
- 2Oncology, Sidney Kimmel Comprehensive Cancer Center, School of Medicine, Johns Hopkins University, Baltimore, MD, United States
Since its introduction in the 1970s, endocrine therapy that targets the estrogen receptor alpha (ERα) signaling pathway has had tremendous success in the clinic in estrogen receptor positive (ER+) breast cancer. However, resistance to endocrine therapy eventually develops in virtually all patients with metastatic disease. Endocrine resistance is a primary unaddressed medical need for ER+ metastatic breast cancer patients. It has been shown that tumors become resistant through various mechanisms, converging on the acquisition of genetic alterations of ER, components of the MAP kinase pathway, or transcription factors (TFs). For instance, mutations in the human epidermal growth factor receptor-2 (HER2) lead to complete resistance to all current endocrine therapies including aromatase inhibitors, selective estrogen receptor modulators, and selective estrogen receptor degraders, as well as cross-resistance to CDK4/6 inhibitors (CDK4/6is). Emerging evidence points to an intriguing connection between endocrine-resistant tumors and the HER2-low subtype. Specifically, recent studies and our analysis of a publicly available breast cancer dataset both indicate that metastatic ER+ breast cancer with endocrine resistance conferred through acquired genetic alterations can often be classified as HER2-low rather than HER2-0/HER2-negative. Limited data suggest that acquired endocrine resistance can also be accompanied by a subtype switch. Therefore, we suggest that there is an underappreciated association between the HER2-low subtype and endocrine resistance. In this perspective piece, we explore the evidence linking the HER2-low subtype with the various pathways to endocrine resistance and suggest that there are signaling networks in HER2-low tumors that intersect endocrine resistance and can be effectively targeted.
1 Introduction
Human epidermal growth factor receptor-2 (HER2)-low breast cancer (BC) accounts for between 45% and 55% of all BC cases (1). By definition, they belong to luminal-like [estrogen receptor-positive (ER+)] and triple-negative BC (TNBC) subtypes with a lack of HER2 (ERBB2) amplification (1, 2). At least 80%–90% of HER2-low are ER+ [luminal A (LumA) or B (LumB)] tumors (3), as opposed to ~70% of HER2-0.
LumA or B ER+ BC is classically treated using ER (ESR1)-directed endocrine therapies—aromatase inhibitors (AIs) or the selective ER modulator (SERM) tamoxifen in the first-line setting, and the selective ER degrader (SERD) fulvestrant or the combination of endocrine with CDK4/6 inhibitor (CDK4/6i) palbociclib or ribociclib, which were tested in the PALOMA (4) and MONALEESA (5) trials, or CDK4/6i abemaciclib monotherapy (MONARCH (6) study), in the first-line and endocrine refractory second-line settings (7, 8). Endocrine resistance to ER inhibitors (ERis) is ubiquitous and a primary contributor to BC-associated mortality.
Endocrine resistance has been studied for decades using cell line models cultured to resistance—regardless of the specific cell line or the endocrine treatment, nearly all such models upregulate HER2 and/or HER2 binding partners (9–11). LumB tumors, which are characterized by increased HER2 expression and pathway activity signature compared to LumA, have worse outcomes on endocrine therapy (12). Thus, HER2 activity has a strong inverse correlation with endocrine sensitivity.
In recent years, large-scale sequencing studies have begun to shed light on the mechanisms of endocrine resistance that arise in the clinical setting (13, 14). Overall, these appear to converge on three major classes: 1) alterations in the target ER through mutations or novel oncofusions (15, 16), 2) transcription factor (TF) alterations, and 3) MAP kinase pathway-associated alterations, including upstream receptor tyrosine kinases (RTKs) and downstream components of the MAP kinase or PI3K/AKT/mTOR signaling cascades (14). Up to 60% of endocrine-resistant tumors do not harbor a genetic alteration in any of these classes, and this remains an area of active study.
Of the known mechanisms of clinical endocrine resistance, ER (ESR1) mutations (ERmuts) are the most common, accounting for 18%–50% of resistant tumors depending on patient cohort characteristics (higher prevalence in AI-treated populations) (17–20). ERmuts have been extensively studied (21) and can be clinically targeted with novel oral SERDs, the first of which elacestrant (22) was Food and Drug Administration (FDA) approved for ERmut tumors in 2023. At the same time, the other specific alterations acquired in resistant tumors are incredibly diverse and, with few exceptions, such as HER2muts that our group and others previously identified (23–25), not clinically targetable. As a result, the management of endocrine resistance is still reliant on the ERi+CDK4/6i combination. A major question remaining is whether the HER2-low subtype has relevance in endocrine-resistant disease.
2 Intersection of endocrine therapy resistance and HER2-low
Resistance to endocrine therapies can be caused by a wide variety of molecular alterations, as summarized in Table 1 and depicted in Figure 1. They converge into three classes: 1) ER, 2) TF, and 3) MAPK (14). These include alterations in ESR1, ERBB2, EGFR, KRAS, FGFR1/2/4, and ER transcriptional regulators, for instance, MYC, CTCF, FOXA1, and TBX3. Simultaneously, genomic and transcriptomic analyses of HER2-low tumors indicate that they harbor several of these same molecular alterations (mutations, amplification/deletion, and fusions) in both the primary (2) and metastatic (26) settings. While the exact frequency remains to be determined, the majority of evidence suggests that a non-insignificant proportion of endocrine-resistant tumors may indeed be HER2-low. We summarize here recent evidence linking endocrine resistance-associated molecular alterations with HER2-low status. To facilitate this, we retrieved tumor-level HER2 immunohistochemistry (IHC) and fluorescence in situ hybridization (FISH) data from a comprehensive 2018 study of the genomic landscape of endocrine resistance by Razavi et al. (14) from cBioPortal (27–29) (Supplementary Table 1) to identify HER2-low tumors among those bearing validated endocrine resistance-associated genetic alterations (Supplementary Table 2). As shown in Table 1, a significant proportion of tumors bearing endocrine resistance-associated alterations could be classified as HER2-low (Supplementary Table 3), in most cases at a higher frequency (~63%–70%) than the expected proportion of HER2-low tumors (~45%–55%).
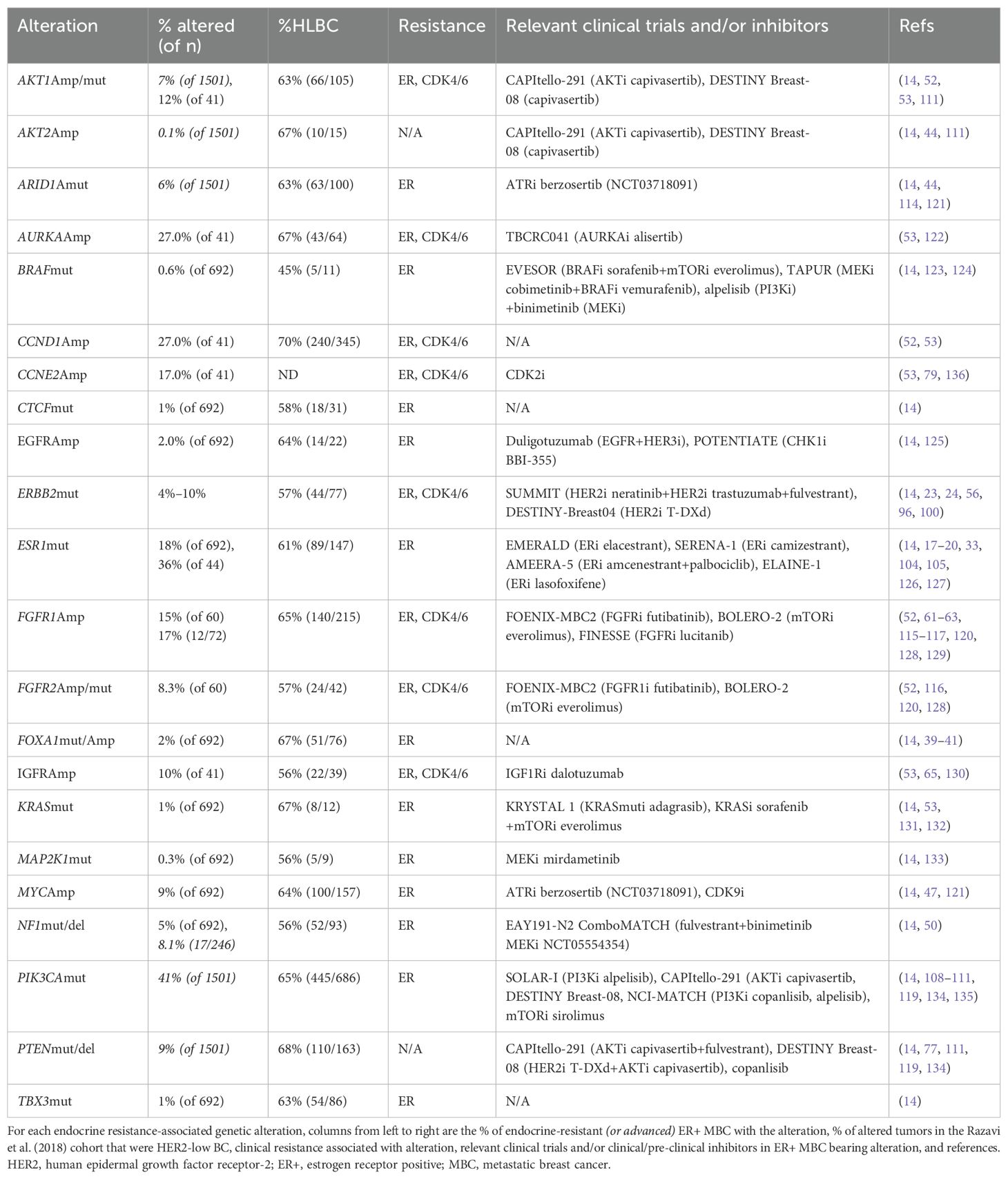
Table 1. Intersection of endocrine resistance mechanisms and HER2-low.
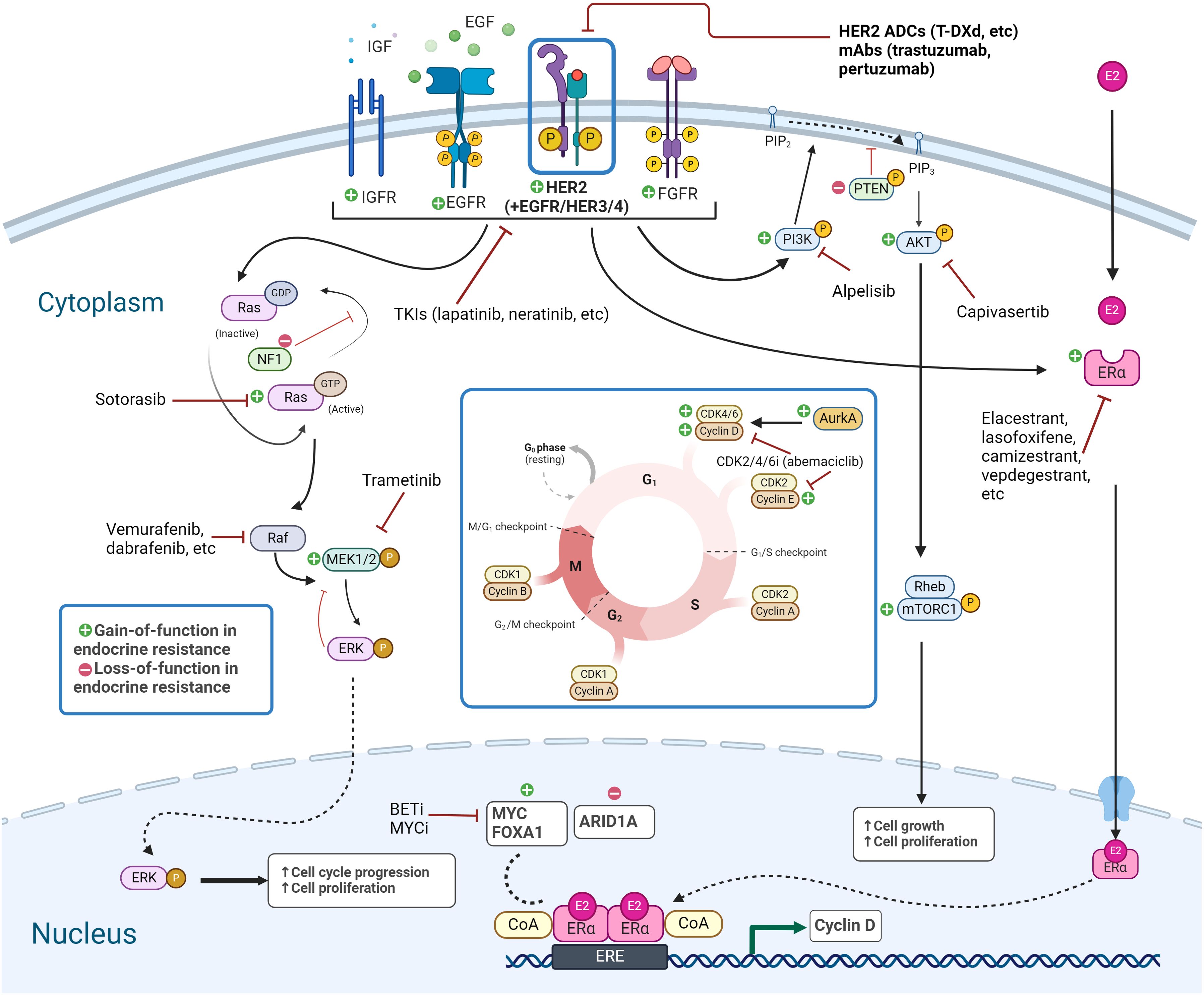
Figure 1. Signaling pathways in endocrine-resistant HER2-low breast cancer. Targetable genetic alterations and pathways in endocrine-resistant HER2-low breast cancer include RTK/MAPK pathway, PI3K/AKT/mTOR pathway, and TFs, along with the ER pathway and transcriptional targets, and alterations related to CDK4/6i resistance. Created with BioRender.com. HER2, human epidermal growth factor receptor-2; TFs, transcription factors; ER, estrogen receptor.
2.1 ER pathway hyperactivation
Acquired activating hotspot ERmuts are the most well-established mechanism of clinical endocrine resistance—they are dramatically enriched in this setting (~18%–50% of tumors) compared to primary BC (~2%) and primarily confer resistance to AIs (14, 17–20). In vitro studies found that ERmuts drive altered (often increased) HER2 downstream signaling (21, 30), thus hinting at potential crosstalk that may be targetable. Some alleles are more active—in the PALOMA-3 study, the Y537S ERmut was associated with resistance to fulvestrant alone and in combination with palbociclib (31, 32). A recent study identified a novel ERmut at F404 in cis with activating ERmuts as an acquired mechanism of resistance to fulvestrant in ~4% of tumors (33). ERmuts have been shown to drive an altered ER cistrome through stabilization of the agonist state (34), enabling ER binding to coactivators in the absence of ligand and enhanced binding to estrogen response elements (EREs) in target gene promoters (21), which maintain ER downstream signaling. ERmuts also have a lower affinity for the inhibitor tamoxifen (34). In addition to mutations, ESR1 overexpression and novel oncofusions (15, 16) can also drive resistance to endocrine therapies.
The recent genomic characterization study of HER2-low metastatic breast cancer (MBC) noted that ERmuts were preferentially enriched in ER+HER2-low tumors over ER+HER2-0 (16% of 370 vs. 11% of 336) (26). In addition, ESR1 amplification was one of only two copy number variations (CNVs) that were significantly enriched in HER2-low tumors regardless of ER status in this study. A related study in HER2-low tumors found that ER expression levels were significantly higher in this subset (3). Thus, endocrine resistance through ER hyperactivation appears to intersect significantly with HER2-low. In the Razavi cohort, 61% (89/147) of ERmut tumors were HER2-low (14).
2.2 Transcription factor alterations
Amplifications and/or hotspot mutations of key transcriptional regulators ARID1A, MYC, CTCF, FOXA1, and TBX3 are candidate mechanisms of resistance in 9% of endocrine-resistant tumors (14).
FOXA1 is a pioneer factor of ER function on chromatin and a known driver of ER+ BC (35–37). FOXA1 and ER co-expressed at high levels in endocrine therapy-resistant MBC (38), and FOXA1Amp contributes to endocrine resistance in preclinical models by reprogramming the ER and FOXA1 cistrome and transcriptome (39) and inducing a pro-metastatic secretome (40). FOXA1muts were also enriched in MBC and associated with worse responses to AIs (41). Increased FOXA1 expression levels through promoter mutation create stronger binding for E2F family TFs and promote cellular tolerance to fulvestrant treatment (42).
ARID1A is the most frequently mutated subunit of the SWI/SNF chromatin remodeling complex in ER+ BC (43) and an essential luminal lineage driver gene. Loss-of-function ARID1Amuts were enriched in the post-endocrine metastatic setting and were shown to mediate endocrine resistance in vitro by changing the chromatin accessibility of TFs playing a role in luminal differentiation as well as binding of the chromatin remodeling complex SWI/SNF at ER-FOXA1-GATA3 sites (14, 44).
MYC is an ER-regulated gene, whose overexpression confers endocrine resistance by promoting cell cycle progression and altered metabolism/unfolded protein response modulation (45), while knockdown of MYC impairs hormone-independent growth of BC cell lines in vitro (46–49).
In HER2-low primary BC, ARID1A, MYC, CTCF, FOXA1, and TBX3 were altered in 15% of tumors, while the frequency in HER2-low MBC remains to be determined (2). FOXA1 expression was significantly higher in HER2-low primary tumors (3). ARID1A-altered tumors accounted for 4% of HER2-low primary BC (all LumB tumors with a HER2 IHC score of 2+) and 8% of ER+HER2-low MBC (2, 26). MYC copy number gain has been identified in HER2-low tumors (2, 26). Analysis of advanced tumors in cBioPortal (Table 1) revealed that of tumors bearing FOXA1muts, MYCAmp, or ARID1Amuts, 67% (51/76), 64% (100/157), and 63% (63/100), respectively, can be classified as HER2-low. Among CTCFmut and TBX3mut tumors, 58% (18/31) and 63% (54/86) were HER2-low.
2.3 MAPK/PI3K-AKT pathway hyperactivation
2.3.1 RAS/RAF/MEK/ERK pathway
Components of the MAPK signaling cascade KRAS, BRAF, and MEK1; the negative regular NF1; and upstream RTKs (EGFR, HER2, FGFR1/2/4) are altered in ~13% of endocrine-resistant MBC (14, 50). Several of these are far more prevalent in heavily pre-treated populations by virtue of their ability to confer cross-resistance to not only second-line endocrine therapies but also CDK4/6is.
KRASmuts were found in 15.4% of AI-resistant tumors by circulating tumor DNA (ctDNA) analysis, were acquired in endocrine-resistant MBC (14), and conferred resistance to fulvestrant in vitro (51, 52). In vitro, the KRAS G12D activating mutation confers resistance to fulvestrant (53). Of KRASmut endocrine-resistant tumors in the Razavi et al. cohort, 67% (8/12) were HER2-low (Table 1) (14).
HER2 (ERBB2) and NF1 were most significantly increased in mutational frequency between pre- and post-hormonal therapy in ER+HER2− MBC (14). NF1 loss-of-function (LOF) mutations (NF1muts) account for 4.6% of endocrine-resistant tumors and drive endocrine resistance in vitro through both ER-dependent MAPK pathway-driven expression of Cyclin D1 and ER-independent S-phase entry (50, 54). Although NF1-altered tumors in Razavi et al. included several with the HER2-low subtype (Table 1, 52/93, 67%), NF1 loss was strongly associated with HER2-0 status in the HER2-low genomic characterization study (26); thus, this association is of uncertain relevance.
Among the RTK family proteins found altered in endocrine-resistant BC, HER2muts represent a mechanism with unique features. We and others previously identified acquired activating somatic hotspot HER2muts that conferred strong endocrine resistance to all clinically approved ER inhibitors and cross-resistance to the CDK4/6i palbociclib by activating MAPK/PI3K downstream pathways and altering the ER transcriptome (23, 55, 56). Transcriptomic analysis of HER2mut tumors revealed they have higher average HER2 expression than non-HER2mut tumors (57). Similarly, the recent HER2-low study determined that HER2mut tumors were slightly increased in ER+HER2-low tumors (26). We found that 57% (44/77) of HER2mut tumors in the Razavi cohort were HER2-low (Table 1). HER2mut tumors were also preferentially enriched in lobular vs. ductal subtypes (58, 59). Lastly, PAM50 analysis of HER2mut tumors appeared to indicate a switch to HER2-E or LumB (23, 60). Thus, ER+HER2mut tumors represent a unique subset of endocrine-resistant tumors that intersect with HER2-low. Mutational landscape analysis of primary HER2-low BC validated HER2muts (8%) (2), while they were also enriched in ER+HER2-low over ER+HER2-0 MBC (26).
2.3.1.1 Other RTKs
FGFR1/2Amp and FGFR2muts have been identified as a clinical mechanism of resistance to endocrine therapy through ER reprogramming and activation of the MAPK pathway in up to 40% of some smaller cohorts and validated in vitro (52, 61–63). FGFR2Amp was much more prevalent in HER2-low MBC compared to HER2-0 (26). As MAPK upstream regulators, EGFRAmp has been reported in 1.7% of endocrine-resistant MBC and demonstrated preclinically to confer endocrine (14) and CDK4/6i resistance (64) via activation of ERK and AKT. Additionally, the IGF1R pathway has been shown as a mechanism of escape from hormone dependence in BC in vitro (65) via crosstalk with the EGFR (66). Significantly, IGF1RAmp was one of only two CNVs that were found to be significantly enriched in HER2-low tumors regardless of the ER level (26). cBioPortal analysis revealed that 64% of MBC with FGFR1/2 gene alterations (164/257) and EGFRAmp (14/22) were HER2-low (Table 1), along with 56% (22/39) of IGF1RAmp.
2.3.2 PI3K/AKT pathway
The PI3K pathway components PIK3CA, AKT1/2, and PTEN are highly mutated in ER+ BC (67, 68). The role of PIK3CAmuts in endocrine resistance is unclear. Clinically, based on the PALOMA-3 study, there was no significant difference in progression-free survival (PFS) between PIK3CAmut and PIK3CAwt tumors (4), and there was no significant enrichment of PIK3CAmuts in endocrine-resistant MBC (14). However, PIK3CAmuts can confer endocrine resistance in vitro (69). Genomic characterization of HER2-low tumors indicated that the frequency of PIK3CAmuts was similar in ER+ tumors regardless of HER2-low or HER2-0 status (26). Of ER+ MBC tumors with PIK3CAmuts, 65% are HER2-low (14) (Table 1), but HER2-low MBC has a lower frequency of PIK3CAmuts overall (26).
AKT is a downstream effector of PI3K, whose overexpression confers endocrine and CDK4/6i resistance in vitro (52, 53). Clinically, AKT1 alterations account for 7% of ER+HER2− MBC (14) and were enriched compared with primary disease (50). Some studies suggested that activated AKT (increased pAKT) levels negatively correlated with the efficacy of endocrine therapy (70) and were associated with worse outcomes among endocrine-treated BC patients (71, 72). AKT1muts were highly enriched in HER2-low MBC when compared with HER2-0 (26).
mTOR activation is highly correlated with a lack of response to ER+CDK4/6i in clinical studies (73) and increased migration and stemness (74). While mTORmuts have not been validated as a mechanism of endocrine resistance in vitro, they have been found to be acquired in ER+CDK4/6i-resistant tumors (60). In a genomic study, MTOR was the top significant gene that was mutated preferentially in HER2-low vs. HER2-0 MBC (26).
PTEN is a negative regulator of the PI3K pathway, and its loss is suggested to contribute to clinical endocrine resistance by activating PI3K signaling (75, 76). In vitro, reduced PTEN levels resulted in endocrine resistance (77), while PTEN loss was enriched in HER2-low vs. HER2-0 MBC (26).
Analysis of advanced tumors in the Razavi cohort (Table 1) revealed that 63% of AKT1-altered (66/105), 67% of AKT2Amp (10/15), 65% of PIK3CAmut (445/686), and 67% of PTEN-altered tumors can be classified as HER2-low.
2.4 Endocrine and CDK4/6i combination therapy resistance
CDK4/6is block the cell cycle transition from the G1 phase to the S phase, leading to cell cycle arrest in ER+ BC (78). However, resistance to CDK4/6is occurs in virtually all patients, leading to high mortality. In ER+ BC, CDK4/6is are usually utilized in combination with ERis—studies of acquired clinical resistance to the combination found that acquired alterations in RB1, CCND1, CDK4/6, CCNE1/2, and AURKA were enriched in resistant tumors (53, 79). Additionally, some established mechanisms of endocrine resistance, specifically those in the “MAPK” class such as ERBB2muts and IGF1RAmp (80), AKT1Amp, and AKT1muts, confer cross-resistance to CDK4/6is. Key recent findings indicate that the HER2-low subtype is independently associated with worse response to ERi+CDK4/6i, while the specific molecular alterations enriched have not been determined. The recent genomic characterization of HER2-low MBC indicated that CCND1 and CDK4 were more frequently amplified in HER2-low over HER2-0 as a potential mechanism of CDK4/6 resistance (26). Thus, ER+HER2-low tumors represent a sizeable fraction of ER+ tumors that are particularly intractable to ER+CDK4/6is in the clinic and for which therapeutic options are desperately needed.
2.5 Subtype switching in endocrine-resistant tumors
The PAM50 intrinsic subtype (IS) was the strongest prognostic factor predicting PFS and OS (LumA > LumB > HER2-E/basal) in a retrospective analysis of ER+ MBC (81), while another analysis of clinical trials in ER+ MBC showed that the HER2-E subtype correlated with response to HER2i and ribociclib, and the basal subtype correlated with response to chemotherapy (82). Endocrine resistance is also linked with enhanced phenotypic plasticity (83), which enables IS switching through downregulation of luminal markers and upregulation of basal markers (84), and transition to a non-luminal subtype (basal or HER2-E). Switching between HER2+ and HER2− is relatively common (~36%) in studies of primary vs. metastatic tumors from the same patient (85), along with IS switching—there is enrichment of HER2-E and decrease of LumA in MBC (86). Gene expression changes and subsequent IS switches were more frequent in LumA MBC than in the other molecular subtypes (87). For example, 40.4% of LumA switched to LumB, and 10%–15% of LumA/B tumors switched to HER2-E in a retrospective study (82), while 90% of LumA tumors switched to LumB or HER2-E in the AURORA study of MBC (88). Another study of paired primary and relapse (endocrine-refractory) tumors found that 19/70 (27.1%) underwent the PAM50 switch, with the HER2-E subtype enriched in the resistant setting (89).
Additionally, there is a switching of HER2-low status in progressive disease—a retrospective study of LumB BC patients showed that there was extensive switching between HER2-low and HER2-0 status at a rate of 34% or 25% in either direction (90). Endocrine-resistant ER+ MBC often undergoes switches between HER2-0 and HER2-low status (90).
While the exact percentage remains to be determined due to a lack of systematic large-scale clinical transcriptomic studies, there is limited evidence that tumors with acquired endocrine resistance-associated genetic alterations undergo subtype switches. For example, a prior study from our group showed that an ER+ MBC tumor that acquired a HER2mut switched from ER+HER2-0 to ER+HER2-low (23), while HER2mut tumors are clinically associated with HER2-E (88). Although transcriptomic characterization of primary tumors linked the HER2-low subtype most closely with a LumA signature (3), these may differ in the endocrine resistance context. Along these lines, a recent small study in ER+ MBC showed that most ERmut, FGFR1Amp, NF1loss, AKT1mut, and CCND1Amp tumors were predominantly LumB, while the HER2mut and BRAFmut tumors in this cohort were HER2-E, rather than LumA (60). FGFRAmp, EGFRAmp, and CCND1Amp were also enriched in HER2-E primary tumors (91). Additionally, all the ERmut tumors in another study of primary HER2-low tumors were classified as the LumB subtype and were characterized by a HER2 IHC score of 2+ (2). While PIK3CAmuts were not associated with endocrine resistance, they were preferentially enriched in MBC undergoing a subtype switch to HER2-E in AURORA (88).
Alterations that lead to subtype switch to basal may be less likely to have relevance to HER2-low signaling, although several of these are significantly associated with HER2-low in the MBC setting (26). For example, ERmut (92) and ARID1Amut (44) tumors demonstrate upregulation of basal genes. GATA3, while not enriched in endocrine resistance, is an important marker of ER dependence and luminal identity (93)—GATA3muts were preferentially enriched in ER+HER2-low tumors over ER+HER2-0 (16% of 370 vs. 9% of 336) (26) while also present in ~18% (18/99) of HER2-low primary BC (2).
Importantly, HER2i was shown to be able to convert HER2-E tumors to less aggressive LumA in HER2+ MBC (94), which may enable re-sensitization to endocrine therapy. Thus, subtype switching in endocrine-resistant HER2-low tumors, particularly to HER2-E, may be able to inform treatment.
3 Potential therapeutic strategies for endocrine-resistant HER2-low tumors
Historical evidence confirmed that HER2 expression or amplification level correlates strongly with response to and benefit from trastuzumab (95), lending credence to HER2 as a biomarker. In contrast, the DESTINY-Breast04 (96) and DAISY (97) trials demonstrated the surprising benefits of treating HER2-low MBC with the HER2 antibody–drug conjugate (ADC) trastuzumab deruxtecan (T-DXd). Deruxtecan is unique in its ability to kill tumor cells nearby, demonstrating a potent bystander effect due to its membrane-permeable payload (98). Patients with refractory ER+ BC who received T-DXd had significantly longer progression-free and overall survival than those who received chemotherapy (96). Surprisingly, this efficacy was independent of HER2 status. Based on this trial, T-DXd was approved in August 2022, demonstrating an improvement in the clinic for those diagnosed with ER+HER2-low MBC.
Endocrine resistance-associated genetic alterations differ widely in their profiles of response to targeted therapies (Table 1). HER2mut tumors were cross-resistant to the CDK4/6i palbociclib (23) and variably sensitive to HER2 tyrosine kinase inhibitors (TKIs) (55), while the irreversible TKI neratinib resensitized HER2muts to fulvestrant in vitro. The combination of neratinib with trastuzumab and fulvestrant was efficacious against HER2mut ER+ BC in the SUMMIT basket trial (23, 25, 99), after progression on CDK4/6is (100), and the fulvestrant+neratinib combination in ER+HER2mut was further supported by the mutHER trial (101). Finally, the DESTINY-Lung02 trial also indicated that T-DXd had surprising activity in HER2mut tumors (102), potentially due to increased mutant receptor internalization (103).
The standard of care treatment for ER+HER2− BC remains the combination of ERi+CDK4/6i in the advanced setting. Patients who developed resistance to therapy by acquiring ERmuts can now benefit from novel oral SERDS, such as elacestrant and camizestrant (104)—the former was FDA-approved specifically for ER+HER2− ERmut MBC in 2023. Newer ER-targeting agents also have been found to have activity against ERmuts—the phase II ELAINE 1 trial demonstrated encouraging antitumor activity of lasofoxifene, a novel SERM/SERD hybrid, in patients with ERmut endocrine-resistant MBC following progression on AI+CDK4/6i (105). Newer ER proteolysis-targeting chimeras (PROTACs) like vepdegestrant also appear promising (106). Aside from newer SERDs, some TF-driven endocrine resistance may continue to respond to other ER-targeting agents. For example, FOXA1muts showed sensitivity to tamoxifen or fulvestrant in vitro (41). Although ERmuts are associated with poor response to CDK4/6is, abemaciclib, which is unique compared to other CDK4/6is, may be of use, particularly with the deployment of a recently developed multigene panel that could predict resistance to abemaciclib in ERmut tumors (107).
Clinically, PIK3CAmuts are targetable through the PI3K inhibitor, alpelisib (108). In ER+HER2− MBC, patients with PIK3CAmut tumors had significantly longer PFS with alpelisib+fulvestrant than fulvestrant alone. The FDA approved alpelisib+ fulvestrant for PIK3CAmut ER+HER2- MBC patients based on overall survival results from the SOLAR-1 trial (109). In the case of endocrine therapy resistance associated with reduced PTEN levels, adding mTOR (110), AKT, or MEK inhibitors to endocrine therapy has been suggested as a promising strategy in vitro (77). PI3K pathway alterations are additionally targetable through the newest approved pan-AKT inhibitor capivasertib (111, 112), which is currently being evaluated in combination with T-DXd for HER2-low BC in the phase III DESTINY-Breast08 trial (113).
Other mechanisms lack clinically approved or defined targeted therapeutic strategies at the moment. These include ARID1A loss, which sensitized ER+ cells to BET inhibition in vitro (114). Targeting FGFR with an irreversible pan-FGFR inhibitor, FIIN-3 reversed the activation of FGFR and resensitized cells expressing FGFR2 to fulvestrant (52). FGFR1 overexpressing ER+ BC cells were highly sensitive to the mTOR inhibitor everolimus (115). The combination of fulvestrant with either everolimus or with the FGFR TKi lucitanib overcame endocrine resistance in vitro (115). Futibatinib, an irreversible FGFR inhibitor, significantly inhibited the growth of PDX models bearing FGFR2 alterations (116). The FOENIX-MBC2 ongoing clinical trial demonstrated the efficacy of futibatinib in patients with locally advanced/metastatic BC harboring FGFR1/2 amplifications who have experienced disease progression after prior therapy for advanced/metastatic disease (117). Other pan-FGFR inhibitors are in clinical trials for FGFR-altered tumors (118).
In most of the abovementioned endocrine resistance-associated altered tumors, classification as HER2-low opens up additional therapeutic strategies that could be advantageous, particularly in cases where dependence on HER2 is borne out by PAM50 subtyping and/or subtype enrichment. Several key clinical trials with relevance to specific genetic alterations are included in Table 1. These include the DESTINY-Breast04 (96) and DESTINY-Breast08 (119) trials, which are in HER2-low MBC and are likely to include endocrine-resistant tumors, as well as others in which specified genetic alterations were inclusion criteria, or in which retrospective subgroup analysis has indicated the significant benefit of the intervention for the altered tumors (22, 100, 105, 109, 111, 117, 120–137).
4 Discussion (perspective)
Overall, the available clinical data indicate that a significant subset of endocrine-resistant tumors may be characterized as HER2-low in opposition to HER2-negative. With the advent of newer HER2-targeting therapies such as T-DXd, the time is ripe to consider whether combination regimens that include HER2-targeting ADCs should be explored in combination with endocrine agents, especially novel oral SERDs, as next-line therapies for patients failing ER and CDK4/6 inhibition. There are two reasons that this approach should be considered: 1) the vast diversity in mechanisms of acquired endocrine and CDK4/6 clinical resistance mechanisms makes it challenging to target tumors effectively, and 2) as the two major molecularly targeted pathways in breast cancer, there is a continuous pipeline of newer ER and HER2 inhibitors that may be potentially viable for endocrine-resistant HER2-low tumors. The major questions that remain to be addressed include 1) what proportion of endocrine-resistant tumors may be classified as HER2-low, 2) which specific genetic alterations are enriched in this context that may be targetable in addition, and 3) whether endocrine-resistant HER2-low tumors tend to be preferentially lobular vs. ductal. The landscape of lobular carcinoma is beginning to be understood as a distinct entity (138), and certain alterations such as HER2muts are enriched in lobular as well as among HER2-low. Thus, our ability to exploit HER2-low status in endocrine resistance will depend on a more comprehensive understanding of the disease context for ER+HER2-low tumors.
Data availability statement
Publicly available datasets were analyzed in this study. This data can be found here: https://www.cbioportal.org/study/summary?id=breast_msk_2018.
Ethics statement
Ethical approval was not required for the study involving humans in accordance with the local legislation and institutional requirements. Written informed consent to participate in this study was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and the institutional requirements.
Author contributions
GY: Conceptualization, Data curation, Formal analysis, Methodology, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. AT: Data curation, Formal analysis, Writing – review & editing. UN: Conceptualization, Data curation, Funding acquisition, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing, Formal Analysis.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Research, authorship, and/or publication of this article was supported by a National Cancer Institute Transition Career Development Award K22CA241377 to UN.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2024.1461190/full#supplementary-material
References
1. Tarantino P, Hamilton E, Tolaney SM, Cortes J, Morganti S, Ferraro E, et al. HER2-low breast cancer: pathological and clinical landscape. J Clin Oncol. (2020) 38:1951–62. doi: 10.1200/JCO.19.02488
2. Berrino E, Annaratone L, Bellomo SE, Ferrero G, Gagliardi A, Bragoni A, et al. Integrative genomic and transcriptomic analyses illuminate the ontology of HER2-low breast carcinomas. Genome Med. (2022) 14:98–. doi: 10.1186/s13073-022-01104-z
3. Schettini F, Chic N, Brasó-Maristany F, Paré L, Pascual T, Conte B, et al. Clinical, pathological, and PAM50 gene expression features of HER2-low breast cancer. NPJ Breast Cancer. (2021) 7:1. doi: 10.1038/s41523-020-00208-2
4. Cristofanilli M, Turner NC, Bondarenko I, Ro J, Im SA, Masuda N, et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. (2016) 17:425–39. doi: 10.1016/S1470-2045(15)00613-0
5. Im SA, Lu YS, Bardia A, Harbeck N, Colleoni M, Franke F, et al. Overall survival with ribociclib plus endocrine therapy in breast cancer. N Engl J Med. (2019) 381:307–16. doi: 10.1056/NEJMoa1903765
6. Sledge GW Jr., Toi M, Neven P, Sohn J, Inoue K, Pivot X, et al. MONARCH 2: abemaciclib in combination with fulvestrant in women with HR+/HER2- advanced breast cancer who had progressed while receiving endocrine therapy. J Clin Oncol. (2017) 35:2875–84. doi: 10.1200/JCO.2017.73.7585
7. Harbeck N, Penault-Llorca F, Cortes J, Gnant M, Houssami N, Poortmans P, et al. Breast cancer. Nat Rev Dis Primers. (2019) 5:66. doi: 10.1038/s41572-019-0111-2
8. Nolan E, Lindeman GJ, Visvader JE. Deciphering breast cancer: from biology to the clinic. Cell. (2023) 186:1708–28. doi: 10.1016/j.cell.2023.01.040
9. Martin LA, Farmer I, Johnston SRD, Ali S, Marshall C, Dowsett M. Enhanced estrogen receptor (ER) alpha, ERBB2, and MAPK signal transduction pathways operate during the adaptation of MCF-7 cells to long term estrogen deprivation. J Biol Chem. (2003) 278:30458–68 (0021-9258 (Print)). doi: 10.1074/jbc.M305226200
10. Knowlden JM, Hutcheson IR, Jones HE, Madden T, Gee JMW, Harper ME, et al. Elevated levels of epidermal growth factor receptor/c-erbB2 heterodimers mediate an autocrine growth regulatory pathway in tamoxifen-resistant MCF-7 cells. Endocrinology. (2003) 144:1032–44 (0013-7227 (Print)). doi: 10.1210/en.2002-220620
11. Frogne T, Benjaminsen RV, Sonne-Hansen K, Sorensen BS, Nexo E, Laenkholm AV, et al. Activation of ErbB3, EGFR and Erk is essential for growth of human breast cancer cell lines with acquired resistance to fulvestrant. Breast Cancer Res Treat. (2009) 114:263–75. doi: 10.1007/s10549-008-0011-8
12. Loi S, Sotiriou C, Haibe-Kains B, Lallemand F, Conus NM, Piccart MJ, et al. Gene expression profiling identifies activated growth factor signaling in poor prognosis (Luminal-B) estrogen receptor positive breast cancer. BMC Med Genomics. (2009) 2:37. doi: 10.1186/1755-8794-2-37
13. Hanker AB, Sudhan DR, Arteaga CL. Overcoming endocrine resistance in breast cancer. Cancer Cell. (2020) 37:496–513. doi: 10.1016/j.ccell.2020.03.009
14. Razavi P, Chang MT, Xu G, Bandlamudi C, Ross DS, Vasan N, et al. The genomic landscape of endocrine-resistant advanced breast cancers. Cancer Cell. (2018) 34:427–38.e6. doi: 10.1016/j.ccell.2018.08.008
15. Li S, Shen D, Shao J, Crowder R, Liu W, Prat A, et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep. (2013) 4:1116–30. doi: 10.1016/j.celrep.2013.08.022
16. Lei JT, Shao J, Zhang J, Iglesia M, Chan DW, Cao J, et al. Functional annotation of ESR1 gene fusions in estrogen receptor-positive breast cancer. Cell Rep. (2018) 24:1434–44.e7. doi: 10.1016/j.celrep.2018.07.009
17. Toy W, Shen Y, Won H, Green B, Sakr RA, Will M, et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet. (2013) 45:1439–45. doi: 10.1038/ng.2822
18. Robinson DR, Wu YM, Vats P, Su F, Lonigro RJ, Cao X, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet. (2013) 45:1446–51. doi: 10.1038/ng.2823
19. Merenbakh-Lamin K, Ben-Baruch N, Yeheskel A, Dvir A, Soussan-Gutman L, Jeselsohn R, et al. D538G mutation in estrogen receptor-α: A novel mechanism for acquired endocrine resistance in breast cancer. Cancer Res. (2013) 73:6856–64. doi: 10.1158/0008-5472.CAN-13-1197
20. Jeselsohn R, Yelensky R, Buchwalter G, Frampton G, Meric-Bernstam F, Gonzalez-Angulo AM, et al. Emergence of constitutively active estrogen receptor-α mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin Cancer Res. (2014) 20:1757–67. doi: 10.1158/1078-0432.CCR-13-2332
21. Jeselsohn R, Bergholz JS, Pun M, Cornwell M, Liu W, Nardone A, et al. Allele-specific chromatin recruitment and therapeutic vulnerabilities of ESR1 activating mutations. Cancer Cell. (2018) 33:173–86.e5. doi: 10.1016/j.ccell.2018.01.004
22. Novel SERD has PFS edge against breast cancer. Cancer Discovery. (2022) 12:281. doi: 10.1158/2159-8290.CD-NB2021-0406
23. Nayar U, Cohen O, Kapstad C, Cuoco MS, Waks AG, Wander SA, et al. Acquired HER2 mutations in ER+ metastatic breast cancer confer resistance to estrogen receptor–directed therapies. Nat Genet. (2019) 51:207–16. doi: 10.1038/s41588-018-0287-5
24. Croessmann S, Formisano L, Kinch LN, Gonzalez-Ericsson PI, Sudhan DR, Nagy RJ, et al. Combined blockade of activating ERBB2 mutations and ER results in synthetic lethality of ER+/HER2 mutant breast cancer. Clin Cancer Res. (2019) 25:277–89. doi: 10.1158/1078-0432.CCR-18-1544
25. Hyman DM, Piha-Paul SA, Won H, Rodon J, Saura C, Shapiro GI, et al. HER kinase inhibition in patients with HER2- and HER3-mutant cancers. Nature. (2018) 554:189–94. doi: 10.1038/nature25475
26. Tarantino P, Gupta H, Hughes ME, Files J, Strauss S, Kirkner G, et al. Comprehensive genomic characterization of HER2-low and HER2-0 breast cancer. Nat Commun. (2023) 14:7496. doi: 10.1038/s41467-023-43324-w
27. de Bruijn I, Kundra R, Mastrogiacomo B, Tran TN, Sikina L, Mazor T, et al. Analysis and visualization of longitudinal genomic and clinical data from the AACR project GENIE biopharma collaborative in cBioPortal. Cancer Res. (2023) 83:3861–7. doi: 10.1158/0008-5472.CAN-23-0816
28. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signaling. (2013) 6:pl1. doi: 10.1126/scisignal.2004088
29. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discovery. (2012) 2:401–4. doi: 10.1158/2159-8290.CD-12-0095
30. Giordano C, Cui Y, Barone I, Ando S, Mancini MA, Berno V, et al. Growth factor-induced resistance to tamoxifen is associated with a mutation of estrogen receptor alpha and its phosphorylation at serine 305. Breast Cancer Res Treat. (2010) 119:71–85. doi: 10.1007/s10549-009-0334-0
31. O'Leary B, Cutts RJ, Liu Y, Hrebien S, Huang X, Fenwick K, et al. The genetic landscape and clonal evolution of breast cancer resistance to palbociclib plus fulvestrant in the PALOMA-3 trial. Cancer Discovery. (2018) 8:1390–403. doi: 10.1158/2159-8290.CD-18-0264
32. Jeselsohn R, Buchwalter G, De Angelis C, Brown M, Schiff R. ESR1 mutations—a mechanism for acquired endocrine resistance in breast cancer. Nat Rev Clin Oncol. (2015) 12:573–83. doi: 10.1038/nrclinonc.2015.117
33. Kingston B, Pearson A, Herrera-Abreu MT, Sim LX, Cutts RJ, Shah H, et al. ESR1 F404 mutations and acquired resistance to fulvestrant in ESR1-mutant breast cancer. Cancer Discovery. (2024) 14:274–89. doi: 10.1158/2159-8290.CD-22-1387
34. Fanning SW, Mayne CG, Dharmarajan V, Carlson KE, Martin TA, Novick SJ, et al. Estrogen receptor alpha somatic mutations Y537S and D538G confer breast cancer endocrine resistance by stabilizing the activating function-2 binding conformation. Elife. (2016) 5:e12792. doi: 10.7554/eLife.12792
35. Carroll JS, Liu XS, Brodsky AS, Li W, Meyer CA, Szary AJ, et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell. (2005) 122:33–43. doi: 10.1016/j.cell.2005.05.008
36. Toska E, Osmanbeyoglu HU, Castel P, Chan C, Hendrickson RC, Elkabets M, et al. PI3K pathway regulates ER-dependent transcription in breast cancer through the epigenetic regulator KMT2D. Science. (2017) 355:1324–30. doi: 10.1126/science.aah6893
37. Hurtado A, Holmes KA, Ross-Innes CS, Schmidt D, Carroll JS. FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nat Genet. (2011) 43:27–33. doi: 10.1038/ng.730
38. Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature. (2012) 481:389–93. doi: 10.1038/nature10730
39. Fu X, Jeselsohn R, Pereira R, Hollingsworth EF, Creighton CJ, Li F, et al. FOXA1 overexpression mediates endocrine resistance by altering the ER transcriptome and IL-8 expression in ER-positive breast cancer. Proc Natl Acad Sci U States A. (2016) 113:E6600–E9. doi: 10.1073/pnas.1612835113
40. Fu X, Pereira R, Liu CC, De Angelis C, Shea MJ, Nanda S, et al. High FOXA1 levels induce ER transcriptional reprogramming, a pro-metastatic secretome, and metastasis in endocrine-resistant breast cancer. Cell Rep. (2023) 42:112821. doi: 10.1016/j.celrep.2023.112821
41. Arruabarrena-Aristorena A, Maag JLV, Kittane S, Cai Y, Karthaus WR, Ladewig E, et al. FOXA1 mutations reveal distinct chromatin profiles and influence therapeutic response in breast cancer. Cancer Cell. (2020) 38:534–50.e9. doi: 10.1016/j.ccell.2020.08.003
42. Rheinbay E, Parasuraman P, Grimsby J, Tiao G, Engreitz JM, Kim J, et al. Recurrent and functional regulatory mutations in breast cancer. Nature. (2017) 547:55–60. doi: 10.1038/nature22992
43. Cheng X, Zhao JX, Dong F, Cao XC. ARID1A mutation in metastatic breast cancer: A potential therapeutic target. Front Oncol. (2021) 11:759577. doi: 10.3389/fonc.2021.759577
44. Xu G, Chhangawala S, Cocco E, Razavi P, Cai Y, Otto JE, et al. ARID1A determines luminal identity and therapeutic response in estrogen-receptor-positive breast cancer. Nat Genet. (2020) 52:198–207. doi: 10.1038/s41588-019-0554-0
45. Shajahan-Haq AN, Cook KL, Schwartz-Roberts JL, Eltayeb AE, Demas DM, Warri AM, et al. MYC regulates the unfolded protein response and glucose and glutamine uptake in endocrine resistant breast cancer. Mol Cancer. (2014) 13:239. doi: 10.1186/1476-4598-13-239
46. Miller TW, Balko JM, Ghazoui Z, Dunbier A, Anderson H, Dowsett M, et al. A gene expression signature from human breast cancer cells with acquired hormone independence identifies MYC as a mediator of antiestrogen resistance. Clin Cancer Res. (2011) 17:2024–34. doi: 10.1158/1078-0432.CCR-10-2567
47. Musgrove EA, Sergio CM, Loi S, Inman CK, Anderson LR, Alles MC, et al. Identification of functional networks of estrogen- and c-Myc-responsive genes and their relationship to response to tamoxifen therapy in breast cancer. PloS One. (2008) 3(8):e2987. doi: 10.1371/journal.pone.0002987
48. Prall OWJ, Rogan EM, Musgrove EA, Watts CKW, Sutherland RL. c-Myc or cyclin D1 mimics estrogen effects on cyclin E-Cdk2 activation and cell cycle reentry. Mol Cell Biol. (1998) 18:4499–508. doi: 10.1128/MCB.18.8.4499
49. Butt AJ, McNeil CM, Musgrove EA, Sutherland RL. Downstream targets of growth factor and oestrogen signalling and endocrine resistance: the potential roles of c-Myc, cyclin D1 and cyclin E. Endocr Relat Cancer. (2005) 12 Suppl 1:S47–59. doi: 10.1677/erc.1.00993
50. Pearson A, Proszek P, Pascual J, Fribbens C, Shamsher MK, Kingston B, et al. Inactivating NF1 mutations are enriched in advanced breast cancer and contribute to endocrine therapy resistance. Clin Cancer Res. (2020) 26:608–22. doi: 10.1158/1078-0432.CCR-18-4044
51. Fribbens C, Garcia Murillas I, Beaney M, Hrebien S, O'Leary B, Kilburn L, et al. Tracking evolution of aromatase inhibitor resistance with circulating tumour DNA analysis in metastatic breast cancer. Ann Oncol. (2018) 29:145–53. doi: 10.1093/annonc/mdx483
52. Mao P, Cohen O, Kowalski KJ, Kusiel JG, Buendia-Buendia JE, Cuoco MS, et al. Acquired FGFR and FGF alterations confer resistance to estrogen receptor (ER) targeted therapy in ER(+) metastatic breast cancer. Clin Cancer Res. (2020) 26:5974–89. doi: 10.1158/1078-0432.CCR-19-3958
53. Wander SA, Cohen O, Gong X, Johnson GN, Buendia-Buendia JE, Lloyd MR, et al. The genomic landscape of intrinsic and acquired resistance to cyclin-dependent kinase 4/6 inhibitors in patients with hormone receptor-positive metastatic breast cancer. Cancer Discovery. (2020) 10:1174–93. doi: 10.1158/2159-8290.CD-19-1390
54. Sokol ES, Feng YX, Jin DX, BaSudan A, Lee AV, Atkinson JM, et al. Loss of function of NF1 is a mechanism of acquired resistance to endocrine therapy in lobular breast cancer. Ann Oncol. (2019) 30:115–23. doi: 10.1093/annonc/mdy497
55. Bose R, Kavuri SM, Searleman AC, Shen W, Shen D, Koboldt DC, et al. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discovery. (2013) 3:224–37. doi: 10.1158/2159-8290.CD-12-0349
56. Hanker AB, Red Brewer M, Sheehan JH, Koch JP, Sliwoski GR, Nagy R, et al. An acquired HER2 T798I gatekeeper mutation induces resistance to neratinib in a patient with HER2 mutant-driven breast cancer. Cancer Discovery. (2017) 9:303. doi: 10.1158/2159-8290.CD-16-1431
57. Hensing WL, Thomas SN, El-Refai S, Mauer E, Ma C, Bose R. Abstract P2-23-10: Gene expression and mutation profiles in HER2-mutated metastatic breast cancer. Cancer Res. (2023) 83:P2–23-10-P2-23-10. doi: 10.1158/1538-7445.SABCS22-P2-23-10
58. Rosa-Rosa JM, Caniego-Casas T, Leskela S, Cristobal E, González-Martínez S, Moreno-Moreno E, et al. High frequency of ERBB2 activating mutations in invasive lobular breast carcinoma with pleomorphic features. Cancers (Basel). (2019) 11:74. doi: 10.3390/cancers11010074
59. Kalra R, Chen CH, Wang J, Salam AB, Dobrolecki LE, Lewis A, et al. Poziotinib inhibits HER2-mutant–driven therapeutic resistance and multiorgan metastasis in breast cancer. Cancer Res. (2022) 82:2928–39. doi: 10.1158/0008-5472.CAN-21-3106
60. Gómez Tejeda Zañudo JA-O, Barroso-Sousa R, Jain E, Jin QA-O, Li T, Buendia-Buendia JE, et al. Exemestane plus everolimus and palbociclib in metastatic breast cancer: clinical response and genomic/transcriptomic determinants of resistance in a phase I/II trial. Nat Commun. (2024) 15:2041–1723 (Electronic). doi: 10.1038/s41467-024-45835-6
61. Giltnane JM, Hutchinson KE, Stricker TP, Formisano L, Young CD, Estrada MV, et al. Genomic profiling of ER+ breast cancers after short-term estrogen suppression reveals alterations associated with endocrine resistance. Sci Trans Med. (2017) 9(402):eaai7993. doi: 10.1126/scitranslmed.aai7993
62. Formisano L, Lu Y, Servetto A, Hanker AB, Jansen VM, Bauer JA, et al. Aberrant FGFR signaling mediates resistance to CDK4/6 inhibitors in ER+ breast cancer. Nat Commun. (2019) 10:1–14. doi: 10.1038/s41467-019-09068-2
63. Turner N, Pearson A, Sharpe R, Lambros M, Geyer F, Lopez-Garcia MA, et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res. (2010) 70:2085–94. doi: 10.1158/0008-5472.CAN-09-3746
64. Belli S, Esposito D, Ascione CM, Messina F, Napolitano F, Servetto A, et al. EGFR and HER2 hyper-activation mediates resistance to endocrine therapy and CDK4/6 inhibitors in ER+ breast cancer. Cancer letters. (2024) 593:216968. doi: 10.1016/j.canlet.2024.216968
65. Fox EM, Miller TW, Balko JM, Kuba MG, Sánchez V, Smith RA, et al. A kinome-wide screen identifies the insulin/IGF-I receptor pathway as a mechanism of escape from hormone dependence in breast cancer. Cancer Res. (2011) 71:6773–84. doi: 10.1158/0008-5472.CAN-11-1295
66. Knowlden JM, Hutcheson IR, Barrow D, Gee JM, Nicholson RI. Insulin-like growth factor-I receptor signaling in tamoxifen-resistant breast cancer: a supporting role to the epidermal growth factor receptor. Endocrinology. (2005) 146:4609–18. doi: 10.1210/en.2005-0247
67. Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discovery. (2014) 13:140–56. doi: 10.1038/nrd4204
68. López-Knowles E, O'Toole SA, McNeil CM, Millar EK, Qiu MR, Crea P, et al. PI3K pathway activation in breast cancer is associated with the basal-like phenotype and cancer-specific mortality. Int J Cancer. (2010) 126:1121–31. doi: 10.1002/ijc.24831
69. Huang D, Tang L, Yang F, Jin J, Guan X. PIK3CA mutations contribute to fulvestrant resistance in ER-positive breast cancer. Am J Trans Res. (2019) 11:6055–.
70. Tokunaga E, Kataoka A, Kimura Y, Oki E, Mashino K, Nishida K, et al. The association between Akt activation and resistance to hormone therapy in metastatic breast cancer. Eur J Cancer. (2006) 42:629–35. doi: 10.1016/j.ejca.2005.11.025
71. Stål O, Pérez-Tenorio G, Akerberg L, Olsson B, Nordenskjöld B, Skoog L, et al. Akt kinases in breast cancer and the results of adjuvant therapy. Breast Cancer Res. (2003) 5:R37–44. doi: 10.1186/bcr569
72. Pérez-Tenorio G, Stål O. Activation of AKT/PKB in breast cancer predicts a worse outcome among endocrine treated patients. Br J Cancer. (2002) 86:540–5. doi: 10.1038/sj.bjc.6600126
73. Abu-Khalaf MM, Alex Hodge K, Hatzis C, Baldelli E, El Gazzah E, Valdes F, et al. AKT/mTOR signaling modulates resistance to endocrine therapy and CDK4/6 inhibition in metastatic breast cancers. NPJ Precis Oncol. (2023) 7:18. doi: 10.1038/s41698-023-00360-5
74. Rodriguez MJ, Perrone MC, Riggio M, Palafox M, Salinas V, Elia A, et al. Targeting mTOR to overcome resistance to hormone and CDK4/6 inhibitors in ER-positive breast cancer models. Sci Rep. (2023) 13:2710. doi: 10.1038/s41598-023-29425-y
75. Tanic N, Milovanovic Z, Tanic N, Dzodic R, Juranic Z, Susnjar S, et al. The impact of PTEN tumor suppressor gene on acquiring resistance to tamoxifen treatment in breast cancer patients. Cancer Biol Ther. (2012) 13:1165–74. doi: 10.4161/cbt.21346
76. Shoman N, Klassen S, McFadden A, Bickis MG, Torlakovic E, Chibbar R. Reduced PTEN expression predicts relapse in patients with breast carcinoma treated by tamoxifen. Mod Pathol. (2005) 18:250–9. doi: 10.1038/modpathol.3800296
77. Fu X, Creighton CJ, Biswal NC, Kumar V, Shea M, Herrera S, et al. Overcoming endocrine resistance due to reduced PTEN levels in estrogen receptor-positive breast cancer by co-targeting mammalian target of rapamycin, protein kinase B, or mitogen-activated protein kinase kinase. Breast Cancer Res. (2014) 16:1–17. doi: 10.1186/s13058-014-0430-x
78. Sobhani N, D'Angelo A, Pittacolo M, Roviello G, Miccoli A, Corona SP, et al. Updates on the CDK4/6 inhibitory strategy and combinations in breast cancer. Cells. (2019) 8:321. doi: 10.20944/preprints201902.0245.v1
79. Milioli HH, Alexandrou S, Lim E, Caldon CE. Cyclin E1 and cyclin E2 in ER+ breast cancer: prospects as biomarkers and therapeutic targets. Endocr Relat Cancer. (2020) 27:R93–r112. doi: 10.1530/ERC-19-0501
80. Wander SA, Mao P, Lloyd MR, Johnson GN, Kowalski K, Nayar U, et al. Abstract PD7-08: Igf1r mediates cdk4/6 inhibitor (cdk4/6i) resistance in tumor samples and in cellular models. Cancer Res. (2021) 81:PD7–08-PD7-. doi: 10.1158/1538-7445.SABCS20-PD7-08
81. Prat A, Cheang MC, Galván P, Nuciforo P, Paré L, Adamo B, et al. Prognostic value of intrinsic subtypes in hormone receptor-positive metastatic breast cancer treated with letrozole with or without lapatinib. JAMA Oncol. (2016) 2:1287–94. doi: 10.1001/jamaoncol.2016.0922
82. Canino F, Piacentini F, Omarini C, Toss A, Barbolini M, Vici P, et al. Role of intrinsic subtype analysis with PAM50 in hormone receptors positive HER2 negative metastatic breast cancer: A systematic review. Int J Mol Sci. (2022) 23:7079. doi: 10.3390/ijms23137079
83. Cohen O, Mao P, Nayar U, Buendia-Bue JE, Kim D, Jain E, et al. Abstract GS2-02: Acquired activating mutations in RTKs confer endocrine resistance in ER+ metastatic breast cancer through ER-reprogramming, MAPK signaling, and an induced stem-like cell state. Cancer Res. (2020) 80:GS2–02-GS2-. doi: 10.1158/1538-7445.SABCS19-GS2-02
84. Bi M, Zhang Z, Jiang YZ, Xue P, Wang H, Lai Z, et al. Enhancer reprogramming driven by high-order assemblies of transcription factors promotes phenotypic plasticity and breast cancer endocrine resistance. Nat Cell Biol. (2020) 22:701–15. doi: 10.1038/s41556-020-0514-z
85. Guarneri V, Giovannelli S, Ficarra G, Bettelli S, Maiorana A, Piacentini F, et al. Comparison of HER-2 and hormone receptor expression in primary breast cancers and asynchronous paired metastases: impact on patient management. Oncologist. (2008) 13:838–44. doi: 10.1634/theoncologist.2008-0048
86. Falato C, Schettini F, Pascual T, Brasó-Maristany F, Prat A. Clinical implications of the intrinsic molecular subtypes in hormone receptor-positive and HER2-negative metastatic breast cancer. Cancer Treat Rev. (2023) 112:102496. doi: 10.1016/j.ctrv.2022.102496
87. Klebe M, Fremd C, Kriegsmann M, Kriegsmann K, Albrecht T, Thewes V, et al. Frequent molecular subtype switching and gene expression alterations in lung and pleural metastasis from luminal A-type breast cancer. JCO Precis Oncol. (2020) 4:848–59. doi: 10.1200/PO.19.00337
88. Aftimos P, Oliveira M, Irrthum A, Fumagalli D, Sotiriou C, Gal-Yam EN, et al. Genomic and transcriptomic analyses of breast cancer primaries and matched metastases in AURORA, the breast international group (BIG) molecular screening initiative. Cancer Discovery. (2021) 11:2796–811. doi: 10.1158/2159-8290.CD-20-1647
89. Schagerholm C, Robertson S, Karlsson E, Sifakis E, Hartman J. Abstract PO3-16-04: Distribution of intrinsic subtypes in endocrine-resistant and endocrine-sensitive breast cancer. Cancer Res. (2024) 84:PO3–16-04-PO3-16-04. doi: 10.1158/1538-7445.SABCS23-PO3-16-04
90. Schettini F, Nucera S, Brasó-Maristany F, Santo ID, Pascual T, Bergamino M, et al. Unraveling the clinicopathological and molecular changes induced by neoadjuvant chemotherapy and endocrine therapy in hormone receptor-positive/HER2-low and HER2-0 breast cancer. ESMO Open. (2024) 9:103619–. doi: 10.1016/j.esmoop.2024.103619
91. The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. (2012) 490:61–70. doi: 10.1038/nature11412
92. Li Z, McGinn O, Wu Y, Bahreini A, Priedigkeit NM, Ding K, et al. ESR1 mutant breast cancers show elevated basal cytokeratins and immune activation. Nat Commun. (2022) 13:2011. doi: 10.1038/s41467-022-29498-9
93. Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW, et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature. (2012) 486:353–60. doi: 10.1038/nature11143
94. Brasó-Maristany F, Griguolo G, Pascual T, Paré L, Nuciforo P, Llombart-Cussac A, et al. Phenotypic changes of HER2-positive breast cancer during and after dual HER2 blockade. Nat Commun. (2020) 11:385. doi: 10.1038/s41467-019-14111-3
95. Paik S, Kim C, Wolmark N. HER2 status and benefit from adjuvant trastuzumab in breast cancer. N Engl J Med. (2008) 358:1409–11. doi: 10.1056/NEJMc0801440
96. Modi S, Jacot W, Yamashita T, Sohn J, Vidal M, Tokunaga E, et al. Trastuzumab deruxtecan in previously treated HER2-low advanced breast cancer. New Engl J Med. (2022) 387:9–20. doi: 10.1056/NEJMoa2203690
97. Mosele F, Deluche E, Lusque A, Le Bescond L, Filleron T, Pradat Y, et al. Trastuzumab deruxtecan in metastatic breast cancer with variable HER2 expression: the phase 2 DAISY trial. Nat Med. (2023) 29:2110–20. doi: 10.1038/s41591-023-02478-2
98. Ogitani Y, Hagihara K, Oitate M, Naito H, Agatsuma T. Bystander killing effect of DS-8201a, a novel anti-human epidermal growth factor receptor 2 antibody–drug conjugate, in tumors with human epidermal growth factor receptor 2 heterogeneity. Cancer Science. (2016) 107:1039–46. doi: 10.1111/cas.2016.107.issue-7
99. Ma CX, Bose R, Gao F, Freedman RA, Telli ML, Kimmick G, et al. Neratinib efficacy and circulating tumor DNA detection of HER2 mutations in HER2 nonamplified metastatic breast cancer. Clin Cancer Res. (2017) 3:5687–95. doi: 10.1158/1078-0432.CCR-17-0900
100. Jhaveri K, Eli LD, Wildiers H, Hurvitz SA, Guerrero-Zotano A, Unni N, et al. Neratinib + fulvestrant + trastuzumab for HR-positive, HER2-negative, HER2-mutant metastatic breast cancer: outcomes and biomarker analysis from the SUMMIT trial. Ann Oncol. (2023) 34:885–98. doi: 10.1016/j.annonc.2023.08.003
101. Ma CX, Luo J, Freedman RA, Pluard TJ, Nangia JR, Lu J, et al. The phase II mutHER study of neratinib alone and in combination with fulvestrant in HER2-mutated, non-amplified metastatic breast cancer. Clin Cancer Res. (2022) 28:1258–67. doi: 10.1158/1078-0432.CCR-21-3418
102. Goto K, Goto Y, Kubo T, Ninomiya K, Kim SW, Planchard D, et al. Trastuzumab deruxtecan in patients with HER2-mutant metastatic non-small-cell lung cancer: primary results from the randomized, phase II DESTINY-lung02 trial. J Clin Oncol. (2023) 41:4852–63. doi: 10.1200/JCO.23.01361
103. Li BT, Michelini F, Misale S, Cocco E, Baldino L, Cai Y, et al. HER2-mediated internalization of cytotoxic agents in ERBB2 amplified or mutant lung cancers. Cancer Discovery. (2020) 10:674–87. doi: 10.1158/2159-8290.CD-20-0215
104. Lawson M, Cureton N, Ros S, Cheraghchi-Bashi A, Urosevic J, D'Arcy S, et al. The next-generation oral selective estrogen receptor degrader camizestrant (AZD9833) suppresses ER+ Breast cancer growth and overcomes endocrine and CDK4/6 inhibitor resistance. Cancer Res. (2023) 83:3989–4004. doi: 10.1158/0008-5472.CAN-23-0694
105. Goetz MP, Bagegni NA, Batist G, Brufsky A, Cristofanilli MA, Damodaran S, et al. Lasofoxifene versus fulvestrant for ER+/HER2– metastatic breast cancer with an ESR1 mutation: results from the randomized, phase II ELAINE 1 trial. Ann Oncol. (2023) 34:1141–51. doi: 10.1016/j.annonc.2023.09.3104
106. Gough SM, Flanagan JJ, Teh J, Andreoli M, Rousseau E, Pannone M, et al. Oral estrogen receptor PROTAC® vepdegestrant (ARV-471) is highly efficacious as monotherapy and in combination with CDK4/6 or PI3K/mTOR pathway inhibitors in preclinical ER+ breast cancer models. Clin Cancer Res. (2024) 30:3549–63. doi: 10.1158/1078-0432.c.7403517.v1
107. Brett JO, Dubash TD, Johnson GN, Niemierko A, Mariotti V, Kim LSL, et al. A gene panel associated with abemaciclib utility in ESR1-mutated breast cancer after prior cyclin-dependent kinase 4/6-inhibitor progression. JCO Precis Oncol. (2023) 7:e2200532. doi: 10.1200/PO.22.00532
108. André F, Ciruelos E, Rubovszky G, Campone M, Loibl S, Rugo HS, et al. Alpelisib for PIK3CA-mutated, hormone receptor-positive advanced breast cancer. N Engl J Med. (2019) 380:1929–40. doi: 10.1056/NEJMoa1813904
109. André F, Ciruelos EM, Juric D, Loibl S, Campone M, Mayer IA, et al. Alpelisib plus fulvestrant for PIK3CA-mutated, hormone receptor-positive, human epidermal growth factor receptor-2-negative advanced breast cancer: final overall survival results from SOLAR-1. Ann Oncol. (2021) 32:208–17. doi: 10.1016/j.annonc.2020.11.011
110. Elkabets M, Vora S, Juric D, Morse N, Mino-Kenudson M, Muranen T, et al. mTORC1 inhibition is required for sensitivity to PI3K p110α inhibitors in PIK3CA-mutant breast cancer. Sci Transl Med. (2013) 5:196ra99. doi: 10.1126/scitranslmed.3005747
111. Turner NC, Oliveira M, Howell SJ, Dalenc F, Cortes JA-O, Gomez Moreno HL, et al. Capivasertib in hormone receptor-positive advanced breast cancer. N Engl J Med. (2023) 388:1533–4406 (Electronic). doi: 10.1056/NEJMoa2214131
112. Turner NC, Kingston B, Kilburn LS, Kernaghan S, Wardley AM, Macpherson IR, et al. Circulating tumour DNA analysis to direct therapy in advanced breast cancer (plasmaMATCH): a multicentre, multicohort, phase 2a, platform trial. Lancet Oncol. (2020) 21:1296–308. doi: 10.1016/S1470-2045(20)30444-7
113. Andre F, Hamilton EP, Loi S, Im S-A, Sohn J, Tseng L-M, et al. Dose-finding and -expansion studies of trastuzumab deruxtecan in combination with other anti-cancer agents in patients (pts) with advanced/metastatic HER2+ (DESTINY-Breast07 [DB-07]) and HER2-low (DESTINY-Breast08 [DB-08]) breast cancer (BC). J Clin Oncol. (2022) 40:3025–. doi: 10.1200/JCO.2022.40.16_suppl.3025
114. Nagarajan S, Rao SV, Sutton J, Cheeseman D, Dunn S, Papachristou EK, et al. ARID1A influences HDAC1/BRD4 activity, intrinsic proliferative capacity and breast cancer treatment response. Nat Genet. (2020) 52:187–97. doi: 10.1038/s41588-019-0541-5
115. Drago JZ, Formisano L, Juric D, Niemierko A, Servetto A, Wander SA, et al. FGFR1 amplification mediates endocrine resistance but retains TORC sensitivity in metastatic hormone receptor-positive (HR(+)) breast cancer. Clin Cancer Res. (2019) 25:6443–51. doi: 10.1158/1078-0432.CCR-19-0138
116. Saridogan T, Akcakanat A, Zhao M, Evans KW, Yuca E, Scott S, et al. Efficacy of futibatinib, an irreversible fibroblast growth factor receptor inhibitor, in FGFR-altered breast cancer. Sci Rep. (2023) 13:20223. doi: 10.1038/s41598-023-46586-y
117. Damodaran S, André F, Unni N, Ferreira M, Giridhar K, Daniel B, et al. Abstract RF01-04: Final results from the phase 2, open-label FOENIX-MBC2 study: efficacy and safety of futibatinib in adult patients with locally advanced/metastatic HR+/HER2– breast cancer harboring high-level FGFR1 gene amplification. Cancer Res. (2024) 84:RF01–4-RF-4. doi: 10.1158/1538-7445.SABCS23-RF01-04
118. Coombes RC, Badman PD, Lozano-Kuehne JP, Liu X, Macpherson IR, Zubairi I, et al. Results of the phase IIa RADICAL trial of the FGFR inhibitor AZD4547 in endocrine resistant breast cancer. Nat Commun. (2022) 13:3246. doi: 10.1038/s41467-022-30666-0
119. Jhaveri K, André F, Hamilton E, Schmid P, Anders C, Testa L, et al. Abstract RF02-03: Trastuzumab deruxtecan (T-DXd) in combination with anastrozole or fulvestrant in patients with HER2-low HR+ advanced/metastatic breast cancer: a Phase 1b, open-label, multicenter, dose-expansion study (DESTINY-Breast08). Cancer Res. (2024) 84:RF02–3-RF-3. doi: 10.1158/1538-7445.SABCS23-RF02-03
120. Hortobagyi GN. Everolimus plus exemestane for the treatment of advanced breast cancer: a review of subanalyses from BOLERO-2. Neoplasia. (2015) 17:279–88. doi: 10.1016/j.neo.2015.01.005
121. Yap TA, O'Carrigan B, Penney MS, Lim JS, Brown JS, de Miguel Luken MJ, et al. Phase I trial of first-in-class ATR inhibitor M6620 (VX-970) as monotherapy or in combination with carboplatin in patients with advanced solid tumors. J Clin Oncol. (2020) 38:3195–204. doi: 10.1200/JCO.19.02404
122. Haddad TC, Suman VJ, D'Assoro AB, Carter JM, Giridhar KV, McMenomy BP, et al. Evaluation of alisertib alone or combined with fulvestrant in patients with endocrine-resistant advanced breast cancer: the phase 2 TBCRC041 randomized clinical trial. JAMA Oncol. (2023) 9:815–24. doi: 10.1001/jamaoncol.2022.7949
123. Meric-MBernstam F, Rothe M, Garrett-Mayer E, Gutierrez R, Ahn ER, Cannon TL, et al. Cobimetinib plus vemurafenib (C+V) in patients (Pts) with solid tumors with BRAF V600E/d/k/R mutation: Results from the targeted agent and profiling utilization registry (TAPUR) study. J Clin Oncol. (2022) 40:3008–. doi: 10.1200/JCO.2022.40.16_suppl.3008
124. Juric D, Soria J-C, Sharma S, Banerji U, Azaro A, Desai J, et al. A phase 1b dose-escalation study of BYL719 plus binimetinib (MEK162) in patients with selected advanced solid tumors. J Clin Oncol. (2014) 32:9051–. doi: 10.1200/jco.2014.32.15_suppl.9051
125. Liao D, Hansen R, Chowdhry S, Holmes E, Wiese J, Steffy A, et al. Abstract 3631: Preclinical and clinical pharmacodynamic characterization of BBI-355, a novel, orally bioavailable, and selective CHK1 inhibitor being evaluated in the first-in-human Phase 1/2 POTENTIATE clinical trial of patients with cancer harboring oncogene amplifications. Cancer Res. (2024) 84:3631–. doi: 10.1158/1538-7445.AM2024-3631
126. Hamilton E, Oliveira M, Turner N, García-Corbacho J, Hernando C, Ciruelos EM, et al. A phase I dose escalation and expansion trial of the next-generation oral SERD camizestrant in women with ER-positive, HER2-negative advanced breast cancer: SERENA-1 monotherapy results. Ann Oncol. (2024) 35:707–17. doi: 10.1016/j.annonc.2024.04.012
127. Bardia A, Cortes J, Hurvitz SA, Delaloge S, Iwata H, Shao ZM, et al. AMEERA-5: a randomized, double-blind phase 3 study of amcenestrant plus palbociclib versus letrozole plus palbociclib for previously untreated ER+/HER2- advanced breast cancer. Ther Adv Med Oncol. (2022) 14:17588359221083956. doi: 10.1177/17588359221083956
128. Turner NC, Krop IE, Bardia A, Damodaran S, Martin M, Benhadji KA, et al. Abstract OT2-07-01: A phase 2 study of futibatinib (TAS-120) in metastatic breast cancers harboring fibroblast growth factor receptor (FGFR) amplifications (FOENIX-MBC2). Cancer Res. (2020) 80:OT2-07-1-OT2–1. doi: 10.1158/1538-7445.SABCS19-OT2-07-01
129. Hui R, Pearson A, Cortes J, Campbell C, Poirot C, Azim HA Jr., et al. Lucitanib for the treatment of HR(+)/HER2(-) metastatic breast cancer: results from the multicohort phase II FINESSE study. Clin Cancer Res. (2020) 26:354–63. doi: 10.1158/1078-0432.CCR-19-1164
130. Rugo HS, Trédan O, Ro J, Morales SM, Campone M, Musolino A, et al. A randomized phase II trial of ridaforolimus, dalotuzumab, and exemestane compared with ridaforolimus and exemestane in patients with advanced breast cancer. Breast Cancer Res Treat. (2017) 165:601–9. doi: 10.1007/s10549-017-4375-5
131. Pant S, Yaeger R, Spira AI, Pelster M, Sabari JK, Hafez N, et al. KRYSTAL-1: Activity and safety of adagrasib (MRTX849) in patients with advanced solid tumors harboring a KRASG12C mutation. J Clin Oncol. (2023) 41:425082–. doi: 10.1200/JCO.2023.41.36_suppl.425082
132. Nogova L, Mattonet C, Scheffler M, Taubert M, Gardizi M, Sos ML, et al. Sorafenib and everolimus in patients with advanced solid tumors and KRAS-mutated NSCLC: A phase I trial with early pharmacodynamic FDG-PET assessment. Cancer Med. (2020) 9:4991–5007. doi: 10.1002/cam4.v9.14
133. Rosen EY, Patel P, Sarapa N, Edris B, Iasonos A, Jhaveri K, et al. Abstract OT2-23-01: A Phase 1b/2a, open-label platform study to evaluate mirdametinib in combination with fulvestrant in ER+ metastatic breast cancers harboring MAPK-activating mutations. Cancer Res. (2022) 82:OT2–23-01-OT2-23-01. doi: 10.1158/1538-7445.SABCS21-OT2-23-01
134. Damodaran S, Zhao F, Deming DA, Mitchell EP, Wright JJ, Gray RJ, et al. Phase II study of copanlisib in patients with tumors with PIK3CA mutations: results from the NCI-MATCH ECOG-ACRIN trial (EAY131) subprotocol Z1F. J Clin Oncol. (2022) 40:1552–61. doi: 10.1200/JCO.21.01648
135. Yi Z, Liu B, Sun X, Rong G, Wang W, Li H, et al. Safety and efficacy of sirolimus combined with endocrine therapy in patients with advanced hormone receptor-positive breast cancer and the exploration of biomarkers. Breast. (2020) 52:17–22. doi: 10.1016/j.breast.2020.04.004
136. Caldon CE, Sergio CM, Kang J, Muthukaruppan A, Boersma MN, Stone A, et al. Cyclin E2 overexpression is associated with endocrine resistance but not insensitivity to CDK2 inhibition in human breast cancer cells. Mol Cancer Ther. (2012) 11:1488–99. doi: 10.1158/1535-7163.MCT-11-0963
137. Sengupta S, Biarnes MC, Jordan VC. Cyclin dependent kinase-9 mediated transcriptional de-regulation of cMYC as a critical determinant of endocrine-therapy resistance in breast cancers. Breast Cancer Res Treat. (2014) 143:113–24. doi: 10.1007/s10549-013-2789-2
Keywords: endocrine resistance, combination therapy, HER2 mutations, ER-HER2 crosstalk, subtype switch
Citation: Yayli G, Tokofsky A and Nayar U (2024) The intersection of the HER2-low subtype with endocrine resistance: the role of interconnected signaling pathways. Front. Oncol. 14:1461190. doi: 10.3389/fonc.2024.1461190
Received: 08 July 2024; Accepted: 29 October 2024;
Published: 22 November 2024.
Edited by:
Sharon R. Pine, University of Colorado Anschutz Medical Campus, United StatesReviewed by:
Yu Xiao, Shenzhen Second People’s Hospital, ChinaCopyright © 2024 Yayli, Tokofsky and Nayar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Utthara Nayar, dW5heWFyQGpodS5lZHU=