 Priyanka Vijay Sonar
Priyanka Vijay Sonar Anuj Kumar Singh1,2†
Anuj Kumar Singh1,2† Nilesh Kumar Sharma
Nilesh Kumar Sharma- 1Cancer and Translational Research Lab, Dr. D.Y. Patil Biotechnology & Bioinformatics Institute, Dr. D.Y. Patil Vidyapeeth, Pune, Maharashtra, India
- 2Ichnos Glenmark Innovation, Glenmark Pharmaceuticals Limited, Navi Mumbai, Maharashtra, India
Background: Tumor displays various forms of tumor heterogeneity including immune heterogeneity that allow cancer cells to survive during conventional anticancer drug interventions. Thus, there is a strong rationale for overcoming anticancer drug resistance by employing the components of immune cells. Using the immune system to target tumor cells has revolutionized treatment. Recently, significant progress has been achieved at preclinical and clinical levels to benefit cancer patients.
Approach: A review of literature from the past ten years across PubMed, Scopus, and Web of Science focused on immunotherapy strategies. These include immune checkpoint inhibitors (ICIs), tumor-infiltrating lymphocyte therapy, antibody-drug conjugates (ADCs), cancer vaccines, CAR T-cell therapy, and the role of the gut microbiome.
Conclusion: While immunotherapy outcomes have improved, particularly for tumor types such as melanoma and non-small cell lung cancer (NSCLC), challenges persist regarding predictive biomarker identification and better management. Ongoing research on modifiers of immune function like gut microbiome-derived metabolites, next-generation ADCs, and new classes of biologics is warranted. Overall, continued investigation toward optimizing synergistic immunotherapeutic combinations through strategic drug delivery systems is imperative for preclinical and clinical success in cancer patients.
1 Introduction
For a long time, cancer has presented a significant obstacle to medical science, and conventional treatment methods frequently fail to offer long-term benefits. Recently, cancer immunotherapy has emerged as a breakthrough approach to treating various malignancies (1–4). Cancer immunotherapy, which uses the body’s immune system to combat cancer, has significantly improved patient outcomes. Cancer immunotherapy includes living medicines such as chimeric antigen receptor (CAR)-T cells, tumor-infiltrating lymphocytes (TILs), and non-living drugs such as monoclonal antibodies, and immune checkpoint inhibitors (ICIs). Cancer immunotherapy is an essential clinical strategy to improve anti-tumor immune responses (5–9). Nevertheless, cancers may become resistant to immune monitoring, which could result in low response rates and ineffective treatment (10–14). Research on the role of gut microbiome in cancer immunotherapy is encouraging. Certain gut bacteria may improve the body’s response to ICIs, potentially converting non-responders into responders (15–18).
Tumor cells altered signaling pathways, genetic modifications, and the patient’s microbial signature are the main causes of this resistance, which makes them less receptive to immunotherapeutic treatments (15–21). Tumors are capable of creating an immunosuppressive environment by releasing molecules and attracting cells that block immune cell infiltration and function (9, 22–31).
Various types of immunotherapy strategies have been developed, each with distinct mechanisms and targets (30–41). Checkpoint inhibitors are drugs that release the brakes on the immune system, allowing it to recognize and attack cancer cells more effectively. By blocking certain proteins, checkpoint inhibitors help unleash the immune system’s full potential. In CAR-T cell treatment, a patient’s T cells are engineered to express a particular receptor capable of identifying cancer cells (42–52). These systems are designed to restore or enhance immune cell activity and amplify immune responses. The modified T cells are then reintroduced into the patient, where they become more effective at recognizing and eliminating cancer cells. Monoclonal antibodies, which are lab-produced molecules, can specifically identify and bind to targets on cancer cells. By doing so, they can either directly destroy cancer cells or stimulate an immune system response against them (53–63).
The gut microbiome, comprising the diverse community of microorganisms residing in the gastrointestinal tract, has garnered recognition as a crucial factor in immunotherapy (64–70). Researchers have suggested that the gut microbiome holds potential as both biomarkers and targets for manipulation to predict and augment the effectiveness of antitumor immunotherapy across various cancer types (71–77). Efforts to modify the gut microbiome using probiotics, prebiotics, antibiotics, or fecal microbiota transplantation (FMT) are being explored to improve immunotherapy outcomes (75–77).
Immunotherapy has altered the way that cancer is treated, but there are still some issues. Multidisciplinary cooperation, ongoing research, and creative thinking are needed to overcome these obstacles. This paper explores the potential of immunotherapy in cancer treatment, focusing on overcoming resistance, identifying predictive biomarkers, managing immune-related adverse events (irAEs), improving affordability, optimizing combination therapies, enhancing smart drug delivery systems (SDDSs), and leveraging the gut microbiome (GM).
2 Tumor immune heterogeneity
Tumor cells acquire several hallmark capabilities that enable their malignant growth and spread. These include sustaining proliferative signaling, evading growth suppressors, resisting cell death, enabling replicative immortality, inducing angiogenesis, and activating invasion and metastasis (15–17). Tumors also exhibit genome instability and inflammation, which facilitate tumor progression. Most critically, tumors evolve mechanisms to avoid immune destruction, known as immune evasion. This capability allows tumors to suppress, inactivate, or avoid detection by the immune system (18–21). Immunosuppressive cells, inhibitory receptors, cytokines, and disrupted antigen presentation weaken anti-tumor immune responses. Overcoming these barriers is crucial for successful immunotherapy.
A key feature underlying immune evasion is marked by heterogeneity within the tumor and its microenvironment. This heterogeneity spans multiple dimensions at genetic, epigenetic, phenotypic, functional, and microenvironmental levels. Intra-tumoral immune heterogeneity involves spatial, temporal, and genomic variations in the abundance, composition, functional orientation, and organization of immune cells (19–21). Tumor heterogeneity allows resistant subclones to grow, helping the tumor evade treatment. These differences determine whether a tumor is inflamed with immune cells or suppressed by regulatory components. Monitoring immune profiles over time and space is crucial to understanding resistance. Advanced profiling of tumor-infiltrating lymphocytes and myeloid cells has identified immune subtypes linked to different responses. Understanding the causes and patterns of heterogeneity can help overcome treatment failure (9, 22–24).
The immune landscape of tumors critically impacts their susceptibility to immunotherapies like immune checkpoint blockade. However, the development of resistance remains a key limitation. Tumors exploit numerous tactics to evade immune attack and blunt immunotherapeutic efficacy (9, 15–24). These include the loss of immunogenic antigens, defects in antigen presentation, increased expression of alternative immune checkpoints, recruitment of immunosuppressive cells, and the release of inhibitory cytokines. Therapies initially effective at unleashing anti-tumor immunity eventually confront resistant subclones or a recalibrated microenvironment (9, 19–24). Even inflamed tumors with high T-cell infiltration can acquire resistance. Tracking the evolution of the immune contexture and escaping subclones is essential to therapeutic durability. Strategies to overcome resistance include disrupting suppressive networks, boosting T cell function, eliminating immunosuppressive cells, converting them into immune-activating cells, and targeting non-immunogenic tumor niches (9, 24). Ultimately, deciphering and redirecting immune heterogeneity in space and time provides a path to more effective and sustained immunotherapy responses.
3 Cancer immunotherapy
Cancer immunotherapy, also referred to as biologic therapy, is a ground-breaking treatment modality that aims to enhance the body’s immune system to recognize and destroy cancer cells (9, 21–24). Unlike chemotherapy, radiation, or targeted therapy, which directly attack cancer cells, immunotherapy boosts the immune system to fight cancer (9, 25–27). The major plus point of immunotherapy lies in the fact that it damages only the cancerous cells and the healthy cells remain minimally unaffected. This reduces the side effects of cancer treatment as well it puts patients’ lives at a lesser risk of death by cancer (9, 25–31).
Immunotherapy has revolutionized cancer treatment. The idea of using the immune system to fight disease dates back centuries. In the late 19th century, William Coley noticed that some cancer patients went into remission after bacterial infections. This led to the foundation of immunotherapy by stimulating the immune response against cancer (9). Researchers in the early 20th century, including Paul Ehrlich and Emil von Behring, explored immune stimulation by developing serums and antibodies to combat diseases like diphtheria and tetanus. These studies demonstrated the potential of immune-based interventions (25).
The field of tumor immunology emerged in the mid-20th century when Lewis Thomas and others began investigating the interaction between the immune system and cancer cells. They observed the presence of immune cells within tumors and recognized the potential for immune responses against cancer. In the 1960s, interferons were discovered as natural proteins with potent antiviral and antitumor properties. Interferon therapy represented one of the earliest attempts at immunotherapy for cancer treatment (26). In 1975, Georges Köhler and César Milstein pioneered the hybridoma technique, enabling the production of large quantities of specific mAbs (27, 28). Rituximab, the first FDA-approved mAb for treatment, marked a significant discovery. James Allison and Tasuku Honjo’s discoveries of cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed cell death protein 1 (PD-1), respectively, opened new avenues for cancer immunotherapy (29, 30). Immune checkpoint inhibitors, such as PD-1 inhibitors (nivolumab, pembrolizumab, cemiplimab), CTLA-4 inhibitor (ipilimumab), and PD-L1 inhibitors (atezolizumab, durvalumab, avelumab), have transformed cancer treatment by helping the immune system recognize and attack cancer cells (9, 15–24). Adoptive cell transfer (ACT) therapy modifies immune cells-specifically, T cells-in patients to improve their capacity to combat cancer. Carl June and associates invented the chimeric antigen receptor (CAR) T-cell therapy, which has shown promise in the treatment of several blood malignancies (31). A summarized flow model on the rationale of cancer immunotherapies is presented (Figure 1).
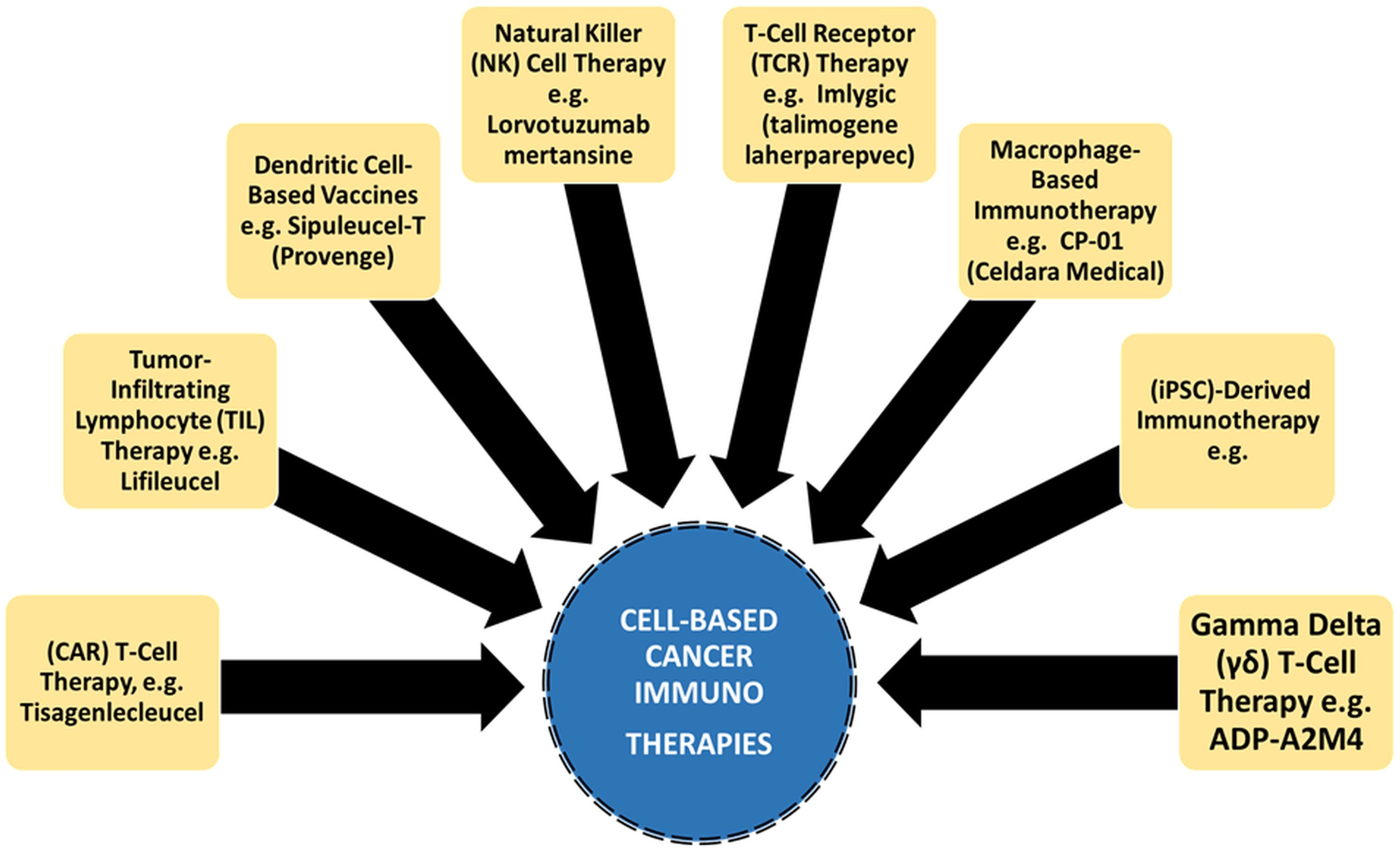
Figure 1. Cancer immunotherapies are attributed with targeted and enhanced anti-tumor response over traditional cancer therapy.
4 Types of immunotherapies
Immunotherapy includes different types, each targeting specific aspects of the immune response to cancer. These treatments offer new hope for lasting responses, better survival rates, and improved quality of life for cancer patients. This section explores their mechanisms and clinical impact.
4.1 Immune checkpoint inhibitors
Immune checkpoint proteins are known to suppress the immune system through various pathways that can prevent harm to healthy tissues. However, cancer cells exploit immune checkpoint proteins such as CTLA4 and PD1 to evade immune attack. Encouraging preclinical and clinical developments have been reported on immune checkpoint inhibitors (ICIs) that can block these proteins and in turn allowing immune cells to recognize and attack cancer cells (17, 32–41). Two key checkpoint proteins, CTLA-4 and PD-1, play crucial roles in regulating the function of T cells. CTLA-4 dampens T-cell activation by engaging with its ligands, while PD-1 modulates T-cell activity in peripheral tissues through its interaction with PD-L1 and PD-L2 (32). ICIs have displayed promise as therapeutic interventions for cancer by reversing the immune system’s suppressive effects on tumor immunity (33, 34).
Early investigations showcased the effectiveness of anti-CTLA-4 therapy in mouse models of tumors, whereas blocking PD-1 or PD-L1 bolstered anti-tumor immune responses. Consequently, antibodies targeting CTLA-4, PD-1, and PD-L1 were developed to hinder their interaction with respective ligands. Clinical trials involving antibodies such as ipilimumab and nivolumab, aimed at CTLA-4 and PD-1/PD-L1 respectively, demonstrated lasting responses in melanoma and other cancers. However, they were also linked to severe irAEs like colitis, dermatitis, thyroiditis, pneumonitis, and hepatitis (35). Efforts have been made to explore combination therapies targeting multiple immune checkpoints, leading to increased response rates albeit with a higher occurrence of severe irAEs. Besides the CTLA-4 and PD-1/PD-L1 pathways, other inhibitory receptors like LAG-3, TIM-3, and TIGIT have been targeted in combination with anti-PD-1 therapy. Nevertheless, the potential severity and lethality of ICI-associated toxicities have become apparent. Fatalities, occurring sporadically and early during treatment, exhibit different patterns of organ involvement between anti-CTLA-4 and anti-PD-1/PD-L1 therapies (36, 37). Colitis is more frequently associated with anti-CTLA-4 treatment, while pneumonitis, hepatitis, and neurotoxicity are more observed with anti-PD-1/PD-L1 treatment.
ICIs have revolutionized cancer treatment, including malignancies previously deemed untreatable (38). These agents can incite inflammation and tissue damage in various organs. Myocarditis, a rare yet severe toxicity associated with ICIs, has garnered attention due to its often swift and fatal progression. The underlying mechanisms and specific antigens that trigger ICI-associated myocarditis remain poorly understood (39–41). Deaths related to combination therapy frequently stem from colitis or myocarditis, with the latter exhibiting the highest fatality rate among irAEs (40-50%). It is imperative to develop strategies that effectively manage ICI-associated toxicities without compromising their anticancer efficacy. While ICIs have shown encouraging responses in some cancer patients, however, develop resistance over time is a concern driven by various alterations in the tumor microenvironment.
Primarily, tumors fail to respond from the start due to poor immune cell infiltration or lack of antigens for T-cell recognition (38–40). At the same time, acquired forms of resistance due to mutations reduce antigen presentation or increase immune checkpoint molecules. Also, immune-mediated resistance is observed with the decline of T-cell functions and the emergence of immunosuppressive cells. In the context of the pro-tumor microenvironment, various non-cellular components such as immunosuppressive molecules or cellular components in the stromal cells that hinder the immune response (39, 40). A summary of the various classes of cancer immunotherapies with suitable examples of clinical drugs is provided (Figure 2).
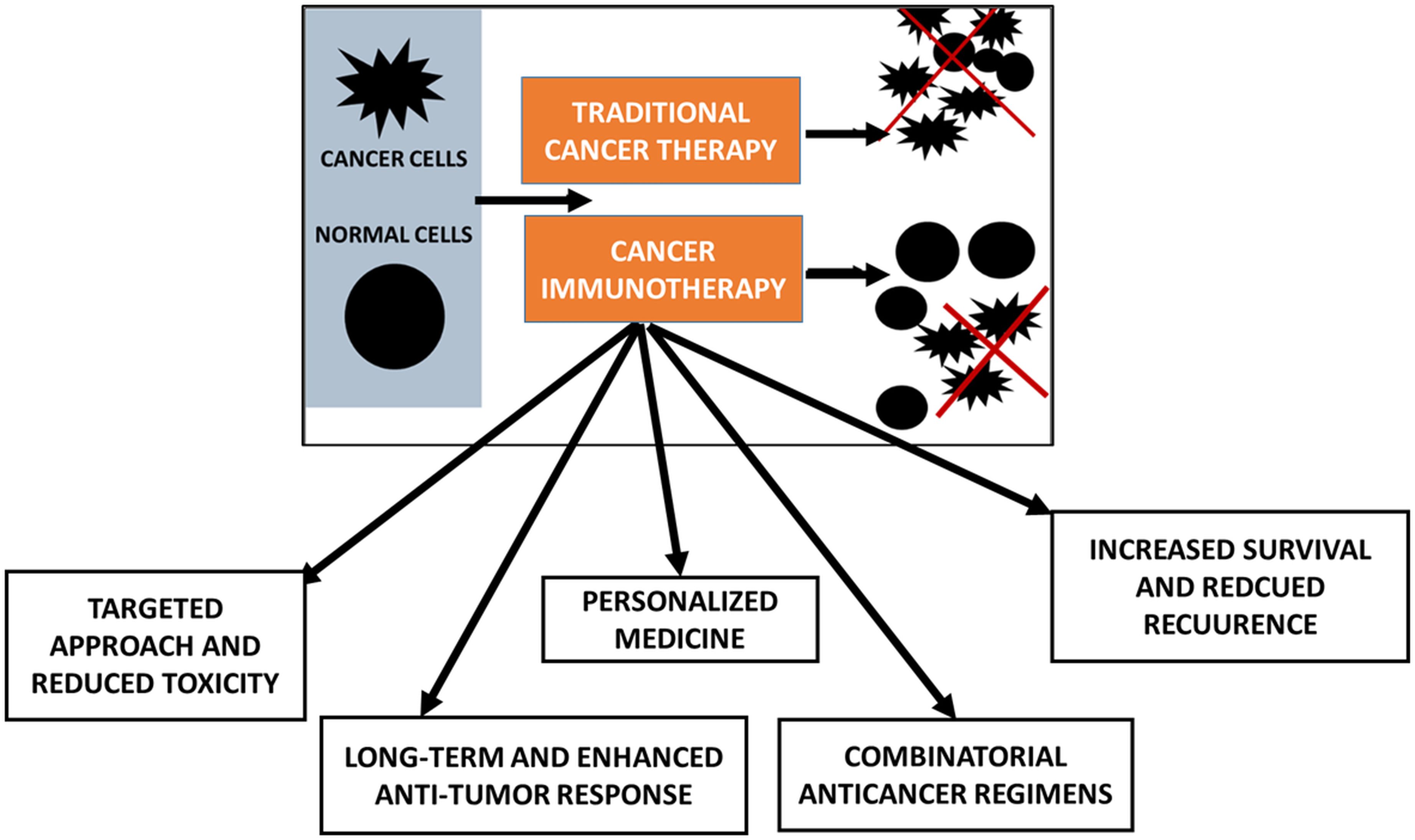
Figure 2. Types of immune checkpoint inhibitors (ICIs) with clinically approved drugs.
4.2 Chimeric antigen receptor T-cell therapy
Beyond antibody-based techniques, the use of autologous T cells modified by gene transfer to express receptors specific to chemicals found on malignant cells offers promise in cancer therapies (42, 43). This technique entails inserting genes encoding antigen receptors into the patient’s T cells, which are then grown in vitro to produce memory and effector lymphocytes capable of vigorous proliferation in the body, ultimately exerting potent anti-tumor effects (44, 45). Using chimeric antigen receptors (CARs) to genetically reroute and reprogram T cells is a noteworthy method of bypassing cancer tolerance. Synthetic receptors enable precise targeting of surface antigens without requiring Major Histocompatibility Complex (MHC) molecules. They combine T-cell functions with the antigen-recognition ability of antibodies (46). Liquid cancers are better suited for this kind of treatment.
More than thirty years ago, the first synthetic immunoglobulin/TCR chimeric molecule with antibody-like specificity was expressed, sparking the start of the development of CARs. Later research showed that T cells may be activated by a CAR made up of CD8 and the CD3ζ chain without the need for their native TCR (47). CARs typically comprise an external domain that identifies tumor antigens and one or more intracellular signaling regions that cause T-cell activation (48, 49). A major advantage of CAR-based immunotherapy is that scFv (Single-chain variable fragments) from antibodies have a much higher affinity than TCRs. CAR T cells can directly target intact surface proteins without needing antigen processing or MHC presentation, reducing the risk of tumor escape (50–52). Moreover, CARs make it possible to recognize antigens that TCRs may miss or find difficult to recognize, such as glycolipids, abnormally glycosylated proteins, and conformational epitopes.
4.3 Tumor-infiltrating lymphocyte therapy
As a type of adoptive cellular therapy, TIL therapy comprises harvesting lymphocytes that have penetrated tumors, growing and amplifying them in vitro, and then infusing the cells back into patients to treat them (53). TIL therapy offers unique advantages over other adoptive cell therapies for solid tumors. It has better tumor-targeting ability, lower off-target toxicity, and diverse T-cell receptor (TCR) clonality (54). TIL therapy was developed by extracting TIL from several mouse tumor models for the use of TIL therapy in human advanced cancers (55).
TIL treatment was tried clinically for the first time, and in patients with metastatic melanoma, it had an objective response rate of 60%. However, the features of solid tumors present serious obstacles to the creation of successful adoptive cellular therapies (56). Solid tumors are highly diverse, making it hard to find a universal target, unlike blood cancers with clear markers. Targeting a single antigen can lead to resistant clones or antigen loss. Even with many adoptively transferred T cells, solid tumors often show limited infiltration (57). The immunosuppressive tumor microenvironment (TME) contains regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), and tumor-associated macrophages (TAMs). These cells weaken T-cell function by increasing immune-inhibitory molecules, cytokines, and metabolites while reducing co-stimulatory molecules (58, 59). Compared to other adoptive cellular therapies like CAR-T and TCR-T cell therapies, tumor heterogeneity is more efficiently addressed by TIL, which is made up of T cells with various TCR clones that can recognize a variety of tumor antigens.
Notably, in solid tumors with high mutation loading, like melanoma, TIL has proven to be more clinically effective than CAR-T treatment. TIL cells are mostly made up of effector memory T (TEM) cells that express chemokine receptors including CCR5 and CXCR3, having been triggered by tumor antigens in the body (60). After being transferred into patients, TIL can readily move to antigenically different tissues, such as malignancies, because of their receptors. Because TCRs were negatively selected during the early stages of T cell immunity formation, TIL treatment has therefore demonstrated minimal off-target damage (61). On the other hand, if tailored tumor-targeting molecules show cross-reactivity with antigens found in normal tissues, such as single-chain variable segments in CAR-T or affinity-enhanced TCRs in TCR-T cell treatments, it could be harmful. In the treatment of melanoma, this strategy has demonstrated encouraging outcomes, with some patients seeing significant response rates (62, 63). Optimizing TIL treatment and expanding its efficacy to cover additional cancer types are ongoing endeavors. A summary on the comparison between different adoptive cellular therapies, clarifying their advantages and limitations is presented (Table 1).
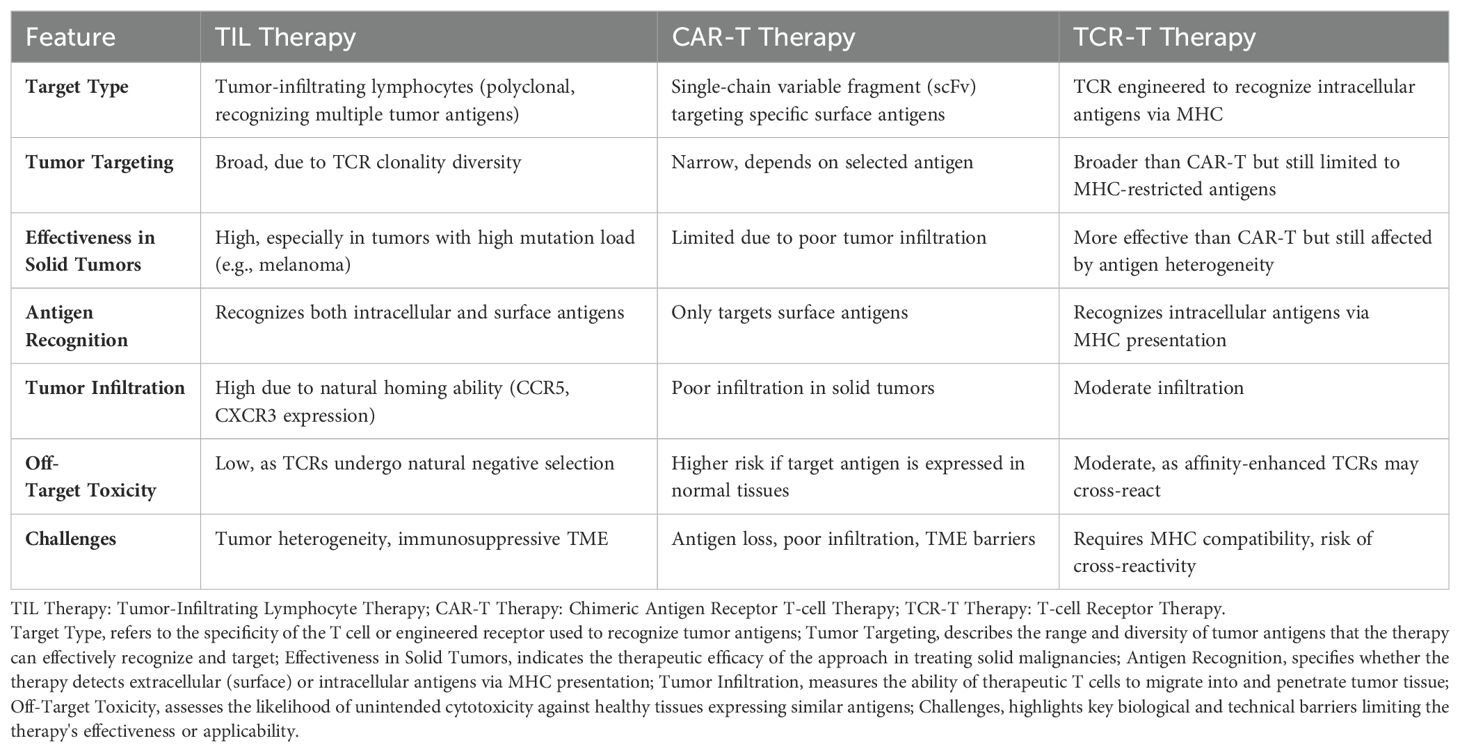
Table 1. A summary on the comparison between different adoptive cellular therapies, clarifying their effectiveness, advantages and limitations are presented.
4.4 Antibody-drug conjugate
The notion of antibody-drug conjugate (ADC) is a revolutionary one that combines the powerful efficacy of cytotoxic medications with the targeting capability of monoclonal antibodies (mAbs) (64–73, 78–80). ADCs provide accurate targeting and concurrently powerful therapeutic effects by conjugating mAbs to cytotoxic payloads using specific chemical linkers (64). Attaching a large hydrophilic antibody improves the therapeutic index by preventing the toxic payload from entering cells without the target antigen. In 2000, the FDA approved Mylotarg® (gemtuzumab ozogamicin), the first ADC drug for acute myeloid leukemia (AML), marking the beginning of the ADC era in targeted cancer therapy (65, 66).
The three parts of an ADC are the chemical linker, the cytotoxic payload, and the antibody. The antibody has minimal immunogenicity and a long plasma half-life, and it selectively binds to the target antigen produced on tumor cells, facilitating effective internalization (67). The linker, which connects the antibody to the cytotoxic drug, determines ADC stability and payload release. ADCs bind to tumor cells, get internalized, and release their toxic payload into lysosomes. This triggers cell death or apoptosis through mechanisms like targeting microtubules or DNA (68). Because ADCs have a low oral bioavailability and are easily broken down by digestive enzymes, they are usually given intravenously. ADCs are anticipated to revolutionize cancer therapy by offering a focused and effective therapeutic strategy, maybe taking the place of traditional chemotherapies, thanks to their growing targets and indications. ADCs are a potentially beneficial tactic in the fight against cancer due to their accurate targeting, efficient killing of cancer cells, improvement of the therapeutic window, and reduction of off-target side effects. ADCs’ lethal effects are further amplified by the bystander effect and possible influence on the tumor microenvironment (69, 70).
As of present, the European Medicines Agency (EMA) has approved four antibody-drug conjugates (ADCs), while the FDA has given regulatory approval for nine ADCs in the US (78, 79). Kadcyla® is an ADC approved by the FDA in 2013 and used to treat metastatic HER2-positive breast cancer. It combines the cytotoxic payload DM1 (Derivative of Maytansine 1) with the HER2-targeting monoclonal antibody trastuzumab (80). In 2011, the FDA approved Adcetris® for treating systemic anaplastic large-cell lymphoma and Hodgkin lymphoma. This ADC consists of the CD30-targeting antibody brentuximab and the antimitotic drug MMAE. Its strong clinical results have provided patients with more treatment options (71, 72). The FDA approved Padcev® in 2019 to treat metastatic urothelial carcinoma. It comprises MMAE, a chemical that disrupts microtubules, and enfortumab, a monoclonal antibody that targets nectin-4. Patients who have previously had immune checkpoint inhibitors and platinum-based chemotherapy have shown improvement with Padcev® (73). In 2019, the FDA approved Enhertu® to treat metastatic HER2-positive breast cancer. In 2019, the FDA approved Enhertu® for metastatic HER2-positive breast cancer. This ADC combines trastuzumab, a HER2-targeting antibody, with deruxtecan, a topoisomerase I inhibitor. It has shown strong clinical responses, especially in patients resistant to previous HER2 therapies (74). Patients with restricted therapy options now have treatment options thanks to these ADCs, which have shown extraordinary success in treating a variety of cancers and having safety profiles (including colorectal, breast, and lymphoma cancer). We may expect the creation of new ADCs and the extension of their uses in the fight against cancer as long as research in this area continues.
4.5 Cancer vaccines
The advancement of cancer immunotherapy has given rise to several instruments and methods for thwarting the immune-evading processes employed by cancer cells. These resources include immunocompetent cells such as T and dendritic cells, as well as antibodies, peptides, proteins, and nucleic acids (75–77, 81–86). Based on their structure and substance, cancer vaccines developed with these methods can be divided into three primary classes. Cell vaccines fall within the first group; they are a type of immunotherapy that employs immune or tumor cells (77). Tumor cell vaccines use modified tumor cells to trigger an immune response against cancer-specific antigens. In contrast, immune cell vaccines enhance and modify immune cells, like T and dendritic cells, to boost their ability to recognize and attack cancer (81). Provenge (sipuleucel-T; Dendreon Corporation) received FDA approval in 2010 as a prostate cancer vaccine. Since then, it has attracted a lot of interest in the field of autologous immune cell-based immunotherapy (82). Vaccines based on particular proteins or peptides derived from tumor-associated antigens fall into the second category. The ability of these antigens to trigger an immune response against cancer cells is the basis for their selection. The immune system may identify and target cancer cells that express the matching antigens by delivering these protein/peptide vaccines, which trigger an anti-tumor immune response (83). An immunostimulatory adjuvant-based strategy has been developed to improve the immunogenicity and effectiveness of peptide vaccines.
Nucleic acid vaccines, which include DNA, RNA, and viral vectors used to transfer genetic material encoding tumor-associated antigens, make up the third category. The goal of this strategy is to ingest the genetic instructions necessary for the patient’s cells to produce these antigens directly (84–86). By doing this, the antigens can be produced internally by the patient’s cells, which will then mount an immunological defense against cancer cells that express those antigens.
A summarized flow model is depicted to highlight the cell-based cancer immunotherapies with suitable clinically approved anticancer drugs (Figure 3). Also, a tabular summary of various cancer immunotherapy approaches with comparisons of mechanisms, efficacy, and advantages is given (Table 2).
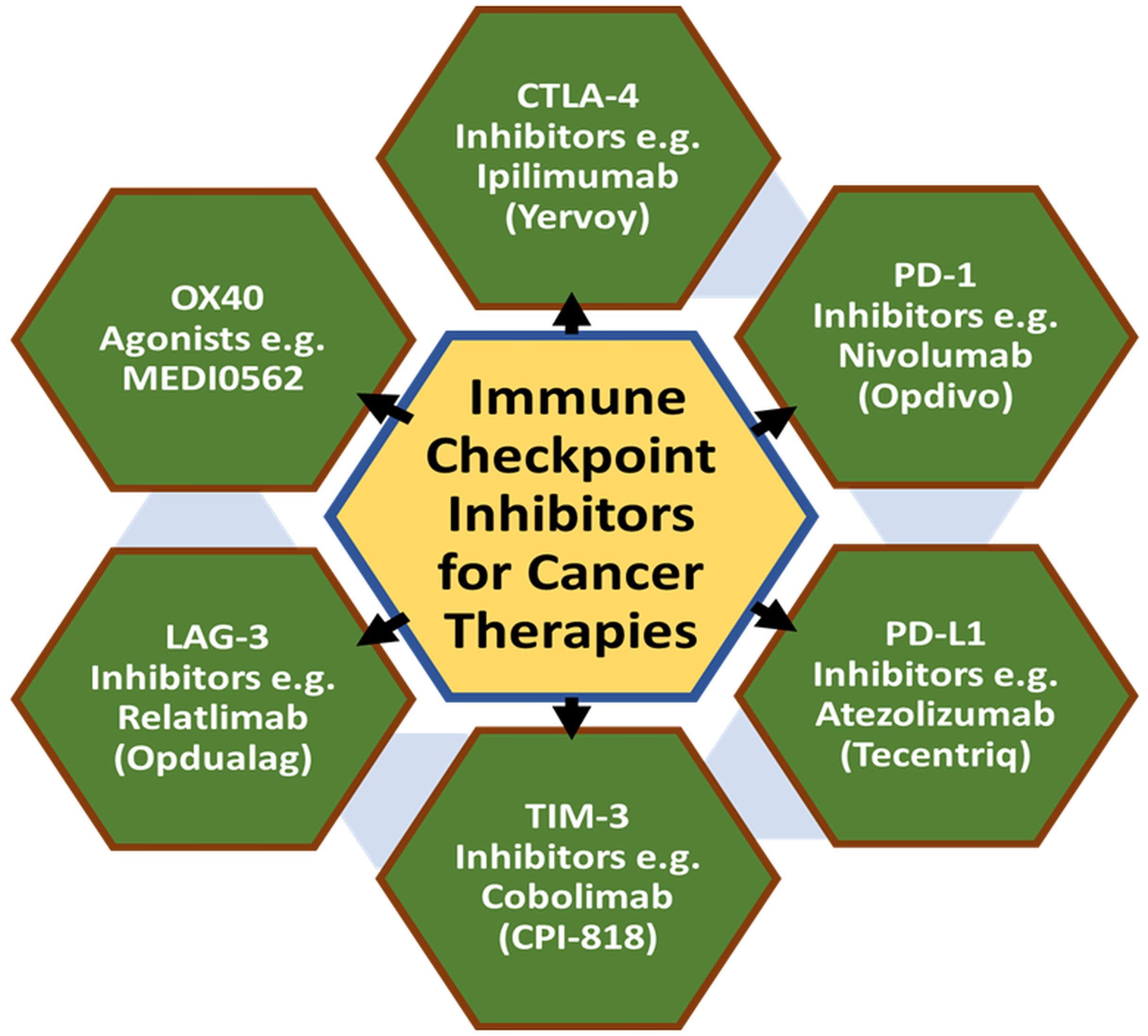
Figure 3. Types of cell-based cancer immunotherapies with suitable examples of clinical drugs.
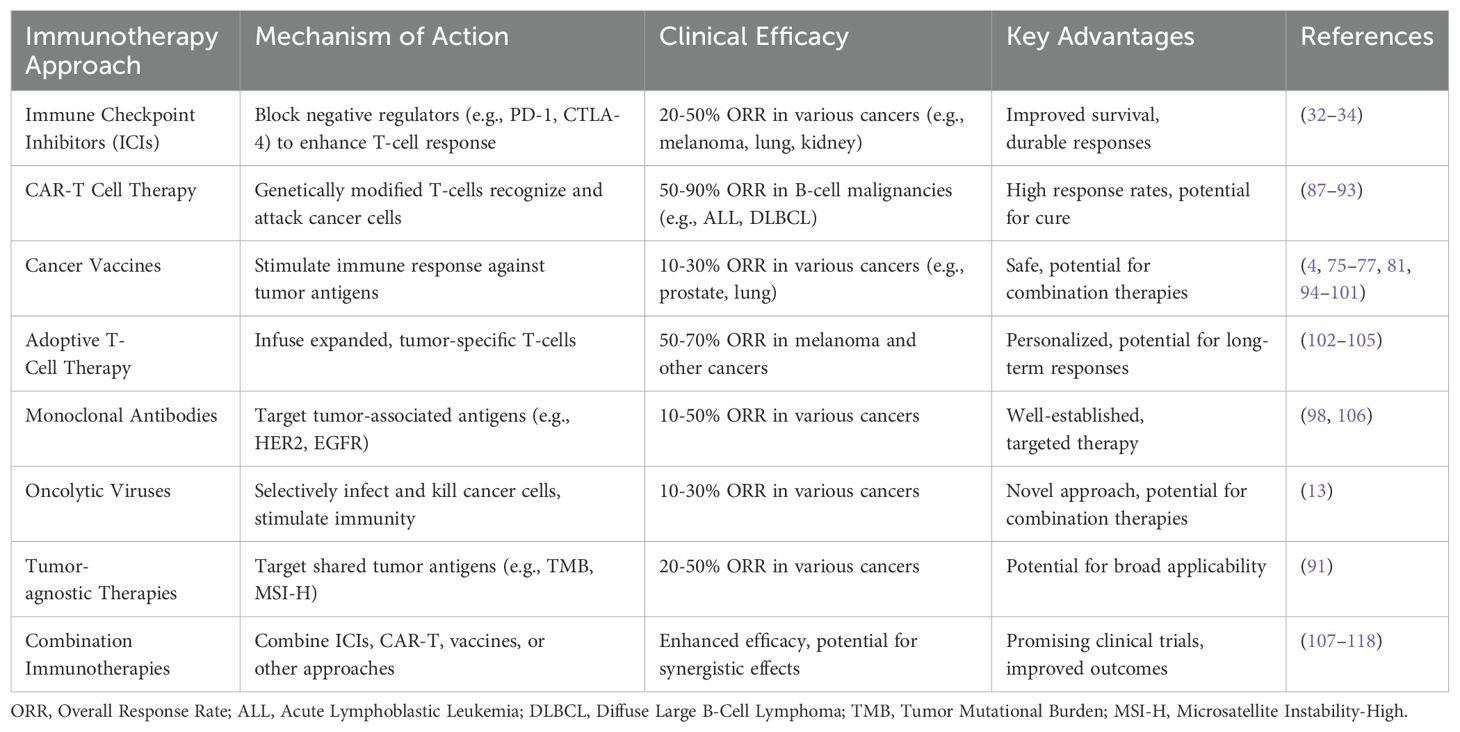
Table 2. Cancer Immunotherapy Approaches: Comparisons on mechanisms, efficacy, and advantages.
5 Preclinical and clinical trial advances
Immunotherapy has witnessed remarkable success in the treatment of various cancers. Checkpoint inhibitors have revolutionized the management of melanoma, lung cancer, and renal cell carcinoma. Clinical trials have shown that patients treated with these inhibitors experience improved survival rates and prolonged progression-free periods compared to conventional treatments (87–93, 107, 119–124). CAR-T cell therapy has shown outstanding success in blood cancers like acute lymphoblastic leukemia and diffuse large B-cell lymphoma. It has led to complete remission in patients who did not respond to traditional treatments (108–117).
Polymerase Proofreading-Associated Polyposis (PPAP) or POLE-mutated and Microsatellite Instability (MSI) endometrial cancers have high tumor-infiltrating lymphocytes and neoantigens. This suggests they may respond well to immunotherapy, especially PD-1/PD-L1 checkpoint inhibitors (88–93, 122–124). In a phase 2, pembrolizumab, an anti-PD-1 antibody, was tested in MMR-deficient tumors identified by PCR, regardless of origin. The FDA later granted accelerated approval for pembrolizumab to treat MSI or MMR-deficient solid tumors, including endometrial cancer (88–91). Although MSI or POLE-mutated status is commonly associated with favorable responses to immunotherapy, some endometrial cancers without these specific alterations have also shown responses (90–92). Ongoing clinical trials in MSS endometrial cancers aim to enhance the efficacy of immune checkpoint inhibitors by combining them with various agents, such as upfront chemotherapy, poly (ADP-ribose) polymerase (PARP) inhibitors, or antiangiogenic drugs. Additionally, the investigation of combination therapies with other immunotherapeutic agents and radiation therapy is of interest (93). Notable ongoing studies include the double-blind, placebo-controlled phase 3 AtTEnd trial (ClinicalTrials.gov identifier NCT03603184) that randomizes patients with metastatic or inoperable uterine carcinoma or carcinosarcoma to receive carboplatin and paclitaxel plus placebo or carboplatin, paclitaxel, and atezolizumab with placebo continued until disease progression (122).
Research is in the early preclinical stage on using natural killer T (NKT) cells to treat glioblastoma multiforme (GBM). Studies show NKT cells can kill GBM cell lines and reduce tumor burden in GBM xenograft mouse models (107, 123, 124). When NKT cells are administered along with α-GalCer (KRN7000), a synthetic glycosphingolipid, the survival of mice with intracranial tumors is enhanced. Type I NKT cells have been shown to exhibit killing activity against CD1d-positive GBM cell lines or patient-derived GBM cells after expansion with IL-2 and α-GalCer. The production of IFN-γ, TNF-α, granzyme B, and IL-4 is significantly increased. In an orthotopic GBM model, co-injecting human type I NKT cells with α-GalCer into tumor-bearing mice with CD1d-positive U251 cells significantly extends survival. However, in an intracranial injection model, type I NKT cells do not inhibit the growth of CD1d-negative U87 cells. This suggests that human-type I NKT cells specifically target CD1d-expressing GBM cells (108, 109).
With the FDA approval of pembrolizumab, immunotherapy has developed as a prominent treatment approach for triple-negative breast cancer (TNBC) (125). Although atezolizumab initially showed promise, its approval for advanced TNBC was withdrawn due to the lack of benefit observed in the IMpassion131 trial (110, 111). Further analysis of this trial is crucial to evaluate potential confounders, such as tumor-infiltrating lymphocytes (TILs), and determine if they were imbalanced between treatment arms, as observed in the NeoTRIPaPDL1 trial (112). In the curative setting, several studies have demonstrated a pathological complete response (pCR) benefit with the addition of immunotherapy to neoadjuvant chemotherapy regimens for early-stage TNBC (113). KEYNOTE-522 trial, pembrolizumab is now approved as part of neoadjuvant treatment and as a single-agent adjuvant treatment for high-risk early-stage TNBC (114). Ongoing trials such as IMpassion030 (NCT03498716) will provide further confirmation if this benefit extends to patients receiving solely adjuvant chemotherapy, who might potentially have a lower volume of immunogenic tumor antigens (115). Additionally, the value of adjuvant immunotherapy alone in patients with residual disease after neoadjuvant chemotherapy would be addressed by the SWOG 1418 trial (NCT02954874) (116).
There are increasing preclinical and clinical reports to evaluate various combinations of ICIs and cell-based therapies such as CAR-T cells and TILs (117, 118, 126–141). A promising evaluation of CHECKMATE-9ER is reported that combined nivolumab (Opdivo) and ipilimumab (Yervoy) in renal cell carcinoma (142). Notable ICIs for combinatorial anticancer therapy trials such as KEYNOTE-671 to evaluate pembrolizumab (Keytruda) + chemotherapy in triple-negative breast cancer (143–147). Also, JULIET is under evaluation as tisagenlecleucel (Kymriah) to treat relapsed/refractory diffuse large B-cell lymphoma. Another clinical trial on JCAR017 (lisocabtagene maraleucel) is reported in relapsed/refractory B-cell lymphomas (144). Clinical trials of IMPOWER150 are reported on the combined effects of atezolizumab (Tecentriq), carboplatin, and paclitaxel in non-squamous non-small cell lung cancer (146).
Besides ICIs in cancer immunotherapies, evaluation of CAR-T therapy as ZUMA-2 is presented to test the effects of axicabtagene ciloleucel (Yescarta) in refractory/relapsed follicular lymphoma (148). The combination of nivolumab, ipilimumab, and radiation therapy for glioblastoma is being explored. Additionally, an overview of clinical trials on cancer immunotherapy combined with other treatments is provided (142–152) (Table 3).
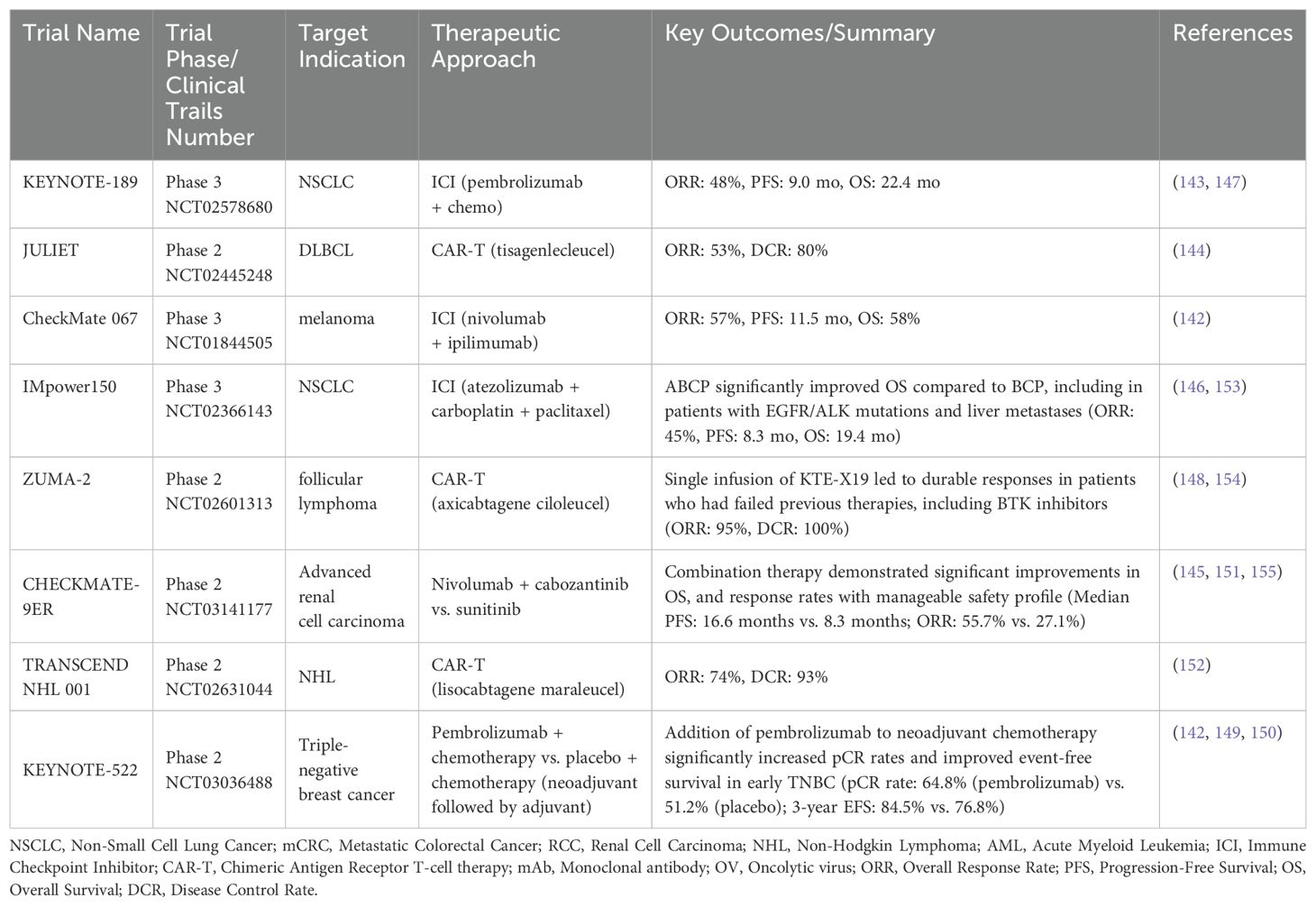
Table 3. A summary on clinical trials of cancer immunotherapies in combinatorial drug treatments for cancer patients.
In recent, significant strides have been made to develop nucleic acid vaccines including various forms of DNA and RNA vaccines as a new form of arsenal in cancer immunotherapy (94–101, 156–158). The progressive intent behind the development of these cancer vaccines could be attributed to precision, high efficacy, and fewer side effects notable clinically approved drugs such as Cavatak (CF33) as DNA vaccines and mRNA-4157, BNT111 as RNA vaccines (97–101, 106, 157, 158). However, the effectiveness of DNA and mRNA cancer vaccines is limited by the evolving complexity and heterogeneity of the tumor immune microenvironment at both intra-tumoral and inter-tumoral levels. A summary table presents various nucleic acid vaccines for cancer immunotherapy, including their mechanisms and outcomes (97–101, 106, 157, 158) (Table 4).
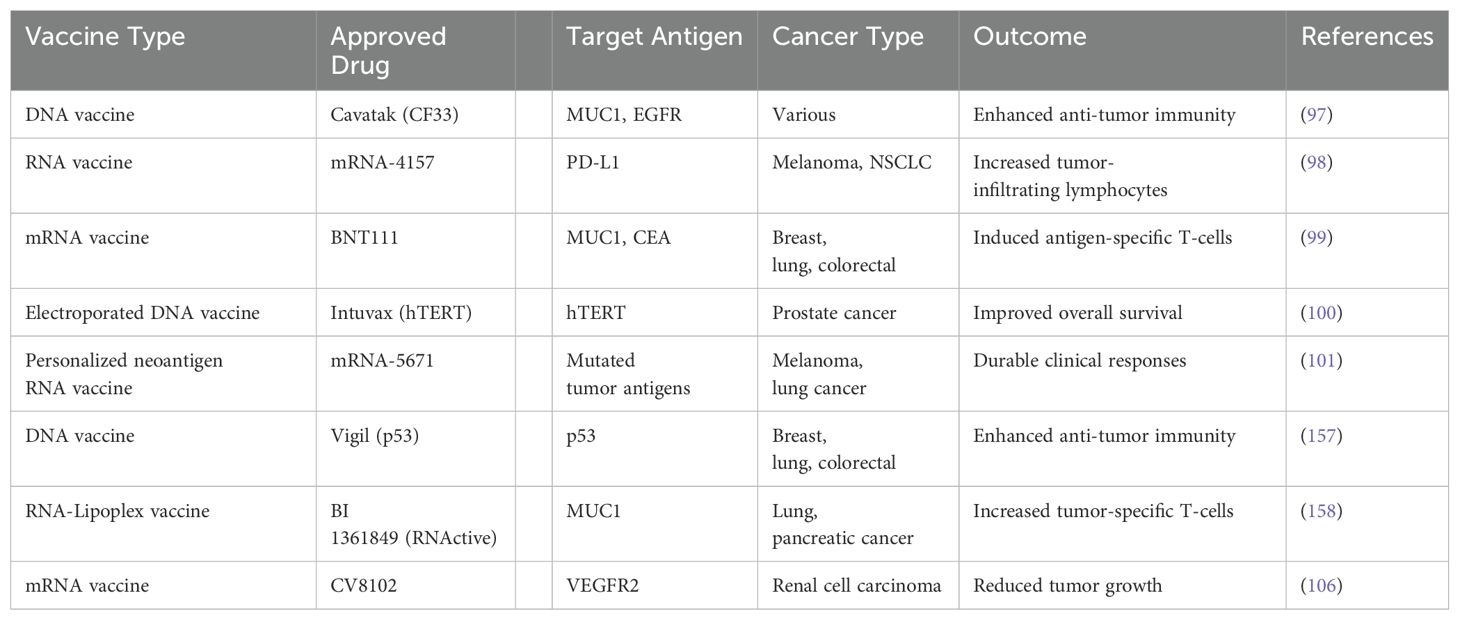
Table 4. Summary on nucleic acid vaccines for cancer immunotherapies, with mechanisms and outcomes.
5.1 Microbiome and immunotherapy
In terms of innate immunity, the immune system actively shapes the GM composition from an early age, and the GM, in turn, influences the immune system’s development (102–104, 159–164). Key interactions between the gut microbiome (GM) and the immune system influence the development of a balanced GM and proper immune function from birth. Disruptions in GM composition, such as antibiotic use, can contribute to immune-related disorders later in life, including asthma and inflammatory bowel disease (104, 105, 165–168). Dysbiosis has the potential to influence both tumor development and the failure of ICI-based therapies overall. A balanced and diverse gut microbiome can activate the immune system to combat cancer and facilitate a robust response to anti-cancer immunotherapies, particularly ICIs (169–181). Several studies have established a correlation between gut microbiota (GM) composition and immunotherapy efficacy (102–105, 162–181).
Clinical data showed that tumors in antibiotic-treated or germ-free mice did not respond to anti-CTLA-4 immunotherapy. The anti-cancer effect of anti-CTLA-4 relied on Bacteroides species, particularly Bacteroides fragilis (160). Additionally, non-responder mice to anti-PD-L1 therapy showed improved responses when given feces from responder mice or orally administered Bifidobacterium. Another clinical finding pointed out that a higher proportion of Bacteroidetes was associated with reduced colitis risk in melanoma (MM) patients treated with immune checkpoint inhibitors (ICIs) (161, 180).
Frankel et al. (163) identified GM signatures associated with ICI efficacy in MM patients using metagenomic and metabolomic profiling. Responders had enriched Bacteroides caccae, Faecalibacterium prausnitzii, Bacteroides thetaiotamicron, Holdemania filiformis, and Dorea formicogenerans, with anacardic acid as a consistently enriched metabolite. Chaput et al. (164) found that MM patients with Bacteroides-driven baseline microbiota had longer progression-free survival (PFS) than those with Faecalibacterium-driven microbiota.
Matson et al. (102) identified enrichment of Bifidobacterium longum, Collinsella aerofaciens, and Enterococcus faecium in responders. Gopalakrishnan et al. (103) observed higher alpha diversity, Ruminococcaceae abundance, and enriched anabolic pathways in responders. Tanoue et al. (166) isolated an 11-strain bacterial consortium that enhanced CD8+ T-cell levels and MHC-I expression in dendritic cells (DCs). Xu et al. (167) showed Prevotella spp. and Akkermansia spp. can affect glycerol-lipid metabolism and anti-PD-1 efficacy. Mager et al. (168) found that Bifidobacterium pseudolongum produces inosine, which activates anti-tumor CD8+ T cells via the A2A receptor. Si W et al. (169) and Shi Y et al. showed that Lactobacillus rhamnosus GG and Bifidobacterium spp. enhance immunotherapy through the cGAS/STING pathway in dendritic cells. A summary of cancer immunotherapy and gut microbiome combinations, supported by preclinical and clinical evidence, is presented (168–181) (Table 5).
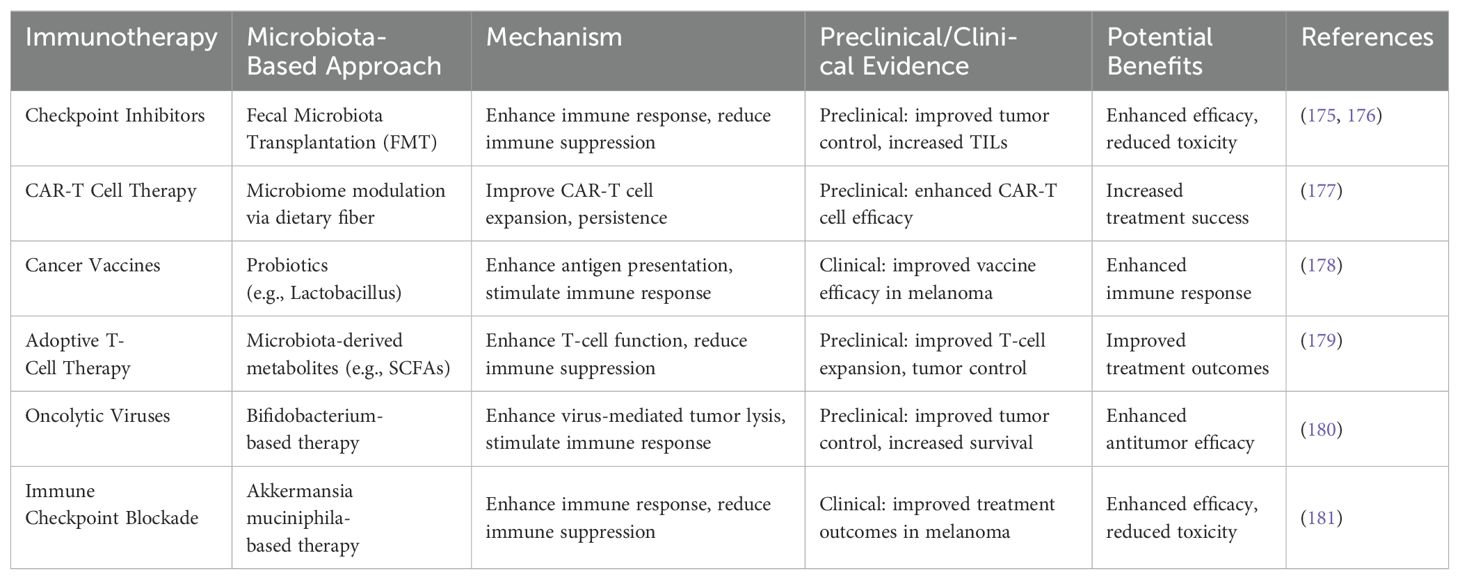
Table 5. Summary on the combined use of cancer immunotherapies and microbiotas for combinatorial therapies with preclinical and clinical evidences.
6 Challenges and future directions
Despite its success, cancer immunotherapy faces challenges that limit its effectiveness and widespread use. Cancer cells adapt by evading immune detection, downregulating target antigens, altering antigen presentation, and creating immunosuppressive microenvironments. Understanding these strategies is a key to improving treatments. Another major challenge is selecting the right patients for therapy. Not all patients respond equally to immunotherapy, and currently, we struggle to predict who will benefit most. The complex interplay between a patient’s immune system, their unique tumor characteristics, and even their gut microbiome make it difficult to design regimes and create combinations of medical, radiation and surgical oncology interventions. This heterogeneity makes identifying reliable predictive biomarkers challenging. When we activate the immune system against cancer, we sometimes trigger immune responses against healthy tissues as well. These irAEs can affect virtually any organ system and may force treatment interruptions or discontinuation. Finding the balance between maintaining therapeutic efficacy while minimizing these side effects remains a delicate challenge.
The high cost of immunotherapies, especially ICIs and CAR-T cell therapy, creates access disparities and strains healthcare systems. Addressing this economic barrier is crucial. Combining immunotherapy with other treatments like chemotherapy or radiation shows promise for better outcomes. However, identifying the optimal combination strategies and determining the optimal sequencing of therapies is complex. Comprehensive studies are needed to elucidate the most effective combinations and treatment sequences to maximize therapeutic synergies and minimize toxicity. SDDSs achieve this by co-delivering multiple therapeutic agents to tumor cells or immunosuppressive cells, thereby increasing drug concentration at the desired site and improving efficacy. They are used to reverse the immunosuppressive microenvironment created by tumors. Furthermore, SDDSs modulate the interferon signaling pathway and enhance the effectiveness of cell therapies. Understanding the microbiome at strain-level along with species-level in gut and TME can open up a new level of personalized medicine. Having that extra resolution is crucial if we are to understand what is happening in the human body and the interplay between cancer treatment and the microbiome. Being able to test the specific mechanisms of this relationship between specific strains and response is the next horizon in this research, and one that could benefit human health in a multitude of ways (94). Rare cancers are challenging to study and treat. While immunotherapy can be highly effective, its outcomes can be unpredictable. Research shows that the microbiome influences responses to combination immunotherapy, while monotherapy yields different results. This highlights the need to consider the microbiome when developing treatments. Additionally, live biotherapeutic products could provide beneficial bacteria to enhance immunotherapy, improving patient responses.
7 Conclusion
Immunotherapy has transformed the field of oncology and given patients fresh hope, revolutionizing the treatment of cancer. In several cancer types, it has demonstrated notable success by enabling the immune system to target cancer cells. To improve the outcomes of cancer immunotherapies in combinatorial treatments, a cohesive approach could involve developing strategies to address tumor cell resistance. There may be need for accessible and reliable data on predictive biomarkers that may be helpful for the selection of the right cancer immunotherapies for the right cancer patients.
The use of AI and machine learning can be adopted to decide the optimal dose and treatment modalities of combinatorial therapies involving various cancer immunotherapies.
For personalized medicines, concerted and comprehensive efforts are needed at the level of microbiome and outcome of cancer immunotherapies. Also, the expanded domain of cancer immunotherapies may be converged for rare cancers and, the influence of circadian rhythm on the success and failure of cancer patients.
In the future, oncolytic viruses, vaccines based on neoantigens, live biotherapeutics, immune engineering methodologies, and combinatorial avenues with engineered microbiomes can be explored as new forms of cancer therapeutics. Achieving this requires AI-driven predictive tools to optimize the type, dose, and timing of immunotherapies, alone or in combination.
An approach incorporating predictive biomarkers, AI-driven treatment personalization, and microbiome-based strategies will be instrumental in refining cancer immunotherapies. Biomarkers will help identify patients who will benefit the most, AI will optimize prognosis treatment selection, dosage, and response prediction, while microbiome-based interventions could enhance immunotherapy efficacy. Cancer immunotherapy is not just a promising frontier—it is a transformative force reshaping oncology. With strategic integration of advanced technologies, it holds the potential to shift cancer from a life-threatening disease to a manageable condition. To improve cancer treatment, CAR-T cells, mRNA, and DNA vaccines should be prioritized for greater efficacy, lower toxicity, personalized care, better accessibility, and long-term monitoring of side effects.
Author contributions
PS: Conceptualization, Writing – original draft, Writing – review & editing. AS: Writing – original draft, Writing – review & editing. SM: Writing – original draft, Writing – review & editing. NS: Conceptualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
Authors AS and SM were employed by the company Glenmark Pharmaceuticals Limited.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript. For the references and citations. Formatting and style and better clarity of language.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
CAR, chimeric antigen receptor; engineered receptors on T cells; TILs, tumor-infiltrating lymphocytes; immune cells that penetrate tumors; ICIs, immune checkpoint inhibitors; drugs that release immune system brakes; FMT, fecal microbiota transplantation; transfer of gut bacteria; irAEs, immune-related adverse events; treatment side effects; SDDSs, smart drug delivery systems; targeted delivery technologies; GM, gut microbiome; microbial community in intestines; CTLA-4 and PD-1/PD-L1; immune checkpoint proteins; MHC, major histocompatibility complex; proteins for antigen presentation; TCR, T-cell receptor; antigen recognition complex; Tregs, regulatory T cells; immunosuppressive T cells; MDSCs, myeloid-derived suppressor cells; TAMs, tumor-associated macrophages; immunosuppressive myeloid cells; TME, tumor microenvironment; cellular surroundings of tumor; TEM, effector memory T cells; antigen-experienced T cells; ADC, antibody-drug conjugate; targeted drug delivery; mAbs, monoclonal antibodies; engineered antibodies; FDA, Food and Drug Administration; US regulatory agency; MSI, Microsatellite Instability; genomic instability marker; PARP, poly ADP-ribose polymerase; DNA repair enzyme; NKT, natural killer T cells; specialized immune cells; GBM, glioblastoma multiforme; aggressive brain cancer; TNBC, triple-negative breast cancer; aggressive breast cancer type; pCR, pathological complete response; absence of cancer after treatment; DCs, dendritic cells; antigen-presenting cells.
References
1. Esfahani K, Roudaia L, Buhlaiga N, Del Rincon SV, Papneja N, Miller WH. A review of cancer immunotherapy: from the past, to the present, to the future. Curr Oncol (Toronto Ont). (2020) 27:S87–97. doi: 10.3747/co.27.5223
2. Szeto GL, Finley SD. Integrative approaches to cancer immunotherapy. Trends Cancer. (2019) 5:400–10. doi: 10.1016/j.trecan.2019.05.010
3. Arina A, Gutiontov SI, Weichselbaum RR. Radiotherapy and immunotherapy for cancer: From “systemic” to “multisite”. Clin Cancer Res. (2020) 26:2777–82. doi: 10.1158/1078-0432.CCR-19-2034
4. Igarashi Y, Sasada T. Cancer vaccines: Building a bridge over troubled waters. J Immunol Res. (2020) 2020:5825401. doi: 10.1155/2020/5825401
5. Chauvin JM, Zarour HM. TIGIT in cancer immunotherapy. J Immunother Cancer. (2020) 8:e000957. doi: 10.1136/jitc-2020-000957
6. Sharma NK, Sarode SC, Sarode GS, Patil S. CD8+ T cell dysfunction by TOX intoxication: a protumorigenic event in the tumor microenvironment. Future Oncol. (2021) 17:5129–34. doi: 10.2217/fon-2020-0532
7. Karagiannis P, Kim SI. iPSC-derived natural killer cells for cancer immunotherapy. Mol Cells. (2021) 44:541–8. doi: 10.14348/molcells.2021.0078
8. Yi W, Yan D, Wang D, Li Y. Smart drug delivery systems to overcome drug resistance in cancer immunotherapy. Cancer Biol Med. (2023) 20:248–67. doi: 10.20892/j.issn.2095-3941.2023.0009
9. Riley RS, June CH, Langer R, Mitchell MJ. Delivery technologies for cancer immunotherapy. Nat Rev Drug Discovery. (2019) 18:175–96. doi: 10.1038/s41573-018-0006-z
10. Chen C, Liu X, Chang CY, Wang HY, Wang RF. The interplay between T cells and cancer: The basis of immunotherapy. Genes. (2023) 14:1008. doi: 10.3390/genes14051008
11. Kumar AR, Devan AR, Nair B, Vinod BS, Nath LR. Harnessing the immune system against cancer: current immunotherapy approaches and therapeutic targets. Mol Biol Rep. (2021) 48:8075–95. doi: 10.1007/s11033-021-06752-9
12. Brahmer JR, Abu-Sbeih H, Ascierto PA, Brufsky J, Cappelli LC, Cortazar FB, et al. Society for Immunotherapy of Cancer (SITC) clinical practice guideline on immune checkpoint inhibitor-related adverse events. J Immunother Cancer. (2021) 9:e002435. doi: 10.1136/jitc-2021-002435
13. Tian Y, Xie D, Yang L. Engineering strategies to enhance oncolytic viruses in cancer immunotherapy. Signal Transduction Targeted Ther. (2022) 7:117. doi: 10.1038/s41392-022-00951-x
14. Roy D, Gilmour C, Patnaik S, Wang LL. Combinatorial blockade for cancer immunotherapy: targeting emerging immune checkpoint receptors. Front Immunol. (2023) 14:1264327. doi: 10.3389/fimmu.2023.1264327
15. Gutschner T, Diederichs S. The hallmarks of cancer: a long non-coding RNA point of view. RNA Biol. (2012) 9:703–19. doi: 10.4161/rna.20481
16. Fares J, Fares MY, Khachfe HH, Salhab HA, Fares Y. Molecular principles of metastasis: a hallmark of cancer revisited. Signal Transduction Targeted Ther. (2020) 5:28. doi: 10.1038/s41392-020-0134-x
17. Morad G, Helmink BA, Sharma P, Wargo JA. Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell. (2021) 184:5309–37. doi: 10.1016/j.cell.2021.09.020
18. Senga SS, Grose RP. Hallmarks of cancer-the new testament. Open Biol. (2021) 11:200358. doi: 10.1098/rsob.200358
19. Sun L, Zhang H, Gao P. Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell. (2022) 13:877–919. doi: 10.1007/s13238-021-00846-7
20. Lian X, Yang K, Li R, Li M, Zuo J, Zheng B, et al. Immunometabolic rewiring in tumorigenesis and anti-tumor immunotherapy. Mol Cancer. (2022) 21:27. doi: 10.1186/s12943-021-01486-5
21. Li J, Ma X, Chakravarti D, Shalapour S, DePinho RA. Genetic and biological hallmarks of colorectal cancer. Genes Dev. (2021) 35:787–820. doi: 10.1101/gad.348226.120
22. Zhu S, Zhang T, Zheng L, Liu H, Song W, Liu D, et al. Combination strategies to maximize the benefits of cancer immunotherapy. J Hematol Oncol. (2021) 14:156. doi: 10.1186/s13045-021-01164-5
23. Bockamp E, Rosigkeit S, Siegl D, Schuppan D. Nano-enhanced cancer immunotherapy: Immunology encounters nanotechnology. Cells. (2020) 9:2102. doi: 10.3390/cells9092102
24. Tandon I, Sharma NK. Macrophage flipping from foe to friend: A matter of interest in breast carcinoma heterogeneity driving drug resistance. Curr Cancer Drug Targets. (2019) 19:189–98. doi: 10.2174/1568009618666180628102247
25. Blanco B, Domínguez-Alonso C, Alvarez-Vallina L. Bispecific immunomodulatory antibodies for cancer immunotherapy. Clin Cancer Res. (2021) 27:5457–64. doi: 10.1158/1078-0432.CCR-20-3770
26. Ahmed A, Tait SWG. Targeting immunogenic cell death in cancer. Mol Oncol. (2020) 14:2994–3006. doi: 10.1002/1878-0261.12851
27. Kaufmann SHE. Immunology’s coming of age. Front Immunol. (2019) 10:684. doi: 10.3389/fimmu.2019.00684
28. Nandangiri R, T NS, Raj AK, Lokhande KB, Khunteta K, Hebale A, et al. Secretion of sphinganine by drug-induced cancer cells and modified mimetic sphinganine (MMS) as c-Src kinase inhibitor. Asian Pacific J Cancer Prev. (2024) 25:433–46. doi: 10.31557/APJCP.2024.25.2.433
29. Gálvez NMS, Bohmwald K, Pacheco GA, Andrade CA, Carreño LJ, Kalergis AM. Type I natural killer T cells as key regulators of the immune response to infectious diseases. Clin Microbiol Rev. (2020) 34:e00232–20. doi: 10.1128/cmr.00232-20
30. Mitra S, Tomar PC. Hybridoma technology; advancements, clinical significance, and future aspects. J Genet Eng Biotechnol. (2021) 19:159. doi: 10.1186/s43141-021-00264-6
31. Finck AV, Blanchard T, Roselle CP, Golinelli G, June CH. Engineered cellular immunotherapies in cancer and beyond. Nat Med. (2022) 28:678–89. doi: 10.1038/s41591-022-01765-8
32. He X, Xu C. Immune checkpoint signaling and cancer immunotherapy. Cell Res. (2020) 30:660–9. doi: 10.1038/s41422-020-0343-4
33. Stirling ER, Bronson SM, Mackert JD, Cook KL, Triozzi PL, Soto-Pantoja DR. Metabolic implications of immune checkpoint proteins in cancer. Cells. (2022) 11:179. doi: 10.3390/cells11010179
34. Tang Q, Chen Y, Li X, Long S, Shi Y, Yu Y, et al. The role of PD-1/PD-L1 and application of immune-checkpoint inhibitors in human cancers. Front Immunol. (2022) 13:964442. doi: 10.3389/fimmu.2022.964442
35. Rotte A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J Exp Clin Cancer Res. (2019) 38:255. doi: 10.1186/s13046-019-1259-z
36. Willsmore ZN, Coumbe BGT, Crescioli S, Reci S, Gupta A, Harris RJ, et al. Combined anti-PD-1 and anti-CTLA-4 checkpoint blockade: Treatment of melanoma and immune mechanisms of action. Eur J Immunol. (2021) 51:544–56. doi: 10.1002/eji.202048747
37. Zhang H, Dai Z, Wu W, Wang Z, Zhang N, Zhang L, et al. Regulatory mechanisms of immune checkpoints PD-L1 and CTLA-4 in cancer. J Exp Clin Cancer Res. (2021) 40:184. doi: 10.1186/s13046-021-01987-7
38. Sharma NK, Sarode SC, Bahot A, Sekar G. Secretion of acetylated amino acids by drug-induced cancer cells: perspectives on metabolic-epigenetic alterations. Epigenomics. (2023) 15:983–90. doi: 10.2217/epi-2023-0251
39. Hu JR, Florido R, Lipson EJ, Naidoo J, Ardehali R, Tocchetti CG, et al. Cardiovascular toxicities associated with immune checkpoint inhibitors. Cardiovasc Res. (2019) 115:854–68. doi: 10.1093/cvr/cvz026
40. Moslehi J, Lichtman AH, Sharpe AH, Galluzzi L, Kitsis RN. Immune checkpoint inhibitor-associated myocarditis: manifestations and mechanisms. J Clin Invest. (2021) 131:e145186. doi: 10.1172/JCI145186
41. Patel RP, Parikh R, Gunturu KS, Tariq RZ, Dani SS, Ganatra S, et al. Cardiotoxicity of immune checkpoint inhibitors. Curr Oncol Rep. (2021) 23:79. doi: 10.1007/s11912-021-01070-6
42. Tsimberidou AM, Baysal MA, Chakraborty A, Andersson BS. Autologous engineered T cell receptor therapy in advanced cancer. Hum Vaccines Immunotherapeut. (2023) 19:2290356. doi: 10.1080/21645515.2023.2290356
43. Aparicio C, Acebal C, González-Vallinas M. Current approaches to develop “off-the-shelf” chimeric antigen receptor (CAR)-T cells for cancer treatment: a systematic review. Exp Hematol Oncol. (2023) 12:73. doi: 10.3389/fonc.2024.1433432
44. Magnani CF, Gaipa G, Lussana F, Belotti D, Gritti G, Napolitano S, et al. Sleeping beauty-engineered CAR T cells achieve antileukemic activity without severe toxicities. J Clin Invest. (2020) 130:6021–33. doi: 10.1172/JCI138473
45. Doglio M, Alexander T, Del Papa N, Snowden JA, Greco R. New insights in systemic lupus erythematosus: From regulatory T cells to CAR-T-cell strategies. J Allergy Clin Immunol. (2022) 150:1289–301. doi: 10.1016/j.jaci.2022.08.003
46. Sivori S, Pende D, Quatrini L, Pietra G, Della Chiesa M, Vacca P, et al. NK cells and ILCs in tumor immunotherapy. Mol Aspects Med. (2021) 80:100870. doi: 10.1016/j.mam.2020.100870
47. Morgan MA, Büning H, Sauer M, Schambach A. Use of cell and genome modification technologies to generate improved “off-the-shelf” CAR T and CAR NK cells. Front Immunol. (2020) 11:1965. doi: 10.3389/fimmu.2020.01965
48. Razeghian E, Nasution MKM, Rahman HS, Gardanova ZR, Abdelbasset WK, Aravindhan S, et al. A deep insight into CRISPR/Cas9 application in CAR-T cell-based tumor immunotherapies. Stem Cell Res Ther. (2021) 12:428. doi: 10.1186/s13287-021-02510-7
49. Bulliard Y, Andersson BS, Baysal MA, Damiano J, Tsimberidou AM. Reprogramming T cell differentiation and exhaustion in CAR-T cell therapy. J Hematol Oncol. (2023) 16:108. doi: 10.1186/s13045-023-01504-7
50. Echeverry G, Fischer GW, Mead E. Next generation of cancer treatments: Chimeric antigen receptor T-cell therapy and its related toxicities: A review for perioperative physicians. Anesth Analgesia. (2019) 129:434–41. doi: 10.1213/ANE.0000000000004201
51. Etxeberria I, Glez-Vaz J, Teijeira Á, Melero I. New emerging targets in cancer immunotherapy: CD137/4-1BB costimulatory axis. ESMO Open. (2020) 4:e000733. doi: 10.1136/esmoopen-2020-000733
52. Meng X, Jing R, Qian L, Zhou C, Sun J. Engineering cytoplasmic signaling of CD28ζ CARs for improved therapeutic functions. Front Immunol. (2020) 11:1046. doi: 10.3389/fimmu.2020.01046
53. Lin B, Du L, Li H, Zhu X, Cui L, Li X. Tumor-infiltrating lymphocytes: Warriors fight against tumors powerfully. Biomed Pharmacother. (2020) 132:110873. doi: 10.1016/j.biopha.2020.110873
54. Kazemi MH, Sadri M, Najafi A, Rahimi A, Baghernejadan Z, Khorramdelazad H, et al. Tumor-infiltrating lymphocytes for treatment of solid tumors: It takes two to tango? Front Immunol. (2022) 13:1018962. doi: 10.3389/fimmu.2022.1018962
55. Cappell KM, Sherry RM, Yang JC, Goff SL, Vanasse DA, McIntyre L, et al. Long-term follow-up of anti-CD19 chimeric antigen receptor T-cell therapy. J Clin Oncol. (2020) 38:3805–15. doi: 10.1200/JCO.20.01467
56. Goff SL, Smith FO, Klapper JA, Sherry R, Wunderlich JR, Steinberg SM, et al. Tumor-infiltrating lymphocyte therapy for metastatic melanoma: Analysis of tumors resected for TIL. J Immunother. (2010) 33:840–7. doi: 10.1097/CJI.0b013e3181f05b91
57. Petitprez F, Meylan M, de Reyniès A, Sautès-Fridman C, Fridman WH. The tumor microenvironment in the response to immune checkpoint blockade therapies. Front Immunol. (2020) 11:784. doi: 10.3389/fimmu.2020.00784
58. Raskov H, Orhan A, Christensen JP, Gögenur I. Cytotoxic CD8+ T cells in cancer and cancer immunotherapy. Br J Cancer. (2021) 124:359–67. doi: 10.1038/s41416-020-01048-4
59. Mao X, Xu J, Wang W, Liang C, Hua J, Liu J, et al. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: New findings and future perspectives. Mol Cancer. (2021) 20:131. doi: 10.1186/s12943-021-01428-1
60. Christofides A, Strauss L, Yeo A, Cao C, Charest A, Boussiotis VA. The complex role of tumor-infiltrating macrophages. Nat Immunol. (2022) 23:1148–56. doi: 10.1038/s41590-022-01267-2
61. Chu YT, Liao MT, Tsai KW, Lu KC, Hu WC. Interplay of chemokine receptors, toll-like receptors, and host immunological pathways. Biomedicines. (2023) 11:2384. doi: 10.3390/biomedicines11092384
62. Leko V, Rosenberg SA. Identifying and targeting human tumor antigens for T cell-based immunotherapy of solid tumors. Cancer Cell. (2020) 38:454–72. doi: 10.1016/j.ccell.2020.07.013
63. Montisci A, Vietri MT, Palmieri V, Sala S, Donatelli F, Napoli C. Cardiac toxicity associated with cancer immunotherapy and biological drugs. Cancers. (2021) 13:4797. doi: 10.3390/cancers13194797
64. Fu Z, Li S, Han S, Shi C, Zhang Y. Antibody-drug conjugate: The “biological missile” for targeted cancer therapy. Signal Transduction Targeted Ther. (2022) 7:93. doi: 10.1038/s41392-022-00947-7
65. Joubert N, Beck A, Dumontet C, Denevault-Sabourin C. Antibody-drug conjugates: The last decade. Pharmaceuticals. (2020) 13:245. doi: 10.3390/ph13090245
66. Tong JTW, Harris PWR, Brimble MA, Kavianinia I. An insight into FDA-approved antibody-drug conjugates for cancer therapy. Molecules. (2021) 26:5847. doi: 10.3390/molecules26195847
67. Maecker H, Jonnalagadda V, Bhakta S, Jammalamadaka V, Junutula JR. Exploration of the antibody-drug conjugate clinical landscape. mAbs. (2023) 15:2229101. doi: 10.1080/19420862.2023.2229101
68. Hafeez U, Parakh S, Gan HK, Scott AM. Antibody-drug conjugates for cancer therapy. Molecules. (2020) 25:4764. doi: 10.3390/molecules25204764
69. Conilh L, Sadilkova L, Viricel W, Dumontet C. Payload diversification: A key step in the development of antibody-drug conjugates. J Hematol Oncol. (2023) 16:3. doi: 10.1186/s13045-022-01397-y
70. Nguyen TD, Bordeau BM, Balthasar JP. Mechanisms of ADC toxicity and strategies to increase ADC tolerability. Cancers. (2023) 15:713. doi: 10.3390/cancers15030713
71. Horwitz S, O’Connor OA, Pro B, Trümper L, Iyer S, Advani R. The ECHELON-2 trial: 5-year results of a randomized, phase III study of brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma. Ann Oncol. (2022) 33:288–98. doi: 10.1016/j.annonc.2021.12.002
72. Sehn LH, Herrera AF, Flowers CR, Kamdar MK, McMillan A. Polatuzumab vedotin in relapsed or refractory diffuse large B-cell lymphoma. J Clin Oncol. (2020) 38:155–65. doi: 10.1200/JCO.19.00172
73. Rosenberg JE, O’Donnell PH, Balar AV, McGregor BA, Heath EI. Pivotal trial of enfortumab vedotin in urothelial carcinoma after platinum and anti-PD-1/PD-L1 therapy. J Clin Oncol. (2019) 37:2592–600. doi: 10.1200/JCO.19.01140
74. Modi S, Jacot W, Yamashita T, Sohn J, Vidal M, Tokunaga E. Trastuzumab deruxtecan in previously treated HER2-low advanced breast cancer. New Engl J Med. (2022) 387:9–20. doi: 10.1056/NEJMoa2203690
75. Sobhani N, Scaggiante B, Morris R, Chai D, Catalano M. Therapeutic cancer vaccines: From biological mechanisms and engineering to ongoing clinical trials. Cancer Treat Rev. (2022) 109:102429. doi: 10.1016/j.ctrv.2022.102429
76. Liu J, Fu M, Wang M, Wan D, Wei Y. Cancer vaccines as promising immunotherapeutics: Platforms and current progress. J Hematol Oncol. (2022) 15:28. doi: 10.1186/s13045-022-01247-x
77. Paston SJ, Brentville VA, Symonds P, Durrant LG. Cancer vaccines, adjuvants, and delivery systems. Front Immunol. (2021) 12:627932. doi: 10.3389/fimmu.2021.627932
78. Gogia P, Ashraf H, Bhasin S, Xu Y. Antibody-drug conjugates: A review of approved drugs and their clinical level of evidence. Cancers. (2023) 15:3886. doi: 10.3390/cancers15153886
79. Riccardi F, Dal Bo M, Macor P, Toffoli G. A comprehensive overview on antibody-drug conjugates: From conceptualization to cancer therapy. Front Pharmacol. (2023) 14:1274088. doi: 10.3389/fphar.2023.1274088
80. Li BT, Smit EF, Goto Y, Nakagawa K, Udagawa H, Mazières J. Trastuzumab deruxtecan in HER2-mutant non-small-cell lung cancer. New Engl J Med. (2022) 386:241–51. doi: 10.1056/NEJMoa2112431
81. Sellars MC, Wu CJ, Fritsch EF. Cancer vaccines: Building a bridge over troubled waters. Cell. (2022) 185:2770–88. doi: 10.1016/j.cell.2022.06.035
82. Sinha M, Zhang L, Subudhi S, Chen B, Marquez J. Pre-existing immune status associated with response to combination of sipuleucel-T and ipilimumab in patients with metastatic castration-resistant prostate cancer. J ImmunoTher Cancer. (2021) 9:e002254. doi: 10.1136/jitc-2020-002254
83. Livingston PO. Construction of cancer vaccines with carbohydrate and protein (peptide) tumor antigens. Curr Opin Immunol. (1992) 4:624–9. doi: 10.1016/0952-7915(92)90038-G
84. Abbasi S, Uchida S. Multifunctional immunoadjuvants for use in minimalist nucleic acid vaccines. Pharmaceutics. (2021) 13:644. doi: 10.3390/pharmaceutics13050644
85. Lorentzen CL, Haanen JB, Met Ö, Svane IM. Clinical advances and ongoing trials on mRNA vaccines for cancer treatment. Lancet Oncol. (2022) 23:e450–8. doi: 10.1016/S1470-2045(22)00372-2
86. Gulley JL, Borre M, Vogelzang NJ, Ng S, Agarwal N. Phase III trial of PROSTVAC in asymptomatic or minimally symptomatic metastatic castration-resistant prostate cancer. J Clin Oncol. (2019) 37:1051–61. doi: 10.1200/JCO.18.02031
87. Liu X, Hogg GD, DeNardo DG. Rethinking immune checkpoint blockade: “Beyond the T cell. J ImmunoTher Cancer. (2021) 9:e001460. doi: 10.1136/jitc-2020-001460
88. O’Malley DM, Bariani GM, Cassier PA, Marabelle A, Hansen AR, De Jesus Acosta A, et al. Pembrolizumab in patients with microsatellite instability-high advanced endometrial cancer: Results from the KEYNOTE-158 study. J Clin Oncol. (2022) 40:752–61. doi: 10.1200/JCO.21.01874
89. Riedinger CJ, Esnakula A, Haight PJ, Suarez AA, Chen W, Gillespie J, et al. Characterization of mismatch-repair (MMR)/microsatellite instability (MSI)-discordant endometrial cancers. Cancer. (2023) 129:1789–99. doi: 10.1002/cncr.35030
90. Han S, Chok AY, Peh DYY, Ho JZ, Tan EKW, Koo SL, et al. The distinct clinical trajectory, metastatic sites, and immunobiology of microsatellite-instability-high cancers. Front Genet. (2022) 13:933475. doi: 10.3389/fgene.2022.933475
91. Oaknin A, Gilbert L, Tinker AV, Brown J, Mathews C, Press J, et al. Safety and antitumor activity of dostarlimab in patients with advanced or recurrent DNA mismatch repair deficient/microsatellite instability-high (dMMR/MSI-H) or proficient/stable (MMRp/MSS) endometrial cancer: Interim results from GARNET-a phase I, single-arm study. J ImmunoTher Cancer. (2022) 10:e003777. doi: 10.1136/jitc-2021-003777
92. Reddy AV, Sehgal S, Hill CS, Zheng L, He J. Upfront chemotherapy followed by stereotactic body radiation therapy with or without surgery in older patients with localized pancreatic cancer: A single institution experience and review of the literature. Curr Oncol. (2022) 29:308–20. doi: 10.3390/curroncol29010028
93. Hurvitz SA, Hegg R, Chung WP, Im SA, Jacot W, Ganju V, et al. Trastuzumab deruxtecan versus trastuzumab emtansine in patients with HER2-positive metastatic breast cancer: Updated results from DESTINY-Breast03, a randomized, open-label, phase 3 trial. Lancet. (2023) 401:105–17. doi: 10.1016/S0140-6736(22)02420-5
94. Chehelgerdi M, Chehelgerdi M. The use of RNA-based treatments in the field of cancer immunotherapy. Mol Cancer. (2023) 22:106. doi: 10.1186/s12943-023-01807-w
95. Pan S, Fan R, Han B, Tong A, Guo G. The potential of mRNA vaccines in cancer nanomedicine and immunotherapy. Trends Immunol. (2024) 45:20–31. doi: 10.1016/j.it.2023.11.003
96. Mendez-Gomez HR, DeVries A, Castillo P, von Roemeling C, Qdaisat S, Fernandez Zapico ME, et al. RNA aggregates harness the danger response for potent cancer immunotherapy. Cell. (2024) 187(10):2521–35.e21. doi: 10.1016/j.cell.2024.04.003
97. Medina-Echeverz J, Fioravanti J, Zabala M, Ardaiz N, Prieto J, Berraondo P. Phase I trial of Cavatak (CF33) in patients with advanced cancer. J Clin Oncol. (2019) 37:1926–36.
98. Zhao K, Chen Y, Jiang Y, Zhao L, Zheng X, Zhu J, et al. mRNA vaccine targeting PD-L1 enhances antitumor immunity. Nat Commun. (2020) 11:1–12.
99. Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, Reuter KC, et al. BNT111, a mRNA vaccine targeting MUC1 and CEA. Cancer Res. (2020) 80:2685–95.
100. Kim YC, Choi JH, Yoo DH, Park JH, Kim DH, Chung MJ, et al. Electroporated DNA vaccine targeting hTERT induces immune responses. J Immunother. (2019) 42:161–70.
101. Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Löwer M, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity. Nature. (2017) 547:222–6. doi: 10.1038/nature23003
102. Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre ML, et al. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science. (2018) 359:104–8. doi: 10.1126/science.aao3290
103. Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science. (2018) 359:97–103. doi: 10.1126/science.aan4236
104. Jin Y, Dong H, Xia L, Yang Y, Zhu Y, Shen Y, et al. The diversity of gut microbiome is associated with favorable responses to anti-programmed death 1 immunotherapy in Chinese patients with NSCLC. J Thorac Oncol. (2019) 14:1378–89. doi: 10.1016/j.jtho.2019.04.007
105. Zheng Y, Wang T, Tu X, Huang Y, Zhang H, Tan D, et al. Gut microbiome affects the response to anti-PD-1 immunotherapy in patients with hepatocellular carcinoma. J Immunother Cancer. (2019) 7:193. doi: 10.1186/s40425-019-0650-9
106. Li Y, Su Z, Zhao W, Zhang X, Momin N, Zhang C, et al. CV8102, an mRNA vaccine targeting VEGFR2. J Clin Oncol. (2020) 38:1558–66.
107. Wang G, Wang W. Advanced cell therapies for glioblastoma. Front Immunol. (2022) 13:904133. doi: 10.3389/fimmu.2022.904133
108. Sullivan BA, Kronenberg M. Activation or anergy: NKT cells are stunned by alpha-galactosylceramide. J Clin Invest. (2005) 115:2328–9. doi: 10.1172/JCI26297
109. Morimoto T, Nakazawa T, Maeoka R, Nakagawa I, Tsujimura T, Matsuda R. Natural killer cell-based immunotherapy against glioblastoma. Int J Mol Sci. (2023) 24:2111. doi: 10.3390/ijms24032111
110. Schmid P, Salgado R, Park YH, Muñoz-Couselo E, Kim SB. Pembrolizumab plus chemotherapy as neoadjuvant treatment of high-risk, early-stage triple-negative breast cancer: Results from the phase 1b open-label, multicohort KEYNOTE-173 study. Ann Oncol. (2020) 31:569–81. doi: 10.1016/j.annonc.2020.01.072
111. Miles D, Gligorov J, André F, Cameron D, Schneeweiss A. Primary results from IMpassion131, a double-blind, placebo-controlled, randomized phase III trial of first-line paclitaxel with or without atezolizumab for unresectable locally advanced/metastatic triple-negative breast cancer. Ann Oncol. (2021) 32:994–1004. doi: 10.1016/j.annonc.2021.05.801
112. Ademuyiwa FO, Gao F, Street CR, Chen I, Northfelt DW, Wesolowski R, et al. A randomized phase 2 study of neoadjuvant carboplatin and paclitaxel with or without atezolizumab in triple-negative breast cancer (TNBC) – NCI 10013. NPJ Breast Cancer. (2022) 8:134. doi: 10.1038/s41523-022-00500-3
113. Rizzo A, Cusmai A, Massafra R, Bove S, Comes MC. Pathological complete response to neoadjuvant chemoimmunotherapy for early triple-negative breast cancer: An updated meta-analysis. Cells. (2022) 11:1857. doi: 10.3390/cells11121857
114. Takahashi M, Cortés J, Dent R, Pusztai L. Pembrolizumab plus chemotherapy followed by pembrolizumab in patients with early triple-negative breast cancer: A secondary analysis of a randomized clinical trial. JAMA Network Open. (2023) 6:e2342107. doi: 10.1001/jamanetworkopen.2023.42107
115. McArthur HL, Ignatiadis M, Guillaume S, Bailey A, Martinez JL, Brandao M, et al. ALEXANDRA/IMpassion030: A phase III study of standard adjuvant chemotherapy with or without atezolizumab in early-stage triple-negative breast cancer. J Clin Oncol. (2019) 37:TPS598. doi: 10.1200/JCO.2019.37.15_suppl.TPS598
116. MacDonald I, Nixon NA, Khan OF. Triple-negative breast cancer: A review of current curative intent therapies. Curr Oncol. (2022) 29:4768–78. doi: 10.3390/curroncol29070378
117. Sharma NK, Sarode SC. Artificial intelligence vs. evolving super-complex tumor intelligence: Critical viewpoints. Front Artif Intell. (2023) 6:1220744. doi: 10.3389/frai.2023.1220744
118. Vu SH, Vetrivel P, Kim J, Lee MS. Cancer resistance to immunotherapy: Molecular mechanisms and tackling strategies. Int J Mol Sci. (2022) 23:10906. doi: 10.3390/ijms231810906
119. Piro G, Carbone C, Carbognin L, Pilotto S, Ciccarese C. Revising PTEN in the era of immunotherapy: New perspectives for an old story. Cancers. (2019) 11:1525. doi: 10.3390/cancers11101525
120. Schirrmacher V, van Gool S, Stuecker W. Counteracting immunosuppression in the tumor microenvironment by oncolytic Newcastle disease virus and cellular immunotherapy. Int J Mol Sci. (2022) 23:13050. doi: 10.3390/ijms232113050
121. He S, Huang Q, Cheng J. The unfolding story of dying tumor cells during cancer treatment. Front Immunol. (2023) 14:1073561. doi: 10.3389/fimmu.2023.1073561
122. Colombo N, Barretina-Ginesta MP, Beale PJ, Harano K, Hudson E, Marmé F, et al. ENGOT-en7: A multicenter phase III double-blind randomized controlled trial of atezolizumab in combination with paclitaxel and carboplatin in women with advanced/recurrent endometrial cancer. J Clin Oncol. (2019) 37:TPS5608. doi: 10.1200/JCO.2019.37.15_suppl.TPS5608
123. Brettschneider EES, Terabe M. The role of NKT cells in glioblastoma. Cells. (2021) 10:1641. doi: 10.3390/cells10071641
124. Li Y, Sharma A, Maciaczyk J, Schmidt-Wolf IGH. Recent developments in NKT-based immunotherapy of glioblastoma: From bench to bedside. Int J Mol Sci. (2022) 23:1311. doi: 10.3390/ijms23031311
125. Schmid P, Cortes J, Pusztai L, McArthur H, Kümmel S, Bergh J, et al. Pembrolizumab for early Triple-Negative breast cancer. New Engl J Med. (2020) 382:810–21. doi: 10.1056/NEJMoa1910549
126. Tan S, Li D, Zhu X. Cancer immunotherapy: Pros, cons and beyond. Biomed Pharmacother. (2020) 124:109821. doi: 10.1016/j.biopha.2020.109821
127. Frega G, Cossio FP, Banales JM, Cardinale V, Macias RIR, Braconi C, et al. Lacking immunotherapy biomarkers for biliary tract cancer: A comprehensive systematic literature review and meta-analysis. Cells. (2023) 12:2098. doi: 10.3390/cells12162098
128. Liu YH, Zang XY, Wang JC, Huang SS, Xu J, Zhang P. Diagnosis and management of immune related adverse events (irAEs) in cancer immunotherapy. Biomed Pharmacother. (2019) 120:109437. doi: 10.1016/j.biopha.2019.109437
129. Robles-Alonso V, Martínez-Valle F, Borruel N. Co treatment with biologic agents and immunotherapy in the setting of irAEs of difficult management. Front Med. (2022) 9:906098. doi: 10.3389/fmed.2022.906098
130. Chen Y, Chen Z, Chen R, Fang C, Zhang C, Ji M, et al. Immunotherapy-based combination strategies for treatment of EGFR-TKI-resistant non-small-cell lung cancer. Future Oncol. (2022) 18:1757–75. doi: 10.2217/fon-2021-0862
131. Venkatachalam S, McFarland TR, Agarwal N, Swami U. Immune checkpoint inhibitors in prostate cancer. Cancers. (2021) 13:2187. doi: 10.3390/cancers13092187
132. Ferro M, Lucarelli G, Crocetto F, Dolce P, Verde A, La Civita E, et al. First line systemic therapy for metastatic castration sensitive prostate cancer: An updated systematic review with novel findings. Crit Rev Oncol/Hematol. (2021) 157:103198. doi: 10.1016/j.critrevonc.2020.103198
133. De Castro V, Galaine J, Loyon R, Godet Y. CRISPR-Cas gene knockouts to optimize engineered T cells for cancer immunotherapy. Cancer Gene Ther. (2024) 31:1124–34. doi: 10.1038/s41417-024-00771-x
134. Lim SH, Beers SA, Al-Shamkhani A, Cragg MS. Agonist antibodies for cancer immunotherapy: History, hopes, and challenges. Clin Cancer Res. (2024) 30:1712–23. doi: 10.1158/1078-0432.CCR-23-1014
135. Craig AW, Frieboes HB, Videira PA. Advancing cancer immunotherapy: From innovative preclinical models to clinical insights. Sci Rep. (2024) 14:1205. doi: 10.1038/s41598-024-51704-5
136. Cao LL, Kagan JC. Targeting innate immune pathways for cancer immunotherapy. Immunity. (2023) 56:2206–17. doi: 10.1016/j.immuni.2023.07.018
137. Sun Z, Zhao H, Ma L, Shi Y, Ji M, Sun X, et al. The quest for nanoparticle-powered vaccines in cancer immunotherapy. J Nanobiotechnol. (2024) 22:61. doi: 10.1186/s12951-024-02311-z
138. Rui R, Zhou L, He S. Cancer immunotherapies: Advances and bottlenecks. Front Immunol. (2023) 14:1212476. doi: 10.3389/fimmu.2023.1212476
139. Pérez-Herrero E, Lanier OL, Krishnan N, D’Andrea A, Peppas NA. Drug delivery methods for cancer immunotherapy. Drug Deliv Trans Res. (2024) 14:30–61. doi: 10.1007/s13346-023-01405-9
140. Al-Taie A, Özcan Bülbül E. A paradigm use of monoclonal antibodies-conjugated nanoparticles in breast cancer treatment: Current status and potential approaches. J Drug Targeting. (2024) 32:45–56. doi: 10.1080/1061186X.2023.2295803
141. Zhang J, Huang Y, Han Y, Dong D, Cao Y, Chen X, et al. Immune microenvironment heterogeneity of concurrent adenocarcinoma and squamous cell carcinoma in multiple primary lung cancers. NPJ Precis Oncol. (2024) 8:55. doi: 10.1038/s41698-024-00548-3
142. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. New Engl J Med. (2015) 373:23–34. doi: 10.1056/NEJMoa1504030
143. Gandhi L, Rodríguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, et al. Pembrolizumab plus chemotherapy for squamous non–small-cell lung cancer. New Engl J Med. (2018) 378:2078–92. doi: 10.1056/NEJMoa1801005
144. Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak Ö, et al. Tisagenlecleucel in adult relapsed/refractory diffuse large B-cell lymphoma. New Engl J Med. (2017) 377:2578–88. doi: 10.1056/NEJMoa1708566
145. Hellmann MD, Paz-Ares L, Bernabe Caro R, Zurawski B, Kim SW, Carcereny Costa E, et al. Nivolumab plus ipilimumab in lung cancer. New Engl J Med. (2019) 381:825–35. doi: 10.1056/NEJMoa1910231
146. Socinski MA, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, Nogami N, et al. Atezolizumab plus carboplatin and paclitaxel in non–small-cell lung cancer. New Engl J Med. (2019) 380:2004–15. doi: 10.1056/NEJMoa1716948
147. Patel JD, Socinski MA, Garon EB, Reynolds CH, Spigel DR, Olsen MR, et al. Pembrolizumab plus pemetrexed and platinum as first-line therapy for advanced non–small-cell lung cancer. J Clin Oncol. (2018) 36:1935–43.
148. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel (CAR-T therapy) for refractory B-cell lymphoma. New Engl J Med. (2017) 377:2629–37. doi: 10.1056/NEJMoa1707447
149. Wong JKM, Dolcetti R, Rhee H, Simpson F, Souza-Fonseca-Guimaraes F. Weaponizing natural killer cells for solid cancer immunotherapy. Trends Cancer. (2023) 9:111–21. doi: 10.1016/j.trecan.2022.10.009
150. Baker DJ, Arany Z, Baur JA, Epstein JA, June CH. CAR T therapy beyond cancer: The evolution of a living drug. Nature. (2023) 619:707–17. doi: 10.1038/s41586-023-06243-w
151. Larkin J, Minor D, D’Angelo S, Neyns B, Smylie M, Miller WH Jr., et al. Combination of vemurafenib, cobimetinib, and nivolumab in advanced melanoma. J Clin Oncol. (2018) 36:9559–9.
152. Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang M, Arnason J, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): A multicentre seamless design study. Lancet. (2020) 396:839–52. doi: 10.1016/S0140-6736(20)31366-0
153. Socinski MA, Nishio M, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, et al. IMPOWER150 Final Overall Survival Analyses for atezolizumab plus bevacizumab and Chemotherapy in First-Line Metastatic Nonsquamous NSCLC. J Thorac Oncol. (2021) 16:1909–24. doi: 10.1016/j.jtho.2021.07.009
154. Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, et al. KTE-X19 CAR T-Cell therapy in relapsed or refractory Mantle-Cell lymphoma. New Engl J Med. (2020) 382:1331–42. doi: 10.1056/nejmoa1914347
155. Choueiri TK, Powles T, Burotto M, Escudier B, Bourlon MT, Zurawski B, et al. Nivolumab plus Cabozantinib versus Sunitinib for Advanced Renal-Cell Carcinoma. New Engl J Med. (2021) 384:829–41. doi: 10.1056/NEJMoa2026982
156. Taibi T, Cheon S, Perna F, Vu LP. mRNA-based therapeutic strategies for cancer treatment. Mol Ther. (2024) 7(1):166. doi: 10.1016/j.ymthe.2024.04.035
157. Ferraro DA, Gaborit N, Maron R, Cohen-Dvashi H, Porat Z, Pareja F, et al. A phase I trial of Vigil (p53) in patients with advanced breast cancer. Clin Cancer Res. (2019) 25:3371–9.
158. Kallen KJ, Heidenreich R, Schnee M, Petsch B, Schlake T, Thess A, et al. BI 1361849 (RNActive), an RNA-lipoplex vaccine. Oncotarget. (2013) 11:2511–23. doi: 10.4161/hv.25181
159. Baruch EN, Youngster I, Ben-Betzalel G, Ortenberg R, Lahat A, Katz L, et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science. (2021) 371:602–9. doi: 10.1126/science.abb5920
160. Vétizou M, Pitt JM, Daillère R, Lepage P, Waldschmitt N, Flament C, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science. (2015) 350:1079–84. doi: 10.1126/science.aad1329
161. Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science. (2015) 350:1084–9. doi: 10.1126/science.aad1329
162. Dubin K, Callahan MK, Ren B, Khanin R, Viale A, Ling L, et al. Intestinal microbiome analyses identify melanoma patients at risk for checkpoint-blockade-induced colitis. Nat Commun. (2016) 7:10391. doi: 10.1038/ncomms10391
163. Frankel AE, Coughlin LA, Kim J, Froehlich TW, Xie Y, Frenkel EP, et al. Metagenomic shotgun sequencing and unbiased metabolomic profiling identify specific human gut microbiota and metabolites associated with immune checkpoint therapy efficacy in melanoma patients. Neoplasia. (2017) 19:848–55. doi: 10.1016/j.neo.2017.08.004
164. Chaput N, Lepage P, Coutzac C, Soularue E, Le Roux K, Monot C, et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann Oncol. (2017) 28:1368–79. doi: 10.1093/annonc/mdx108
165. Salgia NJ, Bergerot PG, Maia MC, Dizman N, Hsu J, Gillece JD, et al. Stool microbiome profiling of patients with metastatic renal cell carcinoma receiving anti-PD-1 immune checkpoint inhibitors. Eur Urol. (2020) 78:498–502. doi: 10.1016/j.eururo.2020.07.011
166. Tanoue T, Morita S, Plichta DR, Skelly AN, Suda W, Sugiura Y, et al. A defined commensal consortium elicits CD8 T cells and anti-cancer immunity. Nature. (2019) 565:600–5. doi: 10.1038/s41586-019-0878-z
167. Xu X, Lv J, Guo F, Li J, Jia Y, Jiang D, et al. Gut microbiome influences the efficacy of PD-1 antibody immunotherapy on MSS-type colorectal cancer via metabolic pathway. Front Microbiol. (2020) 11:814. doi: 10.3389/fmicb.2020.00814
168. Mager LF, Burkhard R, Pett N, Cooke NCA, Brown K, Ramay H, et al. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science. (2020) 369:1481–9. doi: 10.1126/science.abc3421
169. Si W, Liang H, Bugno J, Xu Q, Ding X, Yang K, et al. Lactobacillus rhamnosus GG induces cGAS/STING-dependent type I interferon and improves response to immune checkpoint blockade. Gut. (2022) 71:521–33. doi: 10.1136/gutjnl-2020-323426
170. Davar D, Dzutsev AK, McCulloch JA, Rodrigues RR, Chauvin JM, Morrison RM, et al. Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science. (2021) 371:595–602. doi: 10.1126/science.abf3363
171. Shiao SL, Kershaw KM, Limon JJ, You S, Yoon J, Ko EY, et al. Commensal bacteria and fungi differentially regulate tumor responses to radiation therapy. Cancer Cell. (2021) 39:1202–13. doi: 10.1016/j.ccell.2021.07.002
172. Gunjur A, Sholl LM, Gopalakrishnan V, Andrews MC, Spencer CN, Wargo JA. A gut microbial signature for combination immune checkpoint blockade across cancer types. Nat Med. (2024) 30:680–91. doi: 10.1038/s41591-024-02823-z
173. Daillère R, Derosa L, Bonvalet M, Segata N, Routy B, Zitvogel L. Trial watch: The gut microbiota as a tool to boost the clinical efficacy of anticancer immunotherapy. Oncoimmunology. (2020) 9:1774298. doi: 10.1080/2162402X.2020.1774298
174. Du P, Wang HY, Liu JY, Lin X, Li YJ. Microbiota and their metabolites potentiate cancer immunotherapy: Therapeutic target or resource for small molecule drug discovery? Front Pharmacol. (2022) 13:1091124. doi: 10.3389/fphar.2022.1091124
175. Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. (2018) 359:91–7. doi: 10.1126/science.aan3706
176. Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre ML, et al. The gut microbiome modulates the efficacy of anti-PD-1 therapy in melanoma patients. Science. (2018) 359:97–103. doi: 10.1126/science.aao3290
177. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-Y M, et al. Dietary fiber promotes CAR-T cell expansion and persistence. Cancer Res. (2020) 80:2651–62.
178. Zitvogel L, Daillère R, Roberti MP, Routy B, Kroemer G. Lactobacillus-derived metabolites enhance cancer vaccine efficacy. Cancer Res. (2018) 78:2881–91.
179. Wang Y, Wiesnoski DH, Helmink BA, Gopalakrishnan V, Choi K, DuPont HL, et al. Microbiota-derived SCFAs enhance T-cell function and antitumor immunity. Nat Commun. (2019) 10:1–12.
180. Zhang L, Yu X, Zheng L, Zhang Y, Li Y, Fang Q, et al. Bifidobacterium-based therapy enhances oncolytic virus-mediated tumor lysis. Cancer Res. (2020) 80:3023–34.
Keywords: cancer drug resistance, gut microbiome, immune checkpoint inhibitors, immunotherapy, neoplasms
Citation: Sonar PV, Singh AK, Mandadi S and Sharma NK (2025) Expanding horizons of cancer immunotherapy: hopes and hurdles. Front. Oncol. 15:1511560. doi: 10.3389/fonc.2025.1511560
Received: 15 October 2024; Accepted: 31 March 2025;
Published: 25 April 2025.
Edited by:
Arunasree M Kalle, University of Hyderabad, IndiaReviewed by:
Georgia Damoraki, National and Kapodistrian University of Athens, GreeceThai Minh Duy Le, Hanyang University, Republic of Korea
Copyright © 2025 Sonar, Singh, Mandadi and Sharma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nilesh Kumar Sharma, bmlsZXNoLnNoYXJtYUBkcHUuZWR1Lmlu
†ORCID: Priyanka Vijay Sonar, orcid.org/0009-0005-3649-2259
Anuj Kumar Singh, orcid.org/0009-0006-2208-9183
Sravan Mandadi, orcid.org/0009-0009-4730-7679
Nilesh Kumar Sharma, orcid.org/0000-0002-8774-3020