 Yu-Ting Ma
Yu-Ting Ma Chan Li
Chan Li Ying Shen2
Ying Shen2 Wan-Hui You
Wan-Hui You Feng-Juan Han
Feng-Juan Han- 1Department of Obstetrics and Gynecology, Heilongjiang University of Chinese Medicine, Harbin, Heilongjiang, China
- 2Department of Obstetrics and Gynecology, First Affiliated Hospital, Heilongjiang University of Chinese Medicine, Harbin, Heilongjiang, China
Ovarian cancer (OC) is the most lethal malignancy in the female reproductive system, and chemotherapy drug resistance is the main cause of treatment failure. The Mitogen-Activated Protein Kinases (MAPK) pathway plays a pivotal role in regulating cell proliferation, migration, and invasive capacity in response to extracellular stimuli. This review focuses on the mechanisms and therapeutic strategies related to the JNK/p38 MAPK signaling pathway in OC resistance. The JNK/p38 MAPK pathway plays a dual role in OC chemoresistance. This review examines its role in mediating OC treatment resistance by exploring the mechanisms of action of the JNK/p38 MAPK signaling pathway, particularly its involvement in several key biological processes, including apoptosis, autophagy, DNA damage response, the tumor microenvironment (TME), and drug efflux. Additionally, the review investigates the timing of activation of this pathway and its crosstalk with other signaling pathways such as PI3K/AKT and NF-κB. Targeting JNK/p38 MAPK signaling has shown promise in reversing chemoresistance, with several inhibitors and natural compounds demonstrating potential in preclinical studies. Regulating JNK/p38 MAPK may transform what was once a terminal obstacle into a manageable challenge for OC patients with chemotherapy resistance, ultimately improving survival and quality of life.
1 Introduction
Ovarian cancer (OC) is the gynecologic malignancy with the highest mortality rate, with approximately 19,680 new cases of OC diagnosed in the United States in 2024 (1). OC accounts for 5.1% of all female cancer deaths, with a 5-year survival rate of 50.8% (2). The global incidence of OC is anticipated to rise by 47% and the number of deaths by 58% by 2045 (3). OC is divided into type I and type II, with type I including Low-Grade Serous Ovarian Carcinoma (LGSOC), clear cell carcinoma, mucinous carcinoma, and endometrioid carcinoma. Type II includes high-grade serous ovarian carcinoma (HGSOC), undifferentiated carcinoma, and malignant mixed mesodermal carcinosarcoma.
First-line treatment for OC includes surgery and platinum- and paclitaxel-based chemotherapy, but approximately 70% of patients relapse and develop platinum and paclitaxel resistance (4). The main mechanisms contributing to chemotherapy resistance in OC include dysregulation of drug transport, alterations of apoptosis, DNA damage repair, alteration of TME, as well as gene mutation and signaling pathway abnormalities (5). Mutations in four genes—Tp53, KRAS, BRCA1/2 and PIK3CA—are strongly associated with the etiology of OC and drug resistance (6).
The Mitogen-Activated Protein Kinases (MAPK) signaling pathway plays a pivotal role in several biological processes, including cell proliferation, differentiation, survival, and stress response (7). It is a fundamental signaling pathway within cells, and its dysregulation is closely associated with the onset and development of OC and the emergence of drug resistance (8). Extracellular Signal-Regulated Kinases (ERK) are extensively researched MAPK signaling pathways that are closely associated with cell growth and developmental divisions. Their transduction pathways adhere to a three-stage enzymatic cascade. In the delivery pathway of ERK, RAS acts as an upstream activating protein, RAF acts as a MAPK Kinase Kinase (MAPKKK), which further activates MEK1/2 through its kinase activity, and MEK1/2 acts as a MAPK Kinase (MAPKK), which can phosphorylate and activate ERK or MAPK. These are activated by adenosine triphosphate (ATP) phosphorylation to form the RAS/RAF/MEK/ERK pathway (9).
A substantial body of evidence indicates that high-frequency mutations in RAS/RAF, which result in aberrant activation of RAS/RAF/MEK/ERK signaling, frequently contribute to drug resistance and poor prognoses in patients with OC (10). Extensive research on the RAS/RAF/MEK/ERK pathway in the context of OC resistance has facilitated the development of numerous MAPK inhibitors, such as Trametinib, Cobimetinib, and Binimetinib. These have been applied to a small subgroup of OC patients to control tumor growth, particularly in LGSOC. For instance, the MEK inhibitor, binimetinib, has been demonstrated to confer clinical benefit to patients with epithelial OC exhibiting MAPK pathway alterations, enhancing the efficacy of paclitaxel-induced apoptosis (11). Additionally, trametinib has emerged as a new standard treatment option for patients with recurrent LGSOC (12).
OC treatment resistance is a complex process involving many molecular mechanisms and signaling pathways. Previous studies have demonstrated that c-Jun N-terminal kinase (JNK) and p38 MAPK impede the progression of OC by regulating apoptosis in response to stressors such as carcinogens, reactive oxygen species (ROS), or oncogenes (13). Fawzy et al. (14) proposed that elevated JNK activity, which contributes to OC chemoresistance, exhibits partial overlap with other pathways, including phosphoinositide 3-kinase (PI3K), nuclear factor-κB (NF-κB), and multiple mechanisms mediating cell transformation and apoptosis.
However, the role of the JNK/p38 MAPK pathway in OC resistance has not been extensively investigated. This review focuses on the mechanism of the JNK/p38 MAPK signaling pathway in OC chemoresistance and provides an overview of potential therapeutic strategies targeting these pathways. The findings will direct researchers’ attention to the mechanism of OC resistance and offer novel research avenues.
2 Overview of MAPK signaling pathway
MAPK consists of four major cascades: the ERK1/2, ERK5, JNK, and p38 MAPK signaling pathways. The ERK signaling pathway is closely related to cell proliferation and differentiation, while the JNK and p38 MAPK signaling pathways are predominantly linked to cellular stress and apoptosis (Figure 1).
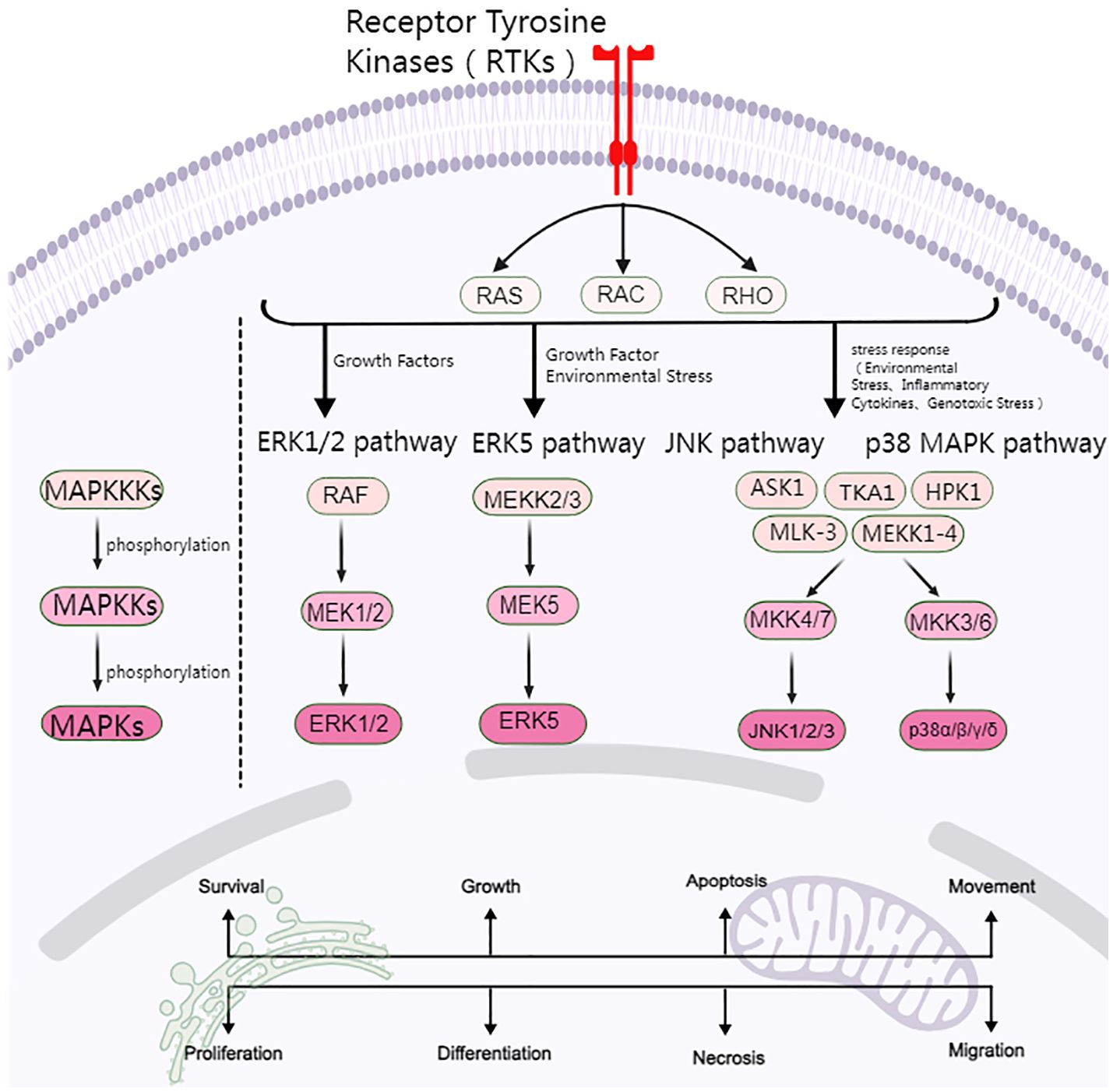
Figure 1. Overview of the MAPK signaling pathway.
2.1 ERK signaling pathway
The RAS/RAF/MEK/ERK pathway represents a highly conserved signaling cascade that plays a pivotal role in the survival and development of tumor cells. OC cisplatin resistance is regulated between p53 and RAS signaling networks through RAS/MAPK pathway activation, which mediates apoptosis and autophagy (15). Synuclein is a small neuron protein, and some studies have found that γ-synuclein is significantly upregulated in OC (16). Pan et al. (17) demonstrated that the overexpression of γ-synuclein activated ERK and downregulated JNK, collectively preventing apoptosis. This cell apoptosis is inhibited by 2–3 times, resulting in resistance to paclitaxel and Vincristine chemotherapy. However, the use of MEK1/2 inhibitors has been demonstrated to partially mitigate this resistance to paclitaxel.
Evidence indicates that high-frequency mutations in RAS/RAF result in aberrant RAS/RAF/MEK/ERK signaling activation, leading to platinum-based chemotherapy resistance in OC cells. Manning-Geist et al. found that in type I OC, 60% of LGSOC have alterations in the MAPK pathway (18), with 30–50% and 15–40% carrying RAS and RAF mutations, respectively (19). Additionally, 88% of junctional tumors (20) and 75% of mucinous ovarian tumors also have RAS or RAF mutations (21). Type II tumors almost universally have p53 mutations, and nearly half have BRCA1/BRCA2 mutations (22). Approximately 12% of HGSOC tumors, the most common histological subtype of OC, have been found to have RAS mutations. In OC, particularly in HGSOC, the percentage of RAS gene mutations is generally lower than 10%. However, the transcription of RAF and ERK1/2 can still activate the RAS/RAF/MEK/ERK pathway, which is closely related to the prognosis of OC (23). Consequently, targeting the ERK signaling pathway with RAF, MEK, and ERK inhibitors in OC patients (particularly those with LGSOC) has yielded promising preliminary clinical outcomes compared to the established standard of care (24–27) (Table 1).
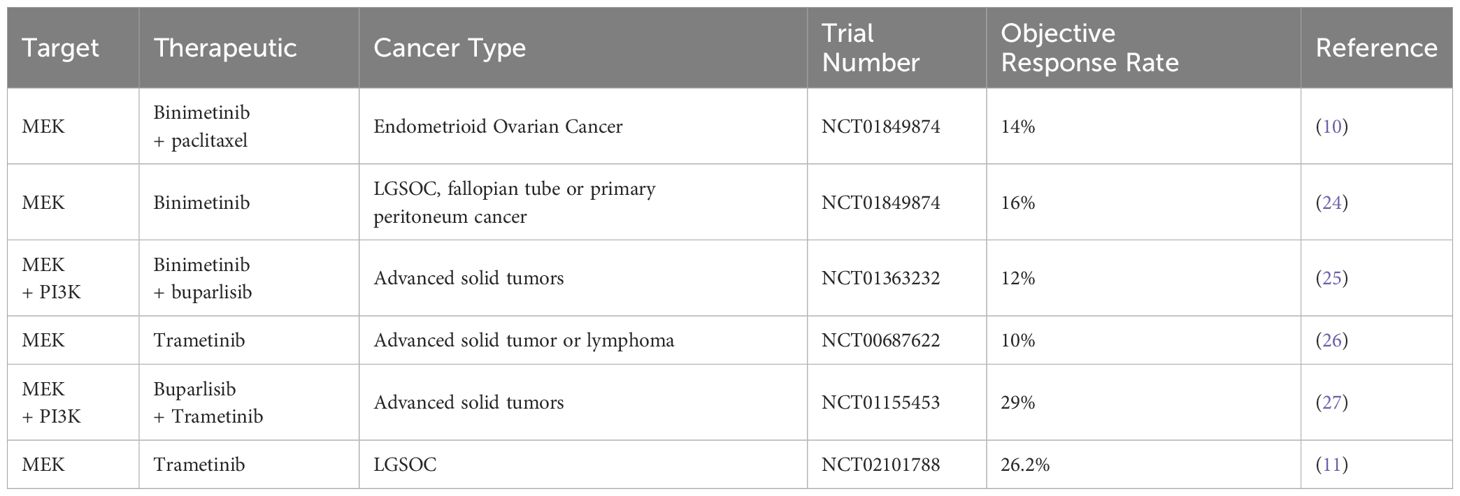
Table 1. Targeting the RAS/RAF/MEK/ERK signaling pathway for OC treatment.
2.2 JNK/p38 MAPK signaling pathway
The JNK and p38 MAPK pathways are primarily activated by environmental and genotoxic stressors and regulate cellular responses to various stimuli, including cytokines, inflammatory factors, and growth factors. Consequently, they are often called Stress-Activated Protein Kinases, and they are primarily involved in cellular stress and apoptosis (28).
2.2.1 JNK signaling pathway
The JNK pathway represents a significant branch of the MAPK signaling family, characterized by its capacity to phosphorylate and activate c-Jun transcription factors (29). The JNK pathway is activated by external stimuli, including ultraviolet light, inflammatory factors, and oxidative stress, and regulates the expression of a series of downstream genes. The JNK family comprises three major isoforms: JNK1, JNK2, and JNK3 (30). JNK1 and JNK2 are expressed in most tissues, whereas JNK3 is expressed in a more limited set of tissues, including the brain, heart, and testis (31).
MAPKKKs (e.g., ASK1, HPK1, MLK-3) are the initial kinases to be activated following cell stimulation by stress. These kinases then phosphorylate MAPKKs (MKK4 and MKK7), triggering a signaling cascade (32). MKK4 and MKK7 activate JNK proteins by phosphorylating threonine (Thr183) and tyrosine (Tyr185) at the Thr-Pro-Tyr (TPY) motif of JNK (33, 34). ROS are also involved in JNK activation (33, 34).
Activated JNK protein spreads in the cytoplasm and enters the nucleus. Its phosphorylation leads to the activation or modulation of a range of non-nuclear and nuclear proteins, including c-Jun, Activator Protein-1 (AP-1), and FBJ murine osteosarcoma viral oncogene homolog (FOS). Transcription Factor 2 (ATF-2) and p53, as well as B-cell lymphoma-2 (Bcl-2) and Bcl-2-associated X protein (Bax), are involved in regulating cell proliferation, apoptosis, autophagy, and DNA repair, and they also play a role in influencing OC resistance (35, 36).
2.2.2 p38 MAPK signaling pathway
p38 MAPK is primarily activated by extracellular inflammatory factors, environmental stress, oxidative stress, and DNA-damaging agents (e.g., cisplatin and adriamycin) and is crucial for maintaining cellular homeostasis. The p38 MAPK family comprises four members: p38α, p38β, p38γ, and p38 (35, 36). p38α is universally expressed in most tissues, whereas p38β, p38γ, and p38δ are specifically expressed only in a small number of tissues (37).
Similarly, upstream MAPKKK (TAK1, ASK1, MEKK1–4) is initially activated by dual phosphorylation of Thr and Tyr at the TGY motif, which is then followed by the activation of MAPKKs (MKK3 and MKK6). These MAPKKs subsequently activate p38 by phosphorylating specific serine and Thr residues of p38. It is noteworthy that MKK4 can also phosphorylate p38 MAPK (38). p38 phosphorylates a range of substrates, including the transcription factors ATF2, CHOP, the kinases MK2, MK3, and other effector proteins, which regulate cell cycle progression, cell survival, apoptosis, stress responses, and inflammatory responses. Consequently, p38 plays a role in influencing OC resistance.
JNK/p38 MAPK is often regarded as a critical mediator of cell death and can act as a tumor suppressor (14). However, it can also act as a tumor promoter under certain conditions. Evidence suggests that JNK may play a role in counteracting apoptosis and promoting cancer cell survival under low oxygen or other forms of cellular stress (39). Accordingly, this study will concentrate on the particular mechanisms of JNK and p38 MAPK signaling pathways in drug-resistant OC to offer new insights for future clinical treatment.
3 Mechanisms of JNK and p38 MAPK-mediated drug resistance in ovarian cancer
Due to the rapid development of chemoresistance, OC remains a significant challenge in oncology. In this section, we summarize the mechanisms involved in JNK/p38 MAPK and OC resistance. These include apoptosis, autophagy, DNA damage response, the tumor microenvironment, drug metabolism, microRNA, activation time, and crosstalk between cell signaling pathways (Figure 2).
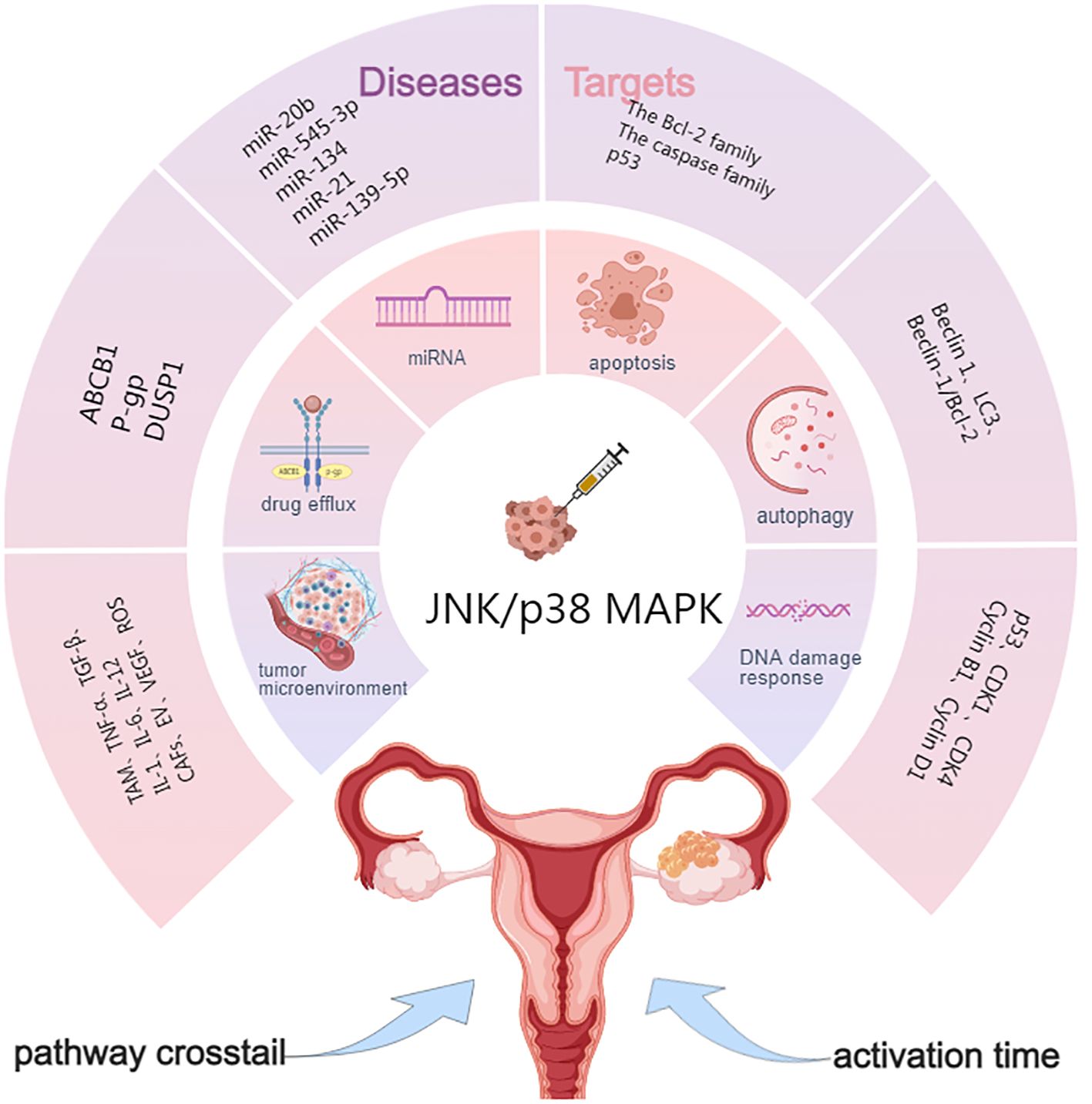
Figure 2. Targets of JNK/p38 MAPK signaling pathway in OC drug resistance. ABCB1, ATP-binding cassette sub-family B member 1; P-gp, P-glycoprotein; DUSP1, Dual specificity phosphatase 1; Beclin 1, LC3, Genes involved in autophagy; TME, Tumor microenvironment; CAFs, Cancer-associated fibroblasts; VEGF, Vascular endothelial growth factor; ECM, Extracellular matrix; EVs, Extracellular vesicles.
3.1 Apoptosis of cells
Apoptosis, a natural process of programmed cell death, is essential for regulating cellular homeostasis. Dysregulation of apoptosis is a key factor in tumorigenesis and cancer progression. Chemotherapeutic agents exert their anti-tumor effects mainly by inducing apoptosis in cancer cells. The JNK/p38 MAPK signaling pathway modulates the expression and activity of key apoptotic regulators, including the Bcl-2 family, caspase family, and p53, thereby orchestrating critical decisions in apoptotic machinery (37).
3.1.1 The Bcl-2 family
Several factors are responsible for the failure of apoptosis, and the Bcl-2 family, a core factor of apoptosis, is closely associated with chemoresistance in OC (38). The Bcl-2 family is a critical regulator of apoptosis, comprising anti-apoptotic proteins such as Bcl-2, Bcl-extra large (Bcl-xL), and myeloid cell leukemia 1 (Mcl-1), as well as pro-apoptotic proteins including Bcl-2-associated X protein (Bax), Bcl-2-associated death promoter (Bad), and a subgroup of “BH3-only” proteins (e.g., BH3-interacting domain death agonist [Bid], Bcl-2-interacting mediator of cell death [Bim], p53 upregulated modulator of apoptosis [Puma], and phorbol-12-myristate-13-acetate-induced protein 1 [Noxa]). These members orchestrate mitochondrial outer membrane permeabilization through dynamic interactions, ultimately determining cellular fate. Anti-apoptotic proteins prevent apoptosis by stabilizing mitochondrial membrane integrity, while pro-apoptotic proteins promote the increase of mitochondrial outer membrane permeability. JNK/p38 MAPK was shown to induce apoptosis of OC-resistant cells by phosphorylating and inactivating Bcl-2 (39, 40).
BH3-only is a subfamily of the Bcl-2 family, which is considered to promote mitochondrial apoptosis. “BH3-mimetic drugs” inhibit the expression of specific pro-survival Bcl-2 family proteins, promote cell apoptosis, and overcome OC chemotherapy resistance (41). Naftopidil, an α1-adrenergic receptor antagonist, has been shown to overcome resistance to the MEK inhibitor trametinib in OC by activating the JNK signaling pathway and inducing BH3-only protein expression (42). Additionally, arsenic trioxide has been shown to promote apoptosis in OC platinum-resistant cells (43). Yuan et al. (44) found that arsenic trioxide induces apoptosis in OC cells by activating Bim signaling, a member of the BH3-only family, through JNK signaling. Furthermore, they determined the threshold for overcoming cisplatin resistance. Sab is a mitochondrial outer membrane scaffolding protein, and high levels of the protein are associated with decreased Bcl-2 protein and increased BH3-only proteins, thereby restoring the sensitivity of OC cells to chemotherapeutic agents (45). Further studies found that subchronic JNK inhibition induces changes in the concentration of Bcl-2 and Bim proteins by altering Sab expression, leading to increased OC resistance.
3.1.2 The caspase family
The cysteinyl aspartate-specific proteinase (caspase) family also plays a central role in the executive phase of apoptosis. Caspases can also reverse chemotherapy resistance by cutting specific proteins, leading to the destruction of cell structure and function and triggering endogenous or exogenous cell death events. Among them, caspase-3 is a central effector of apoptosis (46). Pan et al. (17) found that overexpression of γ-synuclein can inhibit the activation of the JNK pathway, reduce the phosphorylation of downstream caspase-3, prevent cell apoptosis, and lead to chemotherapy resistance to paclitaxel and vinblastine. Yan et al. (47) showed that prostaglandin could regulate JNK activity and prevent OC cell apoptosis by reducing caspase-3 activity. MT-4, a derivative of mescalin, has been shown to activate p38 MAPK phosphorylation, reduce the expression of heat shock protein 27, and promote the expression of caspase-3 activity, thereby overcoming OC resistance (48). Tang et al. (49) found that cross-reactive material 197 (CRM197), a specific HB-EGF inhibitor, could enhance caspase-3 activity and OC apoptosis via the JNK/p38 MAPK pathway and reverse paclitaxel resistance.
3.1.3 p53
JNK can also promote apoptosis by regulating p53, which is a transcription factor involved in apoptosis, cycle regulation, DNA repair, and other processes (50). The p53 protein plays a key role in Bcl-mediated apoptosis by regulating the pro-apoptotic BH3-only proteins PUMA and NOXA to induce apoptosis (38). It can also prevent the binding of Bcl-2 to pro-apoptotic proteins such as BAX, thereby deregulating the anti-apoptotic function of Bcl-2 and promoting apoptosis (38). Yuan et al. (51) found that PUMA, a p53-upregulated regulator of apoptosis, can sensitize SKOV3 cells to chemotherapy by enhancing caspase activation and downregulating Bcl-xL and Mcl-1, resulting in apoptosis. Studies have shown that p53 mutations lose their cancer suppressor function and promote tumor initiation and progression (52). Potapova et al. (53) demonstrated that inhibiting the expression of JNK1/2 using highly specific JNK antisense oligonucleotides suppressed the growth of cells with mutated p53 (e.g., HGSOC) but did not affect the growth of cells with normal p53 function. This suggests that JNK exerts its pro-apoptotic function only in P53-deficient tumor cells. Using in vitro studies, Zhao et al. (54) demonstrated that insulin and cisplatin can activate the JNK signaling pathway, increase p53 expression and the percentage of S-phase cells, promote apoptosis, and enhance the efficacy of cisplatin (Table 2).
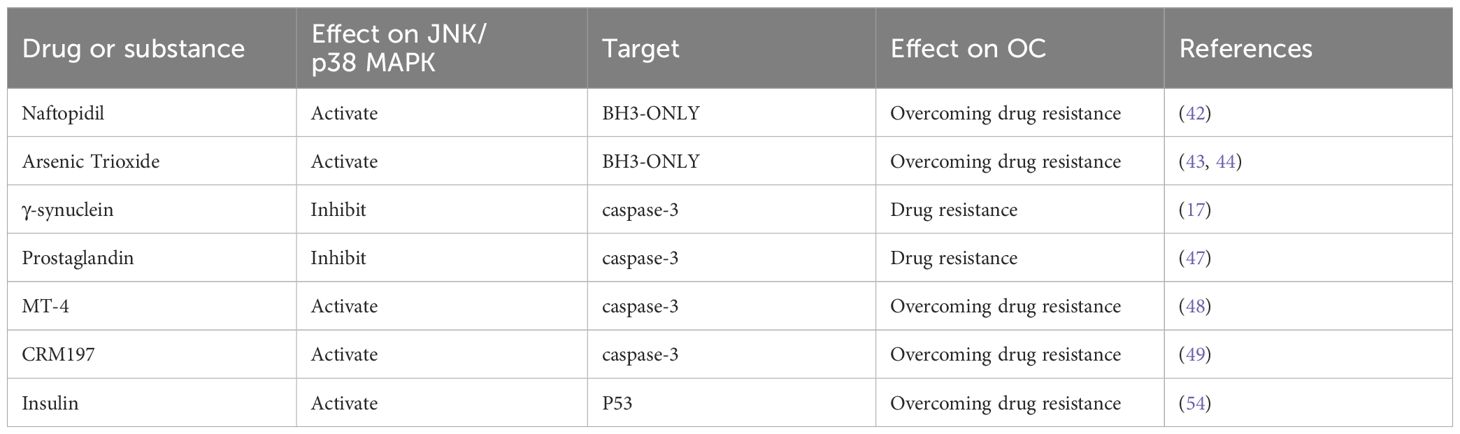
Table 2. Substances affecting OC resistance through apoptosis.
3.2 Autophagy
Autophagy is a cellular process of self-degradation and self-protection. Under stress conditions such as chemotherapy, hypoxia, and radiation, autophagosomes are formed to encapsulate damaged organelles and misfolded proteins in the cytoplasm and transport them to lysosomes for degradation, thereby maintaining cellular homeostasis (55). Therefore, autophagy is activated when OC cells are stimulated by chemotherapy drugs (56). Autophagosome formation and increased autophagy flux enable tumor cells to clear the cell damage caused by chemotherapy drugs so that these cells can survive under drug pressure, leading to the generation of drug resistance (57). However, excessive or sustained autophagy can directly mediate or contribute to cell death and reverse OC resistance (57, 58).
When cells are exposed to commonly used chemotherapy drugs such as cisplatin or paclitaxel, JNK/p38 MAPK activates and regulates the expression of autophagy-associated proteins, promoting autophagosome formation and enhancing autophagy flow. Beclin 1 and Microtubule-associated protein 1 light chain 3 (LC3) are crucial in autophagy. The JNK/p38 MAPK signaling pathway induces autophagy in OC cells and increases autophagosome formation and autophagic flux by promoting the conversion of LC3-I to LC3-II and the formation of autophagosomes (59). Zhu (60) et al. analyzed the mutation profiles of 62 HGSOC and found that overexpression of FGF19 in the fibroblast growth factor family promotes the phosphorylation of p38 to induce autophagy, upregulates the expression of LC3 and Beclin-1, and promotes cisplatin resistance in OC cells. Conversely, the knockdown of FGF19 downregulates the phosphorylation of p38 to reverse cisplatin chemoresistance.
The Unfolded Protein Response (UPR) is a pro-survival mechanism that is activated when unfolded or misfolded proteins accumulate in the endoplasmic reticulum (ER), leading to JNK/Ap-1 activation and increased autophagy (61). Yang et al. (62) found that JNK3 activation through the UPR pathway promoted acid/lysosomal compartment accumulation, blocked autophagy flux during ER stress, and reduced ROS levels in OC-resistant cells to promote OC cell survival.
Sustained activation of the JNK/p38 MAPK pathway can lead to apoptosis or excessive autophagy, resulting in a state of “autophagy cell death” or “autophagy addiction.” JNK/p38 MAPK can also reverse OC resistance by regulating the balance between apoptosis and autophagy (63). Zhao et al. (64) found that propranolol can upregulate the expression of LC3-II and caspase-3 by activating the ROS/JNK signaling pathway, induce apoptosis and autophagy of OC cells, and restore cell sensitivity to chemotherapy. Guo et al. (65) demonstrated that MAP2K6-FP, a genetically engineered fusion protein targeting the MAPK kinase pathway, enhances paclitaxel sensitivity in OC by inducing autophagy. This chimeric protein contains three functional domains (1): a HO8910 OC cell-specific binding peptide (2), the TAT protein transduction domain for cellular internalization, and (3) the MKK6(E) effector domain with anti-tumor activity. Mechanistically, MAP2K6-FP upregulated p38 expression, elevated LC-3 and Beclin-1/Bcl-2 levels, and amplified autophagic flux, thereby overcoming chemoresistance in OC models. Mishra et al. (66) showed that photothermal therapy can induce autophagy to overcome drug resistance by activating JNK signaling and UPR signaling pathways in OC chemoresistant cells.
While autophagy serves as a self-protection mechanism that enables resistance of OC cells to chemotherapy, sustained activation of autophagy by JNK/p38 MAPK signaling can trigger the “autophagic cell death process.” Autophagic cell death can override cellular survival mechanisms by excessive autophagy or modulating the balance between apoptosis and autophagy. This process re-sensitizes OC cells to chemotherapeutic agents and reverses OC drug resistance.
3.3 DNA damage response
Platinum chemotherapeutic agents, such as cisplatin and carboplatin, impede DNA replication and transcription by twisting the DNA Double helix, causing single-strand breaks, double-strand breaks (DSB), and chromosome rearrangements, ultimately resulting in cell death. When DNA damage occurs, abnormal activation of the DNA damage response (DDR) is responsible for pausing the cell cycle and initiating DNA repair. Cell cycle checkpoints regulate cell cycle transitions by acting on the cell’s G1, S, and G2/M phases. This process can provide time for DNA repair when cell cycle arrest occurs, leading to chemotherapy resistance (67). However, in some cases, inducing cell cycle arrest at specific stages and increasing the accumulation of DNA damage can promote apoptosis and reverse chemoresistance in OC cells.
3.3.1 DNA damage repair
JNK/p38 MAPK is activated when stimulated by chemotherapeutic agents to regulate apoptosis and enhance the effect of chemotherapeutic agents. However, in DDR, JNK/P38 MAPK can be activated in response to DNA damage, participating in the DNA damage repair process and leading to treatment resistance (68, 69). Cell division cycle 2 (Cdc2)-like kinase plays a role in RNA splicing, cell cycle regulation, DDR, and other cellular functions. Jiang et al. (70) found that p38 can enhance DNA damage repair through phosphorylation of Cdc2-like kinase, resulting in OC resistance. Erlotinib exerts its anti-tumor effects by inducing DSBs in cancer cells, triggering cell death by the accumulation of unrepaired DNA damage (71). However, p38 MAPK activation can initiate the DDR mechanism, which repairs the DNA damage and regulates cell cycle checkpoints. This repair process reduces DNA damage-induced apoptosis, ultimately contributing to erlotinib resistance (72). In p53-deficient tumor cells, the p38/MK2 pathway is activated and reactivates cell cycle checkpoints to repair the damage, thereby inducing chemotherapy resistance (73, 74). However, when combined with platinum or paclitaxel, RNA peptide nano complexes blocked cell cycle inspection of p38/MK2, sensitized pp53 deficient HGSOC mice to chemotherapeutic agents, and improved overall survival by 37% (75). Seino et al. (76) demonstrated that the high activity of JNK would promote cisplatin resistance through various phosphokinase assays, and the combination of cisplatin and JNK inhibitor (SP600125) could enhance DNA damage to reverse cisplatin resistance. The longevity gene SIRT6 has been identified as a critical factor in stimulating DSB repair. Studies have shown that JNK activates DNA DSB repair by stimulating the phosphorylation of SIRT6 during oxidative stress conditions, leading to OC resistance (77).
3.3.2 Mechanisms of cell cycle regulation
JNK/p38 MAPK can reverse chemoresistance by inducing cell cycle arrest and amplifying DNA damage accumulation. Cyclin D1 and CDK4/6, critical regulators of the G1/S transition, form complexes that drive cell cycle progression (78). However, p38 MAPK inhibits this process by degrading Cyclin D1 and disrupting Cyclin D1/CDK4 complexes, thereby arresting cells in the G1/S phase (79). Cannell et al. (80) demonstrated that p38 could bind to growth arrest and DNA damage-inducible 45α (Gadd45α), control G1/S and G2/M cell cycle checkpoints, and influence OC resistance. Similarly, tocotrienols activate JNK/p38 MAPK signaling to induce G1/S arrest, leading to OC cell death and re-sensitization to cisplatin (81).
In the G2/M phase, Cyclin B1/CDK1 complexes promote cell cycle progression (82), but p38 MAPK exerts checkpoint control by degrading Cyclin B1. This regulatory mechanism is exploited by therapeutic agents such as Chaetoglobosin K (ChK), a fungal metabolite that induces G2 arrest through p53 activation and p38-dependent Cyclin B1 phosphorylation, selectively inhibiting cisplatin-resistant OC cells (83). Conversely, cell cycle and apoptosis regulator 2 promote cisplatin resistance by suppressing p38 signaling and sustaining Cyclin B1/CDK1 activity (84).
Notably, pharmacological modulation of this checkpoint demonstrates clinical potential. The synthetic compound MPT0G066 (13.28-fold more potent than paclitaxel) activates JNK to induce G2/M arrest, thereby re-sensitizing resistant OC cells to cisplatin (85). LB100, a PP2A inhibitor, eliminates cisplatin-induced G2/M arrest by prolonging JNK activation, effectively overriding checkpoint-mediated survival and restoring chemosensitivity (86).
3.4 The tumor microenvironment
TME consists of immune cells, Cancer-associated Fibroblasts (CAFs), Vascular Endothelial Growth Factor (VEGF), adipocytes, Extracellular Matrix (ECM), and extracellular vesicles (EVs), which play a crucial role in OC treatment resistance. Due to the unique characteristics of OC metastasis in the peritoneal cavity, OC cells can survive in ascites as floating unicellular or multicellular spheres, forming a characteristic OC TME (87). Increasing evidence suggests that targeting the TME may be a promising strategy for reversing OC chemoresistance.
Tumor-associated macrophages, an essential component of immune cells in TME, can secrete many pro-inflammatory cytokines, TNF-α, TGF-β, IL-1, IL-6, and IL-12, to enhance the immune response. JNK can be activated by TGF-β, interferon-γ, and other mediated immune evasion mechanisms that affect OC resistance (76, 88). Mitra et al. (89) found that TGFβ1 can induce stemness and chemoresistance in OC cells by activating ERK1/2 and JNK/p38 MAPK pathways and targeting them to participate in epithelial-mesenchymal transition (EMT). Bioinformatic analysis of Gene Expression Omnibus and The Cancer Genome Atlas databases revealed that CAFs affect OC chemoresistance through the p53, cell cycle, PI3K-AKT, and MAPK pathways (90). Izar et al. (91) found that stimulating the expression of CAFs stimulated the formation of OC-specific TME, which increased HGSOC aggressiveness and drug resistance. IL-6 secreted by CAFs is one of the essential pro-inflammatory cytokines and plays a crucial role in OC resistance (92). Alspach et al. (93) reported that p38 promotes IL-6 expression by affecting the RNA-binding protein AUF1, but inhibition of p38 blocks the tumor-promoting ability of CAFs. Curtis et al. (94) found that p38 can provide nutrients through glycogen phosphorylation to alter the metabolic state in OC TEM and support the survival of drug-resistant OC cells. Samuel et al. (95) found that administering chemotherapeutic agents to OC cells induced EV release via JNK/p38 MAPK signaling, increasing drug resistance in OC cells. Octreotide, a somatostatin analogue, in combination with paclitaxel, can reverse paclitaxel resistance by inhibiting p38 and decreasing the expression of VEGF (96).
ROS, a critical regulator of TME, is closely related to the occurrence of OC resistance (97). Elevated levels of ROS can induce oxidative DNA damage and stimulate long-term activation of JNK, which plays an essential role in reversing drug resistance (98). ROS can mitigate OC resistance by oxidizing cysteine residues of ASK1, thereby activating the downstream JNK/p38 MAPK pathway (99). Li et al. (100) found that BH3-only protein mimic ABT-737 enhanced the activation of JNK and ASK1 by increasing ROS levels in cells in a time-dependent manner, overcoming cisplatin resistance in A2780/DDP cells. Ovendazole can inhibit the proliferation of drug-resistant OC cells through ROS-mediated activation of the JNK/p38 MAPK signaling pathways, leading to G1/S or G2/M cell cycle arrest (99).
Thus, JNK/p38 MAPK activation interacts with immune cells and pro-inflammatory factors in the TME to support the survival of OC cells, leading to chemoresistance. However, during persistently elevated levels of ROS, sustained activation of JNK/p38 MAPK can lead to apoptosis of otherwise drug-resistant cells. This finding offers insights into the development of novel therapeutic strategies for treating OC resistance.
3.5 Drug effluent
Multidrug Resistance (MDR) leads to a lack of sensitivity to multiple chemotherapy drugs, which is the main reason for chemotherapy failure. The overexpression of ATP-binding cassette (ABC) transporters is the leading cause of MDR in chemotherapy. P-glycoprotein (P-gp), the most studied protein of the ABC transporter family, is encoded by the ABCB1 gene encodes and affects the uptake of chemotherapy drugs by pumping them out of tumor cells (101).
Evidence suggests that the p38 signaling pathway can enhance drug efflux and participate in OC resistance formation by inducing the upregulation of ABC transporter protein expression. p38 can increase the expression of ABC transporter proteins by activating the downstream transcription factor NF-κB, which binds to and upregulates the ABCB1 promoter. Afatinib, an ATP-competitive aniline-quinazoline compound, can inhibit ABCB1 transcriptional expression by downregulating p38-dependent activation of NF-κB, thereby reversing ABCB1-mediated MDR (102). Dual specificity phosphatase 1 (DUSP1), also known as MKP-1, is a class of phosphokinase capable of dephosphorylating MAPK substrates. Overexpression of DUSP1 leads to increased phosphorylation of p38. Kang et al. (103) found that activation of p38 by DUSP1 enhanced the expression of P-gp, but did not alter the activation of ERK1/2 and JNK1/2, ultimately affecting the efflux of anticancer drugs from the OC.
3.6 MicroRNA
MicroRNA (miRNA) is a class of small non-coding RNAs, which are 18–25 nucleotides long, and alter tumor sensitivity to treatment by regulating gene expression and apoptosis (104). Dysregulation of miRNA expression is an important cause of OC chemotherapy resistance (105). Therefore, correcting miRNA dysregulation is a promising therapeutic strategy to reverse chemoresistance (106).
Inhibition of the JNK/p38 MAPK signaling pathway was found to downregulate the expression levels of miRNAs involved in regulating chemotherapeutic drug sensitivity-related miRNAs and mediated OC chemoresistance. Kumar et al. (107) showed that the downregulation of miR-20b targets genes such as DUSP8 and inhibits p38 in A2780/CP70 drug-resistant OC cell lines, leading to cisplatin resistance. Yin et al. (108) showed that downregulating miR-545-3p leads to cisplatin resistance by reducing the activity of the JNK signaling pathway. Fos-related antigen-1 (Fra-1), a part of the AP-1 transcription factor complex, can amplify ERK/JNK signaling and reduce chemosensitivity in OC cells by promoting miR-134 expression (109). Activation of JNK-1/c-Jun pathway was found to upregulate its miR-21 expression, leading to decreased levels of the tumor suppressor gene programmed cell death protein 4 (PDCD4), increased cell proliferation, and development of OC cisplatin resistance (110). Jiang et al. (111) showed that restoring the expression of miR-139-5p in drug-resistant cells can inhibit the expression of c-Jun, increase the expression of Bcl-xl, activate caspase-9 and caspase-3, and reverse cisplatin resistance in OC (Table 3).
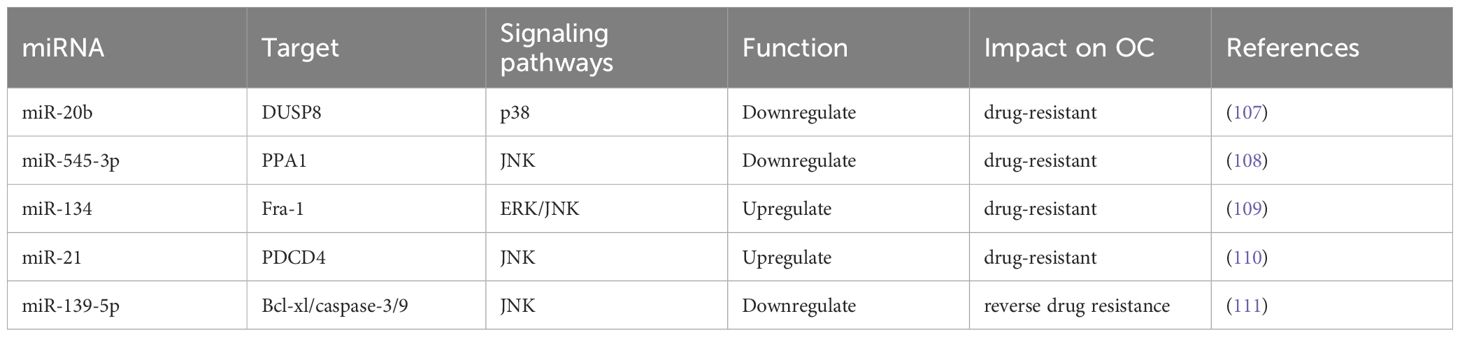
Table 3. Effect of miRNAs on OC drug resistance.
3.7 Activation time
Further study of the JNK/p38 MAPK pathway has revealed that different activation times may lead to different cellular responses and biological effects. The JNK/p38 MAPK pathway is usually activated during the initial phase of chemotherapy to induce apoptosis or other cell death mechanisms. However, in drug-resistant cells, activation of the pathway may be delayed, attenuated, or shortened in duration.
Different activation timings may determine the fate of OC cells. Transient JNK/p38 MAPK activation is usually associated with pro-survival effects, whereas sustained JNK/p38 MAPK activation may be pro-apoptotic (112). Davis et al. (112) found that the activation time of JNK/p38 MAPK persisted for 12 hours in cisplatin-sensitive cells, while it was limited to 1–3 hours in cisplatin-resistant cells. LB100 can eliminate cisplatin-induced cell cycle arrest and sensitize OC cells to cisplatin by altering the temporal order and persistence of JNK activation (91). Elevated expression of HB-EGF (an EGFR ligand) contributes to cancer cell resistance to paclitaxel, and shedding the extracellular domain of HB-EGF can induce sustained activation of JNK/p38 MAPK to overcome this resistance (113). Mansouri et al. (114) showed that the JNK/p38 MAPK pathway plays a central role in cisplatin-induced apoptosis of OC cells by activating the expression of the downstream target pro-apoptotic cytokine FasL. Furthermore, the duration of sustained activation is a critical early determinant of cisplatin-induced apoptosis, as transient activation fails to maintain c-Jun and ATF-2 phosphorylation and upregulate FasL expression.
Thus, successful treatment depends not only on the combination and dosage of drugs but also on the timing, duration, and order of administration (115). A better understanding of the dynamic process of OC resistance will facilitate the clinical development of more effective therapeutic strategies for OC resistance.
3.8 Crosstalk between signaling pathways
The mechanism of drug resistance in OC is complex and multifactorial, involving multiple pathways. The PI3K/AKT pathway engages in bidirectional crosstalk with JNK/p38 MAPK, creating a dynamic equilibrium that dictates OC cell survival under therapeutic stress. Mechanistically, AKT suppresses JNK-mediated apoptosis by phosphorylating and inactivating ASK1, a key upstream kinase in the JNK pathway (116). Conversely, p38 inhibition prevents AKT dephosphorylation and inhibits the AKT pathway, thereby reinforcing PI3K/AKT-dependent survival signaling—a paradoxical interaction that underscores the complexity of pathway interdependencies (117). This stress-adaptation mechanism contributes to the development of OC resistance. For example, the ER protein HERPUD1 activates both PI3K/AKT/mTOR and p38 pathways to maintain autophagy and block apoptosis, thereby driving platinum resistance in OC models (118). Clinically, compensatory pathway activation poses a significant challenge. Resistance to PI3K/AKT inhibitors in OC patients is frequently associated with ERK upregulation and p38 suppression, suggesting a survival mechanism where ERK compensates for inhibited AKT. Notably, dual inhibition of ERK and AKT disrupts this cytoprotective feedback, synergistically eliminating cisplatin-resistant OC cells (119, 120). Growth factor signaling further amplifies this network, with FGF2 overexpression activating p38 and AKT to upregulate anti-apoptotic proteins (Bcl-2/Bcl-xL) and cell cycle drivers (Cyclin D1). Co-targeting FGF2-activated p38 and PI3K/AKT pathways reverses adriamycin resistance, highlighting the therapeutic potential of combinatorial approaches (121).
NF-κB acts as a key sink node to promote inflammatory responses and cellular survival under stress by blocking DNA damage-induced apoptosis through the inhibition of sustained activation of JNK (122, 123). Meanwhile, p38 upregulates anti-apoptotic proteins (such as Bcl-xL) and inflammatory factors (such as IL-6) through NF-κB activation. Hence, p38 promotes OC cell survival (124, 125).
Additionally, Wnt/β-catenin and HIF-1α pathways are involved in JNK/p38MAPK pathway crosstalk. Phosphorylation of β-catenin by JNK contributes to OC chemotherapy resistance by promoting cancer stem cell survival and inducing EMT (126–128). Furthermore, glycolysis mediated by the p38-HIF1α axis promotes the survival of chemoresistant OC cells in a hypoxic environment. Modulating HIF-1α activity by inhibiting p38, in combination with glucose analogues and platinum compounds, could enhance the efficacy of chemotherapy and represents a promising therapeutic strategy for OC (129).
In summary, the intricate crosstalk between JNK/p38 MAPK, PI3K/AKT, NF-κB, Wnt/β-catenin, and metabolic pathways forms a robust network that sustains OC cell survival under therapeutic stress. While this complexity poses significant challenges, it also offers multiple therapeutic vulnerabilities. Future research should focus on combinatorial therapies: Simultaneously targeting multiple nodes (e.g., PI3K/AKT + JNK/p38 + NF-κB) to overcome compensatory pathway activation.
4 JNK/p38 MAPK inhibitors and natural compounds to treat OC resistance
Targeting JNK and p38 MAPK may be a promising strategy to reverse OC resistance due to their important roles in chemoresistance. Several potent and specific inhibitors of JNK and p38 MAPK have been developed to restore cancer cells’ sensitivity to chemotherapy drugs and reverse chemotherapy resistance (Table 4).
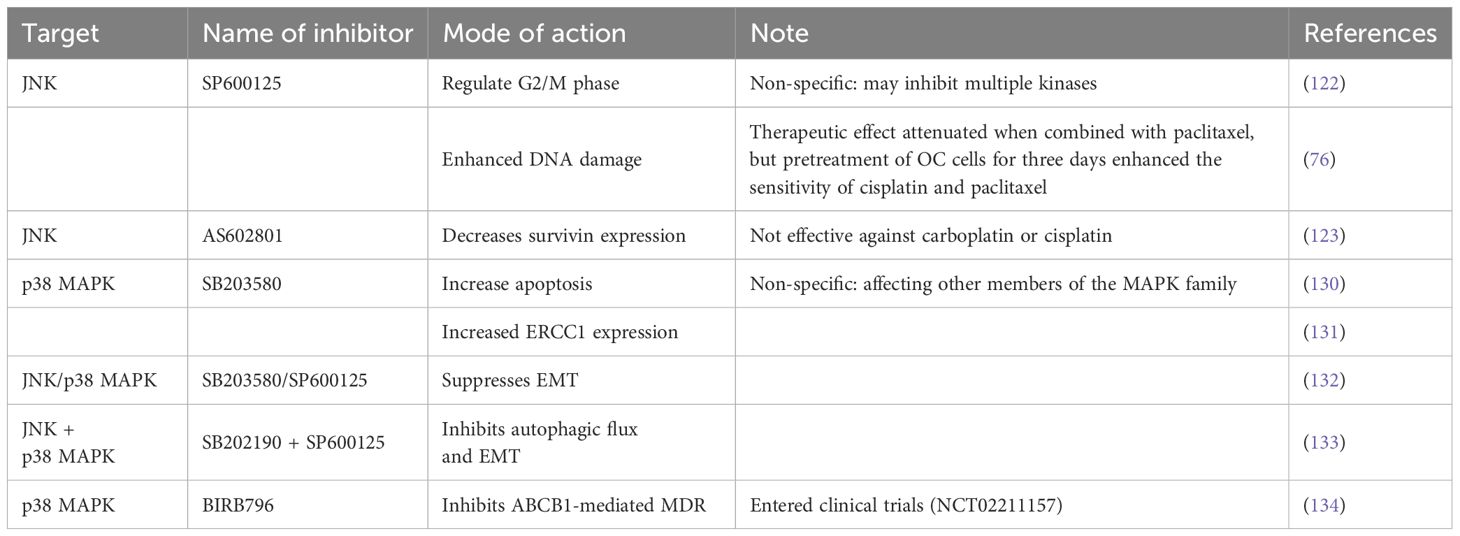
Table 4. Summary of inhibitors targeting the JNK/p38 MAPK signaling pathway.
4.1 JNK inhibitors
SP600125 is a JNK inhibitor that inhibits JNK kinase activity and the JNK signaling pathway by specifically blocking the ATP-binding site of the JNK protein. There is growing evidence that SP600125 can mediate autophagy and apoptosis to overcome drug resistance in several malignant tumors and reverse OC resistance by regulating the cell cycle in the G2/M phase (122). Seino et al. (76) showed that combining cisplatin and SP600125 enhanced DNA damage to reverse cisplatin resistance, but the therapeutic effect was diminished when SP600125 was combined with paclitaxel. Notably, pretreating OC cells with SP600125 for three days before administrating cisplatin or paclitaxel inhibited basal JNK activity and reduced chemotherapy resistance without increasing toxicity. Thus, this time-staggered inhibition of JNK effectively enhances the sensitivity of cisplatin and paclitaxel. However, the clinical application of SP600125 is limited because it is a broad-spectrum JNK reversible inhibitor that is non-specific and can inhibit multiple kinases. AS602801 is an oral, selective JNK inhibitor and anticancer stem cell candidate, which has been shown to reduce the expression of survivin, making OC-resistant cells sensitive to paclitaxel but not carboplatin or cisplatin (123). In the future, AS602801 can be combined with other inhibitors to inhibit the growth and survival of OC cells more effectively.
4.2 p38 inhibitor
Evidence suggests that p38 inhibitors can increase the sensitivity of OC cells to cisplatin and reverse drug resistance. SB203580, a widely used p38 inhibitor, showed a synergistic effect of cisplatin and SB203580 in OC Cisplatin IGROV-1/Pt1 cells, resulting in greater apoptosis than cisplatin alone (130). Xie et al. (131) found that metformin combined with SB203580 application inhibited Excision Repair Cross-Complementation Group 1 (ERCC1) expression in OC-resistant cells and enhanced the sensitivity of OC cisplatin-resistant cells. Additionally, JNK inhibitors can be used in combination with p38 inhibitors. Zhu et al. (132) found that SB203580 or SP600125 inhibited EMT and reduced metastasis in OC cells. Chen et al. (133) showed that SB202190 combined with SP600125 inhibited autophagy flux and EMT to inhibit drug resistance. Despite the promising evidence from in vivo and in vitro studies demonstrating the effectiveness of JNK/p38 MAPK inhibitors, progress has been slow in clinical trials. The p38 inhibitor BIRB 796 has been shown to inhibit ABCB1-mediated MDR147 and is currently undergoing clinical trials (NCT02211157) (134).
Research on JNK/p38 MAPK inhibitors in OC is still in the initial stage. JNK/p38 MAPK inhibitors may cause side effects on normal cells, affecting their efficacy and safety in vivo. Additionally, many inhibitors lack specificity and may interact by targeting other kinases, affecting drug selectivity and side effects. Furthermore, effective drug delivery remains a crucial challenge because different isoforms have different functions in a cellular environment-dependent manner. Further in-depth studies on its mechanism of action and clinical application are needed to develop new strategies and methods for treating OC.
4.3 Natural compounds targeting JNK/p38 MAPK to reverse ovarian cancer chemoresistance
Natural compounds derived from medicinal plants and traditional formulations have emerged as promising agents to overcome chemoresistance in OC by modulating JNK/p38 MAPK signaling. Compared with traditional chemotherapy, these compounds exhibit multi-target effects, including apoptosis induction, DNA repair suppression, and cell cycle arrest, while demonstrating lower systemic toxicity than conventional chemotherapy (135) (Table 5).
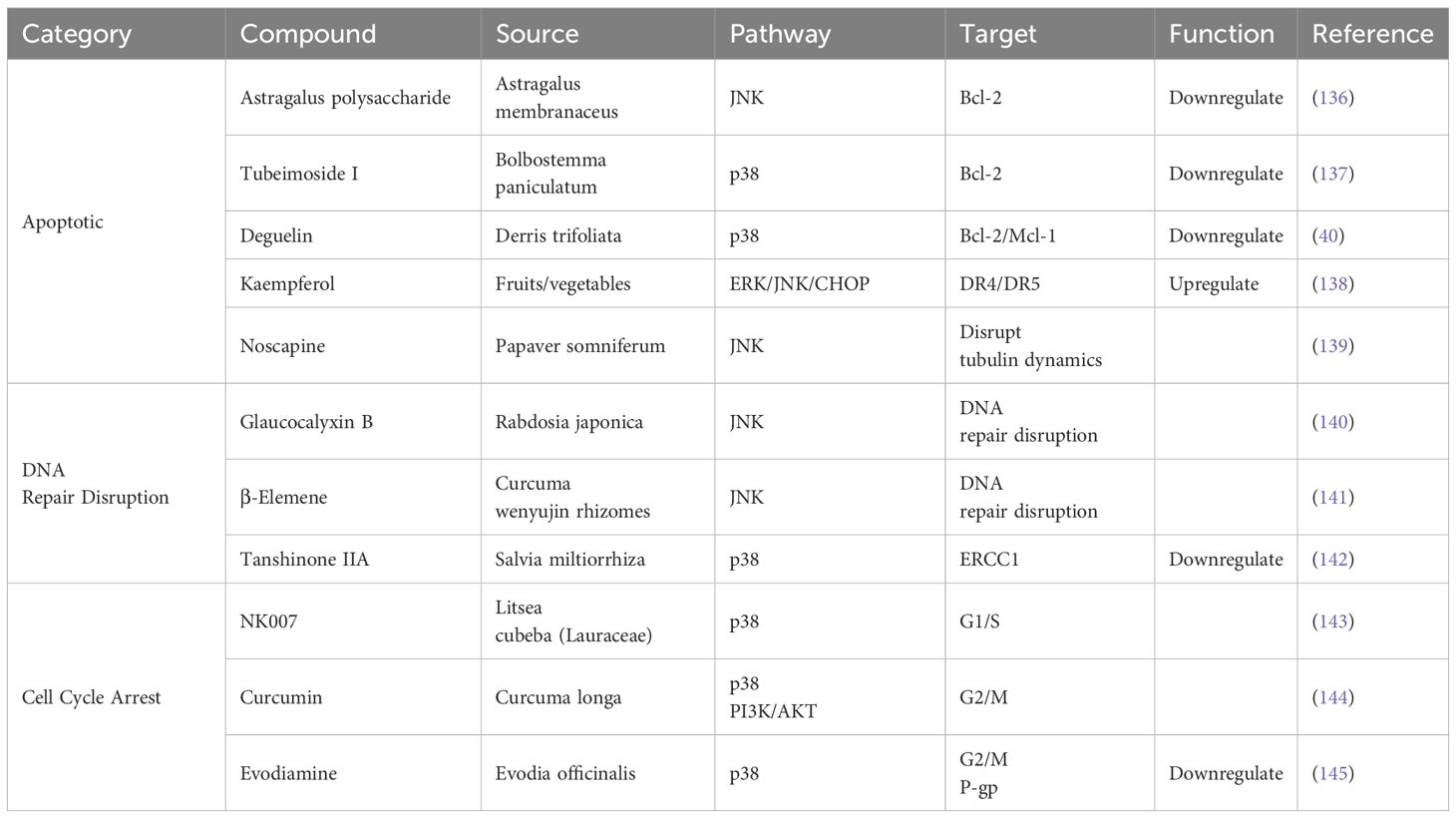
Table 5. Natural compounds reverse OC resistance via JNK/p38 MAPK.
4.3.1 Apoptotic pathway activation
Astragalus polysaccharide, derived from Astragalus membranaceus, activates JNK to downregulate Bcl-2 and upregulate Bax/caspase-3, restoring cisplatin sensitivity in OC cells (136). Tubeimoside I, derived from Bolbostemma paniculatum, enhances p38-mediated Bax activation while suppressing Bcl-2, effectively reversing platinum resistance (137). Deguelin, a flavonoid from Derris trifoliata, synergizes with paclitaxel to inhibit p38, downregulating Bcl-2 and Mcl-1 in SKOV3-TR cells (40). Kaempferol, a flavonoid from fruits/vegetables, upregulates ERK/JNK/CHOP signaling and increases death receptors (DR4/DR5) expression, overcoming OC resistance (138). Noscapine, derived from Papaver somniferum, activates JNK to induce apoptosis in paclitaxel-resistant cells by disrupting tubulin dynamics (139).
4.3.2 DNA repair disruption
Glaucocalyxin B, derived from Rabdosia japonica, overcomes OC cisplatin resistance by mediating JNK pathway activation and increasing DNA damage (140). β-Elemene, derived from Curcuma wenyujin rhizomes, downregulates DNA repair activity and inhibits JNK activation, sensitizing OC cells to cisplatin (141). Tanshinone IIA, derived from Salvia miltiorrhiza, activates p38 to downregulate ERCC1, a critical mediator of platinum adduct removal (142).
4.3.3 Cell cycle arrest
NK007, derived from Litsea cubeba, induces G1/S arrest via p38 upregulation, overcoming paclitaxel resistance (143). Curcumin, derived from Curcuma longa, induces p38-dependent G2/M arrest and inhibits PI3K/AKT survival signaling in cisplatin-resistant OC cells (144). Evodiamine, derived from Nvidia officinal, induces G2/M phase arrest by activating p38 and inhibits P-glycoprotein (P-gp)-mediated drug efflux (145).
4.3.4 Clinical challenges and emerging strategies
In treating OC, natural compounds have demonstrated significant potential for reversing chemoresistance by modulating the JNK/p38 MAPK signaling pathway. However, clinical application faces challenges, including poor bioavailability, limited pharmacokinetic stability, and batch-to-batch variability in quality control.
Advanced delivery systems have been developed to improve efficacy and reduce off-target toxicity. For instance, triptolide-loaded nanoparticles derived from Tripterygium wilfordii enhance the targeting efficiency of JNK/p38 while reducing hepatotoxicity (146). Similarly, curcumin demonstrates promising effects in drug-resistant human OC cells when delivered via nanocarriers (146). These innovative approaches improve the therapeutic efficacy of natural compounds and mitigate their side effects, enhancing their clinical applicability. Future studies on bioavailability, pharmacokinetics, and quality control of natural compounds are expected to change the treatment landscape for patients with OC resistance.
5 Discussion and conclusion
The JNK/p38 MAPK signaling pathway exhibits a dual role in OC chemoresistance, paradoxically promoting both apoptosis and tumor cell survival. Our analysis reveals that JNK/p38 MAPK activation under chemotherapy-induced stress initially triggers apoptosis and DDR to eliminate compromised cells. However, dysregulated signaling shifts toward pro-survival mechanisms, including enhanced autophagy, DNA repair, and drug efflux, enabling OC cells to evade treatment. This adaptive reprogramming is amplified through crosstalk with PI3K/AKT, NF-κB, Wnt/β-catenin, and HIF-1α pathways, collectively sustaining a therapy-resistant TME. Notably, transient JNK/p38 MAPK activation promotes survival, whereas prolonged signaling induces apoptosis or autophagic cell death. This temporal duality highlights the potential of chronotherapeutic approaches, such as staggered chemotherapy dosing, to exploit cumulative DNA damage in resistant cells.
Targeting JNK/p38 MAPK presents a promising strategy to reverse OC chemoresistance. Preclinical studies demonstrate that inhibitors like SP600125 (JNK) and SB203580 (p38) restore cisplatin sensitivity by augmenting apoptosis and cell cycle arrest, although clinical translation remains limited. Natural compounds, including curcumin, Astragalus polysaccharides, and β-elemene, show multimodal regulation of JNK/p38 MAPK signaling with lower toxicity profiles compared to conventional chemotherapeutics. Nanotechnology innovations (e.g., curcumin-loaded liposomes and triptolide nanoparticles) address bioavailability challenges while improving tumor-specific delivery.
Despite these advances, clinical implementation faces multifaceted challenges. First-generation JNK/p38 MAPK inhibitors exhibit off-target effects and systemic toxicity. While natural products offer multi-target advantages, standardization issues related to pharmacokinetics, stability, and quality control hinder their clinical adoption. Furthermore, OC heterogeneity necessitates biomarker discovery (e.g., p38α overexpression, RAS/RAF mutations) to identify patients most likely to benefit from JNK/p38-targeted therapies.
Future research should focus on the following three aspects. First, exploring the mechanism of action of the JNK/p38 MAPK signaling pathway, developing JNK and p38 MAPK inhibitors with higher specificity and fewer adverse reactions, and exploring new natural compounds will be key to advancing OC treatment. Second, combination therapy targeting multiple nodes in the JNK/p38 MAPK pathway and other interacting pathways helps to overcome compensatory pathway activation. Alternatively, interleaved administration before chemotherapy, combined with a chronotherapy approach, provides a more comprehensive therapeutic strategy. Third, advancing research and the discovery of innovative biomarkers such as p38α overexpression and RAS/RAF mutations will facilitate the stratification of patients according to their gene expression profiles, thereby providing personalized treatment options for patients. Therefore, regulating JNK/p38 MAPK can help mitigate resistance to OC chemotherapy ultimately improving the survival and quality of life of OC patients.
Author contributions
Y-TM: Writing – original draft, Writing – review & editing. CL: Supervision, Writing – review & editing. YS: Supervision, Writing – review & editing. W-HY: Writing – review & editing. M-XH: Writing – review & editing. Y-FM: Writing – review & editing. F-JH: Funding acquisition, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. The work was supported by the National Natural Science Foundation of China (No. 82274566), National Natural Science Foundation of China (No.82074484). Young Elite Scientists Sponsorship Program by Heilongiiang Society of Traditional Chinese Medicine (No. 2022-QNRC1-18).
Acknowledgments
We appreciate the Charlesworth Group for their professional language polishing services, which improved the clarity and quality of our manuscript. We also thank Adobe Illustrator. We thank MedPeer (www.medpeer.cn) for editing Figures 1 and 2.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
AKT: protein kinase B
AMPK: AMP-activated protein kinase
CAF: Cancer-associated fibroblast
OC: Ovarian cancer
MAPK: Mitogen-Activated Protein Kinases
ERK: Extracellular Signal-Regulated Kinases
JNK: c-Jun N-terminal kinase
LGSOC: Low-Grade Serous Ovarian Carcinoma
HGSOC: High-grade serous ovarian carcinoma
ROS: Reactive oxygen species
NF-κB: Nuclear factor-κB
PI3K: Phosphoinositide 3-kinase
TME: Tumor microenvironment
TNF-α: Tumor necrosis factor-α
TGF-β: Transforming growth factor-β
IL-1: Interleukin-1
IL-6: Interleukin-6
IL-12: Interleukin-12
VEGF: Vascular endothelial growth factor
ECM: Extracellular matrix
EVs: Extracellular vesicles
MDR: Multidrug resistance
ABC: ATP-binding cassette
P-gp: P-glycoprotein
DUSP1: Dual specificity phosphatase 1
MKP-1: MAPK phosphatase-1
miRNA: MicroRNA
DSB: Double-strand breaks
DDR: DNA damage response
Gadd45α: Growth arrest and DNA damage-inducible 45α
ChK: Chaetoglobosin K
HB-EGF: Hepatocyte growth factor-regulated tyrosine kinase substrate
FGF2: Fibroblast growth factor 2
HIF-1α: Hypoxia-inducible factor-1α
ERCC1: Excision repair cross-complementation group 1
References
1. Siegel RL, Giaquinto AN, Jemal A. Cancer statistics, 2024. CA Cancer J Clin. (2024) 74:12–49. doi: 10.3322/caac.21820
2. Ovarian Cancer Research Alliance. Ovarian Cancer (2024). Available online at: https://ocrahope.org/for-patients/gynecologic-cancers/ovarian-cancer/ (Accessed November 2, 2024).
3. Bray F, Laversanne M, Sung H, Ferlay J, Siegel RL, Soerjomataram I, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2024) 74:229–63. doi: 10.3322/caac.21834
4. Richardson DL, Eskander RN, O’Malley DM. Advances in ovarian cancer care and unmet treatment needs for patients with platinum resistance: a narrative review. JAMA Oncol. (2023) 9:851–9. doi: 10.1001/jamaoncol.2023.0197
5. Matthews BG, Bowden NA, Wong-Brown MW. Epigenetic mechanisms and therapeutic targets in chemoresistant high-grade serous ovarian cancer. Cancers. (2021) 13:5993. doi: 10.3390/cancers13235993
6. Guo T, Dong X, Xie S. Cellular mechanism of gene mutations and potential therapeutic targets in ovarian cancer. Cancer Manag Res. (2021) 13:3081–100. doi: 10.2147/CMAR.S292992
7. Karoulia Z, Gavathiotis E, Poulikakos PI. New perspectives for targeting RAF kinase in human cancer. Nat Rev Cancer. (2017) 17:676–91. doi: 10.1038/nrc.2017.79
8. Harmych SJ, Kumar J, Bouni ME. Nicotine inhibits MAPK signaling and spheroid invasion in ovarian cancer cells. Exp Cell Res. (2020) 394:112167. doi: 10.1016/j.yexcr.2020.112167
9. Santarpia L, Lippman SM, El-Naggar AK. Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy. Expert Opin Ther Targets. (2012) 16:103–19. doi: 10.1517/14728222.2011.645805
10. Hendrikse CSE, Theelen PMM, van der Ploeg P, Westgeest HM, Boere IA, Thijs AMJ, et al. The potential of RAS/RAF/MEK/ERK (MAPK) signaling pathway inhibitors in ovarian cancer: a systematic review and meta-analysis. Gynecol Oncol. (2023) 171:83–94. doi: 10.1016/j.ygyno.2023.01.038
11. Grisham RN, Moore KN, Gordon MS, Harb W, Cody G, Halpenny DF, et al. Phase Ib study of binimetinib with paclitaxel in patients with platinum-resistant ovarian cancer: final results, potential biomarkers, and extreme responders. Clin Cancer Res. (2018) 24:5525–33. doi: 10.1158/1078-0432.CCR-18-0494
12. Gershenson DM, Miller A, Brady WE, Paul J, Carty K, Rodgers W, et al. Trametinib versus standard of care in patients with recurrent low-grade serous ovarian cancer (GOG 281/LOGS): an international, randomised, open-label, multicentre, phase 2/3 trial. Lancet. (2022) 399:541–53. doi: 10.1016/S0140-6736(21)02175-9
13. Villedieu M, Deslandes E, Duval M. Acquisition of chemoresistance following discontinuous exposures to cisplatin is associated in ovarian carcinoma cells with progressive alteration of FAK, ERK and p38 activation in response to treatment. Gynecol Oncol. (2006) 101:507–19. doi: 10.1016/j.ygyno.2005.11.017
14. Fawzy MA, Abu-Baih RH, Abuo-Rahma GEA. In vitro anticancer activity of novel ciprofloxacin Mannich base in lung adenocarcinoma and high-grade serous ovarian cancer cell lines via attenuating MAPK signaling pathway. Molecules. (2023) 28:1137. doi: 10.3390/molecules28031137
15. Zhang X, Qi Z, Yin H. Interaction between p53 and Ras signaling controls cisplatin resistance via HDAC4- and HIF-1α-mediated regulation of apoptosis and autophagy. Theranostics. (2019) 9:1096–114. doi: 10.7150/thno.29673
16. Zhang J, Liu XH, Li C, Wu XX, Chen YL, Li WW, et al. SNCG promotes the progression and metastasis of high-grade serous ovarian cancer via targeting the PI3K/AKT signaling pathway. J Exp Clin Cancer Res. (2020) 39:79. doi: 10.1186/s13046-020-01589-9
17. Pan ZZ, Bruening W, Giasson BI. Gamma-synuclein promotes cancer cell survival and inhibits stress- and chemotherapy drug-induced apoptosis by modulating MAPK pathways. J Biol Chem. (2002) 277:35050–60. doi: 10.1074/jbc.M201650200
18. Manning-Geist B, Gordhandas S, Liu YL, Zhou Q, Iasonos A, Da Cruz Paula A, et al. MAPK pathway genetic alterations are associated with prolonged overall survival in low-grade serous ovarian carcinoma. Clin Cancer. (2022) 28:4456–65. doi: 10.1158/1078-0432.CCR-21-4183
19. Cobb L, Gershenson D. Novel therapeutics in low-grade serous ovarian cancer. Int J Gynecol Cancer. (2023) 33:377–84. doi: 10.1136/ijgc-2022-003677
20. Ho CL, Kurman RJ, Dehari R. Mutations of BRAF and KRAS precede the development of ovarian serous borderline tumors. Cancer Res. (2004) 64:6915–8. doi: 10.1158/0008-5472.CAN-04-2067
21. Sadłecki P, Walentowicz-Sadłecka M, Grabiec M. Molecular diagnosis in type I epithelial ovarian cancer. Ginekol Pol. (2017) 88:692–7. doi: 10.5603/GP.a2017.0123
22. Vang R, IeM S, Kurman RJ. Ovarian low-grade and high-grade serous carcinoma: pathogenesis, clinicopathologic and molecular biologic features, and diagnostic problems. Adv Anat Pathol. (2009) 16:267–82. doi: 10.1097/PAP.0b013e3181b4fffa
23. Hew KE, Miller PC, El-Ashry D, Sun J, Besser AH, Ince TA, et al. MAPK activation predicts poor outcome and the MEK inhibitor, selumetinib, reverses antiestrogen resistance in ER-positive high-grade serous ovarian cancer. Clin Cancer Res. (2016) 22:935–47. doi: 10.1158/1078-0432.CCR-15-0534
24. Monk BJ, Grisham RN, Banerjee S, Kalbacher E, Mirza MR, Romero I, et al. MILO/ENGOT-ov11: binimetinib versus physician’s choice chemotherapy in recurrent or persistent low-grade serous carcinomas of the ovary, Fallopian tube, or primary peritoneum. J Clin Oncol. (2020) 38:3753–62. doi: 10.1200/JCO.20.01164
25. Bardia A, Gounder M, Rodon J, Janku F, Lolkema MP, Stephenson JJ, et al. Phase Ib study of combination therapy with MEK inhibitor binimetinib and phosphatidylinositol 3-kinase inhibitor buparlisib in patients with advanced solid tumors with RAS/RAF alterations. Oncologist. (2020) 25:e160–9. doi: 10.1634/theoncologist.2019-0297
26. Infante JR, Fecher LA, Falchook GS, Nallapareddy S, Gordon MS, Becerra C, et al. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: a phase 1 dose-escalation trial. Lancet Oncol. (2012) 13:773–81. doi: 10.1016/S1470-2045(12)70270-X
27. Bedard PL, Tabernero J, Janku F, Wainberg ZA, Paz-Ares L, Vansteenkiste J, et al. A phase Ib dose-escalation study of the oral pan-PI3K inhibitor buparlisib (BKM120) in combination with the oral MEK1/2 inhibitor trametinib (GSK1120212) in patients with selected advanced solid tumors. Clin Cancer Res. (2015) 21:730–8. doi: 10.1158/1078-0432.CCR-14-1814
28. Zhang W, Liu HT. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. (2002) 12:9–18. doi: 10.1038/sj.cr.7290105
29. Sabapathy K. Role of the JNK pathway in human diseases. Prog Mol Biol Transl Sci. (2012) 106:145–69. doi: 10.1016/B978-0-12-396456-4.00013-4
30. Yarza R, Vela S, Solas M. C-Jun N-terminal kinase (JNK) signaling as a therapeutic target for Alzheimer’s disease. Front Pharmacol. (2015) 6:321. doi: 10.3389/fphar.2015.00321
31. Bogoyevitch MA, Kobe B. Uses for JNK: the many and varied substrates of the c-Jun N-terminal kinases. Microbiol Mol Biol Rev. (2006) 70:1061–95. doi: 10.1128/MMBR.00025-06
32. Wang X, Destrument A, Tournier C. Physiological roles of MKK4 and MKK7: insights from animal models. Biochim Biophys Acta. (2007) 1773:1349–57. doi: 10.1016/j.bbamcr.2006.10.016
33. Bubici C, Papa S. JNK signalling in cancer: in need of new, smarter therapeutic targets. Br J Pharmacol. (2014) 171:24–37. doi: 10.1111/bph.2014.171.issue-1
34. Gkouveris I, Nikitakis NG. Role of JNK signaling in oral cancer: a mini review. Tumour Biol. (2017) 39:1010428317711659. doi: 10.1177/1010428317711659
35. Zhou YY, Li Y, Jiang WQ. MAPK/JNK signalling: a potential autophagy regulation pathway. Biosci Rep. (2015) 35:e00199. doi: 10.1042/BSR20140141
36. Zhao HF, Wang J, Tony To SS. The phosphatidylinositol 3-kinase/Akt and c-Jun N-terminal kinase signaling in cancer: alliance or contradiction? [review. Int J Oncol. (2015) 47:429–36. doi: 10.3892/ijo.2015.3052
37. Cardenas C, Montagna MK, Pitruzzello M. Adipocyte microenvironment promotes BclxL expression and confers chemoresistance in ovarian cancer cells. Apoptosis Int J Programmed Cell Death. (2017) 22:558–69. doi: 10.1007/s10495-016-1339-x
38. Binju M, Amaya-Padilla MA, Wan G. Therapeutic inducers of apoptosis in ovarian cancer. Cancers. (2019) 11:1786. doi: 10.3390/cancers11111786
39. Srivastava RK, Mi QS, Hardwick JM. Deletion of the loop region of Bcl-2 completely blocks paclitaxel-induced apoptosis. Proc Natl Acad Sci U S A. (1999) 96:3775–80. doi: 10.1073/pnas.96.7.3775
40. Bae S, Bae S, Kim HS, Lim YJ, Kim G, Park IC, et al. Deguelin restores paclitaxel sensitivity in paclitaxel-resistant ovarian cancer cells via inhibition of the EGFR signaling pathway. Cancer Manag Res. (2024) 16:507–25. doi: 10.2147/CMAR.S457221
41. Diepstraten ST, Anderson MA, Czabotar PE. The manipulation of apoptosis for cancer therapy using BH3-mimetic drugs. Nat Rev Cancer. (2022) 22:45–64. doi: 10.1038/s41568-021-00407-4
42. Florent R, Weiswald LB, Lambert B, Brotin E, Abeilard E, Louis MH, et al. Bim, Puma and Noxa upregulation by naftopidil sensitizes ovarian cancer to the BH3-mimetic ABT-737 and the MEK inhibitor trametinib. Cell Death Dis. (2020) 11:380. doi: 10.1038/s41419-020-2588-8
43. Tang S, Shen Y, Wei X. Olaparib synergizes with arsenic trioxide by promoting apoptosis and ferroptosis in platinum-resistant ovarian cancer. Cell Death Dis. (2022) 13:826. doi: 10.1038/s41419-022-05257-y
44. Yuan Z, Wang F, Zhao Z, Zhao X, Qiu J, Nie C, et al. BIM-mediated AKT phosphorylation is a key modulator of arsenic trioxide-induced apoptosis in cisplatin-sensitive and -resistant ovarian cancer cells. PloS One. (2011) 6:e20586. doi: 10.1371/journal.pone.0020586
45. Paudel I, Hernandez SM, Portalatin GM. Sab concentrations indicate chemotherapeutic susceptibility in ovarian cancer cell lines. Biochem J. (2018) 475:3471–92. doi: 10.1042/BCJ20180603
46. Eskandari E, Eaves CJ. Paradoxical roles of caspase-3 in regulating cell survival, proliferation, and tumorigenesis. J Cell Biol. (2022) 221:e202201159. doi: 10.1083/jcb.202201159
47. Yan BX, Ma JX, Zhang J, Guo Y, Mueller MD, Remick SC, et al. Prostasin may contribute to chemoresistance, repress cancer cells in ovarian cancer, and is involved in the signaling pathways of CASP/PAK2-p34/actin. Cell Death Dis. (2014) 5:e995. doi: 10.1038/cddis.2013.523
48. Pai HC, Kumar S, Shen CC. MT-4 suppresses resistant ovarian cancer growth through targeting tubulin and HSP27. PloS One. (2015) 10:e0123819. doi: 10.1371/journal.pone.0123819
49. Tang XH, Li H, Zheng XS. CRM197 reverses paclitaxel resistance by inhibiting the NAC-1/Gadd45 pathway in paclitaxel-resistant ovarian cancer cells. Cancer Med. (2019) 8:6426–36. doi: 10.1002/cam4.v8.14
50. Sancar A, Lindsey-Boltz LA, Unsal-Kaçmaz K. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. (2004) 73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723
51. Yuan Z, Cao K, Lin C, Li L, Liu HY, Zhao XY, et al. The p53 upregulated modulator of apoptosis (PUMA) chemosensitizes intrinsically resistant ovarian cancer cells to cisplatin by lowering the threshold set by Bcl-x(L) and Mcl-1. Mol Med (Camb Mass). (2011) 17:1262–74. doi: 10.2119/molmed.2011.00176
52. Kennedy MC, Lowe SW. Mutant p53: it’s not all one and the same. Cell Death Differ. (2022) 29:983–7. doi: 10.1038/s41418-022-00989-y
53. Potapova O, Gorospe M, Dougherty RH. Inhibition of c-Jun N-terminal kinase 2 expression suppresses growth and induces apoptosis of human tumor cells in a p53-dependent manner. Mol Cell Biol. (2000) 20:1713–22. doi: 10.1128/MCB.20.5.1713-1722.2000
54. Zhao T, Bai J, Zou Q. Insulin in combination with cisplatin induces the apoptosis of ovarian cancer cells via p53 and JNK activation. Mol Med Rep. (2017) 16:9095–101. doi: 10.3892/mmr.2017.7752
55. Feng Y, He D, Yao Z. The machinery of macroautophagy. Cell Res. (2014) 24:24–41. doi: 10.1038/cr.2013.168
56. Debnath J, Gammoh N, Ryan KM. Autophagy and autophagy-related pathways in cancer. Nat Rev Mol Cell Biol. (2023) 24:560–75. doi: 10.1038/s41580-023-00585-z
57. Ferro F, Servais S, Besson P. Autophagy and mitophagy in cancer metabolic remodelling. Semin Cell Dev Biol. (2020) 98:129–38. doi: 10.1016/j.semcdb.2019.05.029
58. Denton D, Nicolson S, Kumar S. Cell death by autophagy: facts and apparent artefacts. Cell Death Differ. (2012) 19:87–95. doi: 10.1038/cdd.2011.146
59. Xu L, Zhang X, Li Y, Lu S, Lu S, Li J, et al. Neferine induces autophagy of human ovarian cancer cells via p38 MAPK/JNK activation. Tumour Biol J Int Soc Oncodev Biol Med. (2016) 37:8721–9. doi: 10.1007/s13277-015-4737-8
60. Zhu W, Huang M, Thakur A. FGF19 promotes cell autophagy and cisplatin chemoresistance by activating MAPK signaling in ovarian cancer. PeerJ. (2023) 11:e14827. doi: 10.7717/peerj.14827
61. Zheng GF, Cai Z, Meng XK, Zhang Y, Zhu W, Pang XY, et al. Unfolded protein response mediated JNK/AP-1 signal transduction, a target for ovarian cancer treatment. Int J Clin Exp Pathol. (2015) 8:6505–11. doi: 10.7717/peerj.14827
62. Yang X, Xiang X, Xia M. Inhibition of JNK3 promotes apoptosis induced by BH3 mimetic S1 in chemoresistant human ovarian cancer cells. Anat Rec Hoboken NJ. (2015) 298:386–95. doi: 10.1002/ar.v298.2
63. Sui X, Kong N, Ye L, Han W, Zhou J, Zhang Q, et al. p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett. (2014) 344:174–9. doi: 10.1016/j.canlet.2013.11.019
64. Zhao S, Fan S, Shi Y, Ren H, Hong H, Gao X, et al. Propranolol induced apoptosis and autophagy via the ROS/JNK signaling pathway in human ovarian cancer. J Cancer. (2020) 11:5900–10. doi: 10.7150/jca.46556
65. Guo Y, Yuan J, Yin S. MAP2K6-FP enhances the sensitiveness of paclitaxel for ovarian cancer via inducing autophagy. Int J Gynecol Cancer. (2017) 27:1082–7. doi: 10.1097/IGC.0000000000001003
66. Mishra SK, Dhadve AC, Mal A, Reddy BPK, Hole A, Chilakapati MK, et al. Photothermal therapy (PTT) is an effective treatment measure against solid tumors which fails to respond conventional chemo/radiation therapies in clinic. Biomater Adv. (2022) 143:213153. doi: 10.1016/j.bioadv.2022.213153
67. Groelly FJ, Fawkes M, Dagg RA. Targeting DNA damage response pathways in cancer. Nat Rev Cancer. (2023) 23:78–94. doi: 10.1038/s41568-022-00535-5
68. Thornton TM, Rincon M. Non-classical p38 map kinase functions: cell cycle checkpoints and survival. Int J Biol Sci. (2009) 5:44–51. doi: 10.7150/ijbs.5.44
69. Pilié PG, Tang C, Mills GB. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat Rev Clin Oncol. (2019) 16:81–104. doi: 10.1038/s41571-018-0114-z
70. Jiang Y, Huang S, Zhang L, Zhou Y, Zhang W, Wan T, et al. Targeting the Cdc2-like kinase 2 for overcoming platinum resistance in ovarian cancer. MedComm. (2024) 5:e537. doi: 10.1002/mco2.v5.4
71. Li L, Wang H, Yang ES. Erlotinib attenuates homologous recombinational repair of chromosomal breaks in human breast cancer cells. Cancer Res. (2008) 68:9141–6. doi: 10.1158/0008-5472.CAN-08-1127
72. Mak VC, Li X, Rao L. p85β alters response to EGFR inhibitor in ovarian cancer through p38 MAPK-mediated regulation of DNA repair. Neoplasia. (2021) 23:718–30. doi: 10.1016/j.neo.2021.05.009
73. Chen YC, Hsieh HH, Chang HC. CDC25B induces cellular senescence and correlates with tumor suppression in a p53-dependent manner. J Biol Chem. (2021) 296:100564. doi: 10.1016/j.jbc.2021.100564
74. Reinhardt HC, Hasskamp P, Schmedding I, Morandell S, van Vugt MATM, Wang X, et al. DNA damage activates a spatially distinct late cytoplasmic cell-cycle checkpoint network controlled by MK2-mediated RNA stabilization. Mol Cell. (2010) 40:34–49. doi: 10.1016/j.molcel.2010.09.018
75. Dreaden EC, Kong YW, Quadir MA, Correa S, Suárez-López L, Barberio AE, et al. RNA-peptide nanoplexes drug DNA damage pathways in high-grade serous ovarian tumors. Bioeng Transl Med. (2018) 3:26–36. doi: 10.1002/btm2.10086
76. Seino M, Okada M, Sakaki H, Takeda H, Watarai H, Suzuki S, et al. Time-staggered inhibition of JNK effectively sensitizes chemoresistant ovarian cancer cells to cisplatin and paclitaxel. Oncol Rep. (2016) 35:593–601. doi: 10.3892/or.2015.4377
77. Van Meter M, Simon M, Tombline G, May A, Morello TD, Hubbard BP, et al. JNK phosphorylates SIRT6 to stimulate DNA double-strand break repair in response to oxidative stress by recruiting PARP1 to DNA breaks. Cell Rep. (2016) 16:2641–50. doi: 10.1016/j.celrep.2016.08.006
78. Huang J, Zheng L, Sun Z. CDK4/6 inhibitor resistance mechanisms and treatment strategies. Int J Mol Med. (2022) 50:128. doi: 10.3892/ijmm.2022.5184
79. Thoms HC, Dunlop MG, Stark LA. p38-mediated inactivation of cyclin D1/cyclin-dependent kinase 4 stimulates nucleolar translocation of RelA and apoptosis in colorectal cancer cells. Cancer Res. (2007) 67:1660–9. doi: 10.1158/0008-5472.CAN-06-1038
80. Cannell IG, Merrick KA, Morandell S, Zhu CQ, Braun CJ, Grant RA, et al. A pleiotropic RNA-binding protein controls distinct cell cycle checkpoints to drive resistance of p53-defective tumors to chemotherapy. Cancer Cell. (2015) 28:831. doi: 10.1016/j.ccell.2015.11.003
81. Fontana F, Marzagalli M, Raimondi M. δ-tocotrienol sensitizes and re-sensitizes ovarian cancer cells to cisplatin via induction of G1 phase cell cycle arrest and ROS/MAPK-mediated apoptosis. Cell Prolif. (2021) 54:e13111. doi: 10.1111/cpr.v54.11
82. Lim S, Kaldis P. Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development. (2013) 140:3079–93. doi: 10.1242/dev.091744
83. Li B, Gao Y, Rankin GO, Rojanasakul Y, Cutler SJ, Tu Y, et al. Chaetoglobosin K induces apoptosis and G2 cell cycle arrest through p53-dependent pathway in cisplatin-resistant ovarian cancer cells. Cancer Lett. (2015) 356:418–33. doi: 10.1016/j.canlet.2014.09.023
84. Wang A, Li J, Zhou T, Li T, Cai H, Shi H, et al. CUEDC2 contributes to cisplatin-based chemotherapy resistance in ovarian serious carcinoma by regulating p38 MAPK signaling. J Cancer. (2019) 10:1800–7. doi: 10.7150/jca.29889
85. Huang HL, Chao MW, Li YC, Chang LH, Chen CH, Chen MC, et al. MPT0G066, a novel anti-mitotic drug, induces JNK-independent mitotic arrest, JNK-mediated apoptosis, and potentiates antineoplastic effect of cisplatin in ovarian cancer. Sci Rep. (2016) 6:31664. doi: 10.1038/srep31664
86. Chang KE, Wei BR, Madigan JP, Hall MD, Simpson RM, Zhuang Z, et al. The protein phosphatase 2A inhibitor LB100 sensitizes ovarian carcinoma cells to cisplatin-mediated cytotoxicity. Mol Cancer Ther. (2015) 14:90–100. doi: 10.1158/1535-7163.MCT-14-0496
87. Yeung TL, Leung CS, Yip KP. Cellular and molecular processes in ovarian cancer metastasis. a review in the theme: cell and molecular processes in cancer metastasis. Am J Physiol Cell Physiol. (2015) 309:C444–56. doi: 10.1158/1535-7163.MCT-14-0496
88. Eckhoff K, Flurschütz R, Trillsch F. The prognostic significance of Jun transcription factors in ovarian cancer. J Cancer Res Clin Oncol. (2013) 139:1673–80. doi: 10.1007/s00432-013-1489-y
89. Mitra T, Prasad P, Mukherjee P. Stemness and chemoresistance are imparted to the OC cells through TGFβ1 driven EMT. J Cell Biochem. (2018) 119:5775–87. doi: 10.1002/jcb.v119.7
90. Han L, Guo X, Du R. Identification of key genes and pathways related to cancer-associated fibroblasts in chemoresistance of ovarian cancer cells based on GEO and TCGA databases. J Ovarian Res. (2022) 15:75. doi: 10.1186/s13048-022-01003-2
91. Izar B, Tirosh I, Stover EH, Wakiro I, Cuoco MS, Alter I, et al. A single-cell landscape of high-grade serous ovarian cancer. Nat Med. (2020) 26:1271–9. doi: 10.1038/s41591-020-0926-0
92. Zhang M, Chen Z, Wang Y. The role of cancer-associated fibroblasts in ovarian cancer. Cancers. (2022) 14:2637. doi: 10.3390/cancers14112637
93. Alspach E, Flanagan KC, Luo X, Ruhland MK, Huang H, Pazolli E, et al. P38MAPK plays a crucial role in stromal-mediated tumorigenesis. Cancer Discov. (2014) 4:716–29. doi: 10.1158/2159-8290.CD-13-0743
94. Curtis M, Kenny HA, Ashcroft B. Fibroblasts mobilize tumor cell glycogen to promote proliferation and metastasis. Cell Metab. (2019) 29:141–155.e9. doi: 10.1016/j.cmet.2018.08.007
95. Samuel P, Mulcahy LA, Furlong F, McCarthy HO, Brooks SA, Fabbri M, et al. Cisplatin induces the release of extracellular vesicles from ovarian cancer cells that can induce invasiveness and drug resistance in bystander cells. Philos Trans R Soc Lond B Biol Sci. (2018) 373:20170065. doi: 10.1098/rstb.2017.0065
96. Fan LL, Chen X, Zhang XY, Li ZM, Fan XM, Shen Y. Octreotide-paclitaxel conjugate reverses paclitaxel resistance by p38 mitogen-activated protein kinase (MAPK) signaling pathway in A2780/Taxol human ovarian cancer cells. Med Sci Monit Int Med J Exp Clin Res. (2020) 26:e922612. doi: 10.12659/MSM.922612
97. Stieg DC, Wang Y, Liu LZ, Jiang BH. ROS and miRNA dysregulation in ovarian cancer development, angiogenesis and therapeutic resistance. Int J Mol Sci. (2022) 23:6702. doi: 10.3390/ijms23126702
98. Shi Y, Nikulenkov F, Zawacka-Pankau J, Li H, Gabdoulline R, Xu J, et al. ROS-dependent activation of JNK converts p53 into an efficient inhibitor of oncogenes leading to robust apoptosis. Cell Death Differ. (2014) 21:612–23. doi: 10.1038/cdd.2013.186
99. Chen Y, Wen C, Zhong S, Huang L, Xiang Y, Ou Y, et al. Oxfendazole induces apoptosis in ovarian cancer cells by activating JNK/MAPK pathway and inducing reactive oxygen species generation. Biol Pharm Bull. (2023) 46:1569–75. doi: 10.1248/bpb.b23-00349
100. Li X, Guo Y, Xing Z, Gong T, Yang L, Yang T, et al. ABT-737 increases cisplatin sensitivity through the ROS-ASK1-JNK MAPK signaling axis in human ovarian cancer cisplatin-resistant A2780/DDP cells. Oncol Rep. (2024) 52:122. doi: 10.3892/or.2024.8781
101. Wang Y, Chen G, Dai F, Zhang L, Yuan M, Yang D, et al. miR-21 induces chemoresistance in ovarian cancer cells via mediating the expression and interaction of CD44v6 and P-gp. Onco Targets Ther. (2021) 14:325–36. doi: 10.2147/OTT.S286639
102. Wang SQ, Liu ST, Zhao BX, Yang FH, Wang YT, Liang QY, et al. Afatinib reverses multidrug resistance in ovarian cancer via dually inhibiting ATP binding cassette subfamily B member 1. Oncotarget. (2015) 6:26142–60. doi: 10.18632/oncotarget.v6i28
103. Kang YS, Seok HJ, Jeong EJ, Kim Y, Yun SJ, Min JK, et al. DUSP1 induces paclitaxel resistance through the regulation of p-glycoprotein expression in human ovarian cancer cells. Biochem Biophys Res Commun. (2016) 478:403–9. doi: 10.1016/j.bbrc.2016.07.035
104. Goodall GJ, Wickramasinghe VO. RNA in cancer. Nat Rev Cancer. (2021) 21:22–36. doi: 10.1038/s41568-020-00306-0
105. Zhang H, Lu B. MicroRNAs as biomarkers of ovarian cancer. Expert Rev Anticancer Ther. (2020) 20:373–85. doi: 10.1080/14737140.2020.1760095
106. Xiao F, Li Y, Wan Y. MircroRNA-139 sensitizes ovarian cancer cell to cisplatin-based chemotherapy through regulation of ATP7A/B. Cancer Chemother Pharmacol. (2018) 81:935–47. doi: 10.1007/s00280-018-3548-1
107. Kumar S, Kumar A, Shah PP. MicroRNA signature of cis-platin resistant vs. cis-platin sensitive ovarian cancer cell lines. J Ovarian Res. (2011) 4:17. doi: 10.1186/1757-2215-4-17
108. Yin Y, Li J, Rong J. Circ_0067934 reduces JNK phosphorylation through a microRNA-545-3p/PPA1 axis to enhance tumorigenesis and cisplatin resistance in ovarian cancer. Immunopharmacol Immunotoxicol. (2022) 44:261–74. doi: 10.1080/08923973.2022.2038193
109. Wu J, Sun Y, Zhang PY, Qian M, Zhang H, Chen X, et al. The Fra-1-miR-134-SDS22 feedback loop amplifies ERK/JNK signaling and reduces chemosensitivity in ovarian cancer cells. Cell Death Dis. (2016) 7:e2384. doi: 10.1038/cddis.2016.289
110. Echevarría-Vargas IM, Valiyeva F, Vivas-Mejía PE. Upregulation of miR-21 in cisplatin resistant ovarian cancer via JNK-1/c-Jun pathway. PloS One. (2014) 9:e97094. doi: 10.1371/journal.pone.0097094
111. Jiang Y, Jiang J, Jia H. Recovery of miR-139-5p in ovarian cancer reverses cisplatin resistance by targeting c-Jun. Cell Physiol Biochem Int J Exp Cell Physiol Biochem Pharmacol. (2018) 51:129–41. doi: 10.1159/000495169
112. Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. (2000) 103:239–52. doi: 10.1016/S0092-8674(00)00116-1
113. Yagi H, Yotsumoto F, Sonoda K. Synergistic anti-tumor effect of paclitaxel with CRM197, an inhibitor of HB-EGF, in ovarian cancer. Int J Cancer. (2009) 124:1429–39. doi: 10.1002/ijc.v124:6
114. Mansouri A, Ridgway LD, Korapati AL, Zhang Q, Tian L, Wang Y, et al. Sustained activation of JNK/p38 MAPK pathways in response to cisplatin leads to Fas ligand induction and cell death in ovarian carcinoma cells. J Biol Chem. (2003) 278:19245–56. doi: 10.1074/jbc.M208134200
115. Feng Y, Cheng Y, Chang Y, Jian H, Zheng R, Wu X, et al. Time-staggered delivery of erlotinib and doxorubicin by gold nanocages with two smart polymers for reprogrammable release and synergistic with photothermal therapy. Biomaterials. (2019) 217:119327. doi: 10.1016/j.biomaterials.2019.119327
116. Kim AH, Yano H, Cho H, Meyer D, Monks B, Margolis B, et al. Akt1 regulates a JNK scaffold during excitotoxic apoptosis. Neuron. (2002) 35:697–709. doi: 10.1016/S0896-6273(02)00821-8
117. Noorolyai S, Shajari N, Baghbani E. The relation between PI3K/AKT signalling pathway and cancer. Gene. (2019) 698:120–8. doi: 10.1016/j.gene.2019.02.076
118. Nie X, Liu D, Zheng M, Li X, Liu O, Guo Q, et al. HERPUD1 promotes ovarian cancer cell survival by sustaining autophagy and inhibit apoptosis via PI3K/AKT/mTOR and p38 MAPK signaling pathways. BMC Cancer. (2022) 22:1338. doi: 10.1186/s12885-022-10248-5
119. Muranen T, Selfors LM, Hwang J, Gallegos LL, Coloff JL, Thoreen CC, et al. ERK and p38 MAPK activities determine sensitivity to PI3K/mTOR inhibition via regulation of MYC and YAP. Cancer Res. (2016) 76:7168–80. doi: 10.1158/0008-5472.CAN-16-0155
120. Yu C, Friday BB, Lai JP, McCollum A, Atadja P, Roberts LR, et al. Abrogation of MAPK and Akt signaling by AEE788 synergistically potentiates histone deacetylase inhibitor-induced apoptosis through reactive oxygen species generation. Clin Cancer Res. (2007) 13:1140–8. doi: 10.1158/1078-0432.CCR-06-1751
121. Huang YS, Wang TX, Lin XM, Wang H, Li RZ, Zeng X, et al. Antitumor effect of a short peptide on p53-null SKOV3 ovarian cancer cells. Anti Cancer Drugs. (2019) 30:973–82. doi: 10.1097/CAD.0000000000000830
122. Kim JH, Chae M, Choi AR. SP600125 overcomes antimitotic drug-resistance in cancer cells by increasing apoptosis with independence of P-gp inhibition. Eur J Pharmacol. (2014) 723:141–7. doi: 10.1016/j.ejphar.2013.11.026
123. Yamamoto M, Suzuki S, Togashi K, Sanomachi T, Seino S, Kitanaka C, et al. AS602801 sensitizes ovarian cancer stem cells to paclitaxel by down-regulating MDR1. Anticancer Res. (2019) 39:609–17. doi: 10.21873/anticanres.13154
124. Saha RN, Jana M, Pahan K. MAPK p38 regulates transcriptional activity of NF-kappaB in primary human astrocytes via acetylation of p65. J Immunol Baltimore Md. (2007) 179:7101–9. doi: 10.1186/1471-2407-14-61
125. Ayyagari VN, Brard L. Bithionol inhibits ovarian cancer cell growth in vitro - studies on mechanism(s) of action. BMC Cancer. (2014) 14:61. doi: 10.1186/1471-2407-14-61
126. Huang L, Jin Y, Feng S, Zou Y, Xu S, Qiu S, et al. Role of Wnt/β-catenin, Wnt/c-Jun N-terminal kinase and Wnt/Ca2+ pathways in cisplatin-induced chemoresistance in ovarian cancer. Exp Ther Med. (2016) 12:3851–8. doi: 10.3892/etm.2016.3885
127. Maurice MM, Angers S. Mechanistic insights into Wnt-β-catenin pathway activation and signal transduction. Nat Rev Mol Cell Biol. (2025). doi: 10.1038/s41580-024-00823-y
128. Samant C, Kale R, Pai KSR, Nandakumar K, Bhonde M. Role of Wnt/β-catenin pathway in cancer drug resistance: insights into molecular aspects of major solid tumors. Biochem Biophys Res Commun. (2024) 729:150348. doi: 10.1016/j.bbrc.2024.150348
129. Malm SW, Hanke NT, Gill A, Carbajal L, Baker AF. The anti-tumor efficacy of 2-deoxyglucose and D-allose are enhanced with p38 inhibition in pancreatic and ovarian cell lines. J Exp Clin Cancer Res. (2015) 34:31. doi: 10.1186/s13046-015-0147-4
130. Alampi DM, Ciusani E, Carenini N. AXL downstream targeting unravels synergistic drug combinations in ovarian carcinoma cells. Anticancer Res. (2019) 39:3803–8. doi: 10.21873/anticanres.13529
131. Xie Y, Peng Z, Shi M. Metformin combined with p38 MAPK inhibitor improves cisplatin sensitivity in cisplatin-resistant ovarian cancer. Mol Med Rep. (2014) 10:2346–50. doi: 10.3892/mmr.2014.2490
132. Zhu J, Zheng Y, Zhang H. Galectin-1 induces metastasis and epithelial-mesenchymal transition (EMT) in human ovarian cancer cells via activation of the MAPK JNK/p38 signalling pathway. Am J Transl Res. (2019) 11:3862–78. doi: 10.1186/1471-2407-14-61
133. Chen X, Chen Y, Lin X, Su S, Hou X, Zhang Q, et al. The drug combination of SB202190 and SP600125 significantly inhibit the growth and metastasis of Olaparib-resistant ovarian cancer cell. Curr Pharm Biotechnol. (2018) 19:506–13. doi: 10.2174/1389201019666180713102656
134. He D, Qin ZX, Gui CX, Fang Y, Singh S, Talele TT, et al. BIRB796, the inhibitor of p38 mitogen-activated protein kinase, enhances the efficacy of chemotherapeutic agents in ABCB1 overexpression cells. PloS One. (2013) 8:e54181. doi: 10.1371/journal.pone.0054181
135. Xi Y, Nie X, Wang J. Overexpression of BMPER in ovarian cancer and the mechanism by which it promotes Malignant biological behavior in tumor cells. BioMed Res Int. (2020) 2020:3607436. doi: 10.1155/2020/3607436
136. Li C, Hong L, Liu C. Astragalus polysaccharides increase the sensitivity of SKOV3 cells to cisplatin. Arch Gynecol Obstet. (2018) 297:381–6. doi: 10.1007/s00404-017-4580-9
137. Liu HZ, Yu C, Yang Z, He JL, Chen WJ, Yin J, et al. Tubeimoside I sensitizes cisplatin in cisplatin-resistant human ovarian cancer cells (A2780/DDP) through down-regulation of ERK and up-regulation of p38 signaling pathways. Mol Med Rep. (2011) 4:985–92. doi: 10.1007/s00404-017-4580-9
138. Zhao Y, Tian B, Wang Y. Kaempferol sensitizes human ovarian cancer cells-OVCAR-3 and SKOV-3 to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis via JNK/ERK-CHOP pathway and up-regulation of death Receptors 4 and 5. Med Sci Monit Int Med J Exp Clin Res. (2017) 23:5096–105. doi: 10.12659/MSM.903552
139. Zhou J, Gupta K, Yao J, Ye K, Panda D, Giannakakou P, et al. Paclitaxel-resistant human ovarian cancer cells undergo c-Jun NH2-terminal kinase-mediated apoptosis in response to noscapine. J Biol Chem. (2002) 277:39777–85. doi: 10.1074/jbc.M203927200
140. Zhang T, Xu C, Zheng P, Zhang X, Qiu C, Wu F, et al. Glaucocalyxin B attenuates ovarian cancer cell growth and cisplatin resistance in vitro via activating oxidative stress. Oxid Med Cell Longev. (2022) 2022:6324292. doi: 10.1155/2022/6324292
141. Li QQ, Lee RX, Liang H, Wang G, Li JM, Zhong Y, et al. β-elemene enhances susceptibility to cisplatin in resistant ovarian carcinoma cells via downregulation of ERCC-1 and XIAP and inactivation of JNK. Int J Oncol. (2013) 43:721–8. doi: 10.3892/ijo.2013.1996
142. Jiao JW, Wen F. Tanshinone IIA acts via p38 MAPK to induce apoptosis and the down-regulation of ERCC1 and lung-resistance protein in cisplatin-resistant ovarian cancer cells. Oncol Rep. (2011) 25:781–8. doi: 10.3892/ijo.2013.1996
143. Li Z, Tang X, Luo Y, Chen B, Zhou C, Wu X, et al. NK007 helps in mitigating paclitaxel resistance through p38MAPK activation and HK2 degradation in ovarian cancer. J Cell Physiol. (2019) 234:16178–90. doi: 10.1002/jcp.v234.9
144. Weir NM, Selvendiran K, Kutala VK, Tong L, Vishwanath S, Rajaram M, et al. Curcumin induces G2/M arrest and apoptosis in cisplatin-resistant human ovarian cancer cells by modulating Akt and p38 MAPK. Cancer Biol Ther. (2007) 6:178–84. doi: 10.4161/cbt.6.2.3577
145. Zhong ZF, Tan W, Wang SP. Anti-proliferative activity and cell cycle arrest induced by evodiamine on paclitaxel-sensitive and -resistant human ovarian cancer cells. Sci Rep. (2015) 5:16415. doi: 10.1038/srep16415
Keywords: ovarian cancer, drug-resistant, JNK, p38, MAPK, apoptosis, natural compounds
Citation: Ma Y-T, Li C, Shen Y, You W-H, Han M-X, Mu Y-F and Han F-J (2025) Mechanisms of the JNK/p38 MAPK signaling pathway in drug resistance in ovarian cancer. Front. Oncol. 15:1533352. doi: 10.3389/fonc.2025.1533352
Received: 23 November 2024; Accepted: 02 April 2025;
Published: 24 April 2025.
Edited by:
Nikolaos Gavalas, National and Kapodistrian University of Athens, GreeceReviewed by:
Germain Gillet, Université Claude Bernard Lyon 1, FranceGraziana Digiacomo, University of Parma, Italy
Copyright © 2025 Ma, Li, Shen, You, Han, Mu and Han. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Feng-Juan Han, aGFuZmVuZ2p1YW4yMDA0QDE2My5jb20=