 Hongzhi Ji
Hongzhi Ji Li Zhang2
Li Zhang2- 1Department of Respiratory, Affiliated Hospital of Shandong Second Medical University, Weifang, Shandong, China
- 2Department of Gastroenterology, Affiliated Hospital of Shandong Second Medical University, Weifang, Shandong, China
- 3Department of Oncology, Shanghai Pulmonary Hospital & Thoracic Cancer Institute, Tongji University School of Medicine, Shanghai, China
Recent studies have promoted new insights into the biology of non-small cell lung cancer (NSCLC) and made considerable progress in the field of treatment, including targeted therapy for driver gene mutations. Immunotherapy (IO) is another breakthrough, which has achieved amazing clinical efficacy. However, the survival status of advanced NSCLC patients is still unsatisfactory. Drug resistance is an urgent problem to be solved in almost all anti-cancer treatment schemes. Nowadays, platinum based chemotherapy remains the standard treatment for patients with driver gene negative advanced NSCLC. Previous studies have shown that the reduction of intracellular accumulation of platinum drugs, DNA damage repair and the enhancement of detoxification effect all lead to platinum resistance. The mechanisms of tyrosine kinase inhibitors (TKIs) resistance include the emergence of secondary mutation, the activation of bypass signal pathways, the abnormality of downstream signal pathways and the transformation of phenotype. The mechanisms of immune checkpoint inhibitors (ICIs) resistance are more complex. A variety of cells, cytokines and metabolites participate in it to form an immunosuppressive microenvironment, resulting in the impairment of effector T cell function. Exosomes are small molecules secreted by a variety of cells. They can carry information such as miRNA, lncRNA, and protein, and play a pivotal role in signal transduction between cells. More and more studies show that exosomes are important transmitters in lung cancer cells, which can transfer drug resistance information from drug-resistant cells to sensitive cells. However, the underling specific mechanisms need to be further explored to find a new breakthrough for overcoming drug resistance of NSCLC.
Introduction
Non-small cell lung cancer (NSCLC) accounts for 80% of the histological types of lung cancer (1). While surgery remains the optimal treatment for early-stage patients, most are diagnosed at advanced stages and thus ineligible for resection (2). Currently, platinum-based chemotherapy serves as the standard systemic therapy for advanced NSCLC (3, 4). However, its efficacy is severely limited by intrinsic and acquired resistance—only 30% of patients respond initially, median overall survival (OS) falls below one year, and nearly all eventually develop refractory disease, culminating in poor prognosis (5, 6).
The advent of targeted therapies marked a paradigm shift. Following the 2004 discovery of epidermal growth factor receptor (EGFR) mutations, the first-generation EGFR tyrosine kinase inhibitor (TKIs) like gefitinib showed surprising efficacy in clinical trial (7, 8). Since then, a variety of driver gene mutations were gradually identified in NSCLC, such as EML4-ALK translocations and KRAS mutations (9), which has dramatically changed the treatment strategy of lung cancer. Compared to traditional chemotherapy, targeted therapy could significantly improve the survival of advanced NSCLC patients with driver gene mutations (10, 11). However, patients treated with TKIs also develop acquired drug resistance, so the duration of their clinical benefits is limited.
Immunotherapy (IO) emerged as another breakthrough in the treatment of advanced lung cancer patients, changing the landscape of NSCLC in different settings (12). Various large randomized clinical trials have shown that immune checkpoint inhibitors (ICIs) could significantly improve the survival of NSCLC patients when compared with conventional chemotherapy (13, 14), several anti-PD1/PD-L1 antibodies have been used for antitumor therapy in clinical (15). Despite remarkable clinical advances have been achieved in immunotherapy, primary and secondary resistance mechanisms restrict the durable responses of tumors to ICIs, leaving most patients with eventual disease progression.
Critically, drug resistance represents the central bottleneck across all NSCLC therapies—a multifaceted process driven by tumor-intrinsic mechanisms, microenvironmental crosstalk, and intercellular communication. Among these, exosomes have emerged as pivotal mediators. These extracellular vesicles not only facilitate tumor progression and metastasis but also orchestrate chemotherapy, targeted therapy, and immunotherapy resistance through cargo transfer (such as miRNAs, proteins) between cancer and stromal cells (16, 17).
This review systematically analyzes the resistance mechanisms underlying conventional NSCLC treatments and the deterministic role of exosomes in fostering therapeutic escape. By elucidating these pathways, we aim to guide future strategies for overcoming resistance through exosome-targeted interventions.
Biogenesis and function of exosomes
Exosomes are 30–150 nm small molecules secreted by both normal cells and cancer cells (18), which can be isolated in many body fluids such as blood, urine, saliva, bronchoalveolar fluid, etc. (19). Exosomes are produced in cells through a variety of dynamic endocytosis pathways (Figure 1). Firstly, membrane proteins sprout inward through the plasma membrane and are endocytosed to form early endosomes. Then, the early endosomes mature into the late endosomes, which are known as multivesicular bodies (MVBs) (20). MVBs eventually enter the lysosome, where hydrolases and other enzymes degrade some components of MVBs and remove toxic substances. MVBs have specific surface proteins, such as CD63, lysosome related membrane proteins Lamp1 and Lamp2, so they can fuse to the plasma membrane and become exosomes to release to the extracellular environment (21, 22). Exosomes can adhere to receptor cells through integrins on the surface, then induce intracellular signals. Recipient cells take up exosomes through phagocytosis, pinocytosis, and macro-pinocytosis (23). In addition, exosomes can fuse with the plasma membrane directly to release their contents, regulating the signal transduction of the recipient cells (24).
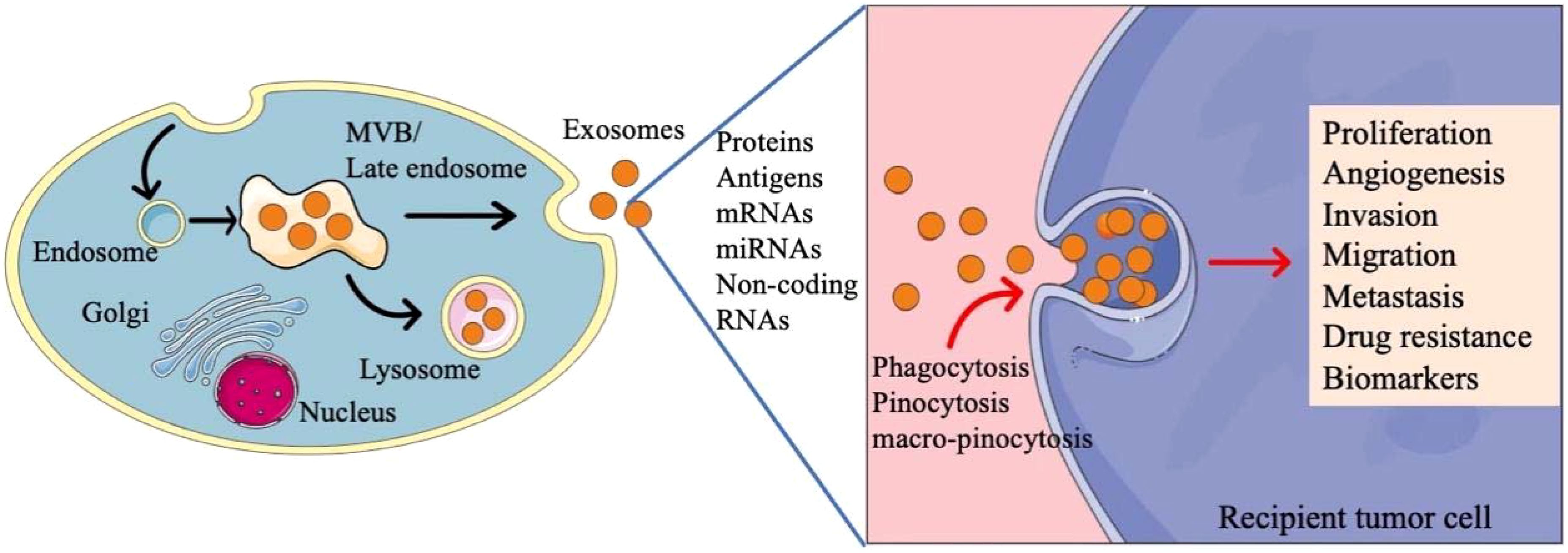
Figure 1. The biogenesis and function of exosomes in tumor cells. Exosomes are produced in cells through a variety of dynamic endocytosis pathways. Tumor derived exosomes contain varying quantities of molecules, participating in the regulation of cell activies.
Exosomes contain varying quantities of molecules such as microRNAs (miRNAs), messenger RNAs (mRNAs) and proteins (25). Tumor derived exosomes in tumor microenvironment have unique contents, mediating interactions between cancer cells and stromal cells, participating in tumor proliferation, angiogenesis, invasion, migration, metastasis and drug resistance (26). In addition, exosomes can also be used as biomarkers for early diagnosis and prognosis evaluation (27). Considering the regulatory role of the components of exosomes in tumor microenvironment, exosomes have been explored for the treatment of cancers. A lot of studies have reported that exosomes can be modified to deliver molecules with therapeutic effects, which will become a promising drug delivery system because of the low toxicity and inherent intercellular communication ability (28, 29).
Role of exosomes in cisplatin resistance of NSCLC
Previous studies have shown that more than 60% of patients with NSCLC were resistant to cisplatin and carboplatin (30). In addition, these patients also showed varying degrees of resistance to other drugs, including docetaxel, gemcitabine, paclitaxel and vinorelbine (30). Therefore, almost all patients with NSCLC will finally develop resistance to chemotherapeutic drugs, even if the initial response is satisfactory. Chemoresistance is a major obstacle in the process of antitumor treatment for all patients and it is one of the main challenges in cancer management. The mechanisms related to chemoresistance are complex and have not been fully understood.
Due to the development of TKIs and ICIs and the diversification of chemotherapy schemes, the treatment options of cancer are increasing. However, cisplatin is still a highly potent anticancer drug, and its resistance mechanism is the most deeply studied in chemotherapeutic drugs at present.
Cisplatin is a kind of cell cycle nonspecific cytotoxic drug. It binds to nucleophilic groups in cells and is selectively distributed in tumor tissues. Cisplatin is hydrolyzed after entering tumor cells and then forms cisplatin-DNA complex with cell DNA. This process can destroy the normal structure of cell DNA, hinder the template effect of DNA, inhibit DNA replication and transcription (31, 32). At the same time, cisplatin affects DNA repair, induces oxidative stress, activates apoptosis, and eventually leads to tumor cell death by activating a variety of signal pathways (33).
The mechanisms of cisplatin resistance of tumor cells are multifaceted. Cisplatin can affect the expression of some transporters, thus reducing their accumulation in cells. Studies have shown that low expression of copper transporter 1 (CTR1) in lung cancer may be related to cisplatin resistance (34). Nucleotide excision repair (NER) system and mismatch repair (MMR) pathway play a key role in the repair of DNA damage caused by chemotherapy (35, 36). Abnormal expression of NER components (such as xeroderma pigmentosum group A (XPA)) and MMR related proteins (such as MutS homologue 2 (MSH2)) can enhance DNA repair and reduce the sensitivity of tumor cells to cisplatin (37, 38). Reduced glutathione (GSH), metallothionein (MT) and other nucleophilic “scavengers” in the cytoplasm can chelate cisplatin, thus reducing the accumulation and cytotoxic activity of cisplatin (39). Cisplatin can inhibit the tumor cells apoptosis and produce drug resistance by regulating PI3K/AKT, Bax/Bcl-2 and other signal pathways (40). In addition, abnormal extracellular matrix and epithelial mesenchymal transition (EMT) can promote the insensitivity of lung cancer cells to cisplatin (41, 42).
In recent years, many studies have explored the mechanisms of exosomes in cisplatin resistance, especially the miRNAs in exosomes, which play an important role in transmitting drug resistance information. The sources of these exosomes are mainly divided into two categories. Some exosomes are derived from tumor cells, while others are secreted by non-tumor cells (Table 1).
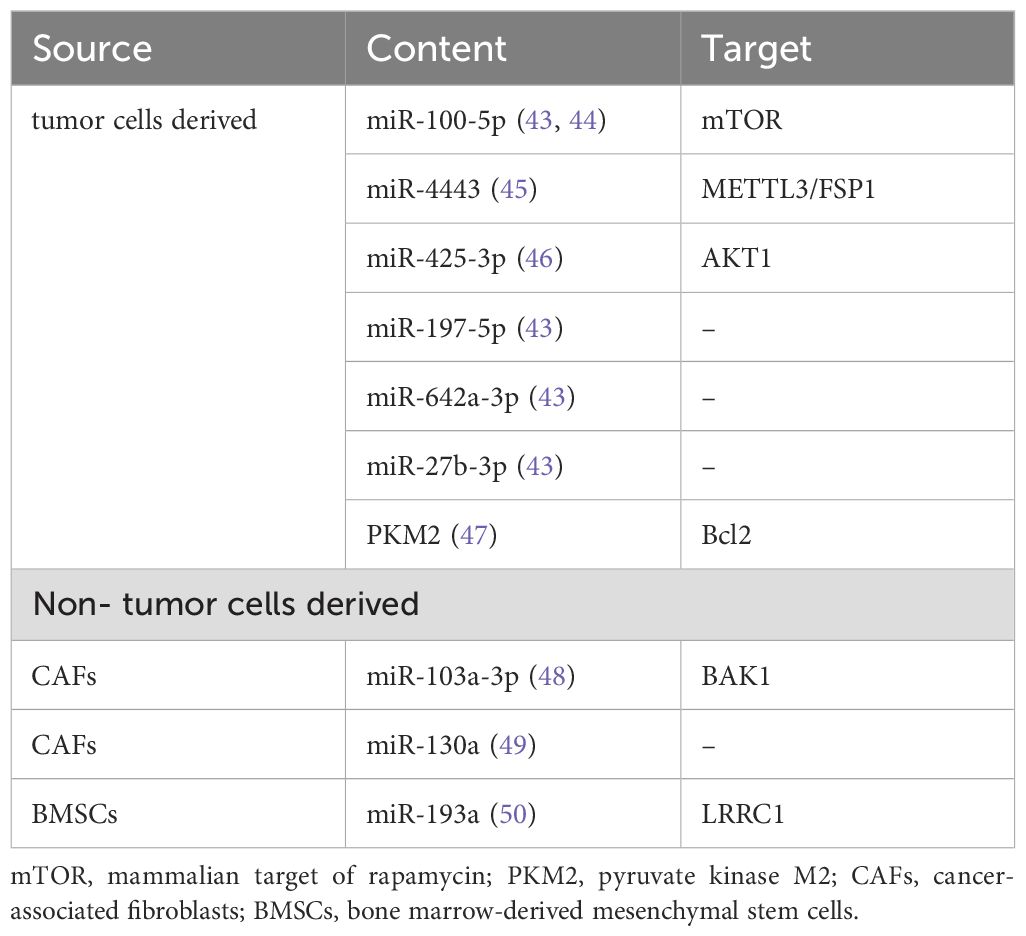
Table 1. Exosomes involved in chemoresistance of NSCLC and their potential targets.
It was found that when lung cancer cells A549 were exposed to cisplatin, they could release more exosomes, and when these exosomes were co-cultured with other A549 cells, their resistance to cisplatin increased. It indicated that A549 cells could release exosomes for intercellular signal communication and realize the transmission of intercellular drug resistance (51). Five microRNAs with the most significant expression difference were found in the exosomes of A549 cells and cisplatin resistance A549 cells (A549/DDP), they were miR-27b-3p, miR-100-5p, miR-197-5p, miR-4443 and miR-642a-3p. Target gene prediction and pathway analysis suggested that these microRNA might be important regulators in the drug resistance of cisplatin (43). There was a study found that compared with A549 sensitive strain, the expression of miR-100-5p in the exosomes of A549/DDP cells was significantly downregulated, while the expression of mammalian target of rapamycin (mTOR) in recipient cells was regulated by miR-100-5p. Therefore, exosomes from cisplatin resistant lung cancer cells could change the sensitivity of other cells by regulating mTOR through miR-100-5p (44). While another study showed that the level of miR-4443 in cisplatin resistant NSCLC tumor tissue-derived exosomes was upregulated. And mechanistic studies indicated that exosomal miR-4443 could participate in the regulation of cisplatin resistance of NSCLC cells, and METTL3/FSP1-mediated ferroptosis might be the potential mechanism (45). After cisplatin stimulation, the expression of miR-425-3p in exosomes was induced by c-Myc-mediated transactivation. Further studies showed that exosomal miR-425-3p promoted the activation of autophagy by targeting AKT1, which eventually reduced the sensitivity of recipient cells to cisplatin (46). A study found that hypoxia induced NSCLC cell-derived exosomal pyruvate kinase M2 (PKM2, a rate-limiting enzyme in glycolysis) could promote the glycolysis of NSCLC cells, reduce cisplatin induced reactive oxygen species (ROS), and inhibit apoptosis through PKM2-Bcl2 pathway. In addition, exosomal PKM2 induced by hypoxia could reprogram cancer-associated fibroblasts (CAFs) to create an acidic environment, thus promoting cisplatin resistance of NSCLC cells (47).
Non-tumor cells derived exosomes are also important transmitters of intercellular drug resistance information. It was found that MiR-103a-3p was highly expressed in CAFs and CAFs exosomes in NSCLC. When CAFs exosomes were added to culture medium of NSCLC cells, the expression of miR-103a-3p in exosomes of NSCLC cells increased. In vitro experiments showed that exosomal miR-103a-3p derived from CAFs could inhibit cisplatin induced apoptosis and promote resistance to cisplatin in NSCLC cells, which has also been confirmed by mouse tumorigenesis assay in vivo. While the RNA-binding protein pumilio homolog 2 (PUM2) could facilitate miR-103a-3p packaging into CAFs derived exosomes in cytoplasm and nucleus. Further experiments showed that miR-103a-3p promoted the resistance of NSCLC cells to cisplatin by downregulating BAK1 (48). In addition, another study showed that miR-130a in CAFs derived exosomes could also promote the resistance of NSCLC cells to cisplatin, and PUM2 was also involved in the packaging process of miR-130a into exosomes (49). The expression of miR-193a in bone marrow-derived mesenchymal stem cells (BMSCs) derived exosomes was increased, which could inhibit the proliferation and migration of NSCLC cisplatin resistant cells. The BMSCs derived exosomal miR-193a could also promote apoptosis and reduce the cisplatin resistance of NSCLC cells. And the downregulation of LRRC1 expression caused by miR-193a played a regulatory role in the mechanism of cisplatin resistance (50).
Role of exosomes in TKIs resistance of NSCLC
Two classic oncogene mutations in NSCLC are epidermal growth factor receptor (EGFR) mutation or anaplastic lymphoma kinase (ALK) chromosome rearrangement (the most common one is EML4-ALK fusion) (52). These two gene mutations have become the standard and routine detection items of advanced lung adenocarcinoma. In various randomized phase III clinical trials, EGFR and ALK TKIs have always shown higher efficacy than chemotherapy, making targeted therapy the standard treatment for advanced NSCLC with such gene mutations (53, 54). However, drug resistance remains a pervasive challenge in clinic, patients will eventually get progress during targeted therapy. Therefore, in this review, we mainly focus on the molecular mechanisms of drug resistance of EGFR and ALK TKIs.
According to the response of the tumor to the initial treatment, drug resistance can be divided into two types. One is primary drug resistance, who have no response at all to the treatment. The other is acquired drug resistance. Such patients may have a complete or partial response at first, but eventually have no response over time (55). The primary resistance of NSCLC to EGFR TKIs is mainly related to wild-type EGFR. The activation mutation of KRAS or BRAF and the loss of function of apoptotic protein BIM will lead to primary drug resistance (56–58). Some rare EGFR mutations can also reduce the sensitivity of tumors to TKIs, such as small insertions or duplications in exon 20 (59). Fibroblast growth factor (FGF), hepatocyte growth factor (HGF) and neuregulin1 (NRG1) in tumor microenvironment are involved in the regulation of primary drug resistance by regulating Ras/MAPK or PI3K/Akt signaling pathway (60). In addition, the activation of NF-κB signaling is also identified as one of the mechanisms of primary resistance to EGFR TKIs (61).
In recent years, extensive efforts have been made to clarifying the mechanisms of acquired drug resistance of TKIs (Figure 2). The acquired resistance of the first-generation EGFR TKIs (gefitinib or erlotinib) is mainly mediated by the development of T790M mutation, which occurs in 50-65% of EFGR mutation and TKIs resistant patients (62), other secondary mutations including T854A, D761Y and L747S (63). The first mutation of resistance to second-generation EGFR-TKI (afatinib) is also T790M mutation, and the secondary mutation of resistance to third generation EGFR-TKI (osimertinib) is EGFR C797S or G796D mutation (64). For the first generation ALK-TKI crizotinib resistant patients, ALK kinase domain mutations are the most common mechanisms, including L1196 M, C1156Y, L1152 R, G1202R, S1206Y and G1269A mutations (65, 66). And G1202R is the most common mutation in patients resistant to second-generation ALK-TKIs (alectinib, ceritinib, brigatinib) (67). Unlike the first or second generation ALK-TKIs, most lorlatinib resistant patients have multiple ALK mutations, such as L1196M/D1203N, F1174L/G1202R and C1156Y/G1269A (68).
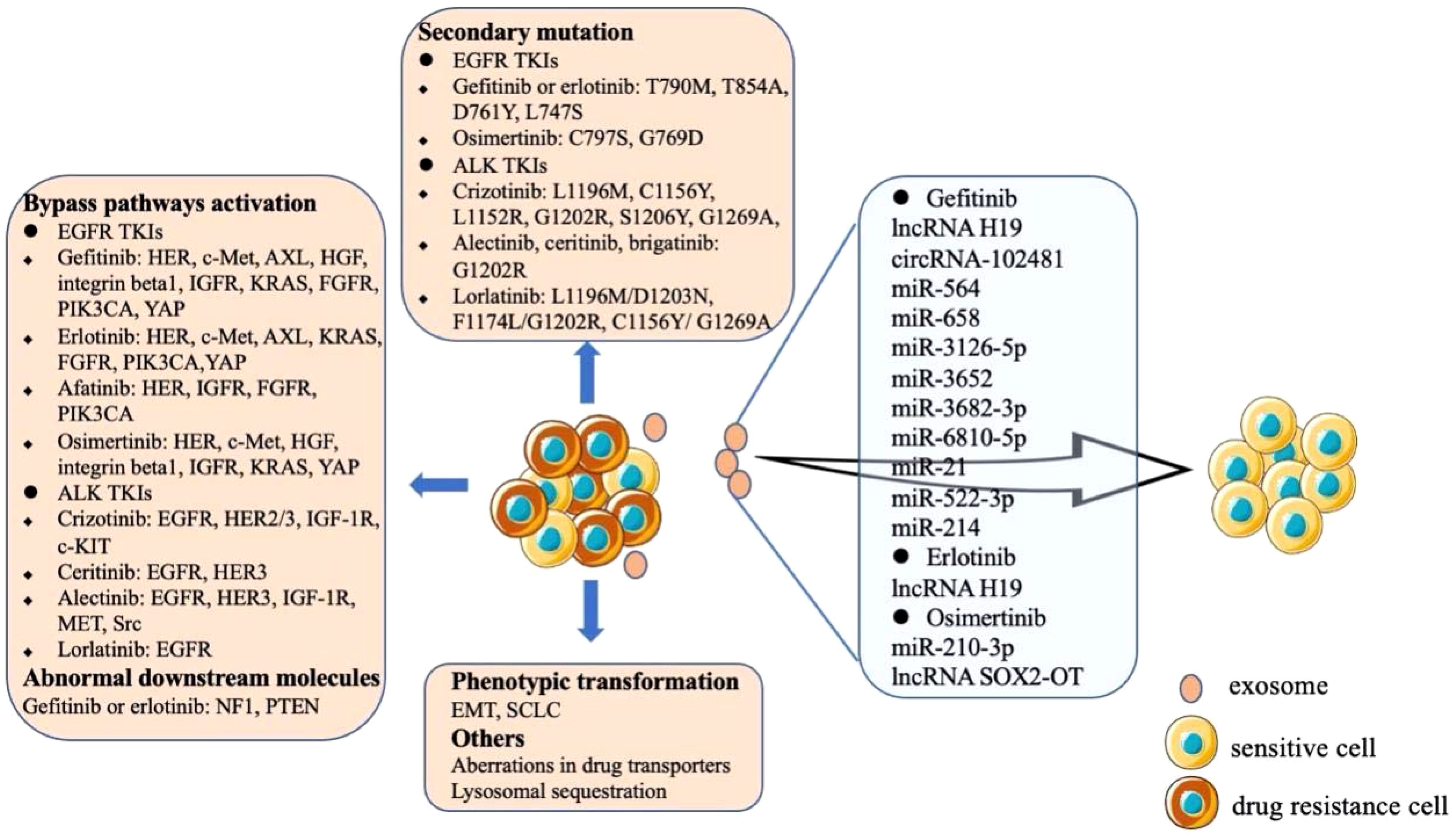
Figure 2. Mechanisms of acquired drug resistance of EGFR and ALK TKIs. The resistance mechanisms of TKIs mainly include the emergence of secondary mutations, bypass activation and phenotypic transformation. The exosomes secreted by drug-resistant cells contain special cytokines, which are transmitted to sensitive cells to make them resistant to TKIs.
Another mechanism of TKIs resistance is the aberrated activation of the bypass pathways (Figure 2). The aberrance of other members of HER family, amplification of c-Met, overexpression of AXL, overexpression of HGF, overexpression and activation of integrin beta1, and the abnormality of IGFR can affect the PI3K/AKT, MAPK, ERK, or NF-κB signaling pathways to induce EGFR TKIs resistance (63). Abnormal expression of downstream molecules may also lead to resistance to gefitinib or erlotinib, such as the aberrant expression of NF1 and the loss of PTEN (63). Bypass signaling tracks associated with ALK-TLIs resistance including the abnormal of EGFR, HER2/3, IGF-1R, c-KIT, etc. (Figure 2) (69). The other two rare mechanisms of acquired TKIs resistance are the phenotypic change of NSCLC via EMT and the histological transformation of NSCLC into SCLC (63, 69). Some drug transporters (ABCB1/PGP and ABCG2/BCRP) and lysosomal sequestration may also be involved in erlotinib and gefitinib resistance (64). In addition, tumor heterogeneity is a common phenomenon. Generally, TKIs sensitive cells and acquired drug-resistant tumor cells are mixed. When the sensitive cells are killed, the drug-resistant cells proliferate rapidly and become the dominant cell group, further enhancing the drug resistance (69).
The studies of exosomes in drug resistance of TKIs mainly focus on gefitinib, erlotinib and osimertinib. It was found that the level of lncRNA H19 in gefitinib resistant cells was higher than that in sensitive cells. In addition, knockout of lncRNA H19 increased the sensitivity of cells to gefitinib. Further studies confirmed that lncRNA H19 could be encapsulated in exosomes and transferred to sensitive cells to induce gefitinib resistance, which was specifically mediated by heterogeneous nuclear ribonucleoprotein A2B1 (hnRNPA2B1), an RNA binding protein that controls the loading of RNA into exosomes (70). Tumor derived exosomal circRNA-102481 could enhance the expression of ROR1 and promote gefitinib resistance (71). A microRNA analysis of exosomes showed that miR-564, miR-658, miR-3126-5p, miR-3652, miR-3682-3p and miR-6810-5p were significantly up-regulated in the exosomes of gefitinib resistant cells. Further studies proved that exosomal miR-564 and miR-658 from gefitinib resistant NSCLC cells could induce drug resistance in sensitive cells (72). Studies have shown that tumor-derived exosomal miR-21 and miR-522-3p could participate in gefitinib resistance by regulating PI3K/Akt signaling pathway, while exosomal miR-214 could confer gefitinib resistance via Bax/Bcl2 signaling (73–75). Exosomal lncRNA H19 could also regulate the expression of autophagy-related protein 7(ATG7) by targeting miR-615-3p, thus affecting the drug resistance of NSCLC cells to erlotinib (76). A study demonstrated that exosomes containing wild-type EGFR protein were internalized by EGFR mutant cancer cells through clathrin-dependent endocytosis, and then the downstream PI3K/Akt and MAPK signaling pathways were activated by the wild-type EGFR protein, thus triggering osimertinib resistance (77). It was found that miR-210-3p was highly expressed in the exosomes of osimertinib resistant cells. What’s more, exosomal miR-210-3p could directly promote EMT of tumor cells and resistance to osimertinib (78). In addition, NSCLC cell line H1975 could transfer lncRNA SOX2-OT into macrophages through exosomes. LncRNA SOX2-OT facilitated the expression of Smads by sponging miR-627-3p and induced macrophages M2 polarization to aggravate the drug resistance of cancer cells to osimertinib (79).
To clarify the role of exosomes in NSCLC drug resistance, a comparison with classical mechanisms, such as gene mutations and drug efflux pumps, is instructive. Mutations like EGFR T790M primarily confer resistance by decreasing the binding affinity for tyrosine kinase inhibitors, thus limiting therapeutic efficacy (73). Meanwhile, drug efflux transporters such as ABCB1 and ABCG2 reduce drug accumulation inside tumor cells through active drug export, resulting in treatment failure (80). These traditional resistance mechanisms are typically cell-intrinsic, affecting individual cancer cells, and generally irreversible once established. By comparison, exosomes provide a distinctive mechanism involving intercellular communication. They can transfer resistance-associated molecules—including miRNAs and proteins—from resistant cells to sensitive cells, thus propagating resistance throughout diverse tumor populations (73). Additionally, exosomes modulate the tumor microenvironment and mediate immune evasion, which are distinct processes not typically associated with traditional mutation or efflux-based resistance mechanisms (81). Clinically, the presence of exosomal contents in biofluids allows for non-invasive monitoring of treatment response. Furthermore, exosome secretion, uptake, and cargo packaging are processes amenable to therapeutic targeting. Therefore, exosomes hold promise not only as biomarkers for resistance but also as novel therapeutic targets, underscoring their increasingly recognized and multifaceted role in NSCLC drug resistance.
Role of exosomes in ICIs resistance of NSCLC
Drug resistance of ICIs can also be divided into primary resistance and acquired resistance. Statistics show that less than 30% of NSCLC patients respond to ICIs, and most patients suffer primary resistance (82). The emergence of ICIs resistance is a complex, dynamic and interdependent process, which is closely related to many tumor and host factors (Figure 3).
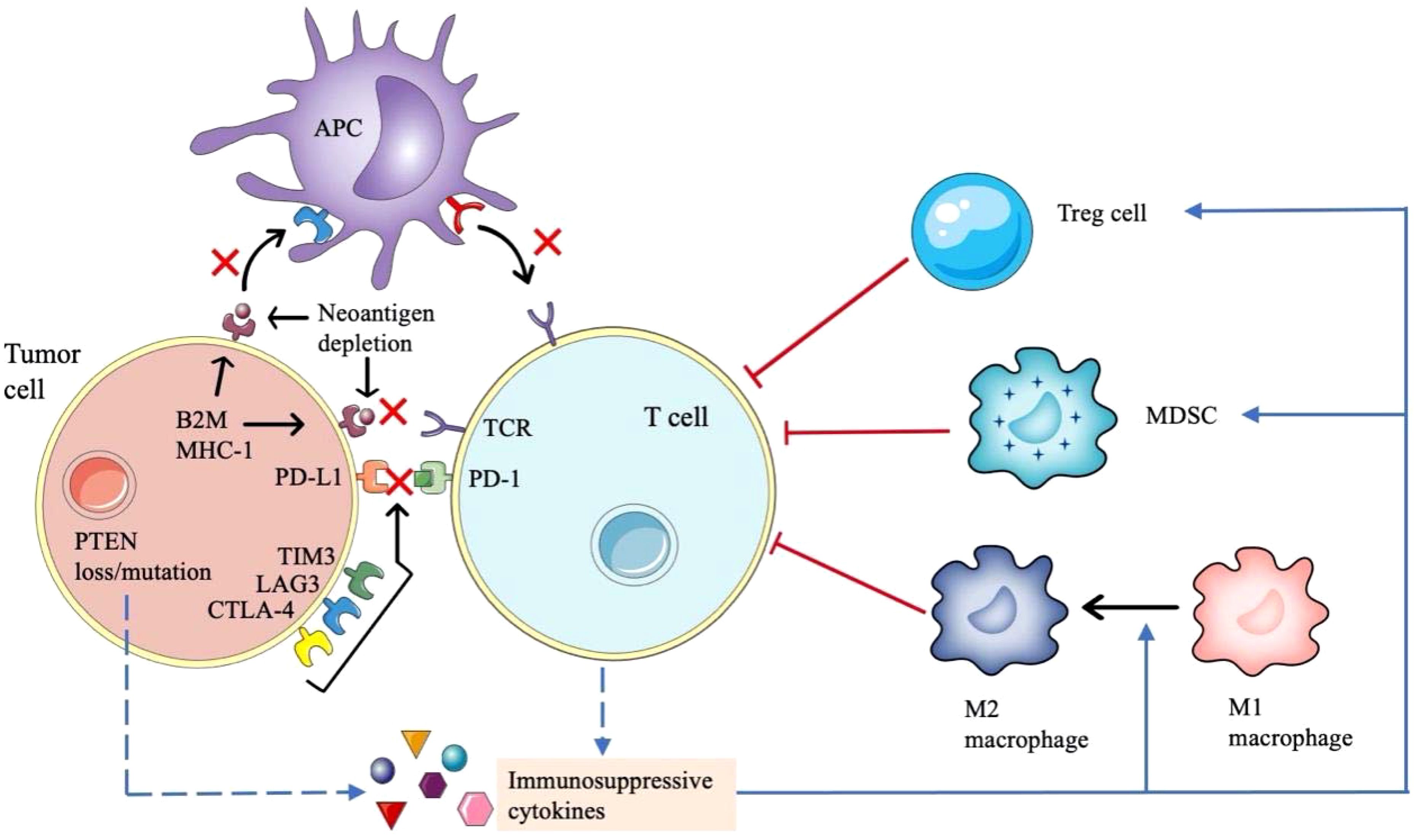
Figure 3. Mechanisms of drug resistance of immune checkpoint inhibitors in NSCLC. The mechanism of immunotherapy resistance is complex. Loss of antigen expression or presentation defect of tumor cells, abnormal signaling pathways in tumor cells and upregulation of non-PD-1 / PD-L1 immune checkpoints will affect the interaction between tumor cells, antigen presenting cells and T cells. In addition, many immunosuppressive cytokines will inhibit the antitumor activity of T cells.
The efficacy of ICIs depends on the formation of tumor new antigens. If the tumor specific antigens are less expressed and their immunogenicity become weak, it will be not enough to activate primitive T cells. And if a neoantigen is structurally like an immune tolerance antigen or an autoantigen, it will be difficult to be recognized by antigen-presenting cells (APCs) and activate T cells, which will lead to the drug resistance to PD-1/PD-L1 inhibitors (83). Class I MHC, β-2 microglobulin (β2M), large multifunctional proteinase (LMP) and transporter associated with antigen processing (TAP) are important components of tumor antigen processing and presentation devices, resistance to PD-1/PD-L1 inhibitors also occurs when the genes encoding them become abnormal. For example, the loss of β2M expression leads to the impaired expression of class I MHC molecules on the surface of APCs, resulting in impaired antigen presentation and finally immune tolerance (84). Furthermore, the downregulation of HLA class I molecules may also be related to ICIs resistance (85).
Several factors lead to the inadequate function of tumor specific T cells and reduce the clinical effect of PD-1/PD-L1 inhibitors. The expression of many co-inhibitory receptors, such as cytotoxic T lymphocyte antigen 4 (CTLA-4), T cell immunoglobulin and mucin domain-3 (TIM3) and lymphocyte activation gene 3 (LAG3) participate in immune escape and affect the antitumor effect of T cells (86). Emerging evidence suggest that T cell exhaustion is associated with epigenetic changes manifested as immune dysfunction and continuous expression of surface inhibitory receptors, making it difficult to function as normal effector T cells (87).
A variety of cells and cytokines in tumor microenvironment (TME) form an immunosuppressive state, which can diminish the therapeutic efficacy of PD-1/PD-L1 inhibitors. Regulatory T cells (Tregs) can inhibit the function of effector T cells by secreting some inhibitory cytokines (88, 89). The removal of Tregs in the TME can enhance the antitumor effect of PD-1/PD-L1 inhibitors (90). MDSCs are the main regulatory factors of immune response under various pathological conditions, which can promote angiogenesis, tumor invasion and metastasis. The presence of MDSCs in TME will reduce the effect of IO (91). Tumor associated macrophages (TAMs), including M1 macrophages and M2 macrophages, another class of cells that regulate the immune environment against tumors (92). Immunosuppressive cytokines such as CCL5, CCL7 and CXCL8, can aggregate MDSCs and Tregs into the TME (93). In addition, indoleamine 2,3-dioxygenase (IDO) produced by tumor or immune cells can improve the production and activity of Tregs and MDSCs, produce immunosuppressive metabolites to influence the function of effector T cells (94). Some gene mutations are associated with immunosuppression. PTEN gene deletion leads to the upregulation of CCL2, hypoxia inhibitory factor 1 (HIF-1) and vascular endothelial growth factor (VEGF), which leads to the transformation of macrophages from M1 type to M2 type, resulting in negative immune regulation (95). KRAS mutation will cause the loss of STK11/LKB1, and then T cell suppressor neutrophils are recruited, resulting in the reduction of T cells infiltration (96, 97).
Tumor cells can produce exosomes rich in cancer promoting components, such as immunosuppressive proteins like PD-L1, regulating immune response and promoting drug resistance (98). PD-L1 derived from tumor exosomes presented both on the surface and within exosomes, and exosomes can transport PD-L1 to other cells with low or no expression of PD-L1 and may bind to PD-1 (99). Insoluble PD-L1 expressed on plasma/serum exosomes is associated with disease progression of NSCLC (100). Previous study found that exosomes containing PD-L1 isolated from NSCLC patients could reduce the production of IFN-γ and interleukin-2 (IL-2), inhibiting the activity of CD8+ T cells, this effect was more significant in exosomes with high level of PD-L1 (101). What’s more, exosomes with high levels of PD-L1 could also induce apoptosis of CD8+ T cells through PD-L1/PD-1 interaction (102). So far, there are few studies on exosomes in ICIs resistance of NSCLC. It is necessary to further explore and clarify their role in immune regulation, to provide new theoretical basis for ICIs resistance, and look for potential therapeutic breakthroughs.
Building upon the mechanistic distinctions outlined above, we further explore the unique clinical implications of exosome-mediated resistance in NSCLC. Exosome-mediated resistance differs from traditional drug resistance mechanisms in NSCLC, such as gene mutations, drug efflux pumps, EMT, and tumor heterogeneity. Generally, mechanisms like gene mutations or drug efflux involve intrinsic cellular changes, often irreversible and restricted to single tumor cells. In contrast, exosomes can deliver resistance-associated molecules such as miRNAs and proteins from resistant cells to sensitive ones, enabling the horizontal spread of resistance throughout tumor populations (103).
Moreover, exosomes have additional roles in modifying the tumor microenvironment and facilitating immune escape, features rarely seen with mutations or efflux pump-related resistance (104). Clinically, exosomes can be detected in patients’ body fluids, providing a non-invasive way to monitor drug resistance and disease progression dynamically. Importantly, various steps involved in exosome biology—including secretion, uptake, and cargo packaging—can be therapeutically targeted, offering novel strategies for overcoming drug resistance beyond conventional approaches (105).
Emerging exosome-based combination strategies in NSCLC
Recent studies suggest that exosome-targeting approaches may complement existing NSCLC therapies by enhancing drug sensitivity, overcoming resistance, or serving as biomarkers. Table 2 provides representative examples of such strategies under preclinical or exploratory investigation (105–108).
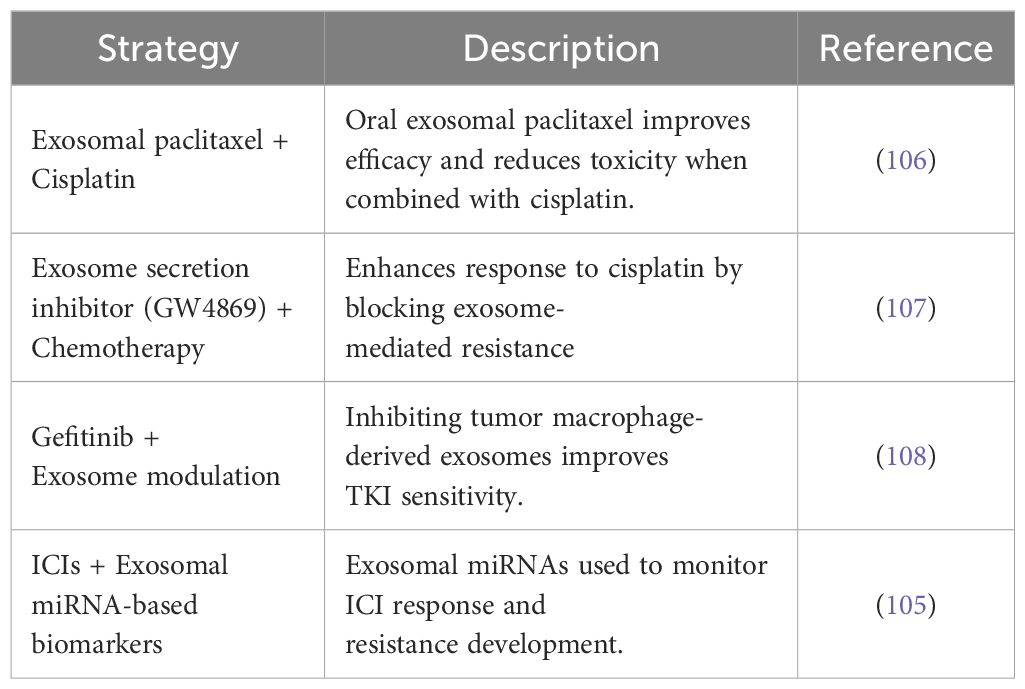
Table 2. Potential combination therapies involving exosome-targeting and standard NSCLC treatments.
Conclusion
Drug resistance is an inevitable thorny problem in the clinical treatment of NSCLC. Whether it is the classical chemotherapy, targeted therapy or immunotherapy, the emergence of drug resistance limits the clinical efficacy, and its molecular mechanisms are multifactorial and complex. There is increasing evidence that exosomes are involved in the drug resistance process of NSCLC, which can transmit information between cells though the miRNAs or proteins in it. However, further exploration of tumor microenvironment after lung cancer treatment is still needed to overcome the issue of drug resistance.
Author contributions
HJ: Writing – original draft, Writing – review & editing. LZ: Conceptualization, Data curation, Writing – review & editing. LY: Conceptualization, Formal analysis, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bray F, Laversanne M, Sung H, Ferlay J, Siegel RL, Soerjomataram I, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2024) 74:229–63. doi: 10.3322/caac.21834
2. Rajaram R, Huang Q, Li RZ, Chandran U, Zhang Y, Amos TB, et al. Recurrence-free survival in patients with surgically resected non-small cell lung cancer: A systematic literature review and meta-analysis. Chest. (2024) 165:1260–70. doi: 10.1016/j.chest.2023.11.042
3. National Comprehensive Cancer Network (NCCN). NCCN Clinical Practice Guidelines in Oncology: Non-Small Cell Lung Cancer, Version 3. Plymouth Meeting, PA: National Comprehensive Cancer Network (2024).
4. Hendriks LE, Kerr KM, Menis J, Mok TS, Nestle U, Passaro A, et al. Non-oncogene-addicted metastatic non-small-cell lung cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann Oncol. (2023) 34:358–76. doi: 10.1016/j.annonc.2022.12.013
5. Umar H, Wahab HA, Attiq A, Amjad MW, Bukhari SNA, and Ahmad W. Platinum-based targeted chemotherapies and reversal of cisplatin resistance in non-small cell lung cancer (NSCLC). Mutat Res. (2024) 828:111856. doi: 10.1016/j.mrfmmm.2024.111856
6. Song TH, Chen XX, Lee CK, Sze SC, Feng YB, Yang ZJ, et al. Dendrobine targeting JNK stress signaling to sensitize chemotoxicity of cisplatin against non-small cell lung cancer cells in vitro and in vivo. Phytomedicine. (2019) 53:18–27. doi: 10.1016/j.phymed.2018.06.018
7. Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. (2004) 350:2129–39. doi: 10.1056/NEJMoa040938
8. Paez JG, Jänne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. (2004) 304:1497–500. doi: 10.1126/science.1099314
9. Barlesi F, Mazieres J, Merlio JP, Debieuvre D, Mosser J, Lena H, et al. Routine molecular profiling of patients with advanced non-small-cell lung cancer: results of a 1-year nationwide programme of the French Cooperative Thoracic Intergroup (IFCT). Lancet. (2016) 387:1415–26. doi: 10.1016/S0140-6736(16)00004-0
10. Liang G, Fan W, Luo H, and Zhu X. The emerging roles of artificial intelligence in cancer drug development and precision therapy. BioMed Pharmacother. (2020) 128:110255. doi: 10.1016/j.biopha.2020.110255
11. Nyen JE, Booth A.Ø, Husby Ø, Bugge C, Engebretsen I, Oteiza F, et al. Targeted treatment and survival in advanced non-squamous non-small cell lung cancer patients – a nationwide and longitudinal study. Front Oncol. (2025) 15:1506041. doi: 10.3389/fonc.2025.1506041
12. Howlader N, Forjaz G, Mooradian MJ, Meza R, Kong CY, Cronin KA, et al. The effect of advances in lung-cancer treatment on population mortality. N Engl J Med. (2020) 383:640–9. doi: 10.1056/NEJMoa1916623
13. Herbst RS, Giaccone G, de Marinis F, Reinmuth N, Vergnenegre A, Barrios CH, et al. Atezolizumab for first-line treatment of PD-L1-selected patients with NSCLC. N Engl J Med. (2020) 383:1328–39. doi: 10.1056/NEJMoa1917346
14. Goulart BHL, Mushti SL, Chatterjee S, Larkins E, Mishra-Kalyani PS, Pazdur R, et al. Correlations of response rate and progression-free survival with overall survival in immunotherapy trials for metastatic non-small-cell lung cancer: an FDA pooled analysis. Lancet Oncol. (2024) 25:455–62. doi: 10.1016/S1470-2045(24)00040-8
15. Marur S, Singh H, Mishra-Kalyani P, Larkins E, Keegan P, Sridhara R, et al. FDA analyses of survival in older adults with metastatic non-small cell lung cancer in controlled trials of PD-1/PD-L1 blocking antibodies. Semin Oncol. (2018) 45:220–5. doi: 10.1053/j.seminoncol.2018.08.007
16. Zhang L and Yu D. Exosomes in cancer development, metastasis, and immunity. Biochim Biophys Acta Rev Cancer. (2019) 1871:455–68. doi: 10.1016/j.bbcan.2019.04.004
17. Mir R, Baba SK, Elfaki I, Algehainy N, Alanazi MA, Altemani FH, et al. Unlocking the secrets of extracellular vesicles: Orchestrating tumor microenvironment dynamics in metastasis, drug resistance, and immune evasion. J Cancer. (2024) 15:6383–415. doi: 10.7150/jca.98426
18. Keller S, Sanderson MP, Stoeck A, and Altevogt P. Exosomes: from biogenesis and secretion to biological function. Immunol Lett. (2006) 107:102–8. doi: 10.1016/j.imlet.2006.09.005
19. Conde-Vancells J, Rodriguez-Suarez E, Embade N, Gil D, Matthiesen R, Valle M, et al. Characterization and comprehensive proteome profiling of exosomes secreted by hepatocytes. J Proteome Res. (2008) 7:5157–66. doi: 10.1021/pr8004887
20. Merchant ML, Rood IM, Deegens JKJ, and Klein JB. Isolation and characterization of urinary extracellular vesicles: implications for biomarker discovery. Nat Rev Nephrol. (2017) 13:731–49. doi: 10.1038/nrneph.2017.148
21. Colombo M, Raposo G, and Théry C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. (2014) 30:255–89. doi: 10.1146/annurev-cellbio-101512-122326
22. Hessvik NP and Llorente A. Current knowledge on exosome biogenesis and release. Cell Mol Life Sci. (2018) 75:193–208. doi: 10.1007/s00018-017-2595-9
23. Chen R, Xu X, Qian Z, Zhang C, Niu Y, Wang Z, et al. The biological functions and clinical applications of exosomes in lung cancer. Cell Mol Life Sci. (2019) 76:4613–33. doi: 10.1007/s00018-019-03233-y
24. Tian T, Wang Y, Wang H, Zhu Z, and Xiao Z. Visualizing of the cellular uptake and intracellular trafficking of exosomes by live-cell microscopy. J Cell Biochem. (2010) 111:488–96. doi: 10.1002/jcb.v111:2
25. Vlassov AV, Magdaleno S, Setterquist R, and Conrad R. Exosomes: current knowledge of their composition, biological functions, and diagnostic and therapeutic potentials. Biochim Biophys Acta. (2012) 1820:940–8. doi: 10.1016/j.bbagen.2012.03.017
26. Wolf-Dennen K and Kleinerman ES. Exosomes: dynamic mediators of extracellular communication in the tumor microenvironment. Adv Exp Med Biol. (2020) 1258:189–97. doi: 10.1007/978-3-030-43085-6_13
27. Li MY, Liu LZ, and Dong M. Progress on pivotal role and application of exosome in lung cancer carcinogenesis, diagnosis, therapy and prognosis. Mol Cancer. (2021) 20:22. doi: 10.1186/s12943-021-01312-y
28. Xu K, Zhang C, Du T, Gabriel ANA, Wang X, Li X, et al. Progress of exosomes in the diagnosis and treatment of lung cancer. BioMed Pharmacother. (2021) 134:111111. doi: 10.1016/j.biopha.2020.111111
29. Wu P, Zhang B, Ocansey DKW, Xu W, and Qian H. Extracellular vesicles: A bright star of nanomedicine. Biomaterials. (2021) 269:120467. doi: 10.1016/j.biomaterials.2020.120467
30. d’Amato TA, Landreneau RJ, McKenna RJ, Santos RS, and Parker RJ. Prevalence of in vitro extreme chemotherapy resistance in resected nonsmall-cell lung cancer. Ann Thorac Surg. (2006) 81:440–6. doi: 10.1016/j.athoracsur.2005.08.037
31. Martin LP, Hamilton TC, and Schilder RJ. Platinum resistance: the role of DNA repair pathways. Clin Cancer Res. (2008) 14:1291–5. doi: 10.1158/1078-0432.CCR-07-2238
32. Rabik CA and Dolan ME. Molecular mechanisms of resistance and toxicity associated with platinating agents. Cancer Treat Rev. (2007) 33:9–23. doi: 10.1016/j.ctrv.2006.09.006
33. Galluzzi L, Vitale I, Michels J, Brenner C, Szabadkai G, Harel-Bellan A, et al. Systems biology of cisplatin resistance: past, present and future. Cell Death Dis. (2014) 5:e1257. doi: 10.1038/cddis.2013.428
34. Sun S, Cai J, Yang Q, Zhao S, and Wang Z. The association between copper transporters and the prognosis of cancer patients undergoing chemotherapy: a meta-analysis of literatures and datasets. Oncotarget. (2017) 8:16036–51. doi: 10.18632/oncotarget.13917
35. Reeves R and Adair JE. Role of high mobility group (HMG) chromatin proteins in DNA repair. DNA Repair (Amst). (2005) 4:926–38. doi: 10.1016/j.dnarep.2005.04.010
36. Tian H, Yan L, Xiao-Fei L, Hai-Yan S, Juan C, and Shan K. Hypermethylation of mismatch repair gene hMSH2 associates with platinum-resistant disease in epithelial ovarian cancer. Clin Epigenetics. (2019) 11(1):153. doi: 10.1186/s13148-019-0748-4
37. Mendoza J, Martínez J, Hernández C, Pérez-Montiel D, Castro C, Fabián-Morales E, et al. Association between ERCC1 and XPA expression and polymorphisms and the response to cisplatin in testicular germ cell tumours. Br J Cancer. (2013) 109:68–75. doi: 10.1038/bjc.2013.303
38. Li B, Xu X, Zheng L, Jiang X, Lin J, and Zhang G. MiR-590-5p promotes cisplatin resistance via targeting hMSH2 in ovarian cancer. Mol Biol Rep. (2023) 50(8):6819–27. doi: 10.1007/s11033-023-08599-8
39. Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. (2003) 22:7265–79. doi: 10.1038/sj.onc.1206933
40. Köberle B, Tomicic MT, Usanova S, and Kaina B. Cisplatin resistance: preclinical findings and clinical implications. Biochim Biophys Acta. (2010) 1806:172–82. doi: 10.1016/j.bbcan.2010.07.004
41. Rintoul RC and Sethi T. The role of extracellular matrix in small-cell lung cancer. Lancet Oncol. (2001) 2:437–42. doi: 10.1016/S1470-2045(00)00421-6
42. Yu M, Zhang C, Li L, Dong S, Zhang N, and Tong X. Cx43 reverses the resistance of A549 lung adenocarcinoma cells to cisplatin by inhibiting EMT. Oncol Rep. (2014) 31:2751–8. doi: 10.3892/or.2014.3163
43. Qin X, Yu S, Xu X, Shen B, and Feng J. Comparative analysis of microRNA expression profiles between A549, A549/DDP and their respective exosomes. Oncotarget. (2017) 8:42125–35. doi: 10.18632/oncotarget.15009
44. Qin X, Yu S, Zhou L, Shi M, Hu Y, Xu X, et al. Cisplatin-resistant lung cancer cell-derived exosomes increase cisplatin resistance of recipient cells in exosomal miR-100-5p-dependent manner. Int J Nanomed. (2017) 12:3721–33. doi: 10.2147/IJN.S131516
45. Song Z, Jia G, Ma P, and Cang S. Exosomal miR-4443 promotes cisplatin resistance in non-small cell lung carcinoma by regulating FSP1 m6A modification-mediated ferroptosis. Life Sci. (2021) 276:119399. doi: 10.1016/j.lfs.2021.119399
46. Ma Y, Yuwen D, Chen J, Zheng B, Gao J, Fan M, et al. Exosomal transfer of cisplatin-induced miR-425-3p confers cisplatin resistance in NSCLC through activating autophagy. Int J Nanomed. (2019) 14:8121–32. doi: 10.2147/IJN.S221383
47. Wang D, Zhao C, Xu F, Zhang A, Jin M, Zhang K, et al. Cisplatin-resistant NSCLC cells induced by hypoxia transmit resistance to sensitive cells through exosomal PKM2. Theranostics. (2021) 11:2860–75. doi: 10.7150/thno.51797
48. Wang H, Huang H, Wang L, Liu Y, Wang M, Zhao S, et al. Cancer-associated fibroblasts secreted miR-103a-3p suppresses apoptosis and promotes cisplatin resistance in non-small cell lung cancer. Aging (Albany NY). (2021) 13:14456–68. doi: 10.18632/aging.103556
49. Zhang T, Zhang P, and Li HX. CAFs-derived exosomal miRNA-130a confers cisplatin resistance of NSCLC cells through PUM2-dependent packaging. Int J Nanomed. (2021) 16:561–77. doi: 10.2147/IJN.S271976
50. Wu H, Mu X, Liu L, Wu H, Hu X, Chen L, et al. Bone marrow mesenchymal stem cells-derived exosomal microRNA-193a reduces cisplatin resistance of non-small cell lung cancer cells via targeting LRRC1. Cell Death Dis. (2020) 11:801. doi: 10.1038/s41419-020-02962-4
51. Xiao X, Yu S, Li S, Wu J, Ma R, Cao H, et al. Exosomes: decreased sensitivity of lung cancer A549 cells to cisplatin. PloS One. (2014) 9:e89534. doi: 10.1371/journal.pone.0089534
52. Thomas A, Liu SV, Subramaniam DS, and Giaccone G. Refining the treatment of NSCLC according to histological and molecular subtypes. Nat Rev Clin Oncol. (2015) 12:511–26. doi: 10.1038/nrclinonc.2015.90
53. Wu YL, Zhou C, Hu CP, Feng J, Lu S, Huang Y, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol. (2014) 15:213–22. doi: 10.1016/S1470-2045(13)70604-1
54. Solomon BJ, Mok T, Kim DW, Wu YL, Nakagawa K, Mekhail T, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. (2014) 371:2167–77. doi: 10.1056/NEJMoa1408440
55. Shien K, Yamamoto H, Soh J, Miyoshi S, and Toyooka S. Drug resistance to EGFR tyrosine kinase inhibitors for non-small cell lung cancer. Acta Med Okayama. (2014) 68:191–200. doi: 10.18926/AMO/52785
56. Massarelli E, Varella-Garcia M, Tang X, Xavier AC, Ozburn NC, Liu DD, et al. KRAS mutation is an important predictor of resistance to therapy with epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. Clin Cancer Res. (2007) 13:2890–6. doi: 10.1158/1078-0432.CCR-06-3043
57. Ohashi K, Sequist LV, Arcila ME, Moran T, Chmielecki J, Lin YL, et al. Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1. Proc Natl Acad Sci U S A. (2012) 109:E2127–33. doi: 10.1073/pnas.1203530109
58. Ng KP, Hillmer AM, Chuah CT, Juan WC, Ko TK, Teo AS, et al. A common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancer. Nat Med. (2012) 18:521–8. doi: 10.1038/nm.2713
59. Arcila ME, Nafa K, Chaft JE, Rekhtman N, Lau C, Reva BA, et al. EGFR exon 20 insertion mutations in lung adenocarcinomas: prevalence, molecular heterogeneity, and clinicopathologic characteristics. Mol Cancer Ther. (2013) 12:220–9. doi: 10.1158/1535-7163.MCT-12-0620
60. Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, Chan E, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. (2012) 487:505–9. doi: 10.1038/nature11249
61. Bivona TG, Hieronymus H, Parker J, Chang K, Taron M, Rosell R, et al. FAS and NF-κB signalling modulate dependence of lung cancers on mutant EGFR. Nature. (2011) 471:523–6. doi: 10.1038/nature09870
62. Ma C, Wei S, and Song Y. T790M and acquired resistance of EGFR TKI: a literature review of clinical reports. J Thorac Dis. (2011) 3:10–8. doi: 10.3978/j.issn.2072-1439.2010.12.02
63. Huang L and Fu L. Mechanisms of resistance to EGFR tyrosine kinase inhibitors. Acta Pharm Sin B. (2015) 5:390–401. doi: 10.1016/j.apsb.2015.07.001
64. Van Der Steen N, Giovannetti E, Carbone D, Leonetti A, Rolfo CD, and Peters GJ. Resistance to epidermal growth factor receptor inhibition in non-small cell lung cancer. Cancer Drug Resist. (2018) 1:230–49. doi: 10.20517/cdr.2018.13
65. Choi YL, Soda M, Yamashita Y, Ueno T, Takashima J, Nakajima T, et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med. (2010) 363:1734–9. doi: 10.1056/NEJMoa1007478
66. Lovly CM and Pao W. Escaping ALK inhibition: mechanisms of and strategies to overcome resistance. Sci Transl Med. (2012) 4:120ps2. doi: 10.1126/scitranslmed.3003728
67. Gainor JF, Dardaei L, Yoda S, Friboulet L, Leshchiner I, Katayama R, et al. Molecular mechanisms of resistance to first- and second-generation ALK inhibitors in ALK-rearranged lung cancer. Cancer Discov. (2016) 6:1118–33. doi: 10.1158/2159-8290.CD-16-0596
68. Recondo G, Mezquita L, Facchinetti F, Planchard D, Gazzah A, Bigot L, et al. Diverse resistance mechanisms to the third-generation ALK inhibitor lorlatinib in ALK-rearranged lung cancer. Clin Cancer Res. (2020) 26:242–55. doi: 10.1158/1078-0432.CCR-19-1104
69. Lin JJ and Shaw AT. Resisting resistance: targeted therapies in lung cancer. Trends Cancer. (2016) 2:350–64. doi: 10.1016/j.trecan.2016.05.010
70. Lei Y, Guo W, Chen B, Chen L, Gong J, and Li W. Tumor−released lncRNA H19 promotes gefitinib resistance via packaging into exosomes in non−small cell lung cancer. Oncol Rep. (2018) 40:3438–46. doi: 10.3892/or.2018.6762
71. Yang B, Teng F, Chang L, Wang J, Liu DL, Cui YS, et al. Tumor-derived exosomal circRNA_102481 contributes to EGFR-TKIs resistance via the miR-30a-5p/ROR1 axis in non-small cell lung cancer. Aging (Albany NY). (2021) 13:13264–86. doi: 10.18632/aging.203011
72. Azuma Y, Yokobori T, Mogi A, Yajima T, Kosaka T, Iijima M, et al. Cancer exosomal microRNAs from gefitinib-resistant lung cancer cells cause therapeutic resistance in gefitinib-sensitive cells. Surg Today. (2020) 50:1099–106. doi: 10.1007/s00595-020-01976-x
73. Jing C, Cao H, Qin X, Yu S, Wu J, Wang Z, et al. Exosome-mediated gefitinib resistance in lung cancer HCC827 cells via delivery of miR-21. Oncol Lett. (2018) 15:9811–7. doi: 10.3892/ol.2018.8604
74. Liu X, Jiang T, Li X, Zhao C, Li J, Zhou F, et al. Exosomes transmit T790M mutation-induced resistance in EGFR-mutant NSCLC by activating PI3K/AKT signalling pathway. J Cell Mol Med. (2020) 24:1529–40. doi: 10.1111/jcmm.14838
75. Zhang Y, Li M, and Hu C. Exosomal transfer of miR-214 mediates gefitinib resistance in non-small cell lung cancer. Biochem Biophys Res Commun. (2018) 507:457–64. doi: 10.1016/j.bbrc.2018.11.061
76. Pan R and Zhou H. Exosomal Transfer of lncRNA H19 Promotes Erlotinib Resistance in Non-Small Cell Lung Cancer via miR-615-3p/ATG7 Axis. Cancer Manag Res. (2020) 12:4283–97. doi: 10.2147/CMAR.S241095
77. Wu S, Luo M, To KKW, Zhang J, Su C, Zhang H, et al. Intercellular transfer of exosomal wild type EGFR triggers osimertinib resistance in non-small cell lung cancer. Mol Cancer. (2021) 20:17. doi: 10.1186/s12943-021-01307-9
78. Hisakane K, Seike M, Sugano T, Yoshikawa A, Matsuda K, Takano N, et al. Exosome-derived miR-210 involved in resistance to osimertinib and epithelial-mesenchymal transition in EGFR mutant non-small cell lung cancer cells. Thorac Cancer. (2021) 12:1690–8. doi: 10.1111/1759-7714.13943
79. Zhou D, Xia Z, Xie M, Gao Y, Yu Q, and He B. Exosomal long non-coding RNA SOX2 overlapping transcript enhances the resistance to EGFR-TKIs in non-small cell lung cancer cell line H1975. Hum Cell. (2021) 34:1478–89. doi: 10.1007/s13577-021-00572-6
80. Wu C-P, Murakami M, Hsiao S-H, Chou A-W, Li Y-Q, Huang Y-H, et al. Overexpression of ATP-binding cassette subfamily G member 2 confers resistance to phosphatidylinositol 3-kinase inhibitor PF-4989216 in cancer cells. Mol Pharm. (2017) 14:2368–77. doi: 10.1021/acs.molpharmaceut.7b00277
81. Patra S, Sahoo RK, Biswal S, Panda SS, and Biswal B. Enigmatic exosomal connection in lung cancer drug resistance. Mol Ther Nucleic Acids. (2024) 35:102177. doi: 10.1016/j.omtn.2024.102177
82. Pu X, Wu L, Su D, Mao W, and Fang B. Immunotherapy for non-small cell lung cancers: biomarkers for predicting responses and strategies to overcome resistance. BMC Cancer. (2018) 18:1082. doi: 10.1186/s12885-018-4990-5
83. Patel SA and Minn AJ. Combination cancer therapy with immune checkpoint blockade: mechanisms and strategies. Immunity. (2018) 48:417–33. doi: 10.1016/j.immuni.2018.03.007
84. Nowicki TS, Hu-Lieskovan S, and Ribas A. Mechanisms of resistance to PD-1 and PD-L1 blockade. Cancer J. (2018) 24:47–53. doi: 10.1097/PPO.0000000000000303
85. Gettinger S, Choi J, Hastings K, Truini A, Datar I, Sowell R, et al. Impaired HLA class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung cancer. Cancer Discov. (2017) 7:1420–35. doi: 10.1158/2159-8290.CD-17-0593
86. Huang RY, Francois A, McGray AR, Miliotto A, and Odunsi K. Compensatory upregulation of PD-1, LAG-3, and CTLA-4 limits the efficacy of single-agent checkpoint blockade in metastatic ovarian cancer. Oncoimmunology. (2017) 6:e1249561. doi: 10.1080/2162402X.2016.1249561
87. Jenkins RW, Barbie DA, and Flaherty KT. Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer. (2018) 118:9–16. doi: 10.1038/bjc.2017.434
88. Wang F, Wan L, Zhang C, Zheng X, Li J, and Chen ZK. Tim-3-Galectin-9 pathway involves the suppression induced by CD4+CD25+ regulatory T cells. Immunobiology. (2009) 214:342–9. doi: 10.1016/j.imbio.2008.10.007
89. Piccirillo CA, d’Hennezel E, Sgouroudis E, and Yurchenko E. CD4+Foxp3+ regulatory T cells in the control of autoimmunity: in vivo veritas. Curr Opin Immunol. (2008) 20:655–62. doi: 10.1016/j.coi.2008.09.006
90. Binnewies M, Mujal AM, Pollack JL, Combes AJ, Hardison EA, Barry KC, et al. Unleashing type-2 dendritic cells to drive protective antitumor CD4(+) T cell immunity. Cell. (2019) 177:556–571.e16. doi: 10.1016/j.cell.2019.02.005
91. Meyer C, Cagnon L, Costa-Nunes CM, Baumgaertner P, Montandon N, Leyvraz L, et al. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol Immunother. (2014) 63:247–57. doi: 10.1007/s00262-013-1508-5
92. Chanmee T, Ontong P, Konno K, and Itano N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers (Basel). (2014) 6:1670–90. doi: 10.3390/cancers6031670
93. Do HTT, Lee CH, and Cho J. Chemokines and their receptors: multifaceted roles in cancer progression and potential value as cancer prognostic markers. Cancers (Basel). (2020) 12(2). doi: 10.3390/cancers12020287
94. Opitz CA, Somarribas Patterson LF, Mohapatra SR, Dewi DL, Sadik A, Platten M, et al. The therapeutic potential of targeting tryptophan catabolism in cancer. Br J Cancer. (2020) 122:30–44. doi: 10.1038/s41416-019-0664-6
95. Li N, Qin J, Lan L, Zhang H, Liu F, Wu Z, et al. PTEN inhibits macrophage polarization from M1 to M2 through CCL2 and VEGF-A reduction and NHERF-1 synergism. Cancer Biol Ther. (2015) 16:297–306. doi: 10.1080/15384047.2014.1002353
96. Arbour KC, Jordan E, Kim HR, Dienstag J, Yu HA, Sanchez-Vega F, et al. Effects of co-occurring genomic alterations on outcomes in patients with KRAS-mutant non-small cell lung cancer. Clin Cancer Res. (2018) 24:334–40. doi: 10.1158/1078-0432.CCR-17-1841
97. Shire NJ, Klein AB, Golozar A, Collins JM, Fraeman KH, Nordstrom BL, et al. STK11 (LKB1) mutations in metastatic NSCLC: Prognostic value in the real world. PloS One. (2020) 15:e0238358. doi: 10.1371/journal.pone.0238358
98. Li I and Nabet BY. Exosomes in the tumor microenvironment as mediators of cancer therapy resistance. Mol Cancer. (2019) 18:32. doi: 10.1186/s12943-019-0975-5
99. Yang Y, Li CW, Chan LC, Wei Y, Hsu JM, Xia W, et al. Exosomal PD-L1 harbors active defense function to suppress T cell killing of breast cancer cells and promote tumor growth. Cell Res. (2018) 28:862–4. doi: 10.1038/s41422-018-0060-4
100. Wang J, Zeng H, Zhang H, and Han Y. The role of exosomal PD-L1 in tumor immunotherapy. Transl Oncol. (2021) 14:101047. doi: 10.1016/j.tranon.2021.101047
101. Kim DH, Kim H, Choi YJ, Kim SY, Lee JE, Sung KJ, et al. Exosomal PD-L1 promotes tumor growth through immune escape in non-small cell lung cancer. Exp Mol Med. (2019) 51:1–13. doi: 10.1038/s12276-019-0295-2
102. Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. (2006) 439:682–7. doi: 10.1038/nature04444
103. Bach DH, Hong JY, Park H, and Lee S. The role of exosomes and miRNAs in drug-resistance of cancer cells. Int J Cancer. (2017) 141:220–30. doi: 10.1002/ijc.30669
104. Steinbichler TM, Dudás J, Riechelmann D, and Skvortsova H. Therapy resistance mediated by exosomes. Mol Cancer. (2019) 18:58. doi: 10.1186/s12943-019-0970-x
105. Pan W, Miao Y, Xie J, Ma H, and Huang L. The role and clinical applications of exosomes in cancer drug resistance. Cancer Drug Resist. (2024) 7:102177. doi: 10.20517/cdr.2024.97
106. Kandimalla R, Moholkar S, Rajendran M, and Gondi S. Exosomal paclitaxel formulation alone and in combination with cisplatin for oral delivery in NSCLC. Cancer Res. (2022) 82:371. doi: 10.1158/1538-7445.AM2022-371
107. Zhang T, Zhang P, Li HX, and Aktas M. CAFs-Derived Exosomal miRNA-130a Confers CisplatinResistance of NSCLC Cells Through PUM2-Dependent Packaging. Int J Nanomedicine. (2021) 16:561–77. doi: 10.2147/IJN.S271976
Keywords: drug resistance, exosomes, chemotherapy, targeted therapy, immunotherapy
Citation: Ji H, Zhang L and Ye L (2025) Exosome, an important transmitter in the drug resistance of non-small cell lung cancer. Front. Oncol. 15:1539047. doi: 10.3389/fonc.2025.1539047
Received: 03 December 2024; Accepted: 28 April 2025;
Published: 15 May 2025.
Edited by:
Raghuram Kandimalla, James Graham Brown Cancer Center, United StatesReviewed by:
M. Teresa Agulló Ortuño, Research Institute Hospital 12 de Octubre, SpainPriya Mondal, Central Food Technological Research Institute (CSIR), India
Copyright © 2025 Ji, Zhang and Ye. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lingyun Ye, eWVsaW5neXVuMTAxOUAxMjYuY29t