 Jun Xia1†Guanqun Yang2†Qingling Yu3†
Jun Xia1†Guanqun Yang2†Qingling Yu3† Ruiyi Zhang4Ge Li4
Ruiyi Zhang4Ge Li4 Man Wang5Hongli Sun1Heng Chen1Lingling Wang1Ping Chen4*Kai-Li Gu4*Chao Sun1*
Man Wang5Hongli Sun1Heng Chen1Lingling Wang1Ping Chen4*Kai-Li Gu4*Chao Sun1*- 1Department of Hematology, Affiliated Wuxi People’s Hospital, Nanjing Medical University, Wuxi, China
- 2Changshu Hospital Affiliated to Soochow University, Changshu NO.1 People’s Hospital, Changshu, Jiangsu, China
- 3Department of Hematology, Affiliated Jianhu Hospital of Nantong University Xinglin College, Yancheng, China
- 4Suzhou Jsuniwell Medical Laboratory, Suzhou, China
- 5Jiangsu Institute of Hematology, National Clinical Research Center for Hematologic Diseases, Key Laboratory of Thrombosis and Hemostasis, The First Affiliated Hospital of Soochow University, Suzhou, China
Acute myeloid leukemia (AML), which is most common in adults, is a challenging hematological malignancy. The occurrence and the progression of AML are often accompanied by various gene fusions and/or mutations. Herein, we report the first case of a TOR1AIP2::ETV6 fusion transcript with a translocation of t(1;12)(q25;p13) in AML progressed from myelodysplastic syndrome (MDS) combined with an FLT3-ITD (internal tandem duplication) mutation. Further studies should focus on the biological functions of these novel chimeric products in disease onset and progression, as well as their potential as monitoring markers in disease regression.
Introduction
Acute myeloid leukemia (AML) is a malignant clonal disease characterized by the impaired proliferation and differentiation of myeloid immature cells, leading to the dysfunction of normal hematopoietic function (1) .There is a lot of evidence showing that the occurrence and the development of AML are often accompanied by the emergence of gene fusions (e.g., PML::RARA, CBFB::MYH11, and RUNX1::RUNX1T1, among others) and mutations such as c-KIT, FLT3-ITD, NPM1, and TP53 (2, 3). Approximately 20%–30% of newly diagnosed AML patients have an FLT3-ITD mutation, which is a representative marker indicating poor overall survival and recurrence of AML (4). These molecular markers provide an extremely important basis for clinical AML diagnosis and treatment, prognosis evaluation, and minimal residual disease (MRD) monitoring (5, 6). Furthermore, the interactions between gene fusions and mutated genes have been reported to be prominent driving factors for the onset of AML (7). With an increasing number of new fusions identified, this provides a promising future for the exploration of pathogenesis and new therapeutic targets for hematopoietic malignancies.
Herein, we report a novel TOR1AIP2::ETV6 transcript that was predicted to express no fusion protein in a patient with AML that progressed from MDS with the FLT3-ITD mutation. Interestingly, the FLT3-ITD mutation in the present case was not detected by a next-generation sequencing (NGS) panel, while the chimeric fusion remained detectable after treatment with the venetoclax and azacitidine combined regimen. In addition, the fragments per kilobase million (FPKM) value of the TRO1AIP2 of this patient appeared to be higher compared with those of other patients with AML, indicating that such fusion transcript could lead to the onset of AML via regulation of the expression of TRO1AIP2. Whether this fusion transcript represents a new tumor subclone and acts as a monitoring marker warrant more sequential studies. This case report might provide new insights into understanding the occurrence and progression of AML.
Case presentation
A 72-year-old man was first diagnosed with myelodysplastic syndrome with excess blasts-2 (MDS-EB2) with protoplasmic cells accounting for 10% in 2016 and received multiple rounds of chemotherapy (Figure 1A). After admission, a complete routine peripheral blood examination showed a white blood cell count of 0.11 × 109/L, a hemoglobin level of 84.0 g/L, and a platelet count of 125 × 109/L. The albumin, globulin, and C-reactive protein (CRP) levels were 26.8 g/L, 37.8 g/L, and 64.1 mg/L, respectively.
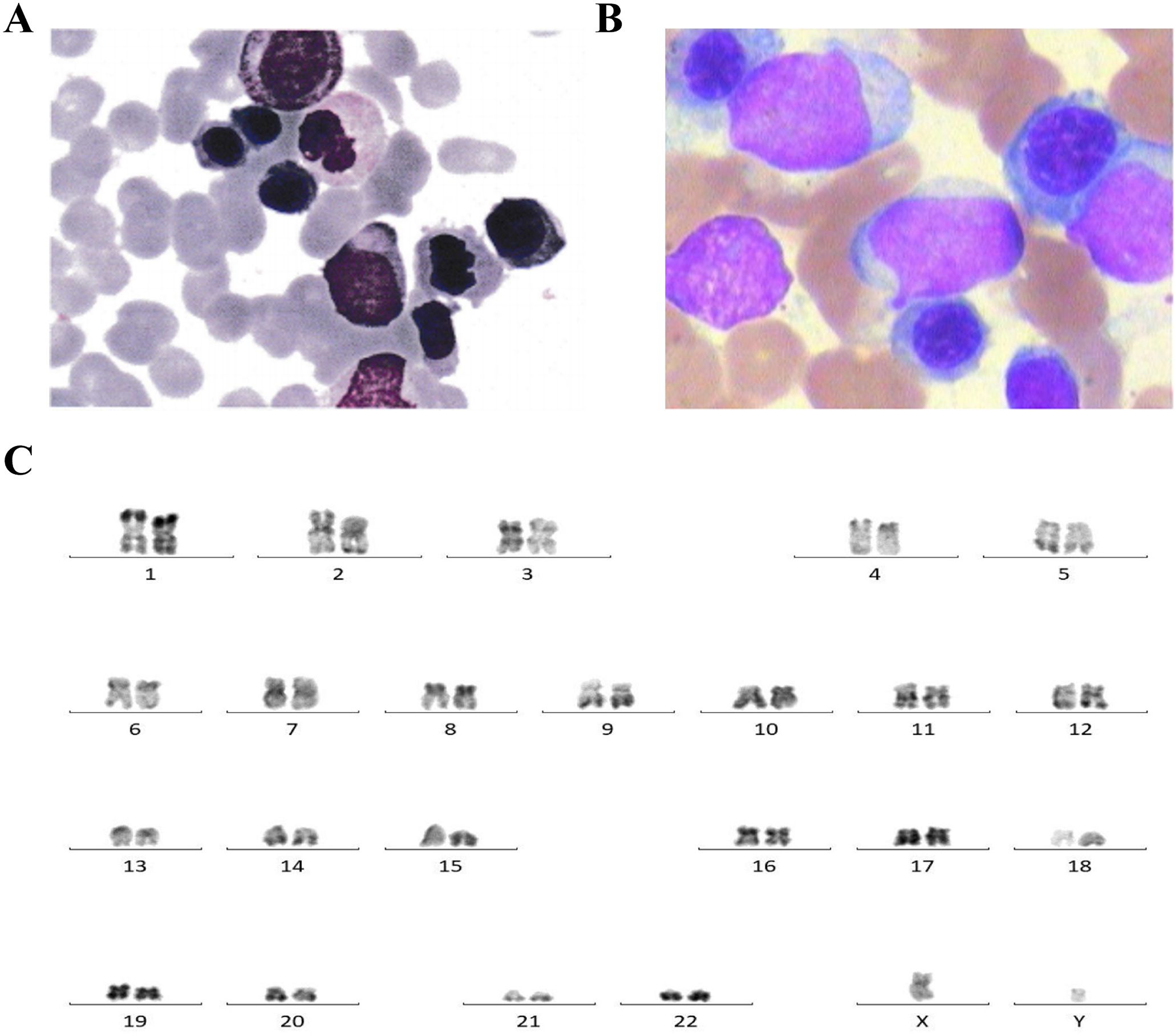
Figure 1. (A) Bone marrow cytology showing protoplasmic cells accounting for 10% in this patient for the newly diagnosed myelodysplastic syndrome with excess blasts-2 (MDS-EB2). (B) Bone marrow cytology showing primitive cells accounting for 40% in acute myeloid leukemia (AML) progressed from myelodysplastic syndrome (MDS). (C) Karyotype analysis showing normal 46,XY [20].
The bone marrow (BM) aspirate smear showed primitive cells accounting for about 40% (Figure 1B). The karyotype analysis showed normal karyotype (Figure 1C). Flow cytometry analysis of the BM showed residual tumor cells, which expressed CD34, CD7, CD33, and CD38; partially expressed CD117; and were negative for CD13, HLA-DR, CD10, CD11b, CD19, and CD56.
Subsequently, NGS was performed using an 82-gene panel to identify gene mutations and a 71-gene panel to carry out a more comprehensive fusion screening through targeted RNA sequencing (RNA-seq). The results showed detection of a 36- and a 123-bp FLT3-ITD. At the same time, the FLT3-ITD mutations were examined using capillary electrophoresis (CE). The results showed that FLT3-ITD had mutated in this patient (Supplementary Figure S1C). NPM1 mutations were not detected. The targeted RNA-seq data were analyzed using STAR-Fusion, which identified a novel fusion transcript of TOR1AIP2::ETV6, fused by TOR1AIP2 (NM_001199260) exon 3 with ETV6 (NM_001987.5) exon 6 through translocation (Figure 2A). To confirm the existence of the chimeric transcript, primers were designed to amplify the fusion product. The forward and reverse primers were TGTCAGCTCCTCTGTTTCACC and AGTTTTCGTACCGGCTGTCA, respectively. Using agarose gel electrophoresis, the targeted fusion product was shown in a band in 249 bp, while the reciprocal fusion TOR1AIP2::ETV6 was not found (Figure 2B). Furthermore, Sanger sequencing was used, which verified the presence of the TOR1AIP2::ETV6 fusion transcript (Figure 2C). Molecular diversity was not assessed at the time of initial diagnosis due to the limited laboratory technology.
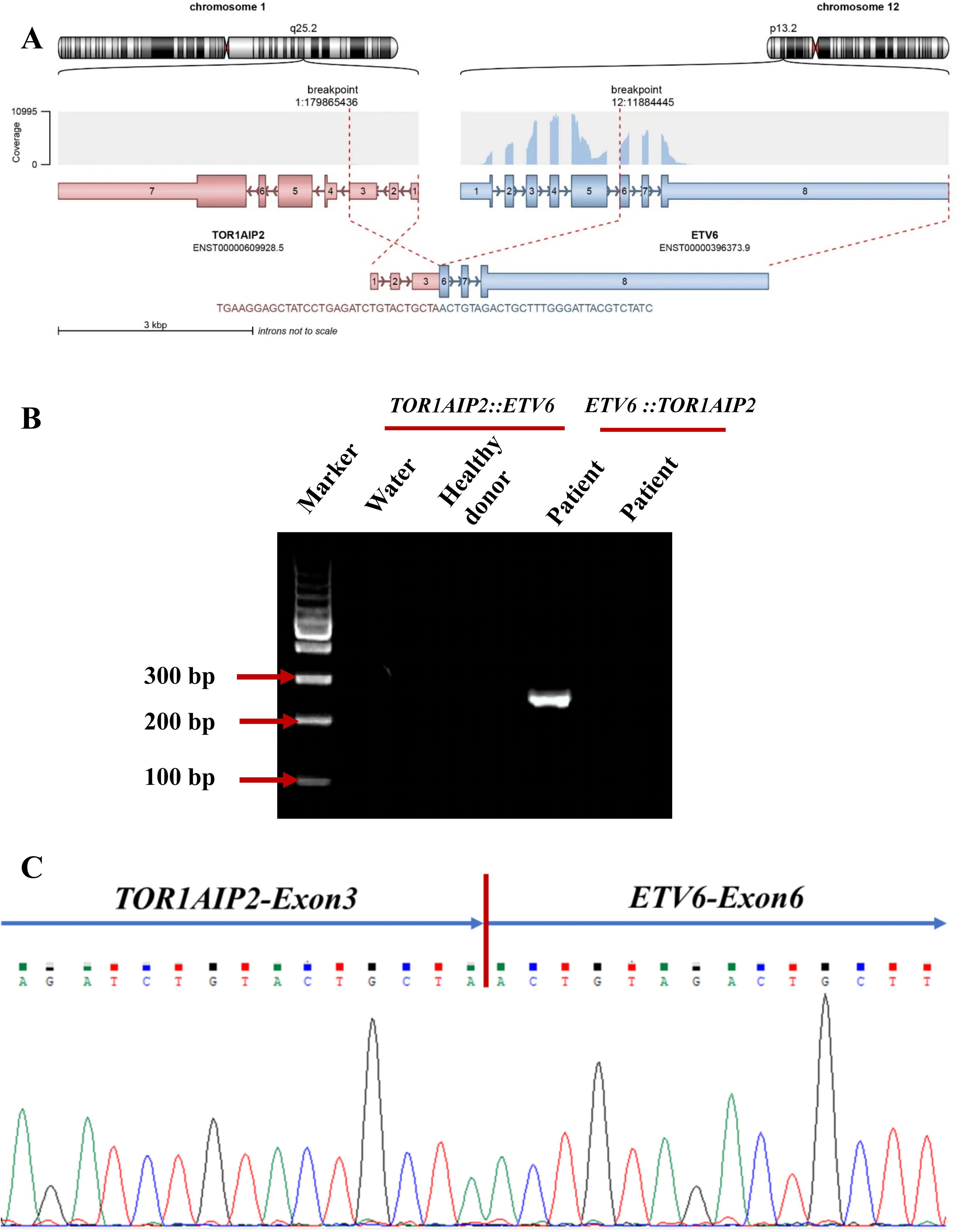
Figure 2. (A) Schematic diagram of the formation mechanism of the TOR1AIP2::ETV6 transcript. (B) Electrophoresis of the RT-PCR products from the patient showing the TOR1AIP2::ETV6 fusion transcript. (C) Partial nucleotide sequences surrounding the junctions of the TOR1AIP2::ETV6 fusion transcript.
The patient was diagnosed with progression of MDS to AML according to the 5th World Health Organization (WHO) classification. Since the patient was a 72-year-old elderly person with lower tolerance, the venetoclax plus azacitidine therapy regimen was chosen (100 mg azacitidine for 7 days, then 100–300 mg venetoclax for half a month, and then 100–200 mg venetoclax for maintenance treatment, and no longer with azacitidine) as an individualized dosing regimen for treatment due to his old age and poor physical condition (Figure 3). The condition of the patient improved after treatment and he was discharged from our hospital.
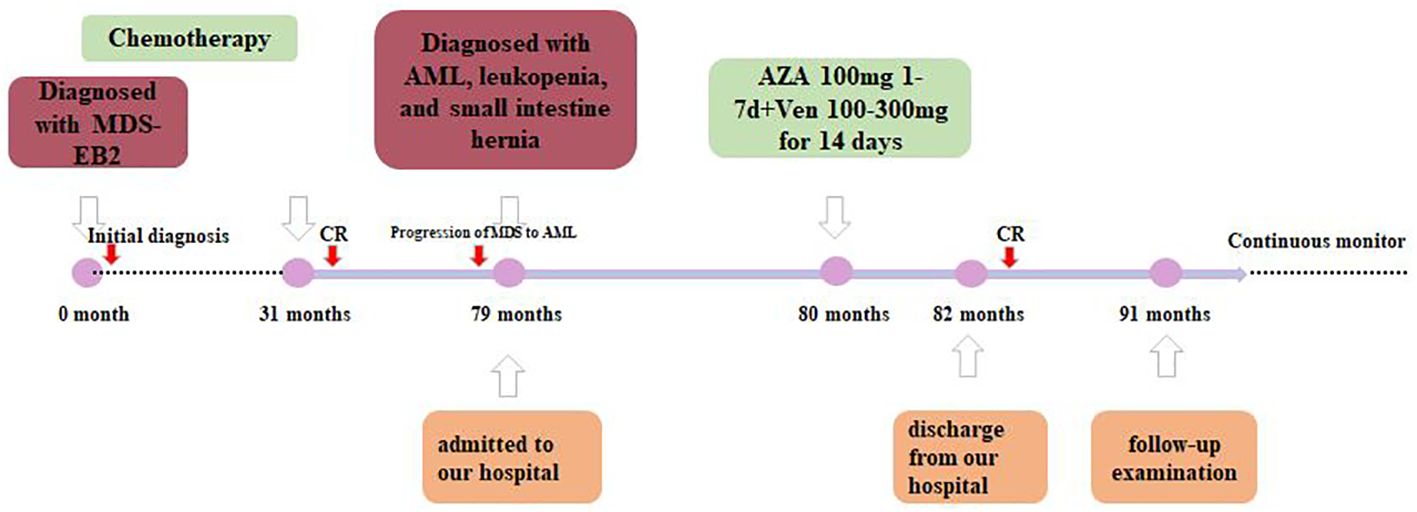
Figure 3. Timeline of therapy.
A year after the recurrence, the patient came back for a follow-up examination. The primitive cells accounted for 4%. The 11 flow cytometry panels for AML-MRD were conducted, with positive results. A total of 239,980 nucleated cells were detected, of which 2,266 were abnormal, accounting for 0.94% of the nucleated cells (the results are not shown in the figure). In addition, the FLT3-ITD mutation was undetectable with NGS and CE using the patient’s BM, with a mutation detection sensitivity of 1%. However, the chimeric fusion transcript was still present, with the relative expression of its transcript being 0.35% by quantitative PCR (qPCR). The timeline of therapy is shown in Figure 3.
Discussion
In the current study, we report the first case of a TOR1AIP2::ETV6 fusion in a patient with AML that progressed from MDS using targeted RNA-seq, underlining its application in the identification of minor chromosomal variations with elevated sensitivity in spite of the karyotype analysis showing a normal karyotype.
ETV6, previously known as TEL, is located on chromosome 12p13 and is a member of the ETS (E-Twenty-Six) family of transcription factors. The ETV6 gene consists of eight exons. The ETV6 protein is composed of two major domains: the helix–loop–helix (HLH) and ETS domains. The HLH domain is encoded by exons 3 and 4 and functions as a homo-oligomerization domain, while the ETS domain, which is encoded by exons 6–8, is responsible for protein–protein interactions. A central domain, called the internal domain, is involved in the recruitment of a repression complex including NCOR1, NCOR2, and SIN3A (9). The ETV6 protein and other ETS proteins work together to affect the process of cell proliferation, differentiation, tumorigenesis, and hematopoietic regulation (10). Multiple chromosomal rearrangements involving ETV6 (such as ETV6::RUNX1, ETV6::ABL1, ETV6::NTRK3, and ETV6::ACSL6) have been demonstrated to play a crucial role in the development of several hematologic diseases such as AML, as well as in their diagnosis and prognosis (11). Thus far, 103 gene fusions involving ETV6 due to translocations, insertions, or inversions have been identified, as shown in the Mitelman database (https://mitelmandatabase.isb-cgc.org-accesssed).
In this paper, we report a brand new fusion partner of ETV6, named torsin 1A interacting protein 2 (TOR1AIP2), which is a protein-coding gene located at 1q25.2. TOR1AIP2 is required for endoplasmic reticulum integrity. It regulates the distribution of TOR1A between the endoplasmic reticulum and the nuclear envelope and induces the ATPase activity of TOR1A, TOR1B, and TOR3A (12). There are only two TOR1AIP2 fusion partners reported: TOR1AIP2::ACBD6 and TOR1AIP2::IFI16 (13). The TOR1AIP2::ETV6 transcript is formed through translocation, with TOR1AIP2 breaking at chr1:179865436:− and ETV6 breaking at chr12:11884445:+. Despite the confirmation of novel fusion transcripts using different methods, no chimeric fusion protein was predicted due to a stop codon being formed in the junction site, which might generate a truncated ETV6 protein. It is possible that the production of ETV6 truncated transcripts could result in a pathogenic role for ETV6 haploinsufficiency in this patient. In addition, no evidence of the reciprocal ETV6::TOR1AIP2 fusion transcript was found. This suggests a more complex translocation process, possibly combined with further transcriptional or posttranscriptional regulation. Interestingly, the expression level of TOR1AIP2 represented by the FPKM of this patient was higher than those of patients with AML without such chimeric fusion, indicating that it could lead to the onset of AML via the upregulation of the expression of TOR1AIP2, with the ETV6 FPKM value remaining unaffected (Supplementary Figure S1).
In our case, FLT3-ITD was also detected, which occurs in 20%–30% of de novo AML patients and is associated with disease relapse, as well as with inferior overall survival (14). In the present study, the patient carried an FLT3-ITD with a low MAF of 2% and was negative for NPM1 mutations, which represent intermediate risk according to the 2022 European Leukemia Network (ELN) (8). Research has shown that the interactions between gene fusions and mutated genes play a crucial role in the prognosis and recurrence of AML (7). Thanasopoulou’s group provided experimental proof that the co-expression of NUP98::NSD1 and FLT3-ITD could collaborate for transformation in vitro and the progression of AML in vivo (15). Schessl et al. reported that AML1::ETO collaborated with FLT3 length mutation and could induce acute leukemia in a murine BM transplantation model (16). Moreover, in vivo, Kim et al. found that FLT3-ITD mutations could cooperate with CBFB::SMMHC in an animal model of inv(16)-accompanied AML (17). More research should focus on whether the co-expression of the ETV6::TOR1AIP2 fusion transcript with FLT3-ITD could act synergistically in the onset and progression of AML.
FLT3 inhibitors (including sorafenib and gilteritinib, among others) and allogeneic hematopoietic cell transplantation (alloHCT) are commonly used as treatments for AML with FLT3-ITD (18). Short et al. found that the combination of azacitidine, venetoclax, and gilteritinib could improve survival in newly diagnosed FLT3-mutated AML (19). After being administered the venetoclax plus azacitidine regimen, the patient was assessed for recurrence. Interestingly, FLT3-ITD was no longer detected using the NGS panel, indicating a decrease in the FLT3-ITD clones with this treatment regimen. However, the fusion transcript was still present. Longitudinal monitoring of the current patient might illustrate whether the fusion transcript represents a leukemia subclone. Furthermore, the FLT3-ITD mutation decreased with a regimen without the FLT3 inhibitor, indicating the possibility that the combination of the current fusion and FLT3-ITD may be more sensitive to chemotherapy.
Conclusion
In summary, to the best of our knowledge, this is the first report of a TOR1AIP2::ETV6 transcript co-expressed with the FLT3-ITD mutation in AML progressed from MDS. This novel fusion combined with FLT3-ITD showed potential as a monitoring marker. We hope that this case report can provide a new perspective for understanding the disease progression of AML, as well as for treatment options for FLT3-positive AML patients.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
The studies involving humans were approved by Research Ethics Committee of Wuxi People’s Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
JX: Investigation, Writing – original draft. GY: Conceptualization, Data curation, Methodology, Software, Writing – review & editing. QY: Conceptualization, Writing – review & editing. RZ: Data curation, Investigation, Methodology, Writing – original draft. GL: Data curation, Methodology, Project administration, Resources, Supervision, Validation, Writing – review & editing. MW: Formal analysis, Methodology, Software, Writing – original draft. HS: Data curation, Writing – original draft. HC: Data curation, Writing – original draft. LW: Data curation, Writing – original draft. PC: Conceptualization, Supervision, Validation, Writing – review & editing. K-LG: Conceptualization, Data curation, Validation, Writing – review & editing. CS: Conceptualization, Formal analysis, Project administration, Validation, Visualization, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2024.1466590/full#supplementary-material
References
1. DiNardo CD, Erba HP, Freeman SD, Wei AH. Acute myeloid leukaemia. Lancet. (2023) 401:2073–86. doi: 10.1016/S0140-6736(23)00108-3
2. Huang BJ, Smith JL, Wang YC, Taghizadeh K, Leonti AR, Ries RE, et al. CBFB-MYH11 fusion transcripts distinguish acute myeloid leukemias with distinct molecular landscapes and outcomes. Blood Adv. (2021) 5:4963–8. doi: 10.1182/bloodadvances.2021004965
3. Makkar H, Majhi RK, Goel H, Gupta AK, Chopra A, Tanwar P, et al. Acute myeloid leukemia: novel mutations and their clinical implications. Am J Blood Res. (2023) 13:12–27.
4. Engen C, Hellesøy M, Grob T, Al Hinai A, Brendehaug A, Wergeland, et al. LFLT3-ITD mutations in acute myeloid leukaemia-molecular characteristics, distribution and numerical variation. Mol Oncol. (2021) 15:2300–17. doi: 10.1002/1878-0261.12961
5. Voso MT, Ottone T, Lavorgna S, Venditti A, Maurillo L, Lo-Coco F, et al. MRD in AML: the role of new techniques. Front Oncol. (2019) 9:655. doi: 10.3389/fonc.2019.00655
6. Kayser S, Levis MJ. The clinical impact of the molecular landscape of acute myeloid leukemia. Haematologica. (2023) 108:308–20. doi: 10.3324/haematol.2022.280801
7. Guan W, Zhou L, Li Y, Yang E, Liu Y, Lv N, et al. Profiling of somatic mutations and fusion genes in acute myeloid leukemia patients with FLT3-ITD or FLT3-TKD mutation at diagnosis reveals distinct evolutionary patterns. Exp Hematol Oncol. (2021) 10:27. doi: 10.1186/s40164-021-00207-4
8. Döhner H, Wei AH, Appelbaum FR, Craddock C, DiNardo CD, Dombret H, et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood. (2022) 140:1345–77. doi: 10.1182/blood.2022016867
9. De Braekeleer E, Douet-Guilbert N, Morel F, Le Bris MJ, Basinko A, De Braekeleer M. ETV6 fusion genes in hematological Malignancies: a review. Leuk Res. (2012) 36:945–61. doi: 10.1016/j.leukres.2012.04.010
10. Zhou F, Chen B. Acute myeloid leukemia carrying ETV6 mutations: biologic and clinical features. Hematology. (2018) 23:608–12. doi: 10.1080/10245332.2018.1482051
11. Nishii R, Baskin-Doerfler R, Yang W, Oak N, Zhao X, Yang W, et al. Molecular basis of ETV6-mediated predisposition to childhood acute lymphoblastic leukemia. Blood. (2021) 137:364–73. doi: 10.1182/blood.2020006164
12. Demircioglu FE, Sosa BA, Ingram J, Ploegh HL, Schwartz TU. Structures of TorsinA and its disease-mutant complexed with an activator reveal the molecular basis for primary dystonia. Elife. (2016) 5:e17983. doi: 10.7554/eLife.17983
13. Gao Q, Liang WW, Foltz SM, Mutharasu G, Jayasinghe RG, Cao S, et al. Driver fusions and their implications in the development and treatment of human cancers. Cell Rep. (2018) 23:227–238.e3. doi: 10.1016/j.celrep.2018.03.050
14. Engen C, Hellesøy M, Grob T, Al Hinai A, Brendehaug A, Wergeland L, et al. FLT3-ITD mutations in acute myeloid leukaemia - molecular characteristics, distribution and numerical variation. Mol Oncol. (2021) 15:2300–17. doi: 10.1002/1878-0261.12961
15. Thanasopoulou A, Tzankov A, Schwaller J. Potent co-operation between the NUP98-NSD1 fusion and the FLT3-ITD mutation in acute myeloid leukemia induction. Haematologica. (2014) 99:1465–71. doi: 10.3324/haematol.2013.100917
16. Schessl C, Rawat VP, Cusan M, Deshpande A, Kohl TM, Rosten PM, et al. The AML1-ETO fusion gene and the FLT3 length mutation collaborate in inducing acute leukemia in mice. J Clin Invest. (2005) 115:2159–68. doi: 10.1172/JCI24225
17. Kim HG, Kojima K, Swindle CS, Cotta CV, Huo Y, Reddy V, et al. FLT3-ITD cooperates with inv (16) to promote progression to acute myeloid leukemia. Blood. (2008) 111:1567–74. doi: 10.1182/blood-2006-06-030312
18. Bystrom R, Levis MJ. An update on FLT3 in acute myeloid leukemia: pathophysiology and therapeutic landscape. Curr Oncol Rep. (2023) 25:369–78. doi: 10.1007/s11912-023-01389-2
Keywords: acute myeloid leukemia, gene fusions, TOR1AIP2::ETV6, FLT3-ITD, mutation
Citation: Xia J, Yang G, Yu Q, Zhang R, Li G, Wang M, Sun H, Chen H, Wang L, Chen P, Gu K-L and Sun C (2024) Case report: Identification of a novel TOR1AIP2::ETV6 transcript with FLT3-ITD mutation in acute myeloid leukemia progressed from myelodysplastic syndrome. Front. Oncol. 14:1466590. doi: 10.3389/fonc.2024.1466590
Received: 18 July 2024; Accepted: 06 November 2024;
Published: 10 December 2024.
Edited by:
Cosimo Cumbo, University of Bari Aldo Moro, ItalyReviewed by:
Shuang Fu, China Medical University, ChinaPamlea Sung, University at Buffalo, United States
Copyright © 2024 Xia, Yang, Yu, Zhang, Li, Wang, Sun, Chen, Wang, Chen, Gu and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chao Sun, NzUwODMxNjc5QHFxLmNvbQ==; Kai-Li Gu, a2FpbGlfZ3VAZnVqaWRhYmlvLmNvbQ==; Ping Chen, cGluZ19jaGVuQGZ1amlkYWJpby5jb20=
†These authors have contributed equally to this work and share first authorship