 Erin E. Crotty
Erin E. Crotty Aimee A. Sato1,2†
Aimee A. Sato1,2†- 1Ben Towne Center for Childhood Cancer and Blood Disorders Research and the Department of Pediatrics, Seattle Children’s Hospital, Fred Hutchinson Cancer Research Center, University of Washington, Seattle, WA, United States
- 2Division of Pediatric Neurology, Department of Neurology, Seattle Children’s Hospital, University of Washington, Seattle, WA, United States
- 3Division of Hematology, Oncology and Bone Marrow Transplant, St. Louis Children’s Hospital, Washington University School of Medicine, St. Louis, MO, United States
Pediatric low-grade gliomas (pLGG) are a group of tumors largely driven by alterations in a single genetic pathway, known as the RAS-RAF-mitogen-activated protein kinase (MAPK) pathway. Recent biologic insights and therapeutic targeting of MAPK-alterations have dramatically shifted the treatment approach in pLGG. While chemotherapy remains front-line therapy for unresectable pLGG in most scenarios (with the notable exception of BRAF V600E-altered tumors), many patients recur following cytotoxic agents and require further treatment. Inhibitors of the MAPK pathway, primarily MEK and RAF kinase inhibitors, have emerged as effective and tolerable second-line or later therapy for pLGG. As familiarity with these targeted agents increases, their indications for use continue to expand and Phase 3 clinical trials investigating their utility in the front-line setting are ongoing. We have adopted mitigation strategies for their associated toxicities; skin toxicity, in particular, is now managed by prevention strategies and early dermatologic intervention. This review highlights current approaches for the clinical implementation of MEK and RAF kinase inhibitors for pLGG, focusing on the practical aspects of drug administration, toxicity management, response monitoring, and distribution to patients experiencing geographic or financial barriers to care. Additionally, we review important considerations for the off-label use of these agents while contemporaneous clinical trials assessing front-line efficacy are ongoing. We discuss the potential for more expansive or histology-agnostic tumor targeting using MEK inhibitors, harnessing their biologic relevance for other RAS-altered conditions.
Introduction
Pediatric low-grade glioma (pLGG) represents the most common brain tumor in children, comprising up to 30-40% of all pediatric central nervous system tumors (1). The umbrella term, pLGG, encompasses a heterogenous group of tumors, with individual entities defined by their unique immunohistopathologic and molecular features. pLGG tumors are classified according to the 2021 World Health Organization (WHO) Central Nervous System (CNS) under 3 distinct categories: pediatric-type diffuse low-grade gliomas, circumscribed astrocytic gliomas, and glioneuronal or neuronal tumors (2). Among these, pilocytic astrocytoma (PA) is the most prevalent, making up 10-15% of all brain tumors in children and 5% of brain tumors in adults (1). The vast majority, nearly 70% of pLGG, harbor genomic driver mutations in the mitogen-activated protein kinase (MAPK) pathway, and therefore, pLGG is often considered a “single pathway disease.” (3) While pLGG can occur in any anatomic location in the CNS, distinct molecular drivers within the MAPK pathway often map to a particular CNS location. For instance, pleomorphic xanthoastrocytomas arise most commonly in the supratentorium and harbor BRAF V600E mutations, while PAs more frequently harbor BRAF fusions and arise in the posterior fossa or optic pathway. pLGGs are also commonly associated with neurofibromatosis type 1 (NF1), where optic pathway gliomas occur in 15-20% percent of patients (4, 5). Patients with NF1 carry a higher risk for the development of other RAS pathway-associated tumors (e.g. plexiform neurofibromas) and glial tumors, including brainstem gliomas and diffuse astrocytomas (6, 7).
Current standard therapy
Surgical resection is the mainstay of front-line therapy for symptomatic pLGG; however, more than 50% of pLGGs occur in locations that are either not amenable to surgery or only amenable to a biopsy or limited resection (e.g. tumors located in the diencephalon, brainstem, optic pathway, or spinal cord) (8). Treatment for inoperable pLGG is largely determined by clinical symptoms, with gliomas in the optic pathway or brainstem often necessitating urgent initiation of therapy for vison preservation or neurologic symptoms.
Chemotherapy remains standard of care for inoperable or residual pLGG, with well-documented efficacy and tolerability in pediatric patients of all ages, even the very young. While the 5-year overall survival (OS) with chemotherapy ranges from 86-94% on recent studies, 5-year progression-free survival (PFS) is less favorable at 42-45% in pLGG, with slightly better PFS reported in patients with NF1 (5-yr PFS 69-85%) (9, 10). Chemotherapy regimens including carboplatin and vincristine or single-agent vinblastine are commonplace today, and while the adverse effect profile of these agents is relatively well tolerated, they carry risks of myelosuppression, gastrointestinal toxicity, and peripheral sensory and motor neuropathy (9–11). Carboplatin allergy or similar adverse reaction has been reported in up to 8-20% of patients with pLGG receiving combination carboplatin and vincristine (9, 10, 12, 13). Cranial irradiation is also very effective against pLGG and has demonstrated improved PFS compared to conventional chemotherapy (62% versus 42% 10-year PFS compared to carboplatin/vincristine on HIT-LGG-1996) (13); however, radiation therapy is largely avoided in most sporadic pLGG and almost all NF1-associated pLGG due to the risk of significant late effects, including secondary malignancy, neurocognitive impairment, hormonal dysfunction, growth delay, vasculopathy and cerebrovascular injury (14–17).
Regardless of the choice of upfront therapy, unresectable pLGG recurs in nearly 50% of patients after first-line therapy, and in many cases, requires episodic treatment throughout childhood and adolescence for periods of clinical progression (13, 18). The approach to multiply recurrent pLGG therapy is therefore reminiscent of a chronic condition, directed at periodic exacerbations and symptoms. Despite the relatively poor PFS of pLGG as a whole, the OS remains excellent (18–20). The current focus of our efforts to improve outcomes in pLGG is to identify agents that lead to a durable response and reduce morbidity. Targeted therapies, namely MAPK inhibitors that target RAF and MEK proteins, may fullfill that need. As the clinical use of MAPK inhibitors expands and duration of exposure increases, it will be critical to understand their side effects within the drug class and as individual agents, along with how to appropriately monitor tumor response. Herein, we review the practical aspects of MAPK inhibitor administration and toxicity management, as well as discuss implications for patients in limited resource settings. These oral agents carry inherent benefits for patients for whom access to a pediatric oncology facility is restricted, and while ongoing studies are designed to answer lingering questions regarding the safest, most effective front-line therapy for pLGG, the use of MAPK pathway inhibitors frontline in limited populations is also discussed.
MAPK inhibitor therapy
Molecular landscape
Alterations in the BRAF oncogene are some of the most described molecular variants found in cancer. Together, oncogenic BRAF aberrations result in constitutive activation of BRAF protein kinase activity, leading to downstream signaling that drives tumor growth and proliferation. All pLGG tumor subgroups have a documented driver mutation in the BRAF oncogene or within a cooperating protein along the MAPK pathway, namely the RAS, RAF, MEK, and ERK kinases. In the case of BRAFV600E hotspot mutations, the Val600Glu activating mutation functions as a monomer to promote ERK signaling. BRAF can also form rearrangements or pair with fusion partners where the N-terminal inhibitory domain of BRAF is replaced and the resulting dimer retains BRAF kinase signaling that drives aberrant expression independent of RAS signaling (21, 22). Among these genomic fusions, KIAA1549::BRAF fusions are the most common and are found in nearly 70-80% of pilocytic astrocytomas and 30-40% of all pLGG (18, 22–25). As such, novel agents targeting this ubiquitous fusion are highly sought after for therapeutic indications (Table 1).
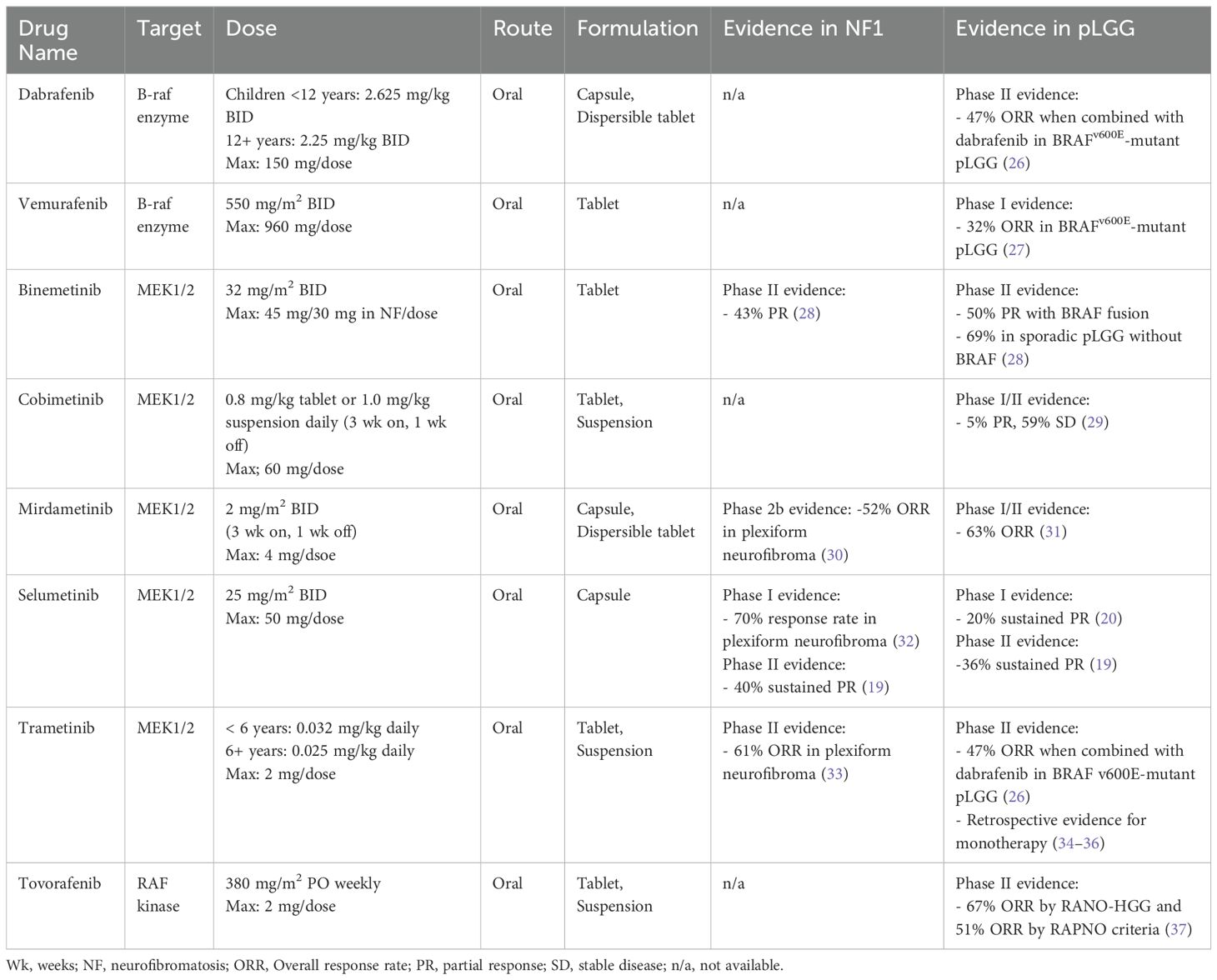
Table 1. Targeted therapy for pediatric low-grade glioma harboring MAPK pathway alterations.
A handful of agents targeting the MAPK pathway have demonstrated tolerability and efficacy in prospective studies of children and adolescents with relapsed or refractory pLGG (19, 20, 26, 30, 32, 34, 37–39). BRAF inhibitors (e.g. vemurafenib, dabrafenib) were among the first agents tested in pLGG in an attempt to replicate early successes seen in adult glioma and melanoma driven by BRAFV600E mutations, in addition to the particularly poor PFS in this pLGG subset (40, 41). While BRAF inhibitors showed remarkable efficacy in BRAFV600E-altered pLGG (42), surprisingly, tumors harboring KIAA1549::BRAF fusions demonstrated paradoxical growth with these agents, a biologic phenomenon that has since been extensively modeled and molecularly described (21). BRAF inhibitors are now exclusively used in patients with BRAFV600E point mutations and are contraindicated in patients with BRAF fusions. Dabrafenib, was later tested in combination with trametinib, a MEK inhibitor, as first-line therapy for BRAFV600E-altered LGG and demonstrated improved PFS compared to standard chemotherapy (PFS 20.1 versus 7.4 months; HR, 0.31) and a superior safety profile (26, 38). These early studies supported trials showing efficacy of this combination in non-CNS solid tumors and led to accelerated FDA approval in 2022 of the combination dabrafenib/trametinib for the treatment of progressive or metastatic solid tumors harboring BRAFV600E mutations. In 2023, the FDA approved the combination for the first-line treatment of pediatric patients 1 year of age and older with pLGG with a BRAFV600E mutation (43).
MEK inhibitors, including selumetinib, trametinib, mirdametinib, binimetinib, and cobimetinib, provide active inhibition of ERK signaling by targeting MEK, a kinase downstream from BRAF in the MAPK pathway. Several have been studied in pLGG, with demonstrated safety and efficacy data available in the recurrent pLGG and NF1-associated pLGG populations, and selumetinib is currently in clinical trials for newly diagnosed patients (NCT03871257, NCT04166409). The following discussion focuses on agents with published phase 2 trial data in pLGG and favorable toxicity profiles, with selumetinib and trametinib available for prescription off-label, leading to frequent real-world use as second-line therapy.
Selumetinib, a selective, oral small molecule inhibitor of MEK1/2, has shown a 40% sustained response rate (complete or partial response) in a phase 2 trial of children with progressive PAs harboring KIAA1549::BRAF fusions or BRAFV600E mutations (19). PFS at 2 years was 70% in BRAF-altered non-NF1 associated pLGG and 96% in patients with germline NF1. Patients with NF1-associated plexiform neurofibromas have similarly shown response rates around 70% to treatment with single-agent selumetinib and it is now FDA-approved for that indication (32). Selumetinib is currently being compared to upfront standard chemotherapy in two phase 3 clinical trials for patients with newly-diagnosed pLGG, both with and without NF1 (NCT03871257, NCT04166409).
Trametinib is another potent, oral small molecule inhibitor of MEK1/2 with non-competitive ATP binding properties. While large, prospective trials are still ongoing (NCT03363217), retrospective analyses show promising objective response rates in recurrent pLGG, supporting drug class efficacy in this population (34–36). Interim results of a phase 2 study of single-agent trametinib demonstrated an overall response rate of around 47% (complete, partial, or minor response) in pLGG and a 60% response rate using volumetric assessments in patients with NF1-associated plexiform neurofibroma (33, 44). Trametinib was FDA approved in 2013 for adult BRAFV600E-altered melanoma and has been used commonly off-label in pediatric patients as molecularly-guided therapy for recurrent or refractory pLGG.
Mirdametinib is another highly brain penetrant inhibitor of MEK1/2 that is currently being investigated as monotherapy in a phase I/II trial to assess safety and preliminary efficacy for relapsed or refractory pLGG (NCT04923126). To date, 12/19 (63%) patients achieved promising objective responses (1 major, 6 partial, 5 minor). The phase 2 component is ongoing to establish safety and efficacy, including in newly diagnosed patients and those with prior exposure to MEK inhibitors (31, 45).
Tovorafenib, in contrast to the MEK inhibitors discussed above, is an oral, highly selective, type II RAF inhibitor that targets both mutant and wild-type A-Raf, B-Raf, and C-Raf protein kinases. Type II RAF inhibitors effectively inhibit RAF dimers, including BRAF fusions, in addition to BRAF monomers for pan-RAF inhibitory function. Among 137 patients enrolled on a phase 1 trial in BRAF-altered relapsed or refractory pLGG, tovorafenib showed an overall response rate (including minor responses) of 51% by RAPNO criteria and 67% by RANO-HGG criteria (primary endpoint), including 12 (17%) patients with a complete response and a median duration of response of 13.8 months (37). Patients entered the trial having received a median of three lines of prior therapy, with 61% previously treated with a MEK and/or BRAF inhibitor. The FDA granted tovorafenib accelerated approval for recurrent or refractory pLGG in 2024 (46). Tovorafenib is currently being compared to standard-of-care chemotherapy regimens, including single-agent vinblastine or carboplatin/vincristine in newly diagnosed RAF-altered pLGG (37).
Toxicity management
MAPK inhibitor toxicities have been well described in pediatric populations (Figure 1). The most common treatment-related adverse events include dermatologic manifestations (rash, paronychia, photosensitivity, hair color change), gastrointestinal toxicity (nausea, vomiting, diarrhea), ophthalmologic toxicity (serous retinopathy, retinal vein occlusion, retinal detachment), cardiac complications (decreased ejection fraction), asymptomatic creatinine kinase elevation, weight gain, and fatigue (47). Recently, cooperative groups have identified a need to develop clinical guidance for practitioners for managing frequently encountered side effects, especially regarding dermatologic recommendations due to the frequency and occasional severity of MAPK inhibitor-associated rashes (48). One such advisory board from the Children’s Tumor Foundation has proposed and implemented recommendations for toxicity management and side effect monitoring for patients with NF1 receiving MEK inhibitors, which has now been widely adopted (49).
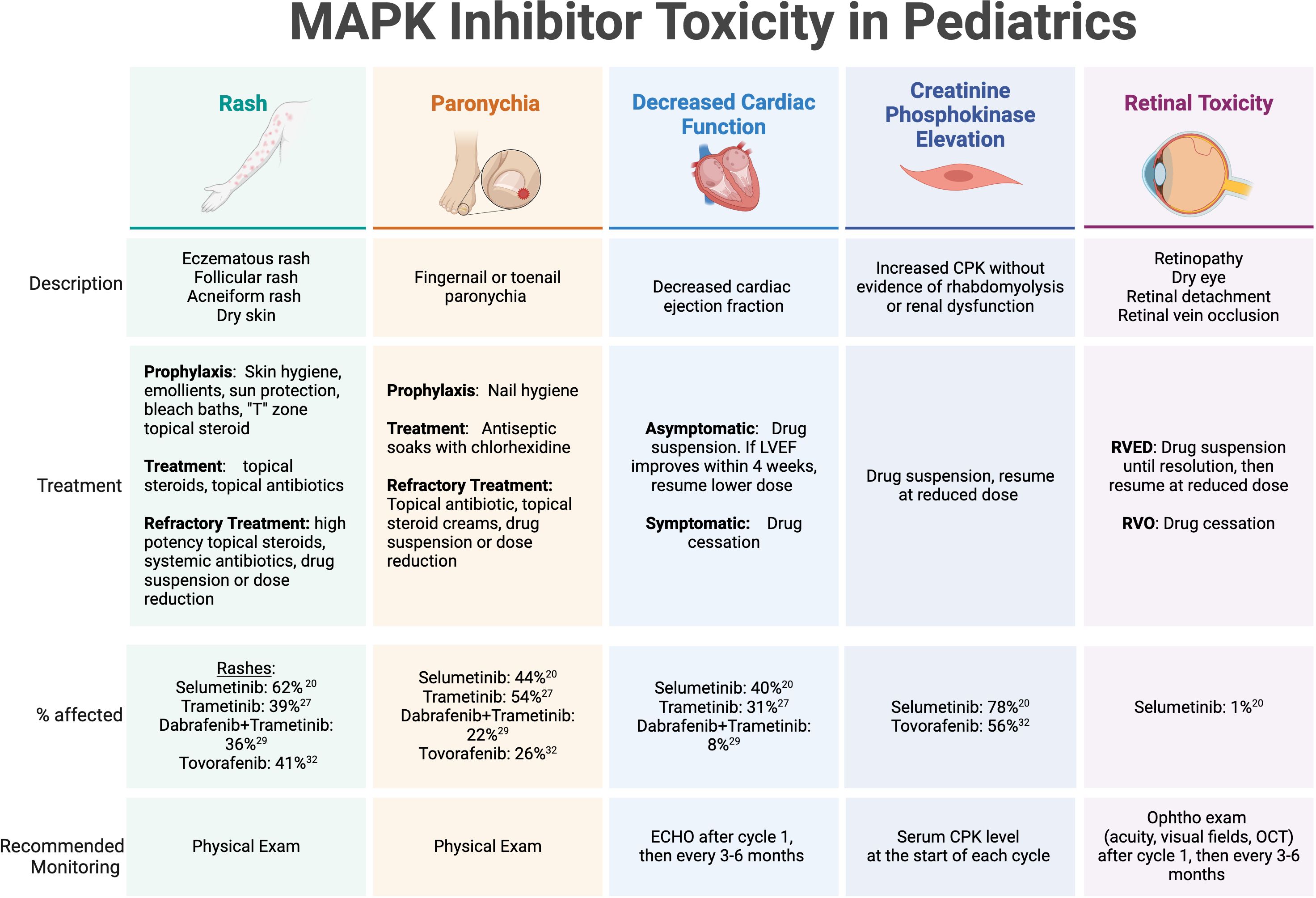
Figure 1. MAPK inhibitor toxicity in pediatrics. Select toxicities with high incidence or potential severity reported on published trials of MAPK inhibitors in children and adolescents with recurrent or refractory pLGG. LVEF, left ventricular ejection fraction; CPK, creatinine phosphokinase; RVED, Retinal Vascular Endothelial Dysfunction; RVO, retinal vein occlusion; Ophtho, ophthalmology.
The dermatologic manifestations of MAPK-targeted therapy can range from eczematous and acneiform rashes to paronychia and wound complications. Recommended strategies for prevention and management include symptom-related measures applied to specific dermatologic conditions. For rashes, consensus guidelines include prophylactic measures, such as applying skin moisturizers/emollients to maintain an optimal skin barrier, sun protection, and bleach baths to minimize skin flora and reduce the risk of infectious complications and pruritis. In adolescent patients, topical antibiotics and low dose topical steroids are recommended for prevention of facial acne in the “T” zone. Once a rash is present, treatments include topical steroids, topical and oral antibiotics, and consideration of drug suspension until resolution or significant improvement. Paronychia is also pervasive in patients on MEK and RAF inhibitors, and mild cases can be addressed with frequent antiseptic soaks using chlorhexidine, topical antibiotics, and topical steroids. Dermatology specialty care is recommended for persistent or severe cases of skin toxicity. Wound healing complications have been less frequently reported, but are a potential cause of morbidity in patients who undergo surgery while on therapy, which may include procedures for the placement of a ventricular shunt catheter for CSF diversion or a central venous access line. Primary incisions or skin ulcerations caused by pressure wounds may have longer healing times and require wound care expertise for appropriate management.
While the rates of cardiac toxicity are low in children receiving MAPK pathway inhibitors in published reports, most guidelines recommend routine echocardiograms (ECHO) to monitor for asymptomatic diminishing ejection fraction (EF) while on MEK inhibitor therapy. Selumetinib, for example, has been associated with a decrease in EF >10% below the normal range in a single pediatric patient with NF1-associated pLGG and 20 (40%) patients with non-NF1 associated pLGG (19, 32). Most of the limited reported cases of EF dysfunction were detected on routine screening, but even for asymptomatic decreases beyond 10% of baseline function, the consensus recommendation is to hold MEK inhibitor therapy and discontinue altogether if no improvement within 4 weeks off therapy since EF decrease is generally reversible after drug suspension or discontinuation. Routine ECHO monitoring is recommended after 1 month of therapy, then at routine intervals (e.g. 3-6 months) thereafter. It is worth noting that tovorafenib has not been associated with cardiac adverse effects.
Ocular adverse events are fortunately rarely reported in pediatric patients on published studies of MAPK inhibitors. However, when present in adult patients, they can result in significant visual complications and even blindness (50, 51). Considered a class effect that affects anywhere from 5-90% of adult patients treated with MEK/BRAF inhibitors based on published reports (51), retinopathy can present with symptomatic blurry vision, floaters, or photophobia, and most severely, may result in retinal detachment or retinal vein occlusion. MEK inhibitor-associated retinopathy (MEKAR) is generally self-limited and may resolve with or without drug cessation (50). Recommendations for monitoring vary considering that visual symptoms are normally present and therefore can be monitored clinically, however most practitioners recommend an ophthalmologic exam early into therapy, usually after 1 month, then at routine intervals (e.g. 3-6 months) thereafter (49). The ophthalmology exam should include visual acuity at a minimum, visual field testing in patients with optic pathway gliomas, and consideration of optical coherence testing (OCT) for detection of small retinal detachments by direct examination of the retina and choroid. Tovorafenib has not been associated with retinal or ocular-related adverse effects.
Interestingly, tovorafenib can lead to growth arrest – a side effect that hasn’t been reported with any of the MEK inhibitors, and it is unknown whether this particular toxicity is considered a class effect. Preliminary data on tovorafenib shows pediatric and adolescent patients demonstrate growth arrest on therapy without advancement of bone age or immature closure of growth plates (37, 52). This adverse effect reverses once drug therapy is stopped and patients have resumed expected annualized growth velocities after therapy, with some exceeding the expected average post-treatment (52).
Data on long-term toxicities or late effects of MAPK inhibitors are immature, owing to the limited amount of time they have been commercially available, which is a disadvantage to these drugs compared to chemotherapy, however, off therapy toxicity data is actively being collected. Organ function, particularly cardiac and ocular function, along with physiological effects on growth, puberty, and visual and neurocognitive outcomes are being monitored.
Response monitoring
The objective criteria used for monitoring treatment response to MAPK inhibitor therapy has been through several iterations. On the phase I trial of selumetinib in patients with recurrent pLGG, objective response was initially measured by including the enhancing tumor components on T1 post-contrast magnetic resonance imaging (MRI). This was later amended to incorporate more widely accepted definitions of response in pLGG, which does not rely on changes in the enhancement pattern over time, as these are known to increase/decrease without intervention, deviate from tumor volume measurements, and are not prognostic (20). Instead, T2/fluid attenuated inversion recovery (FLAIR) sequences were used to more accurately define tumor response using bidimensional measurements and these were carried forward for objective response assessments on the phase 2 selumetinib trial (19).
Response assessment on the phase 2 trial of tovorafenib in recurrent pLGG utilized the Response Assessment in Neuro-Oncology High-Grade Glioma (RANO-HGG) criteria as a primary endpoint, as these were considered the only validated criteria by the FDA at the time of trial initiation (37). To address the same concerns about inclusion of enhancement pattern as a defining feature, secondary assessment of response was performed by Response Assessment in Pediatric Neuro-Oncology Low-Grade Glioma (RAPNO) criteria and Response Assessment in Neuro-Oncology Low-Grade Glioma (RANO-LGG), both of which more accurately characterize disease burden in pLGG. The ongoing phase 3 trial of tovorafenib versus standard chemotherapy utilizes RANO-LGG criteria, with alternative imaging criteria as secondary objectives and endpoints.
In addition to objective tumor response, it is important to understand MAPK therapy-related effects on chronic morbidities associated with pLGG, including functional outcomes like vision and neurocognitive development. While traditional chemotherapy may stabilize vision, published data on MEK inhibitors suggest that these agents may have further benefit by improving visual acuity (19, 26, 37). To determine an accurate risk-benefit ratio of newer agents, ongoing studies are thoughtfully addressing these measures and including quality of life (QOL) and patient-reported outcomes (PRO) assessments, adaptive behavior tools, daily living domain scores, communication assessments, additional visual assessment tools, and neuropathy scores. In addition, the clinical benefit rate, time to response, and duration of response are adding qualifying data to our standard measures of tumor response.
MAPK targeted therapies have an established role in the treatment of relapsed or refractory pLGG following chemotherapy as second-line or later therapy, and may better accommodate episodic use in a way that cytotoxic agents cannot. For example, retreatment with the MEK inhibitor selumetinib has been shown to be effective for tumor control in melanoma and may suggest drug resistance to these agents is reversible, unlike other small molecule inhibitors used in cancer (53). In one study. discontinuing treatment with BRAF inhibitors (dabrafenib or vemurafenib) in BRAFV600E -mutated pLGGs resulted in rapid regrowth in 76.5% of tumors, however 90% of pLGGs responded if rechallenged with BRAF inhibition alone or when combined with MEK inhibition (54). This retreatment phenomenon draws a stark contrast to chemotherapy regimens, which are generally not re-used at progression, and has implications for how duration of response should be measured on targeted therapies.
Administration and access for special populations
Compared to standard first-line chemotherapy, oral agents may have inherent benefits for minimizing the burden of disease in patients with pLGG. The MAPK-targeted therapies described above are administered orally, with most (except selumetinib) available as a liquid formulation or dispersible tablet ideal for young children and patients requiring enteral nutrition. Based on pharmacokinetic studies, MEK and BRAF inhibitors are given daily (once to twice per day), with some MEK inhibitors requiring an empty stomach for optimal absorbtion (45, 55). Tovorafenib can be taken with or without food and is administered once weekly. Beyond convenience, oral MAPK inhibitors may also reduce financial toxicity for patients living in low resource or geographically limited areas, since on therapy monitoring necessitates fewer ambulatory office visits compared to a chemotherapy regimen administered weekly. Additionally, unlike chemotherapy, MAPK inhibitors are not associated with serious infectious complications since they do not cause significant myelosuppression or require central venous access, so unexpected hospital admissions are rare. While few studies have fully characterized the long and short-term treatment-related morbidities associated with pLGG treated with standard chemotherapy and surgery, a recent retrospective study found a surprisingly higher level of healthcare resource utilization (HRU) and symptom burden in this population than has previously been captured (16, 56). These data and the data currently being captured in ongoing clinical trials assessing QOL and PRO will be invaluable in establishing treatment guidelines and assessing whether a front-line oral regimen may substantially improve cost burden, psychosocial impact, and access to care in pLGG.
Ambulatory infusion centers capable of delivering intravenous chemotherapy to pediatric patients are limited, and pediatric neuro-oncology programs with clinical expertise in pLGG even more so. In a recent study evaluating geographic access to pediatric cancer care, the median travel times to a pediatric oncology center were longest for American Indian (AI) or Alaska Native (AN) pediatric populations (46 [range16-104] minutes) and residents of rural areas (95 [range 68-135] minutes) (57). These data provide additional evidence that AI and AN communities continue to endure health disparities and that geographic-based social determinants of health may be a significant contributing factor. A real-world example of this is experienced by patients living in rural Alaska, wherein weather patterns may routinely delay or prevent travel in and out of community villages and a single pediatric oncology center exists to serve the entire state, encompassing a large geographic region. Historically, patients with pLGG requiring therapy beyond surgery have had to relocate, often moving hundreds of miles away for the duration of treatment. As discussed above, chronic episodic therapy for multiply recurrent disease only adds to the immense financial and psychosocial burden of treatment on the family and larger community. Alaska is just one example of many locations in the United States with high Area Deprivation Index (ADI) scores that may benefit from rethinking our current front-line strategy for pLGG in context of a larger goal toward health equity in pediatric cancer care.
With the emergence of telehealth capabilities, providers are better equipped with infrastructure to deliver specialized cancer care remotely and appropriately monitor therapy-related toxicities. Oral regimens may require fewer in-person evaluations and infrequent tests performed by ancillary services (e.g. cardiology, ophthalmology), allowing much broader access to care by reducing travel burden and financial toxicity. This would allow patients to receive therapy while living further away from pediatric brain tumor programs. The difference in financial toxicity between weekly and monthly, or even every 3-month travel to and from a healthcare facility alone is noteworthy. In addition, drugs can now be delivered by mail-in specialty pharmacies, reducing the strain on local healthcare systems. While expanding clinical sites may not be feasible, expansion of existing infrastructure and a thoughtful approach to our clinical care standards for at risk populations may go a long way in improving health outcomes (Figure 2).
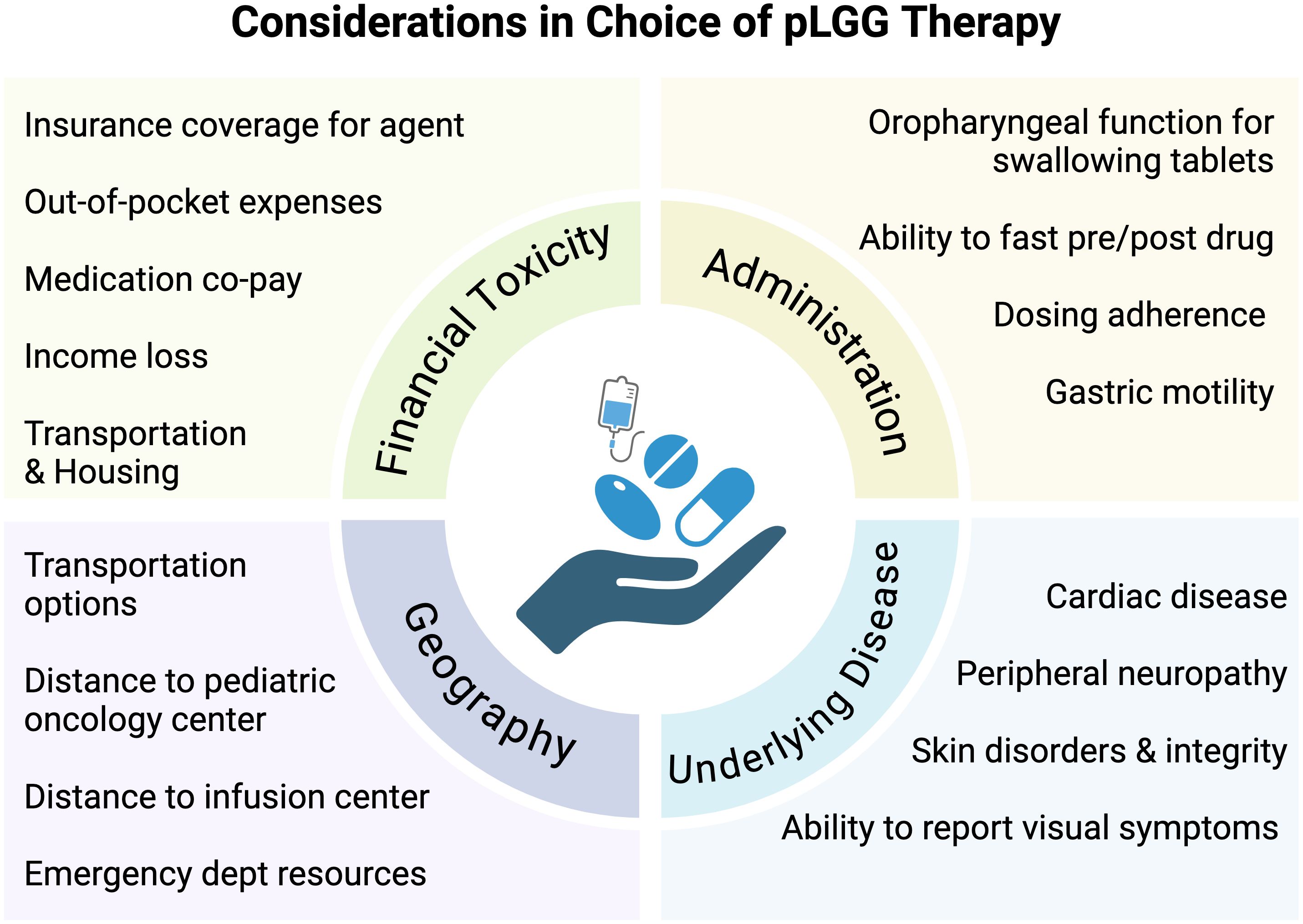
Figure 2. Considerations in choice of pLGG therapy. Schema depicting psychosocial, economic, geographic, financial, and disease-related considerations in determining treatment decisions in pediatric low-grade glioma.
In low- or middle-income countries (LMIC), access to MAPK inhibitors is even more restricted, and is impacted by drug availability and pricing. Targeted therapies are not available in many LMIC countries, and when available, there are significant limitations related to regional drug supply or their very high cost (58, 59). In addition, molecular testing for identifying MAPK alterations is limited in these settings, impacting the identification of patients who are likely to benefit from targeted therapies. In an effort to improve outcomes for children with cancer around the world, the World Health Organization (WHO) launched a Global Initiative for Childhood Cancer (GICC) under its CureAll Framework and included pLGG as one of six “tracer cancers” to monitor success of the program (60). Medicine pricing and procurement is among the pillars of the program, and requires both government and industry partnerships, however it will be equally critical to evaluate its success in co-establishing a infrastructure for molecular diagnosis in LMIC settings.
Considerations for balancing research integrity with off-label use
Off-label use of MAPK pathway inhibitors, namely their use as first-line therapy in newly diagnosed patients with pLGG, is controversial. “Off-label” typically entails the use of cancer therapeutics in unapproved populations and is a common practice in pediatric oncology generally, since pediatric and adolescent patients are almost always excluded from registrational FDA trials and there is often sound scientific evidence and safety data to support their use (61). Practitioner comfort with MAPK inhibitor prescribing has increased with the accelerated FDA approvals of selumetinib, dabrafenib/trametinib, and tovorafenib, along with the publication of phase I/II trial safety and PK data. However, this has coincided with the opening of phase 3 clinical trials assessing front-line efficacy of selumetinib versus carboplatin/vincristine in pediatric patients with NF1 (NCT03871257) and without NF1 (NCT04166409), and tovorafenib versus standard chemotherapy in non-NF1 associated pLGG (NCT05566795). Prior to the results of these trials and maturation of the longer-term data on late effects, the use of MAPK inhibitors as front-line therapy raises important ethical considerations. The research community has prioritized answering critical questions about efficacy (non-inferiority compared to standard of care chemotherapy), long-term safety, and functional outcomes that would not be captured outside the context of a clinical trial. Each of these facets is essential to fully characterize the clinical benefit of MAPK targeted therapy, as compared to our current standard, and will be invaluable in developing much needed treatment guidelines.
While the vast majority of patients with pLGG can undergo study randomization to a chemotherapy regimen or MAPK inhibitor without undue burden, the special populations described above, including patients with geographic or financial stress, may face additional barriers. For these patients, many factors may preclude study enrollment and in order to facilitate patient-centered care, many providers offer front-line, off-label use of MEK or RAF inhibition. The literature on this type of prescribing is lacking and would benefit from dedicated efforts to further characterize which specific patient populations may be candidates for alternative upfront therapy for pLGG.
Future directions
Given the ubiquitous nature of MAPK pathway alterations in disease and chronic syndromes beyond cancer, researchers are now exploring new target populations that may benefit from MAPK targeting. MEK inhibitors are being investigated, and in some cases prescribed as first-line therapy, in NF1 and other RASopathy syndromes, including Noonan syndrome, hypertrophic cardiomyopathy, multifocal atrial tachycardia (MAT), lymphatic anomalies, capillary–arteriovenous malformations, Kaposiform lymphangiomatosis, and cardiofaciocutaneous syndrome, among others. While MEK inhibitors are effective in treating pLGGs in those with NF1, consensus remains that they are best used within a clinical trial or for relapsed disease (62). However, with several applications of MEK inhibitors for complications in NF1, there may be more interest in utilizing MEK inhibitor as first line-therapy in circumstances when multiple disease types may be addressed with single agent therapy, such as a MEK inhibitor for simultaneous pLGG and plexiform neurofibroma treatment.
The lessons learned from our early BRAF inhibitor experience serve as a reminder that not all RAS alterations are alike and target inhibition can have paradoxical effects in certain biologic conditions. As the use of MAPK inhibition expands for rare subsets of patients, it will be critical to report early experiences, including unexpected toxicities. Additionally, in our current molecular area as we discover more genomic alterations that span disparate tumor types, such as BRAFv600E, which can be seen in melanoma, glioma, and colon cancer, there may be an expanded role for tumor-agnostic drug approvals for targeted therapies.
Conclusion
Treatment guidelines are lacking on how best to incorporate targeted therapy into standard practice for pLGG. As familiarity and availability of MEK and RAF inhibitors increases, we have an opportunity to provide early, evidence-based support for practitioners on how and when to use these agents towards the goal of improving outcomes and reducing morbidity. While essential phase 3 clinical trials are ongoing, all patients should be offered opportunities to participate in research and the use of MAPK inhibition as first-line therapy (beyond BRAFv600E-altered tumors) reserved for judicious use in special populations where financial or geographic barriers exist.
Author contributions
EC: Conceptualization, Project administration, Writing – original draft, Writing – review & editing. AS: Writing – original draft, Writing – review & editing. MA: Conceptualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The Seattle Children’s Pediatric Brain Tumor Research Fund.
Acknowledgments
We would like to thank our patients and families for their selfless participation in research and entrusting us with their care. BioRender.com was used to generate high-quality figures.
Conflict of interest
MA is a consultant for DayOne pharmaceuticals and IPSEN pharma. MA has drug supply from Novartis pharmaceuticals for one his clinical trials.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ostrom QT, Price M, Neff C, Cioffi G, Waite KA, Kruchko C, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2016-2020. Neuro Oncol. (2023) 25:iv1–iv99. doi: 10.1093/neuonc/noad149
2. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol. (2021) 23:1231–51. doi: 10.1093/neuonc/noab106
3. Ziegler DS, Lehmann R, Eisenstat DD. A paradigm shift in how we treat pediatric low-grade glioma—Targeting the molecular drivers. Neuro-Oncology. (2024) 26:593–5. doi: 10.1093/neuonc/noae008
4. Packer RJ, Bilaniuk LT, Cohen BH, Braffman BH, Obringer AC, Zimmerman RA, et al. Intracranial visual pathway gliomas in children with neurofibromatosis. Neurofibromatosis. (1988) 1:212–22.
5. Listernick R, Charrow J, Greenwald MJ, Esterly NB. Optic gliomas in children with neurofibromatosis type 1. J Pediatr. (1989) 114:788–92. doi: 10.1016/s0022-3476(89)80137-4
6. Packer RJ, Iavarone A, Jones DTW, Blakeley JO, Bouffet E, Fisher MJ, et al. Implications of new understandings of gliomas in children and adults with NF1: report of a consensus conference. Neuro-Oncology. (2020) 22:773–84. doi: 10.1093/neuonc/noaa036
7. Samples DC, Mulcahy Levy JM, Hankinson TC. Neurosurgery for optic pathway glioma: optimizing multidisciplinary management. Front Surgery. (2022) 9:884250. doi: 10.3389/fsurg.2022.884250
8. Zapotocky M, Stucklin AG, Lassaletta A, Scheinemann K. Low-Grade Glioma in Children and Adolescents. Cham, Switzerland: Springer International Publishing (2024) p. 143–55.
9. Ater JL, Zhou T, Holmes E, Mazewski CM, Booth TN, Freyer DR, et al. Randomized study of two chemotherapy regimens for treatment of low-grade glioma in young children: A report from the children’s oncology group. J Clin Oncol. (2012) 30:2641–7. doi: 10.1200/jco.2011.36.6054
10. Lassaletta A, Scheinemann K, Zelcer SM, Hukin J, Wilson BA, Jabado N, et al. Phase II weekly vinblastine for chemotherapy-naïve children with progressive low-grade glioma: A canadian pediatric brain tumor consortium study. J Clin Oncol. (2016) 34:3537–43. doi: 10.1200/jco.2016.68.1585
11. Packer RJ, Ater J, Allen J, Phillips P, Geyer R, Nicholson HS, et al. Carboplatin and vincristine chemotherapy for children with newly diagnosed progressive low-grade gliomas. J Neurosurg. (1997) 86:747–54. doi: 10.3171/jns.1997.86.5.0747
12. Gnekow AK, Walker DA, Kandels D, Picton S, Giorgio P, Grill J, et al. A European randomised controlled trial of the addition of etoposide to standard vincristine and carboplatin induction as part of an 18-month treatment programme for childhood (≤16 years) low grade glioma - A final report. Eur J Cancer. (2017) 81:206–25. doi: 10.1016/j.ejca.2017.04.019
13. Gnekow AK, Falkenstein F, von Hornstein S, Zwiener I, Berkefeld S, Bison B, et al. Long-term follow-up of the multicenter, multidisciplinary treatment study HIT-LGG-1996 for low-grade glioma in children and adolescents of the German Speaking Society of Pediatric Oncology and Hematology. Neuro Oncol. (2012) 14:1265–84. doi: 10.1093/neuonc/nos202
14. De Blank P, Bandopadhayay P, Haas-Kogan D, Fouladi M, Fangusaro J. Management of pediatric low-grade glioma. Curr Opin Pediatrics. (2019) 31:21–7. doi: 10.1097/mop.0000000000000717
15. Liu APY, Hastings C, Wu S, Bass JK, Heitzer AM, Ashford J, et al. Treatment burden and long-term health deficits of patients with low-grade gliomas or glioneuronal tumors diagnosed during the first year of life. Cancer. (2019) 125:1163–75. doi: 10.1002/cncr.31918
16. Armstrong GT, Conklin HM, Huang S, Srivastava D, Sanford R, Ellison DW, et al. Survival and long-term health and cognitive outcomes after low-grade glioma. Neuro Oncol. (2011) 13:223–34. doi: 10.1093/neuonc/noq178
17. Greenberger BA, Pulsifer MB, Ebb DH, MacDonald SM, Jones RM, Butler WE, et al. Clinical outcomes and late endocrine, neurocognitive, and visual profiles of proton radiation for pediatric low-grade gliomas. Int J Radiat Oncol Biol Phys. (2014) 89:1060–8. doi: 10.1016/j.ijrobp.2014.04.053
18. Ryall S, Zapotocky M, Fukuoka K, Nobre L, Guerreiro Stucklin A, Bennett J, et al. Integrated molecular and clinical analysis of 1,000 pediatric low-grade gliomas. Cancer Cell. (2020) 37:569–583.e5. doi: 10.1016/j.ccell.2020.03.011
19. Fangusaro J, Onar-Thomas A, Young Poussaint T, Wu S, Ligon AH, Lindeman N, et al. Selumetinib in paediatric patients with BRAF-aberrant or neurofibromatosis type 1-associated recurrent, refractory, or progressive low-grade glioma: a multicentre, phase 2 trial. Lancet Oncol. (2019) 20:1011–22. doi: 10.1016/s1470-2045(19)30277-3
20. Banerjee A, Jakacki RI, Onar-Thomas A, Wu S, Nicolaides T, Young Poussaint T, et al. A phase I trial of the MEK inhibitor selumetinib (AZD6244) in pediatric patients with recurrent or refractory low-grade glioma: a Pediatric Brain Tumor Consortium (PBTC) study. Neuro-Oncology. (2017) 19:1135–44. doi: 10.1093/neuonc/now282
21. Sievert AJ, Lang S-S, Boucher KL, Madsen PJ, Slaunwhite E, Choudhari N, et al. Paradoxical activation and RAF inhibitor resistance of BRAF protein kinase fusions characterizing pediatric astrocytomas. Proc Natl Acad Sci. (2013) 110:5957–62. doi: 10.1073/pnas.1219232110
22. Jones DTW, Kocialkowski S, Liu L, Pearson DM, BäCklund LM, Ichimura K, et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res. (2008) 68:8673–7. doi: 10.1158/0008-5472.can-08-2097
23. Penman CL, Faulkner C, Lowis SP, Kurian KM. Current understanding of BRAF alterations in diagnosis, prognosis, and therapeutic targeting in pediatric low-grade gliomas. Front Oncol. (2015) 5:54. doi: 10.3389/fonc.2015.00054
24. Jones DTW, Kieran MW, Bouffet E, Alexandrescu S, Bandopadhayay P, Bornhorst M, et al. Pediatric low-grade gliomas: next biologically driven steps. Neuro-Oncology. (2018) 20:160–73. doi: 10.1093/neuonc/nox141
25. Johnson A, Severson E, Gay L, Vergilio J-A, Elvin J, Suh J, et al. Comprehensive genomic profiling of 282 pediatric low- and high-grade gliomas reveals genomic drivers, tumor mutational burden, and hypermutation signatures. Oncol. (2017) 22:1478–90. doi: 10.1634/theoncologist.2017-0242
26. Bouffet E, Geoerger B, Moertel C, Whitlock JA, Aerts I, Hargrave D, et al. Dabrafenib plus trametinib in pediatric glioma with BRAF V600 mutations. New Engl J Med. (2023) 389:1108–20. doi: 10.1056/nejmoa2303815
27. Nicolaides T, Nazemi KJ, Crawford J, Kilburn L, Minturn J, Gajjar A, et al. Phase I study of vemurafenib in children with recurrent or progressive BRAFV600E mutant brain tumors: Pacific Pediatric Neuro-Oncology Consortium study (PNOC-002). Oncotarget. (2020) 11:1942–52. doi: 10.18632/oncotarget.27600
28. Robison N, Pauly J, Malvar J, Gardner S, Allen J, Margol A, et al. LTBK-04. LATE BREAKING ABSTRACT: MEK162 (binimetinib) in children with progressive or recurrent low-grade glioma: a multi-institutional phase II and target validation study. Neuro Oncol. (2022) 24:i191–2. doi: 10.1093/neuonc/noac079.716
29. Trippett T, Toledano H, Campbell Hewson Q, Verschuur A, Langevin A-M, Aerts I, et al. Cobimetinib in pediatric and young adult patients with relapsed or refractory solid tumors (iMATRIX-cobi): A multicenter, phase I/II study. Targeted Oncol. (2022) 17:283–93. doi: 10.1007/s11523-022-00888-9
30. Moertel CL, Hirbe AC, Shuhaiber HH, Viskochil D, Sidhu A, Bielamowicz KJ, et al. ReNeu: A pivotal phase 2b trial of mirdametinib in children and adults with neurofibromatosis type 1 (NF1)-associated symptomatic inoperable plexiform neurofibroma (PN). J Clin Oncol. (2024) 42:3016–6. doi: 10.1200/JCO.2024.42.16_suppl.3016
31. Robinson GW, Vinitsky A, Bag AK, Chiang J, Li Q, Lin T, et al. LGG-53. Results from the phase 1 and phase 1 expansion cohorts of SJ901: A phase 1/2 trial of single-agent mirdametinib (PD-0325901) in children, adolescents, and young adults with low-grade glioma. Neuro-Oncology. (2024) 26:0–0. doi: 10.1093/neuonc/noae064.443
32. Dombi E, Baldwin A, Marcus LJ, Fisher MJ, Weiss B, Kim A, et al. Activity of selumetinib in neurofibromatosis type 1–related plexiform neurofibromas. New Engl J Med. (2016) 375:2550–60. doi: 10.1056/nejmoa1605943
33. Perreault S, Kiaei DS, Dehaes M, Larouche V, Tabori U, Hawkin C, et al. A phase 2 study of trametinib for patients with pediatric glioma or plexiform neurofibroma with refractory tumor and activation of the MAPK/ERK pathway. J Clin Oncol. (2022) 40:2042–2. doi: 10.1200/JCO.2022.40.16_suppl.2042
34. Manoharan N, Choi J, Chordas C, Zimmerman MA, Scully J, Clymer J, et al. Trametinib for the treatment of recurrent/progressive pediatric low-grade glioma. J Neuro-Oncol. (2020) 149:253–62. doi: 10.1007/s11060-020-03592-8
35. Selt F, van Tilburg CM, Bison B, Sievers P, Harting I, Ecker J, et al. Response to trametinib treatment in progressive pediatric low-grade glioma patients. J Neuro-oncol. (2020) 149:499–510. doi: 10.1007/s11060-020-03640-3
36. Kondyli M, Larouche V, Saint-Martin C, Ellezam B, Pouliot L, Sinnett D, et al. Trametinib for progressive pediatric low-grade gliomas. J Neuro-Oncol. (2018) 140:435–44. doi: 10.1007/s11060-018-2971-9
37. Kilburn LB, Khuong-Quang D-A, Hansford JR, Landi D, van der Lugt J, Leary SES, et al. The type II RAF inhibitor tovorafenib in relapsed/refractory pediatric low-grade glioma: the phase 2 FIREFLY-1 trial. Nat Med. (2024) 30:207–17. doi: 10.1038/s41591-023-02668-y
38. Bouffet E, Geoerger B, Moertel C, Whitlock JA, Aerts I, Hargrave D, et al. Efficacy and safety of trametinib monotherapy or in combination with dabrafenib in pediatric BRAF V600–mutant low-grade glioma. J Clin Oncol. (2023) 41:664–74. doi: 10.1200/jco.22.01000
39. Hargrave DR, Terashima K, Hara J, Kordes UR, Upadhyaya SA, Sahm F, et al. Phase II trial of dabrafenib plus trametinib in relapsed/refractory BRAF V600–mutant pediatric high-grade glioma. J Clin Oncol. (2023) 41:5174–83. doi: 10.1200/jco.23.00558
40. Lassaletta A, Zapotocky M, Mistry M, Ramaswamy V, Honnorat M, Krishnatry R, et al. Therapeutic and prognostic implications of BRAF V600E in pediatric low-grade gliomas. J Clin Oncol. (2017) 35:2934–41. doi: 10.1200/jco.2016.71.8726
41. Robert C, Grob JJ, Stroyakovskiy D, Karaszewska B, Hauschild A, Levchenko E, et al. Five-year outcomes with dabrafenib plus trametinib in metastatic melanoma. New Engl J Med. (2019) 381:626–36. doi: 10.1056/nejmoa1904059
42. Hargrave DR, Bouffet E, Tabori U, Broniscer A, Cohen KJ, Hansford JR, et al. Efficacy and safety of dabrafenib in pediatric patients with BRAF V600 mutation–positive relapsed or refractory low-grade glioma: results from a phase I/IIa study. Clin Cancer Res. (2019) 25:7303–11. doi: 10.1158/1078-0432.CCR-19-2177
43. Barbato MI, Nashed J, Bradford D, Ren Y, Khasar S, Miller CP, et al. FDA approval summary: dabrafenib in combination with trametinib for BRAFV600E mutation-positive low-grade glioma. Clin Cancer Res. (2024) 30:263–8. doi: 10.1158/1078-0432.CCR-23-1503
44. Perreault S, Kiaei DS, Dehaes M, Larouche V, Tabori U, Hawkin C, et al. A phase 2 study of trametinib for patients with pediatric glioma or plexiform neurofibroma with refractory tumor and activation of the MAPK/ERK pathway: TRAM-01. BMC Cancer. (2019) 19:1250. doi: 10.1186/s12885-019-6442-2
45. Vinitsky A, Chiang J, Bag AK, Campagne O, Stewart CF, Dunphy P, et al. LGG-22. SJ901: Phase I/II evaluation of single agent mirdametinib (PD-0325901), a brain-penetrant MEK1/2 inhibitor, for the treatment of children, adolescents, and young adults with low-grade glioma (LGG). Neuro-Oncology. (2022) 24:i92–2. doi: 10.1093/neuonc/noac079.336
46. Dhillon S. Tovorafenib: first approval. Drugs. (2024) 84:985–93. doi: 10.1007/s40265-024-02069-6
47. Welsh SJ, Corrie PG. Management of BRAF and MEK inhibitor toxicities in patients with metastatic melanoma. Ther Adv Med Oncol. (2015) 7:122–36. doi: 10.1177/1758834014566428
48. Azizi AA, Hargrave D, Passos J, Wolkenstein P, Rosenbaum T, Santoro C, et al. Consensus recommendations on management of selumetinib-associated adverse events in pediatric patients with neurofibromatosis type 1 and plexiform neurofibromas. Neuro-Oncol Pract. (2024) 11:515–31. doi: 10.1093/nop/npae038
49. Klesse LJ, Jordan JT, Radtke HB, Rosser T, Schorry E, Ullrich N, et al. The use of MEK inhibitors in neurofibromatosis type 1-associated tumors and management of toxicities. Oncologist. (2020) 25:e1109–16. doi: 10.1634/theoncologist.2020-0069
50. Méndez-Martínez S, Calvo P, Ruiz-Moreno O, Barón NP, Bueno JL, Ruiz MdRG, et al. Ocular adverse events associated with MEK inhibitors. Retina. (2019) 39:1435–50. doi: 10.1097/IAE.0000000000002451
51. Urner-Bloch U, Urner M, Jaberg-Bentele N, Frauchiger AL, Dummer R, Goldinger SM. MEK inhibitor-associated retinopathy (MEKAR) in metastatic melanoma: Long-term ophthalmic effects. Eur J Cancer. (2016) 65:130–8. doi: 10.1016/j.ejca.2016.06.018
52. Kline C, Waanders AJ, Ziegler DS, Kilburn LB, Nysom K, Van Der Lugt J, et al. LGG-40. Type II RAF inhibitor tovorafenib in relapsed/refractory pediatric low-grade glioma (PLGG): reversible decreases in growth velocity in the phase 2 firefly-1 trial. Neuro-Oncology. (2024) 26:0–0. doi: 10.1093/neuonc/noae064.431
53. Rizos H, Menzies AM, Pupo GM, Carlino MS, Fung C, Hyman J, et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: spectrum and clinical impact. Clin Cancer Res. (2014) 20:1965–77. doi: 10.1158/1078-0432.ccr-13-3122
54. Nobre L, Zapotocky M, Ramaswamy V, Ryall S, Bennett J, Alderete D, et al. Outcomes of BRAF V600E pediatric gliomas treated with targeted BRAF inhibition. JCO Precis Oncol. (2020) 4):561–71. doi: 10.1200/PO.19.00298
55. Campagne O, Yeo KK, Fangusaro J, Stewart CF. Clinical pharmacokinetics and pharmacodynamics of selumetinib. Clin Pharmacokinet Mar. (2021) 60:283–303. doi: 10.1007/s40262-020-00967-y
56. Zelt S, Cooney T, Yu S, Daral S, Krebs B, Markan R, et al. Disease burden and healthcare utilization in pediatric low-grade glioma: A United States retrospective study of linked claims and electronic health records. Neuro-Oncol Pract. (2024) 11:583–92. doi: 10.1093/nop/npae037
57. Liu X, Fluchel MN, Kirchhoff AC, Zhu H, Onega T. Geographic access to pediatric cancer care in the US. JAMA Netw Open. (2023) 6:e2251524. doi: 10.1001/jamanetworkopen.2022.51524
58. Chinnaswamy G, Gollamudi VRM, Choudhari A, Prasad M, Dasgupta A, Chatterji A, et al. LMIC-27. MEK inhibitors in pediatric low-grade gliomas (PLGG)- early experience in A resource limited setting. Neuro-Oncology. (2024) 26:0–0. doi: 10.1093/neuonc/noae064.744
59. Abu Laban D, Alsharif A, Al-Hussaini M, Obeidat M, Maraqa B, Alzoubi Q, et al. BRAF/MEK inhibitors use for pediatric gliomas; real world experience from a resource-limited country. Front Oncol. (2024) 14:1417484. doi: 10.3389/fonc.2024.1417484
60. WHO Global Initiative for Childhood Cancer: an overview (2020). World Health Organization. Available online at: https://www.who.int/docs/default-source/documents/health-topics/cancer/who-childhood-cancer-overview-booklet.pdf (Accessed Januray 2025).
61. Lim M, Shulman DS, Roberts H, Li A, Clymer J, Bona K, et al. Off-label prescribing of targeted anticancer therapy at a large pediatric cancer center. Cancer Med. (2020) 9:6658–66. doi: 10.1002/cam4.3349
Keywords: pediatric low grade glioma, trametinib, selumetinib, MEK, MAPK pathways, BRAF, CNS tumors, LMIC
Citation: Crotty EE, Sato AA and Abdelbaki MS (2025) Integrating MAPK pathway inhibition into standard-of-care therapy for pediatric low-grade glioma. Front. Oncol. 15:1520316. doi: 10.3389/fonc.2025.1520316
Received: 31 October 2024; Accepted: 22 January 2025;
Published: 11 February 2025.
Edited by:
Ashley Sloane Margol, Children’s Hospital of Los Angeles, United StatesReviewed by:
Antonella Cacchione, Bambino Gesù Pediatric Hospital (IRCCS), ItalyNaureen Mushtaq, Aga Khan University, Pakistan
Copyright © 2025 Crotty, Sato and Abdelbaki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Erin E. Crotty, ZXJpbi5jcm90dHlAc2VhdHRsZWNoaWxkcmVucy5vcmc=
†These authors have contributed equally to this work