 Shuai Tan1*†
Shuai Tan1*† Huizhen He1†Yuxin Li1†
Huizhen He1†Yuxin Li1† Mingyue Shang1Yaofang Cao1Dongmei Zou1Ronghua Hu1Wuhan Hui1
Mingyue Shang1Yaofang Cao1Dongmei Zou1Ronghua Hu1Wuhan Hui1 Xiaoli Chang1Jing Ni1Qiang Ma1Li Su1Jing Sun1Wanxue He2
Xiaoli Chang1Jing Ni1Qiang Ma1Li Su1Jing Sun1Wanxue He2 Xingmin Feng3*
Xingmin Feng3* Wanling Sun1*
Wanling Sun1*- 1Department of Hematology, Xuanwu Hospital, Capital Medical University, Beijing, China
- 2Department of Pulmonary and Critical Care Medicine, Xuanwu Hospital, Capital Medical University, Beijing, China
- 3National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD, United States
Acquired aplastic anemia (AA) is a bone marrow failure syndrome characterized by pancytopenia and decreased hematopoietic stem and progenitor cells (HSPCs) in the bone marrow, it can be either congenital or acquired, predominantly affecting adolescents and the elderly, with higher incidence in Asia compared to Europe and America. Current treatment options include allogeneic hematopoietic stem cell transplantation or immunosuppressive agents, yet proximately a third of patients fail to reach long-term survival. AA is primarily driven by immune-mediated destruction of HSPCs, initiated by self-activated T cells. Early stages feature a Th1 response, which later shifts to Th17 and effector memory CD8+ T cells. Key cytokines including interferon-gamma (IFN-γ) and tumor necrosis factor-alpha (TNF-α) play crucial roles in this immune dysregulation, influencing HSPCs and contributing to bone marrow failure. Furthermore, bone marrow macrophages (MΦ), particularly M1 subtype, are implicated in AA via the TNF-α/TNF-α receptor pathway, leading to T cell activating and subsequent HSPC damage. Interestingly, MΦ with high expression of IL-27Ra have been demonstrated to contribute to HSPC destruction in AA murine models. Beyond their role in thrombosis, platelets also participate in immune regulation. Some studies suggest that platelet may modulate T cell responses through mechanisms such as Akt-PGC1α-TFAM pathway or PF4-mediated activity, which could play a role in AA. However, direct evidence connecting platelet regulation to T cell-mediated HSPC damage is limited, and current research has largely focuses on CD8+ T cells. Moving forward, it is essential to investigate the interactions between platelets, CD4+ T cells, and mitochondrial energy metabolism. In this review, we propose that platelet-derived factors such as PF4 and TGFβ may activate mitochondrial pathways, influencing T cell activation and leading to HSPC destruction in AA. This hypothesis could provide new insights into the molecular mechanisms of AA and pave the way for novel therapeutic strategies (Highlight).
Highlights
● This review will discuss and explore the pathogenesis of AA from a new perspective, focusing on platelet-regulated T-cell immune response.
● The function of platelets are not only thrombus and hemostasis, but also regulate T-cell immunity. The key mediators involved are PF4 and the mitochondrial energy metabolism signaling pathway.
● We also summarize the research history of platelet drugs in cardiovascular and cerebrovascular diseases, as well as the potential of immunotherapy in the current era.
● Molecular Activity of Platelet-Regulated T Cell Immune Response in AA as below:
Introduction
Aplastic Anemia (AA) may arise from an unknown pathogen infecting hematopoietic stem cells (HSCs) or peripheral cells, leading to the presentation of pathogen particles and either unmodified or chemically/genetically modified components on their cell surfaces. These antigens are subsequently processed by antigen-presenting cells (APCs) and presented to CD4+ T cells. Platelets play a role in this immune regulation by releasing various soluble mediators, such as PF4/CXCL4, TGFβ, PAF, TXA2, NAP, and RANTES. PF4 binds to CXCR3 and CXCR5 receptors on CD4+ or CD8+ T cells, activating downstream mitochondrial energy metabolism signaling pathway (Akt-PGC1α-TFAM). This affects mitochondrial quantity, ATP production, and reactive oxygen species (ROS), ultimately regulating T cell immune response. In CD4+ T cells, this regulation leads to: ①Differentiation into Th1, Th2, Th9, and Th22 phenotypes; ②Simultaneous differentiation into Th17 phenotypes under the stimulation of IL-23 and IL-12; ③Suppression of Treg cells, resulting in weakened immune regulation and imbalance. In CD8+ T cells, the immune regulation involves: ①Direct cytotoxicity through the release of granzyme B (GzmB) and perforin (PFN); ②Paracrine effects via secretion of TNFα, IFNγ, and Fas ligand (Fas-L).
In summary, platelets regulate the immune responses of CD4+ and CD8+ T cells through mitochondrial energy metabolism, contributing to immune dysregulation and increased levels of IFNγ and TNFα, leading to the destruction of HSPCs and potential bone marrow failure.
While platelets are best known for their role in hemostasis and thrombosis, recent evidence suggests that platelets release mediators such as PF4/CXCL4 (platelet factor 4), TGFβ (transforming growth factor β), RANTES, and others through α-particle secretion. These processes, mediated by pathways like mitochondrial Akt-PGC1α and Toll-like receptors (TLR), selectively regulate T-cell recruitment under normal blood flow, affecting the lifespan of naive naïve T cells (TN) and memory T cells (TM). Moreover, platelets influence the dynamics and differentiation of Th (T helper) and Treg (regulatory T) cells, thus playing a critical role in immune regulation (1–6).
AA is characterized by bone marrow failure, pancytopenia, and reduced bone marrow cellularity. It most commonly affects in adolescents and the elderly, with a higher incidence in Asia than in Europe and the United States. Immunosuppressive therapy is the primary treatment, but it has a slow onset and significant side effects, with about a third of patients not surviving long term. Recent advances have seen TPO-RAs (thrombopoietin receptor agonists) combined with IST as a first-line treatment, though whether TPO-RAs can correct immune imbalances while promoting platelet production remains unclear (7–9).
Research shows that patients with AA exhibit abnormal T-cell activation, characterized by elevated levels of IFN-γ and TNF-α, leading to the destruction of HSPCs. Identifying the factors driving excessive T-cell activation and developing effective strategies to correct immune imbalance remain crucial challenges (10–13).
This paper proposes a novel clinical perspective: investigating the role of platelet-mediated T-cell immune function in bone marrow failure, potentially offering innovative approaches to addressing treatment challenges in the future.
Platelet physiology
Platelets are anucleate, discoid cells with dimensions of approximatley (2.0-5.0)×0.5 µm, an average cell volume of 6–10 fl, a blood concentration of 200-300×109/L, and a lifespan of approximately 10 days (14). Though primarily known for their role in hemostasis and thrombosis, platelets are critical in conditions such as myocardial infarction (MI), which is frequently caused by thrombus formation. They are also implicated in immune system disorders, including viral and bacterial infections in patients with idiopathic thrombocytopenic purpura (15). Emerging evidence indicates that platelets also contribute to immune functions through Toll-like receptors (TLR) (1, 16–19).
Upon activation, platelets release α-granules, which are linked to major histocompatibility complex I (MHCI) molecules. MHCI expression can be found on the platelet plasma membrane and in the cytosol, although it is relatively unstable in these areas (20, 21). In addition to MHCI, α-granules release mediators like PF4/CXCL4 and TGFβ, that play essential roles in thrombosis, inflammation, and immune modulation, while also contributing to vascular intimal injury (22). PF4 exhibits a strong chemotactic effect on neutrophils by binding to heparan sulfate on vascular endothelium, promoting thrombin activation. Additionally, circulating TGFβ primarily originate from platelets, serving as a specific marker of in vivo platelet activation (2, 23–25).
Atherosclerosis, a chronic inflammatory process, involves an sustained immune response. Platelets are involved in all stages of thrombus formation, which encompasses four key stages: endothelial dysfunction, fatty streak formation, advanced and complicated lesions, and emergence of unstable fibrous plaques (26). For the prevention and treatment of atherosclerotic diseases, antiplatelet therapy has long been a cornerstone, with a consensus on its benefits (27). Current therapies mainly target direct platelet activation, using agents like COX-inhibitor aspirin and ADP P2Y12 receptor antagonists such as clopidogrel or ticagrelor. These treatments reduce acute coronary syndrome risks by approximately 30-40%. However, efforts to further enhance antiplatelet therapy have reached a therapeutic plateau, as illustrated by ticagrelor’s marginal 2% superior over clopidogrel in primary endpoint protection over one year (28, 29).
This highlights the need for novel antiplatelet strategies. Future therapies may focus on platelet interactions with inflammatory and immune processes. A combination of antiplatelet drugs that target both platelet activation and inflammation could synergistic effects, offering more comprehensive protection against thrombotic and immune-related complications.
Platelets exhibit immunomodulatory potential, as evidenced by their ability to stimulate lymphocyte proliferation, optimize the subpopulation ratio of lymphocytes by regulating the CD3+CD8+ T lymphocytes and bolster the cytotoxic function of lymphocytes in vitro when activated (30). Additionally, platelets inhibit cytolytic function, adhesion ability and cytotoxic properties of NK cells by expressing glucocorticoid-induced TNF-related ligand (GITRL), releasing TGF-β and transferring MHC-I, thereby protecting tumor cells and promoting the formation of immunosuppressive environment (6, 31, 32). Moreover, platelets play a role in modulating the balance of macrophage phenotypes. Studies have shown that platelets can promote the polarization of macrophages to M1 phenotype in a mouse model of septic shock in vitro (33, 34). Meanwhile, related research has also indicated that thrombopoietin receptor agonists induce macrophages to shift towards the M2 phenotype (35). These discoveries not only enhance our understanding of the immune-regulating role of platelets, but also offer fresh perspectives and approaches for the treatment of associated diseases.
One of the characteristics of aplastic anemia is decrease in platelets. Thrombopoietin receptor agonists (TPO-RAs) regulate the differentiation and maturation of megakaryocytes required for platelet production. So thrombopoietin receptor agonists have been used to treat bone marrow failure syndromes, such as aplastic anemia (36). Eltrombopag is one of TPO-RAs that improves the blood platelet level. A two-arm study suggests patients with severe aplastic anemia (SAA) and very severe aplastic anemia (vSAA) who do not have a suitable bone marrow transplant donor need standard immunosuppression treatment. Compared with Group A (the standard immunosuppressive therapy), more participants in group B(the standard immunosuppressive therapy with Eltrombopag) have elevated blood cell levels into the normal range and responded more quickly to treatment, while side effects were similar in both groups. So immunosuppression treatment with eltrombopag benefits participants with SAA and vSAA (37). And the recovery of platelet level plays an important role in the treatment of aplastic anemia.
CD4+ and CD8+ T cells
CD4+ T cells
Antigen-presenting cells (APCs) form a major histocompatibility complex II (MHCII)-peptide complex on their surface. This complex can be recognized and bound by the T cell receptor (TCR) and CD4, which constitutes the first signal for CD4+ T cell activation. The second signal involves costimulation from either cytokines or membrane proteins such as B7 (CD80) on the APC surface interacting with CD28 on the CD4+ T cell surface. Without this costimulation, CD4+ T cells remain energy-deprived. When CD4+ T cells are activated, they start to synthesize and secrete IL-2, leading to their proliferation and differentiation. Most activated CD4+ T cells become effector cells and eventually die, but a few will survive to become memory cells, retaining the long-term memory of the antigen. The first and second signals are critical for T cell survival and proliferation, but they’re insufficient for complete functional differentiation. The third signal, which drives differentiation, comes from cytokines secreted by other cells in the surrounding microenvironment, which, along with TCR recognition, determines the final outcome of CD4+ T cells (38, 39).
CD4+ Th and Treg cells
CD4+ T cells can differentiate into various Th cell subsets, such as Th1, Th2, Th9, Th17, Th22, TFH, TFR, and Treg cells. These subsets play distinct roles in the immune system function of eliminating pathogens and are influenced by specific transcription factors like T-bet, GATA3, RORγt, and FoxP3 (40, 41). Additionally, certain chromatin modifiers can affect T cell function at the gene level (42).
Th1 and Th2
Under normal physiological conditions, Th1 and Th2 cells maintain a balanced state (43, 44). Th1 cells primarily secrete pro-inflammatory cytokines like IFNγ, which mediate cellular immunity and activate other cell types, such as macrophages (45–49). Th2 cells, on the other hand, support the survival and function of B cells, including their proliferation, maturation, antibody production, and mediation of humoral immunity. This is achieved through the secretion of IL-4, IL-5, and IL-13 by Th2 cells. It’s worth noting that these two subsets regulate each other to maintain immune homeostasis; IL-4 and IFNγ can inhibit the functions of Th1 and Th2 cells, respectively (50). There is also a specialized subtype of “Th2/Th1” cells that express both IL-4 and IFNγ, along with GATA3 and T-bet. Intriguingly, antigens like Papain and House Dust Mites (HDM) can induce Th2 cells through non-MHC-II pathways, suggesting an additional role for Th2 cells in adaptive immunity (51, 52).
Th9
Under the influence of IL-2-induced STAT5 activation, along with the regulation by IL-4 and TGF-β, CD4+ T cells can polarize toward the Th9 phenotype (53, 54). Conversely, GFI1 acts as a negative regulator for Th9 polarization. Key downstream transcription factors like PU.1, IRF4 (Interferon Regulatory Factor 4), Id1, and HIF1α are crucial for Th9 differentiation (55–57). The hallmark cytokine produced by Th9 cells is IL-9, which is closely associated with allergies and autoimmunity. TGF-β promotes IL-9 production by activating PU.1 or by facilitating the interaction between Smad2/3 and IRF4 through the TGF-β/Smad axis. Id1, when bound with Tcf3/4 or IRF4, enhances IL-9 expression in Th9 cells by interacting with the IL-9 promoter region (58). Animal and in vitro experiments have shown that DBP can increase IL-9 gene expression, while EB28 does the opposite (59). When IL-9 binds to its receptor, it can activate three distinct STAT proteins: STAT1, STAT3, and STAT5, which play unique roles in gene induction, differentiation, and inhibition of apoptosis (60–65).
Th17
Th17 cells are characterized by their expression of IL-17 (IL-17+ IFNγ–) (66, 67). IL-17 is a key pro-inflammatory factor with multiple functions, such as stimulating neutrophil proliferation and maturation and inducing pro-inflammatory cytokine expression in various cell types. Because of this, Th17 cells can affect the pathophysiology of several conditions, including infections, cancer, autoimmunity, aplastic anemia, rheumatoid arthritis (RA), and others (68–71).
Th22
Th22 cells are marked by their high production of IL-22, which is regulated by the NOTCH-HES-1 axis and the characteristic expression of chemokine receptors CCR4, CCR6, and CCR10 (72–74). IL-22 activates several downstream pathways, including MAPK, PI3K/Akt, and NF-κB, enabling it to perform various functions (75–78). While Th22 cells were initially found in studies of skin pathophysiology, recent research has shown that they are involved in various autoimmune diseases, viral infections, cardiovascular diseases, and tumors.
TFH (CXCR5, CXCR13/BCA-1, ICOS, IcosL, Bcl6, CD40L; CD40, IL-21, PD-1)
TFH (T follicular helper) cells are characterized by the expression of CXCR5 (CXC chemokine receptor 5). Various factors play roles in the multi-stage regulation of TFH differentiation (79). CXCR5, ICOS (Inducible costimulator), IL-12, IL-21, IFNγ, IL-27, and IL-6 positively regulate TFH differentiation, while PD-1 (Programmed Death 1), CTLA-4 (Cytotoxic T Lymphocyte Antigen 4), ubiquitin ligase Peli1, IL-2, and IL-7 have the opposite effects (80–86). TFH cells primarily assist in the formation of germinal centers (GCs) and B cell differentiation and maturation, closely tied to the humoral immune response (87–89).
TFR (CXCR5, ICOS, Bcl6, PD-1; CTLA4, GITR, FOXP3)
In 2011, a specialized subset of regulatory T cells (Tregs), located in the germinal center (GC), was identified as Tfr cells (90). Similar to Tregs, this subset contains key molecules such as FoxP3, CTLA-4, GITR, Prdm1, and Blimp-1. Additionally, Tfr cells express Bcl-6, CXCR5, PD-1, and ICOS, similar to TFH cells, distinguishing them as a unique CD4 T-cell subset (91–98). The primary function of Tfr cells is to balance immune activation and tolerance, which provides insight into autoimmune diseases, allergic reactions, antibody-mediated rejection, viral infections, and type 1 diabetes (99–102).
Treg cells
Treg cells constitute an immunosuppressive T cell subset the production of which is induced by IL-2-stimulated FoxP3 transcriptional activity. This subset plays a crucial role in maintaining peripheral immune tolerance and controlling autoimmune responses (103–115). The balance between Treg and other Th subsets is crucial in preventing autoimmunity by impeding the activation of autoreactive T cells and the expression of cytokines (116, 117). Tregs express IL-10 and TGFβ, which inhibit macrophage function after TLR4 activation (118–120). IL-10 downregulates T cell-mediated immune responses, including repressing the proliferation of Th1 and Th2 cells, while TGFβ regulates the functions of various immune cells (121–124).
In summary, the diagram of CD4+ T cell subsets and immune response mechanism in Figure 1.
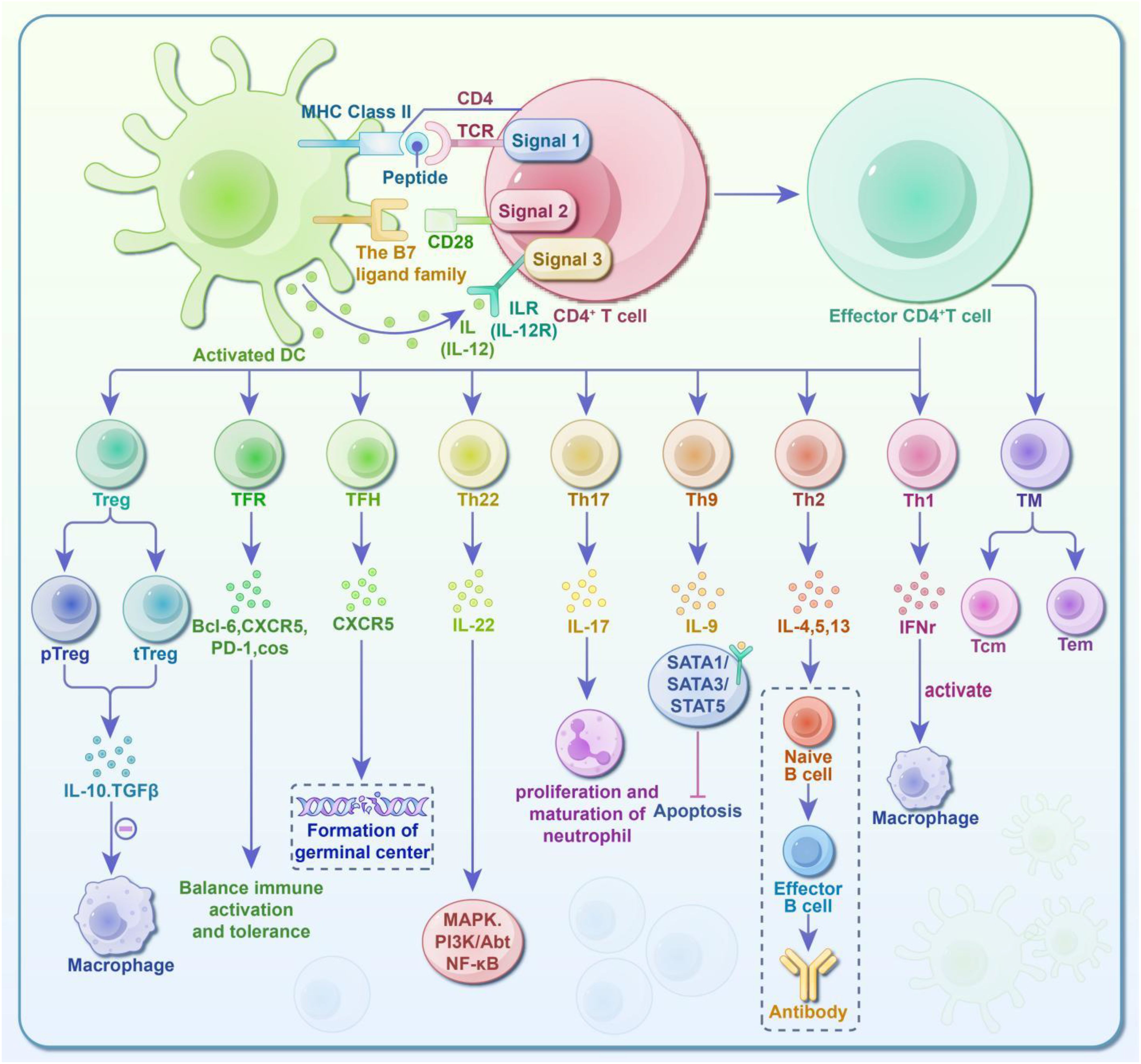
Figure 1. Diagram of CD4+ T cell subsets and immune response activity. CD4+ T cells require three sequential signals to activate and acquire the ability to differentiate and function: ① First Signal: Antigen-specific interactions. The CD4 co-receptor and TCR-CD3 complex recognize the antigen-MHC II complex on antigen-presenting cells (APCs), enabling signal transmission. ② Second Signal: Co-stimulatory molecules. The TCR binds to the MHC II complex, while co-stimulatory molecules on the APC surface bind to ligands on the T cell surface. ③ Third Signal: Instructive cytokines. While the first and second signals activate the T cells (promoting survival and proliferation), they are insufficient for functional differentiation. The third signal, provided by cytokines secreted by surrounding cells, together with the TCR, determines the final differentiation of the CD4+ T cells. Fully activated T cells can differentiate into various subsets: T Helper Cells (Th): These promote immune responses and include Th1, Th2, Th9, Th17, Th22, TFH, and TFR cells. Regulatory T Cells (Treg): These inhibit and regulate immune responses. Their specific roles are depicted in the diagram. Additionally, some effector T cells transition into memory T cells (TM), which are further classified into central memory T cells (Tcm) and effector memory cells (Tem). Upon encountering pathogens again, memory T cells produce rapid and robust immune responses, which are crucial for long-term immunity and vaccine effectiveness.
CD8+ T cells/CTL
Autoreactive cytotoxic CD8+ T cells recognize hematopoietic stem and progenitor cell (HSPC) antigens through major histocompatibility MHC I/II, resulting in secretion of pro-inflammatory cytokines such as IFN-γ. After activation, CD8+ T cells rapidly proliferate, exit the lymph nodes, enter the bloodstream, and migrate to the infection site. They directly kill target cells by releasing perforin and granzyme or induce apoptosis by using the Fas ligand protein on their surface to bind with the Fas protein on target cells (125–127). Similar to CD4+ cells, CD8+ cells can be classified into three subtypes based on their distinct phenotypes and functional heterogeneity (128, 129).
Naïve and memory T cells
T cell subsets serve as indicators of immune function. CD4+ cells play a pivotal role in humoral and cellular immune systems by secreting numerous cytokines and facilitating B cell antibody production (130–132). Notably, T cells can be classified as either Tn (CD45RA+ and CD45RO–) or TM (CD45RA– and CD45RO+) based on the expression of these variants (133). Tn cells mature in the thymus and migrate to peripheral secondary lymphatic organs/tissues such as the spleen and lymph nodes (LNs). For Tn, central memory T cell (Tcm), and effector memory (Tem) cells, APC functional differentiation strength increases gradually, while proliferative capacity and antitumor efficacy decline progressively (134). CD8+ effector T cell subsets identified thus far include Tc1, Tc2, Tc9, Tc17, follicular cytotoxic T (Tfc), follicular helper T (CD8+ Tfh), and regulatory T (CD8+ Treg), holds significant potential in treating tumors, viral infections, allergies, and autoimmune diseases (135, 136).The majority of effector cells undergo apoptosis with small subset persists and differentiates into memory cells. Some effector T cells directly transition into Tcm and Tem, while others are converted from Tcm (41, 137, 138). Compared to Tn, TM cells mount and execute a faster immune response upon re-infection by pathogens (137, 139–141).
Memory T cells can be further classified as Tem and Tcm based on the expression of CD62L and CCR7. CD62L, a lymph node homing receptor, influences cell migration, while CCR7, a chemokine receptor, contributes to T cell recirculation and effector function (130, 142, 143). Tcm localize to secondary lymphoid tissues for recirculation similar to Tn cells, whereas Tem preferentially migrate and distribute throughout non-lymphoid tissues and local immune response sites to mount rapid immune responses (144–149). Additionally, Tem cells are involved in early infection stages, while Tcm cells predominate in later stages due to their robust proliferative potential and prolonged effector function (150–159).
Memory cells are vital in vaccine immunity (139). Tcm cells exhibit high expression of CD45RO, CD62L, CD28, CD44, CD11a, and IL-12R (β1 subunit), displaying a stronger proliferative potential and anti-tumor immunity than Tem cells (134, 144, 160) while Tem cells express low levels of CD62L and CCR7. Due to limited CCR7 expression, Tem cells promptly localize in inflamed tissues via chemotactic gradients and express pro-inflammatory factors like IL-4, IL-5, IFNγ, and perforin. Notably, Tem function is implicated in autoimmune diseases and AS development (161–165), evidenced by the Tem cells in synovial fluid or skin during clinical diagnosis (161) and the decrease in Tn cells and increase in Tem cells during AS (166–171), respectively. Tcm cells, akin to stem cells, possess self-renewal capability, while some Tem cells can originate from Tcm in response to antigen stimulation (166–171).
In summary, the diagram of CD8+ T cell/CTL subsets and immune response mechanism in Figure 2.
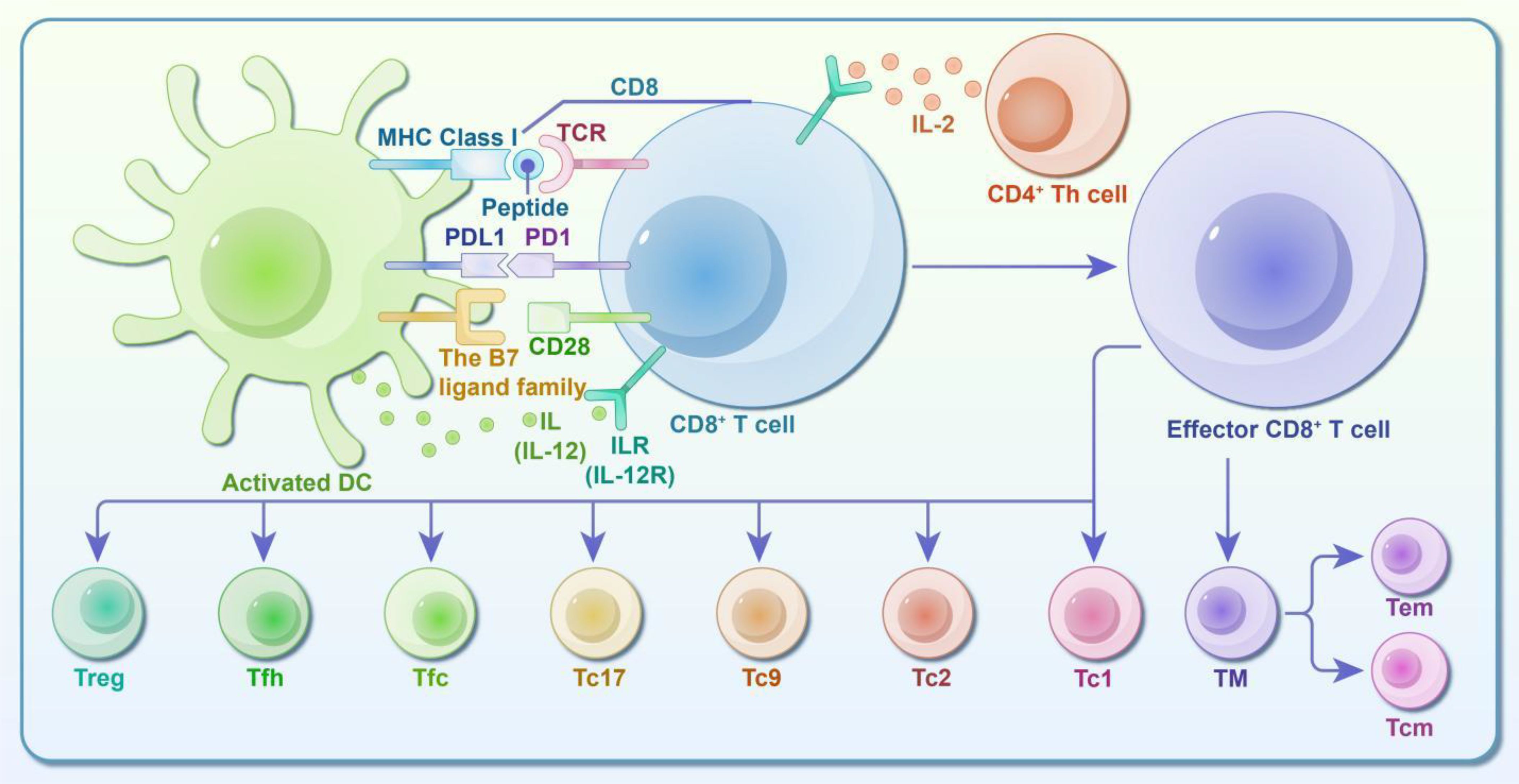
Figure 2. Diagram of CD8+ T Cell/CTL subsets and immune response activity. CD8+ T cells, also known as cytotoxic T lymphocytes (CTLs), are activated through two primary activities: ① Activation via Antigenic Peptide from APCs: The first signal involves the interaction between the antigenic peptide-MHC I complex and TCR/CD3-CD8 molecules on the APC. The second signal comes from the binding of B7 on the APC to CD28 on the CD8+ T cell. Additionally, the T cell expresses the IL-12 receptor, which binds IL-12 secreted by the APC, leading to activation. ② Activation via Antigenic Peptide from Target Cells: In this case, the first signal is provided by the antigenic peptide-MHC I complex binding to TCR/CD3-CD8 molecules. Since target cells lack B7 molecules, CD4+ Th cells provide IL-2 to the corresponding receptor on CD8+ T cells, serving as the second signal. Upon activation, CD8+ T cells gain the ability to differentiate and function. They rapidly proliferate, leave the lymphoid system, enter the bloodstream, and kill target cells. Identified CD8+ effector T cell subsets include Tc1, Tc2, Tc9, Tc17, follicular cytotoxic T cells (Tfc), follicular helper T cells (CD8+ Tfh), and regulatory T cells (CD8+ Treg). These subsets play significant roles in treating tumors, viral infections, allergies, and autoimmune diseases by directly eliminating or inducing apoptosis in target cells. Following the resolution of an infection, most effector cells undergo apoptosis during the contraction phase. However, some CD8+ T cells persist as memory cells (TM), which are further categorized into central memory T cells (Tcm) and effector memory T cells (Tem). TM cells are crucial for tumor immunity, host defense, and other immune responses, and they mediate memory responses following vaccination.
T cell subsets and their associated functions are summarized in Table 1.

Table 1. T cell subsets and their associated functions.
Platelet regulates T cell immune response
T cells play a pivotal role in both physiological and pathological processes by releasing various cytokines that exert paracrine or autocrine effects on different T cell subsets and other cells, thereby regulating immune responses. Notably, the majority of effector T cells (up to 90 to 95%) undergo apoptosis following antigen clearance (172, 173), posing a challenge for the immune system to maintain long-term self-tolerance during rapid expansions and contractions of cellular population (174). The immune system’s ability to maintain immunological specificity and memory ensures that a small population of memory T cells, which have lower proliferation rates and increased resistance to apoptosis, can persist (175–178). These memory T cells are crucial for initiating rapid and robust immune responses upon re-exposure to the same antigen, forming the backbone of anti-tumor specific immunity.
The mechanisms underlying the persistence of memory T cell remain a topic of debate. While some scientists argue about the necessity of antigen stimulation for memory T cell maintenance (179, 180), the shared surface molecules shared by memory and effector T cells suggest that antigen exposure may indeed play a role in promoting long-term memory. Some reports suggest that weak antigen stimulation can contribute to the persistence of memory T cells (181–183).
Moreover, TM cells are associated with cognate antigen-independent cellular turnover in response to IL-7/IL-15 (154, 184, 185), which supports the sustained maintenance of immune memory (186). An alternative perspective suggests that the longevity of T cells might be directly related to immunological memory itself (186, 187). Additionally, there’s evidence that inflammatory signaling favors the production of effector cells, while the absence of inflammation promotes the development of memory T cells (186).
Platelets are recognized not only for their roles in thrombus formation and hemostasis, but also for their significant regulatory role in immune responses. In collaborating with T cells, platelets release bioactive mediators like PF4 and TGFβ, which contribute to immune modulation and can exacerbate vascular intimal injury (22). Other mediators like PAF, TXA2, NAP, and RANTES activate immune cells-such as neutrophils and monocytes/macrophages-promoting chemotaxis and enhancing immune responses through mitochondrial energy metabolism pathways. In concert with CD4+ T cells, Platelets influence immune responses by modulating Th and Treg cell phenotypes, affecting the secretion of cytokines like IFN-γ and TNF-α (3, 4, 26, 188).
PF4/CXCL4, TGFβ, and other mediators are secreted by platelet α-granules and are involved in process related to thrombosis, inflammation, and immunity, contributing to vascular intimal injury (22). PF4 exhibits strong chemotactic properties for neutrophils by binding to heparan sulfate on the vascular endothelium, which attenuates thrombin inactivation. TGFβ, predominantly derived from platelets in circulation, serves as a specific marker for in vivo platelet activation (2, 23–25).
In the context of inflammatory atherosclerosis, specific macrophage subpopulations have been identified, notably those induced by PF4/CXCL4 and labeled as “M4” (PF4/CXCL4-induced plaque macrophage). The PF4/CXCL4-induced upregulation of MMP7 and S100A8 is mitigated by heparin, which binds to PF4/CXCL4 and glycosaminoglycans, potentially representing macrophage receptors for PF4/CXCL4, characterized by CD68+ MMP7+ and S100A8+ expression (189). studies of atherosclerotic plaque and rheumatoid arthritis synovium have shown that macrophages are a major source of PF4/CXCL4 (190, 191). These interactions between PF4 produced by macrophages and receptors on endothelial cells, fibroblasts, and alveolar type 2 cells suggest significant immune responses, particularly involving PTPRC+ immune cells, with PF4 transcripts detected in macrophages based on their expression of CD68, CD163, and MRC1. This indicates that macrophages may be a potential source of PF4/CXCL4 in mouse models of lung and heart fibrosis, as well as in individuals with pulmonary fibrosis (192).
In a study using a pressure overload TAC (transverse aortic constriction) mouse model, PF4 expression was observed in macrophages co-expressing C1q molecules. These macrophages exhibited an M2-like signature, with increased proportions in TAC compared to sham-operated mice after one week (193).
The immune function in chronic inflammatory atherosclerosis involves both innate and adaptive immunity. The innate immunity response is triggered by the accumulation of LDL (low-density lipoprotein) in the arterial wall, which is taken up by macrophages, leading to the formation of microcrystals. This activates inflammasomes, processing of IL-1b, and promoting its secretion, contributing to inflammation and plaque formation. Adaptive immunity plays its part as LDL is transported to arterial draining lymph nodes, where peptide fragments from LDL are presented to T cells, leading to their activation, division, and differentiation. Some T cells stimulate B cells to produce antibodies against LDL, while others contribute to plaque formation and inflammation by activating macrophages, endothelial cells, and smooth muscle cells (194).
Additionally, platelet-thrombocyte aggregates (PTAs) are associated with an increased risk of thrombosis, with cancer patients exhibiting significantly higher percentages of PTAs among CD4+ and CD8+ T lymphocyte populations compared to healthy individuals (195). Platelets significantly inhibit pro-inflammatory cytokines (IL-12, IL-6, TNFα) while promoting the production of anti-inflammatory cytokine (IL-10) in moDCs (monocyte-derived-dendritic cells) primed with both Toll-like receptor (TLR)-dependent and TLR-independent stimuli. Moreover, platelets and their soluble mediators impede T cell priming and differentiation into the IFNg+ Th1 phenotype by moDCs (196).
Interestingly, the absence or inhibition of T cells enhances the antiplatelet effect of clopidogrel by boosting its metabolic activation in the liver, leading to significant production of Cyp2c and Cyp3a in mice. This finding suggests that damage to T cells can enhance the metabolism of drugs that are substrates of Cyp2c or Cyp3a (197).
Platelets possess immunosuppressive properties, releasing anti-inflammatory molecules such as TGF-β and soluble CD40L, which help suppress excessive immune responses and prevent tissue damage. Platelets are the primary source of TGF-β in the human body. This cytokine has been demonstrated to exert deleterious effects on various lymphocytes. Specifically, TGF-β inhibits the differentiation of T cells into cytotoxic T cells while increasing the population of Tregs. Tregs can further inhibit effector T cells and NK cells. TGF-β also directly affects NK cells by impairing their lytic activity and reducing IFN-γ production (198). Therefore, TGF-β released by platelets can inhibit excessive cellular immune responses to prevent tissue damage, its suppression of cellular immunity in tumors can support cancer cell survival.
Platelet-derived CD40 ligand (CD40L) induces production of IL-6 and IL-12 from DCs and enhances their expression of costimulatory molecules such as CD80, CD86 and ICAM-1. Furthermore, CD40L has been shown to enhance DC maturation and their ability to directly kill Staphylococcus aureus, thereby promoting efficient adaptive immunity against the bacterium (199). Besides, platelet CD40L can enhance CD8+ T cell response, hence, functioning as a bridge to the adaptive immune system (200). Additionally, activated platelets can synthesize and secrete IL-1β, a potent pro-inflammatory cytokine. IL-1β up-regulates the expression of adhesion receptors and the secretion of IL-6 and IL-8 in endothelial cells, as well as increases nitric oxide (NO) -induced vascular permeability, thereby playing a significant role in the immune response (201).
SARS-CoV-2 binds to platelet-expressed ACE2/TMPRSS2 through its spike protein, activates the MAPK pathway and enhances platelet activation (aggregation, granule secretion, leukocyte aggregation) and thrombosis, which can be inhibited by recombinant human ACE2 protein and anti-spike monoclonal antibody. The molecular mechanism by which the virus directly drives COVID-19 thromboinflammation is revealed (202). SARS-CoV-2 infection leads to altered platelet gene expression, enhanced activation (manifested as increased P-selectin expression), and increased aggregation with immune cells (neutrophils, monocytes, T cells). Its excessive activation and accelerated aggregation are associated with MAPK pathway activation and increased thromboxane production. In some patients, platelets carry viral mRNA (independent of ACE2), and these abnormalities may exacerbate the pathological process of thrombosis and organ failure in COVID-19 (203).Therefore, platelet immunomodulatory functions reported by studies in SARS-CoV-2 infections, where there has been accumulating evidence that coagulation and complement cascades are deeply interconnected and this can influence immune cell activation.
Platelets regulate T cell immune response and cytokine release by PF4/TGFβ—via mitochondrial Akt PGC1 α-TFAM signaling pathway
Studies indicate that platelets in healthy individuals selectively enhance CD4+ T cell recruitment under normal arterial blood flow conditions. They regulate T effector cell responses through mediators like PF4, TGFβ, and RANTES, which exhibit varying kinetics and effects on the regulation of Th1, Th17, and Treg cells (204).
Mitochondria, the body’s metabolic energy factories, play a crucial role in T cell activation. Within the first 24 to 48 hours after T cell activation, mitochondrial energy metabolism and ATP production are vital for the immune response of T effector cells (205). TFAM (Mitochondrial Transcription Factor A), encoded by nuclear genes, is transported to mitochondria, and serves as a key factor in activating and regulating mitochondrial DNA transcription (3, 4). Additionally, PGC1α (Peroxisome Proliferator-Activated Receptor γ Coactivator 1 α) regulates mitochondrial biosynthesis and proliferation by binding to the downstream target gene NRF-1, which activates its transcription. PGC1α also promotes TFAM expression through co-transcription with NRF-1 and NRF-2, enhancing mitochondrial oxidative function (206).
CXCR3, a functional receptor for PF4, is expressed on CD4+ T cells such as Tem and Tcm (3–5, 207). Single-cell RNA sequencing (scRNA-seq) analysis has revealed significant diversity among CD8 T subgroups, particularlly during the peak of influenza virus load and resolution. An enrichment of CXCR3+ CD8+ T cells correlates with stronger cytotoxic responses. Notably, CXCR3 blockade during late-stage CD8 T cell responses in influenza-cleared lungs can mitigate lung injury without affecting viral clearance, suggesting therapeutic potential for preventing influenza-associated lung injury (208). Moreover, functional CD4+ and CD8+ T cells exhibiting traits of tissue-resident memory T cells (TRM) have been identified in human kidney tissues, indicating a dynamic immune environment (209). CXCR3 can also enhance specific CD8+ T cell activation via plasmacytoid dendritic cells (pDCs) during intracellular pathogen infections (210).
The research highlights that platelets significantly influence T cell subsets (Tem, Tcm, and Tn) and their immune responses, involving mitochondria through PF4 bridging. Key findings include:
1. Platelets significantly impact the Th1/Treg response, with Treg cell response increasing while Th1 responses are inhibited.
2. Platelets undergo PF4-dependent mitochondrial biogenesis and cell proliferation. The Akt-PGC1α-TFAM signaling pathway, initiated by PF4 binding with CXCR3, enhances the responses of Tem and Tcm cells, promoting mitochondrial ATP and ROS production and thus increasing Th1 and Treg responses.
3. Platelets regulate CD4+ Tn cell response through PF4-TGFβ interactions. At mildly elevated concentrations, PF4 combined with TGFBRIII promotes TGFβ presentation and TGFBRII expression, enhancing signal transduction and Tn effector cell response. At excessively high concentrations, PF4 can directly bind TGFβ-TGFBRII, blocking TGFβ signal transduction and disrupting the T cell response. This underscores the complex interplay between platelets and T cell regulation, introducing a novel immune regulatory function and highlighting that the platelet-regulated T effector cell response results from multiple factors (3–5, 204).
In conclusion, while platelets are primarily recoginized for their role in blood clotting (hemostasis), they also play significant roles in the immune response. Their involvement is a dynamic process, influenced by various mechanisms, as their interactions with the immune system can modulate responses depending on the context. Thus, the role of platelets in immune activation and suppression is both complex and dynamic.
Platelet activation triggers an increase in oxidative metabolism to meet energy demands, a process that is more efficient than aerobic glycolysis alone. Additionally, platelets exhibit supplemental mitochondrial oxidative phosphorylation, which may serve as a necessary chemical source for platelet activation (211).
However, some studies suggest that:
1. mitochondria derived from platelets can inhibit the proliferation of PBMCs (peripheral blood mononuclear cells).
2. mitochondria from platelets can modulate anti-CD3/CD28-activated CD4+ T cells by directly targeting CXCR4 and its ligand SDF-1 (stromal cell-derived factor-1), leading to the upregulation of CD4+ Tn and Tcm, while causing a decrease in CD4+ Tem (212).
Platelet-based drug delivery strategies have been explored for targeting primary tumors, circulating tumor cells (CTCs), and circulating malignant tumors like lymphoma. Therapeutic agents have been loaded into platelets via endocytosis (e.g., doxorubicin), cell surface chemistry methods (e.g., anti-PD-1 binding), and gene modification (e.g., TRAIL expression). Drug-antibody conjugates have also targeted to target platelet receptors. Given the unique tumor vascular system and the ability of platelets to adhere to circulating tumor cells—especially through surface receptors such as GP IIb/IIIa—innovative targeting strategies have been designed to leverage platelet accumulation in tumors. However, it is still unknown whether this platelet-based drug delivery strategy will cause excessive accumulation of platelets, thereby leading to thrombocythemia and thrombosis formation. More exploration is needed in the future. Alternative approaches include using ligands cleaved by proteins in the tumor microenvironment and surface structures that provide propulsion (213–217).
Platelet-T cell interaction and impact on AA
AA is a T-cell-mediated bone marrow failure syndrome characterized by the depletion of HSPCs. Research links the activation of T cells to cytokines and chemokines, such as INF-γ, TNF-α, and IL-2, which negatively affect HSPCs, resulting in persistent inhibition of hematopoietic function (218–220). Therefore, IST combined with TPO-RAs (thrombopoietin receptor agonists) or HSCT (hematopoietic stem cell transplantation) is recommended, depending on the patient’s age. Because TPO receptors are expressed on HSPCs, recent research suggests that TPO-RAs can alleviate the inhibitory effects of INF-γ on HSCT in multiple ways, beyond merely raising platelet counts. IFN-γ significantly impacts various T cell subsets, including Th, Treg, and TFH (221, 222). There have been reports of myelofibrosis in patients treated with TPO-RAs. Therefore, patients treated with TPO-RAs should perform bone marrow biopsy once a year/every six months. It is very necessary to be able to discontinue these drugs in a timely manner when grade 2/3 myelofibrosis occurs. Discontinuing TPO-RAs can also prevent the development of clinical manifestations by blocking the progression of grade 2/3 fibrosis (223). Recent study has also shown that Janus kinase (JAK) 1/2 inhibitor ruxolitinib (RUX) can inhibit T cell infiltration activation and inhibit bone marrow cell apoptosis in mice with immune AA. This provides a new idea for the treatment of AA (224).
In children with AA, Treg levels significantly decrease, diminishing their immune suppressive capacity. However, during remission, Treg levels do not show substantial changes (225). New membrane proteins may be identified as platelet sensors for pathogen- or damage-associated molecular patterns (PAMPs and DAMPs), shedding light on novel molecular functions in immunity (226). Furthermore, prior studies have revealed a reduction in platelet-related cytokines in plasma, such as CCL5 and CD40L, reflecting thrombocytopenia in AA. These cytokines are crucial in regulating TH1 and TH2 balance (227).
High levels of PF4 in malignant pleural effusion (MPE) are associated with poor prognosis. The impaired T lymphocyte response caused by PF4 confers an advantage for tumor progression (228).
Platelets and AA
Platelets can either eliminate microorganisms by direct binding or inhibit their transmission by limiting cell division or survival through indirect effects, which trigger host immune responses. However, many invasive microbial pathogens can target host platelets, directly or indirectly altering platelet counts or function. These microbial pathogens can also influence autoimmunity and alloreactivity in immune-mediated diseases such as immune thrombocytopenia, systemic lupus erythematosus, and multiple sclerosis by interacting with platelet antigens. Conditions like autoimmune thrombocytopenia and fetal and neonatal alloimmune thrombocytopenia are examples of such effects (229).
Platelets have been shown to promote the proliferation of acute leukemia (AL) cells, reduce their sensitivity to chemotherapy, and induce apoptosis. However, the role of platelets in AA remains unclear (230).
Mice lacking CD84, a receptor of the SLAM family, on platelets or T cells, exhibit reduced CD4+ T cell infiltration and thrombotic activity in the brain, lowering nerve damage. High platelet CD84 expression is also linked to poorer prognosis in stroke patients. There is overlap in the cytokines and chemokines released by platelets and T cells. Soluble CD48, shed from platelets, can stimulate CD4 T cell migration, and high CD84 expression is associated with an inflammatory immune response (231).
Studies on myeloproliferative neoplasms (MPN) show that PLT interactions with CD8+ T cells reduce the proliferation and cytotoxicity of these T cells (232, 233). In malignant pleural effusion, high levels of platelet-derived PF4 are associated with a more severe T lymphocyte response and poor prognosis (227). Mitochondria from platelets can directly interact with CD4+ T cells through SDF-1/CXCR4, influencing T cell behavior (212).
A study published in Nature Aging in 2024 revealed that the TRMT6/61A complex, involved in m1A (methylation on the first nitrogen atom of mRNA and tRNA adenosine), drives hematopoietic stem cell aging through a non-methyltransferase pathway. Targeted inhibition of this pathway can delay hematopoietic stem cell aging, which has implications for blood diseases and bone marrow failure. The accumulation of TRMT6/61A in aging HSCs due to deactivated CRL4DCAF1 ubiquitin degradation pathway, may disrupt normal blood cell production (234).
Exploring platelet-T cell interactions offers new insights into bone marrow failure treatments. For instance, elevated levels of RANTES and PDPN Mφs have been linked to in severe AA (235). The TPO/Mpl complex regulates megakaryocyte development and platelet production through downstream pathways such as JAK/STAT, Ras/Raf-1/MAPK, and PI3k/Akt. Other regulators, like the interleukin family and IGF-1, can also play supplementary roles in TPO regulation. MicroRNAs, such as miR-9, miR-22, and miR-125a, modulate megakaryocyte and platelet production at various stages. Current treatments like etripopal and romiplimumab aim to boost platelet count by targeting these pathways (236, 237).
T cells, cytokines, and AA
In a mouse model of AA, an increase in β-chemokines was noted, partly depending on IFN-γ. This cytokine is crucial for upregulating the chemokine receptor CCR5 in macrophages. Blocking CCR5 in murine AA models improved survival, correlating with increased platelet counts and enhanced platelet-biased CD41hi HSCs. While T cells are essential in AA pathogenesis, CCR5 expression on T cells and T cell-derived CCL5 are not necessary for disease progression. In fact, CCR5 antagonism reduces bone marrow macrophages and lower the production of TNF and CCL5, correlating with reduced IFN-γ secretion from bone marrow T cells. Further studies revealed that elderly mice and humans exhibit significantly higher CCR5 expression in macrophages, highlighting CCR5’s role in age-related bone marrow failure. CCR5 signaling plays a crucial role in maintaining bone marrow macrophages, particularly in aging individuals (238).
Additionally, CD8+ T cells in AA (aplastic anemia) patients show an activated phenotype characterized by elevated expression of HLA-DR, CD57, and CD27, contributing to hematopoiesis inhibition and bone marrow failure progression. CD38+ CD8+ T cells, also enriched in both AA patients and animal models, display enhanced pro-inflammatory and proliferative abilities (239–241). Additionally, CD8+ GITR+ T cells exhibit increased CTLA-4 expression, resulting in a reduced cytotoxic phenotype (242).
CD4+ Th1 cells play a significant role in the pathogenesis of bone marrow failure by secreting pro-inflammatory cytokines like IFN-γ and TNF-α, which mediate HSPC apoptosis (216). IFN-γ, in particular, is critical in cellular immunity, as it inhibits precursor cell proliferation in vitro, and induces Fas expression on HSPCs. Once Fas is expressed, activated T cells trigger apoptosis through the Fas/FasL pathway, which ultimatly leads to bone marrow failure. In transgenic mice with elevated levels of IFN-γ, early signs of bone marrow aging manifest both in the bone marrow and peripheral blood. These mice exhibit the symptoms characteristic of bone marrow dysfunction and immune dysregulation (243, 244).
Interestingly, despite the significant elevation of IL-18 (a cytokine that is also induced by IFN-γ) in severe AA patients, studies have shown that IL-18 gene knockout in mouse models does not prevent bone marrow failure. This suggests that while IL-18 may be involved in the inflammatory response, it does not directly drive the pathogenesis of AA (243).
Additionally, TNF-α is a key negative regulator of hematopoiesis. It acts through its receptors on T cells and bone marrow CD34+ cells, further contributing to cell damage and the progression of bone marrow failure (245–247).
Thes studies underscore the complex interplay of cytokines like IFN-γ, TNF-α, and IL-18 in the immune-mediated destruction of hematopoietic cells, with IFN-γ playing a central role in inducing bone marrow failure through the Fas/FasL pathway and inhibiting progenitor cell proliferation.
T cell dysfunction is closely related to the physiological and pathological status of bone marrow failure, and there are also many associations between CD4+ T cells and CD8+ T cells
CD4+ Treg cells in the tumor microenvironment (TME) express high levels of PD-1 (programmed cell death protein 1), suggesting that PD-1 blockade might enhance Treg’s immunosuppressive function. Research has indicated that anti-PD-1 monoclonal antibodies (mAb), commonly used in immune checkpoint inhibition therapy, may paradoxically boost Treg-mediated immunosuppressive activity in cancer patients. Moreover, Treg cells deficient in PD-1 deficiency exhibit an even more potent ability to suppress immune responses (248, 249). Conversely, PD-1 expression is also a hallmark of exhausted CD8+ T cells, which limits their cytotoxic function due to chronic TCR (T cell receptor) stimulation. Immune checkpoint inhibitors (ICIs), like anti-PD-1 or anti-PD-L1 mAb, block the interaction between “PD-1—PD-L1 (PD-1 ligand)”, effectively restoring cytotoxic capabilities of CD8+ T cells. This restoration helps in reducing viral load and tumor progression, as demonstrated in clinical practice across various cancer treatments (250–255).
In addition, CXCR5+ CD8+ T cells have emerged as crucial players in immune regulation, particularly within the TME and during chronic infections. These cells exhibit dual functions: they not only assist B cells in germinal centers in a manner similar to CXCR5+ CD4+ T follicular helper (Tfh) cells by promoting antibody production, but they also retain cytotoxic activity, crucial in infection and cancer contexts (254–258). CXCR5+ CD8+ T cells, like their CD4+ counterparts, also express high levels of PD-1, and may exhibit an “exhausted” phenotype in the tumor microenvironment, making them potential targets for ICIs (256–260).
In the context of thromboinflammation, activated platelets expressing integrin αIIbβ3 and P-selectin are known to contribute to platelet aggregation, endothelial damage, and microthrombosis. This interaction increases the formation of neutrophil extracellular traps (NETs), which amplify cytokine release and further inflammation. Given this, future therapeutic strategies targeting thromboinflmmatory responses are focusing on dieases such as thrombosis, sepsis, influenza, and COVID-19 (261). Advances in platelet-T cell immune regulation may also enable novel drug delivery systems, such as the use of platelet-coated gold nanoparticles to modulate the tumor microenvironment (217, 262) (Figure 3).
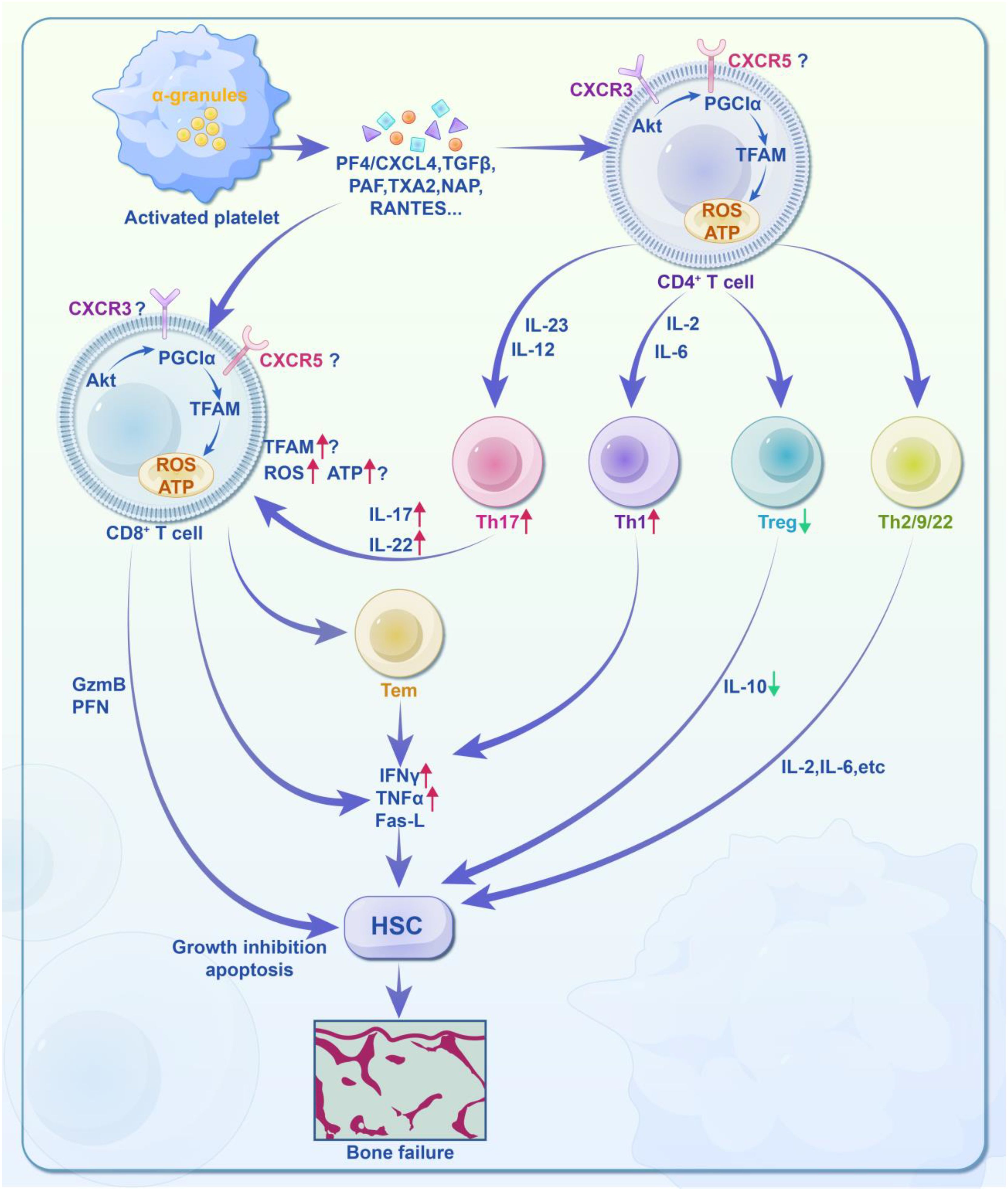
Figure 3. Molecular activity of platelet-regulated T cell immune response in acquired aplastic anemia (AA). AA may be due to an unknown pathogen infecting hematopoietic stem cells (HSCs) or peripheral cells, leading to the presentation of pathogen particles and either unmodified or chemically/genetically modified components on their cell surfaces. These antigens are then presented to antigen-presenting cells (APCs), which process and present them to CD4+ T cells. Platelets play a role in this immune regulation by releasing various soluble mediators, such as PF4/CXCL4, TGFβ, PAF, TXA2, NAP, and RANTES. PF4 binds to CD4+ or CD8+ T cell/CTL membrane receptors CXCR3 and CXCR5, activating the downstream mitochondrial energy metabolism signaling pathway (Akt-PGC1α-TFAM). This affects mitochondrial quantity, ATP production, and reactive oxygen species (ROS), thereby regulating the T cell immune response. For CD4+ T cells: ① Differentiation into Th1, Th2, Th9, and Th22 phenotypes. ② Simultaneous differentiation into Th17 phenotypes under the stimulation of IL-23 and IL-12. ③ Suppression of Treg phenotypes, leading to weakened immune regulation and immune imbalance. For CD8+ T cells: ① Direct cytotoxicity through the release of granzyme B (GzmB) and perforin (PFN). ② Paracrine effects via TNFα, IFNγ, and Fas ligand (Fas-L). In summary, platelets regulate the immune responses of CD4+ and CD8+ T cells through mitochondrial energy metabolism, contributing to immune imbalance and increased levels of IFNγ and TNFα. This results in the attack on HSPCs and may ultimately lead to bone marrow failure.
Summary and future perspective
The development of platelet drugs and antiplatelet therapies has progressed through four key stages: the prehistoric phase, focusing on the conceptualization of platelets; the mono-antiplatelet phase, marked by the introduction of aspirin; the dual-antiplatelet phase, involving the combination of two antiplatelet drugs; and the current new era, characterized by immune targeting and precision medicine (Figure 4). The CHANCE2 study has enabled the selection of appropriate antiplatelet drugs, such as ticagrelor or clopidogrel, based on detecting CYP2C19 gene loss-of-function mutations (263).
Despite advancements, the efficacy of antiplatelet drugs has hit a plateau. Improvements, even with widely used drugs like aspirin, clopidogrel, and ticagrelor, have only reached about 2%. This highlights the need for new targets and directions beyond traditional COX inhibitors or ADP P2Y12 receptor antagonists.
Interestingly, recent studies suggest that platelet function extends beyond their roles in thrombosis and hemostasis, playing significant roles in immune regulation. As the second most abundant blood cell type, platelets engage with T cells through mitochondrial metabolic pathways, influencing T cell development, proliferation, differentiation, survival, and apoptosis.
Platelets activate the downstream mitochondrial energy metabolism signaling pathway (Akt-PGC1α-TFAM) of T cells by releasing various soluble mediators such as PF4/CXCL4, TGFβ, PAF, TXA2, NAP and RANTES, affecting the number of mitochondria, ATP production and reactive oxygen species (ROS), thereby regulating the immune response of T cells. CD4 + T cells differentiate into Th1, Th2, Th9, Th17 and Th22 phenotypes, while the Treg phenotype is inhibited, resulting in weakened immune regulation and immune imbalance. CD8 + T cells, on the one hand, directly produce cytotoxicity by releasing granzyme B (GzmB) and perforin (PFN), and on the other hand, exert paracrine effects through TNFα, IFNγ and Fas ligand (FAS-L). These immune responses promote immune imbalance and elevated levels of IFNγ and TNFα. This leads to an attack on hematopoietic stem cells and may eventually result in bone marrow failure.
In AA, platelets may regulate T cells through several mechanisms: 1) Mitochondria-mediated regulation of T cells via the Akt-PGC1α-TFAM signaling pathway leads to increased mitochondrial biogenesis, boosting ATP and ROS production. This may result in immune imbalance, overactivation of CD4+ Th1, suppression of Tregs, and imbalance in CD8+ T cell phenotypes (e.g., CD38, PD-1, GITR, CTLA-4, HLA-DR, CD57, CD27), contributing to elevated IFN-γ and TNF-α levels and subsequent HSPC damage. 2) Platelet-derived mediators like PF4 and TGFβ can modulate PF4-TGFβ dual signaling. Low PF4 levels increase TGFβ signaling through TGFBRIII, while high levels inhibit this pathway, potentially triggering excessive immune response and damage to hematopoietic stem cells.
Further research is required to understand how telomere dysfunction, hematopoietic microenvironment abnormalities, and other immune cells contribute to AA. Investigating these mechanisms may guide future clinical treatments.
Overall, the emerging understanding of platelet-T cell interactions, combined with platelet-based drug delivery technologies, could lay a strong foundation for immune-inflammatory therapies. Platelets may become a central focus in research on atherosclerosis and immune-related inflammation, garnering significant interest from the scientific community (Figure 4).
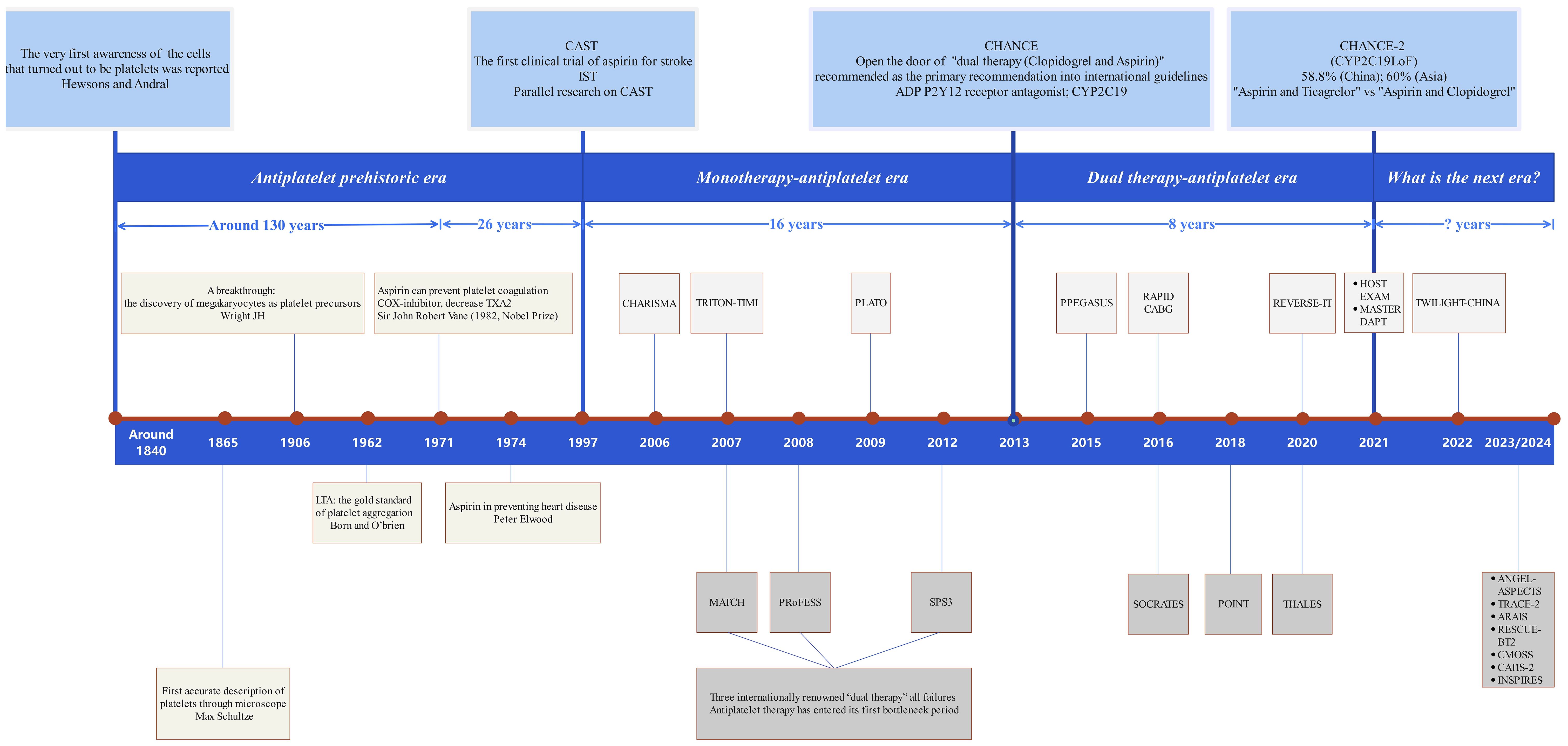
Figure 4. Diagram of the brief history of platelet drugs. The brief history of platelet drugs and antiplatelet therapy spans over 180 years and can be divided into four key stages: ① Initial Development Stage (The Concept of Platelets): This stage marks the initial understanding of platelets and their role in clotting. ② Mono-Antiplatelet Stage (Introduction of Aspirin): The development of aspirin as the first antiplatelet drug represents a significant milestone in antiplatelet therapy. ③ Dual-Antiplatelet Stage (Combination Therapy): This stage involves the combination of two antiplatelet drugs to enhance therapeutic efficacy. ④ Current New Era Stage (Immune Targeting and Precision Medicine): Advances in personalized medicine and immune targeting characterize this latest phase. The CHANCE2 study highlights that suitable antiplatelet drugs, such as ticagrelor or clopidogrel, can be selected by detecting CYP2C19 loss-of-function mutations. Despite ticagrelor providing only 2% greater protection for primary endpoints compared to clopidogrel within one year, it appears we have reached a new bottleneck in antiplatelet therapy. Thus, identifying new targets for platelet drugs remains a crucial goal for future research.
Author contributions
ST: Writing – original draft, Writing – review & editing. HH: Writing – original draft. YL: Writing – original draft. MS: Writing – original draft. YC: Writing – original draft. DZ: Writing – original draft. RH: Writing – original draft. WHu: Writing – original draft. XC: Writing – original draft. JN: Writing – original draft. QM: Writing – original draft. LS: Writing – original draft. JS: Writing – original draft. WHe: Writing – original draft. XF: Writing – original draft, Writing – review & editing. WS: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. The study was supported by grants from the National Natural Science Foundation of China (NSFC) (project no 82300161), the Natural Science Foundation of Beijing Municipality (project no Z200022), the Natural Science Foundation of Beijing Municipality (project no 7242072), Beijing High-level Overseas Returnee Talent Funding Project (project no 2-2-008-0243), Capital Medical University Science and Innovation Elite Plan Project (project no 2024KCJY0405), the “National Natural Science Foundation of Youth Cultivation Project” of “Xuanwu Hospital, Capital Medical University, Beijing, China” (project no QNPY2022014), and the “Person of Outstanding Ability Training Program” of “Xuanwu Hospital, Capital Medical University, Beijing, China” (project no YC20220127).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kapur R, Zufferey A, Boilard E, and Semple JW. Nouvelle cuisine: platelets served with inflammation. J Immunol. (2015) 194:5579–87. doi: 10.4049/jimmunol.1500259
2. Kapur R and Semple JW. The nonhemostatic immune functions of platelets[C]//Seminars in Hematology. WB Saunders. (2016) 53:S2–6. doi: 10.1053/j.seminhematol.2016.04.002
3. Tan S, Li S, Min Y, Gisterå A, Moruzzi N, Zhang J, et al. Platelet factor 4 enhances CD4+ T effector memory cell responses via Akt-PGC1α-TFAM signaling-mediated mitochondrial biogenesis. J Thromb Haemost. (2020) 18:2685–700. doi: 10.1111/jth.15005
4. Tan S, Zhang J, Sun Y, Gisterå A, Sheng Z, Malmström RE, et al. Platelets enhance CD4+ central memory T cell responses via platelet factor 4-dependent mitochondrial biogenesis and cell proliferation. Platelets. (2021) 33(3):360–70. doi: 10.1080/09537104.2021.1936479
5. Min Y, Hao L, Liu X, Tan S, Song H, Ni H, et al. Platelets fine-tune effector responses of naïve CD4+ T cells via platelet factor 4-regulated transforming growth factor β signaling. Cell Mol Life Sci. (2022) 79:247. doi: 10.1007/s00018-022-04279-1
6. Yan C, Wu H, Fang X, He J, and Zhu F. Platelet, a key regulator of innate and adaptive immunity. Front Med (Lausanne). (2023) 10:1074878. doi: 10.3389/fmed.2023.1074878
7. Feng X, Lin Z, Sun W, et al. Rapamycin is highly effective in murine models of immune-mediated bone marrow failure. Haematologica. (2017) 102:1691–703. doi: 10.3324/haematol.2017.163675
8. Kuter DJ. The structure, function, and clinical use of the thrombopoietin receptor agonist avatrombopag. Blood Rev. (2022) 53:100909. doi: 10.1016/j.blre.2021.100909
9. Takasaki K, Kacena MA, Raskind WH, Weiss MJ, and Chou ST. GATA1-Related Cytopenia. In: Adam MP, Everman DB, Mirzaa GM, et al, editors. GeneReviews®. University of Washington, Seattle, Seattle (WA (2006).
11. Hosokawa K, Maranski P, Feng X, Townsley DM, Liu B, Knickelbein J, et al. Memory stem T cells in autoimmune disease: high frequency of circulating CD8+ memory stem T cells in acquired aplastic anemia. J Immunol. (2016) 196:1568–78. doi: 10.4049/jimmunol.1501739
12. Smith JN, Kanwar VS, and MacNamara KC. Hematopoietic stem cell regulation by type I and II interferons in the pathogenesis of acquired aplastic anemia. Front Immunol. (2016) 7:330. doi: 10.3389/fimmu.2016.00330
13. Sun W, Wu Z, Lin Z, Hollinger M, Chen J, Feng X, et al. Macrophage TNF-α licenses donor T cells in murine bone marrow failure and can be implicated in human aplastic anemia. Blood. (2018) 132:2730–43. doi: 10.1182/blood-2018-05-844928
15. Cines DB, Cuker A, and Semple JW. Pathogenesis of immune thrombocytopenia. La Presse Médicale. (2014) 43:e49–59. doi: 10.1016/j.lpm.2014.01.010
16. Semple JW, Italiano JE, and Freedman J. Platelets and the immune continuum. Nat Rev Immunol. (2011) 11:264–74. doi: 10.1038/nri2956
17. Panigrahi S, Ma Y, Hong L, Gao D, West XZ, Salomon RG, et al. Engagement of platelet toll-like receptor 9 by novel endogenous ligands promotes platelet hyper-reactivity and thrombosis. Circ Res. (2012) 112:274241. doi: 10.1161/CIRCRESAHA.112.274241IF
18. Wong CHY, Jenne CN, Petri B, Chrobok NL, and Kubes P. Nucleation of platelets with blood-borne pathogens on Kupffer cells precedes other innate immunity and contributes to bacterial clearance. Nat Immunol. (2013) 14:785–92. doi: 10.1038/ni.2631
19. Sreeramkumar V, Adrover JM, Ballesteros I, Cuartero MI, Rossaint J, Bilbao I, et al. Neutrophils scan for activated platelets to initiate inflammation. Science. (2014) 346:1234–8. doi: 10.1126/science.1256478
20. Gouttefangeas C, Diehl M, Keilholz W, Cuartero MI, Rossaint J, Bilbao I, et al. Thrombocyte HLA molecules retain nonrenewable endogenous peptides of megakaryocyte lineage and do not stimulate direct allocytotoxicity in vitro. Blood. (2000) 95:3168–75. doi: 10.1182/blood.V95.10.3168
21. Zufferey A, Schvartz D, Nolli S, Reny JL, Sanchez JC, and Fontana P. Characterization of the platelet granule proteome: evidence of the presence of MHC1 in alpha-granules. J Proteomics. (2014) 101:130–40. doi: 10.1016/j.jprot.2014.02.008
22. Blair P and Flaumenhaft R. Platelet α-granules: basic biology and clinical correlates. Blood Rev. (2009) 23:177–89. doi: 10.1016/j.blre.2009.04.001
23. Andersson PO, Olsson A, and Wadenvik H. Reduced transforming growth factor-β1 production by mononuclear cells from patients with active chronic idiopathic thrombocytopenic purpura. Br J haematol. (2002) 116:862–7. doi: 10.1046/j.0007-1048.2002.03345.x
24. Alicja KZ. Recovery of platelet factor 4 (PF-4) and beta-thromboglobulin (beta-TG) plasma concentrations during remission in patients suffering from atopic dermatitis. Platelets. (2010) 21:522–4. doi: 10.3109/09537104.2010.493247
25. Thachil J. The prothrombotic potential of platelet factor 4. Eur J Internal Med. (2010) 21:79–83. doi: 10.1016/j.ejim.2009.11.007
26. Hansson GK and Hermansson A. The immune system in atherosclerosis. Nat Immunol. (2011) 12:204–12. doi: 10.1038/ni.2001
27. Michelson AD. Antiplatelet therapies for the treatment of cardiovascular disease. Nat Rev Drug Discov. (2010) 9:154–69. doi: 10.1038/nrd2957
28. Wallentin L, Becker RC, Budaj A, Cannon CP, Emanuelsson H, Held C, et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. (2009) 361:1045–57. doi: 10.1056/NEJMoa0904327
29. Huber K, Bates ER, Valgimigli M, Wallentin L, Kristensen SD, Anderson JL, et al. Antiplatelet and anticoagulation agents in acute coronary syndromes: what is the current status and what does the future hold? Am Heart J. (2014) 168:611–21. doi: 10.1016/j.ahj.2014.06.014
30. Chen C, Leng X, Zhang Y, Hu J, Wei D, Wang P, et al. Effects of platelets on characteristics of lymphocytes cultured in vitro and optimization of adoptive immunotherapy. BIOCELL. (2023) 47:2661–9. doi: 10.32604/biocell.2023.043084
31. Pankowska KA, Będkowska GE, Chociej-Stypułkowska J, Rusak M, Dąbrowska M, and Osada J. Crosstalk of immune cells and platelets in an ovarian cancer microenvironment and their prognostic significance. Int J Mol Sci. (2023) 24:9279. doi: 10.3390/ijms24119279
32. Zhu S, Gokhale S, Jung J, Spirollari E, Tsai J, Arceo J, et al. Multifaceted immunomodulatory effects of the BTK inhibitors ibrutinib and acalabrutinib on different immune cell subsets - beyond B lymphocytes. Front Cell Dev Biol. (2021) 9:727531. doi: 10.3389/fcell.2021.727531
33. Carestia A, Mena HA, Olexen CM, Ortiz Wilczyñski JM, Negrotto S, Errasti AE, et al. Platelets promote macrophage polarization toward pro-inflammatory phenotype and increase survival of septic mice. Cell Rep. (2019) 28:896–908. doi: 10.1016/j.celrep.2019.06.062
34. Anitua E, Troya M, and Alkhraisat MH. Immunoregulatory role of platelet derivatives in the macrophage-mediated immune response. Front Immunol. (2024) 15:1399130. doi: 10.3389/fimmu.2024.1399130
35. Di Paola A, Palumbo G, Merli P, Argenziano M, Tortora C, Strocchio L, et al. Effects of eltrombopag on in vitro macrophage polarization in pediatric immune thrombocytopenia. Int J Mol Sci. (2020) 22:97. doi: 10.3390/ijms22010097
36. Nakamura-Ishizu A. Thrombopoietin-mediated regulation of hematopoietic stem cells. Rinsho Ketsueki. (2024) 65:872–7. doi: 10.11406/rinketsu.65.872
37. de Latour RP, Kulasekararaj A, Iacobelli S, Griffin M, Halkes CJ, Dufour C, et al. Plain language summary of RACE study results: addition of eltrombopag to standard treatment of severe aplastic anemia. Immunotherapy. (2024) 16:135–42. doi: 10.2217/imt-2023-0200
38. DuPage M and Bluestone JA. Harnessing the plasticity of CD4(+) T cells to treat immune-mediated disease. Nat Rev Immunol. (2016) 16:149–63. doi: 10.1038/nri.2015.18
39. Chalmin F, Humblin E, Ghiringhelli F, and Végran F. Transcriptional programs underlying cd4 T cell differentiation and functions. Int Rev Cell Mol Biol. (2018) 341:1–61. doi: 10.1016/bs.ircmb.2018.07.002
40. Ohkura N, Hamaguchi M, Morikawa H, Sugimura K, Tanaka A, Ito Y, et al. T cell receptor stimulation-induced epigenetic changes and FoxP3expression are independent and complementary events required for Treg cell development. Immunity. (2012) 37:785–99. doi: 10.1016/j.immuni.2012.09.010
41. Nguyen QP, Deng TZ, Witherden DA, and Goldrath AW. Origins of CD 4+ circulating and tissue-resident memory T-cells. Immunology. (2019) 157:3–12. doi: 10.1111/imm.2019.157.issue-1
42. Busslinger M and Tarakhovsky A. Epigenetic control of immunity. Cold Spring Harbor Perspect Biol. (2014) 6:a019307. doi: 10.1101/cshperspect.a019307
43. Malmhäll C, Bossios A, Rådinger M, Sjöstrand M, Lu Y, Lundbäck B, et al. Immunophenotyping of circulating T helper cells argues for multiple functions and plasticity of T cells in vivo in humans-possible role in asthma. PloS One. (2012) 7:e40012. doi: 10.1371/journal.pone.0040012
44. Ahlfors H, Morrison PJ, Duarte JH, Li Y, Biro J, Tolaini M, et al. IL-22 fate reporter reveals origin and control of IL-22 production in homeostasis and infection. J Immunol. (2014) 193:4602–13. doi: 10.4049/jimmunol.1401244
45. Wu C, Kirman JR, Rotte MJ, Davey DF, Perfetto SP, Rhee EG, et al. Distinct lineages of TH1 cells have differential capacities for memory cell generation in vivo. Nat Immunol. (2002) 3:852–8. doi: 10.1038/ni832
46. Moser B and Loetscher P. Lymphocyte traffic control by chemokines. Nat Immunol. (2001) 2:123–8. doi: 10.1038/84219
47. Broderick L, Yokota SJ, Reineke J, Mathiowitz E, Stewart CC, Barcos M, et al. Human CD4+ effector memory T cells persisting in the microenvironment of lung cancer xenografts are activated by local delivery of IL-12 to proliferate, produce IFN-γ, and eradicate tumor cells. J Immunol. (2005) 174:898–906. doi: 10.4049/jimmunol.174.2.898
48. Herrera MT, Torres M, Nevels D, Perez-Redondo CN, Ellner JJ, Sada E, et al. ComparTMentalized bronchoalveolar IFN-γ and IL-12 response in human pulmonary tuberculosis. Tuberculosis. (2009) 89:38–47. doi: 10.1016/j.tube.2008.08.002
49. Bonecini-Almeida MG, Chitale S, Boutsikakis I, Geng J, Doo H, He S, et al. Induction of in vitro human macrophage anti-Mycobacterium tuberculosis activity: requirement for IFN-γ and primed lymphocytes. J Immunol. (1998) 160:4490–9. doi: 10.4049/jimmunol.160.9.4490
50. Hansson GK, Libby P, Schönbeck U, Panse I, Fröhlich A, Bergthaler A, et al. Innate and adaptive immunity in the pathogenesis of atherosclerosis. Circ Res. (2002) 91:281–91. doi: 10.1161/01.RES.0000029784.15893.10
51. Hegazy AN, Peine M, Helmstetter C, Hu-Li J, Urban JF Jr, and Paul WE. Interferons direct Th2 cell reprogramming to generate a stable GATA-3+ T-bet+ cell subset with combined Th2 and Th1 cell functions. Immunity. (2010) 32:116–28. doi: 10.1016/j.immuni.2009.12.004
52. Guo L, Huang Y, Chen X, Yu H, Zhu J, Lukacs N, et al. Innate immunological function of TH2 cells in vivo. Nat Immunol. (2015) 16:1051–9. doi: 10.1038/ni.3244
53. Canaria DA, Yan B, Clare MG, Zhang Z, Taylor GA, Boone DL, et al. STAT5 represses a STAT3-independent th17-like program during th9 cell differentiation. J Immunol. (2021) 207:1265–74. doi: 10.4049/jimmunol.2100165
54. Canaria DA, Clare MG, Yan B, Campbell CB, Ismaio ZA, Anderson NL, et al. IL-1β promotes IL-9-producing Th cell differentiation in IL-2-limiting conditions through the inhibition of BCL6. Front Immunol. (2022) 13:1032618. doi: 10.3389/fimmu.2022.1032618
55. Friesen L, Kostlan R, Liu Q, et al. Cutting edge: the expression of transcription inhibitor GFI1 is induced by retinoic acid to rein in th9 polarization. J Immunol. (2022) 209:1237–42. doi: 10.4049/jimmunol.2200328
56. Chen W. TGF-β Regulation of T cells. Annu Rev Immunol. (2023) 41:483–512. doi: 10.1146/annurev-immunol-101921-045939
57. Roy S, Rizvi ZA, Clarke AJ, Macdonald F, Pandey A, Zaiss DMW, et al. EGFR-HIF1α signaling positively regulates the differentiation of IL-9 producing T helper cells. Nat Commun. (2021) 12:3182. doi: 10.1038/s41467-021-23042-x
58. Lee WH, Hong KJ, Li HB, and Lee GR. Transcription factor id1 plays an essential role in th9 cell differentiation by inhibiting tcf3 and tcf4. Adv Sci (Weinh). (2023) 10:e2305527. doi: 10.1002/advs.202305527
59. Park SA, Lim YJ, Ku WL, Zhang D, Cui K, Tang LY, et al. Opposing functions of circadian protein DBP and atypical E2F family E2F8 in anti-tumor Th9 cell differentiation. Nat Commun. (2022) 13:6069. doi: 10.1038/s41467-022-33733-8
60. Gomez-Bris R, Saez A, Herrero-Fernandez B, Rius C, Sanchez-Martinez H, and Gonzalez-Granado JM. CD4 T-cell subsets and the pathophysiology of inflammatory bowel disease. Int J Mol Sci. (2023) 24:2696. doi: 10.3390/ijms24032696
61. Zheng N and Lu Y. Targeting the IL-9 pathway in cancer immunotherapy. Hum Vaccin Immunother. (2020) 16:2333–40. doi: 10.1080/21645515.2019.1710413
62. Benoit-Lizon I, Jacquin E, Rivera Vargas T, Richard C, Roussey A, Dal Zuffo L, et al. CD4 T cell-intrinsic STING signaling controls the differentiation and effector functions of T1 and T9 cells. J Immunother Cancer. (2022) 10:e003459. doi: 10.1136/jitc-2021-003459HH
63. Pei S, Huang M, Huang J, Zhu X, Wang H, Romano S, et al. BFAR coordinates TGFβ signaling to modulate Th9-mediated cancer immunotherapy. J Exp Med. (2021) 218:e20202144. doi: 10.1084/jem.20202144
64. Vinokurova D and Apetoh L. The emerging role of IL-9 in the anticancer effects of anti-PD-1 therapy. Biomolecules. (2023) 13:670. doi: 10.3390/biom13040670
65. Muñoz-Paleta O and Licona-Limón P. JAKing up IL-9 expression in TH9 cells. Nat Immunol. (2023) 24:891–2. doi: 10.1038/s41590-023-01509-x
66. Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. (2005) 201:233–40. doi: 10.1084/jem.20041257
67. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1α–dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. (2011) 208:1367–76. doi: 10.1084/jem.20110278
68. Miossec P. IL-17 and Th17 cells in human inflammatory diseases. Microbes Infect. (2009) 11:625–30. doi: 10.1016/j.micinf.2009.04.003
69. Lowes MA, Russell CB, Martin DA, Towne JE, and Krueger JG. The IL-23/T17 pathogenic axis in psoriasis is amplified by keratinocyte responses. Trends Immunol. (2013) 34:174–81. doi: 10.1016/j.it.2012.11.005
70. Burkett PR, zu Horste GM, and Kuchroo VK. Pouring fuel on the fire: Th17 cells, the environment, and autoimmunity. J Clin Invest. (2015) 125:2211–9. doi: 10.1172/JCI78085
71. Wang C, Yosef N, Gaublomme J, Wu C, Lee Y, Clish CB, et al. CD5L/AIM regulates lipid biosynthesis and restrains Th17 cell pathogenicity. Cell. (2015) 163:1413–27. doi: 10.1016/j.cell.2015.10.068
72. Zeng C, Shao Z, Wei Z, Yao J, Wang W, Yin L, et al. The NOTCH-HES-1 axis is involved in promoting Th22 cell differentiation. Cell Mol Biol Lett. (2021) 26:7. doi: 10.1186/s11658-021-00249-w
73. Chatzileontiadou DSM, Sloane H, Nguyen AT, Gras S, and Grant EJ. The many faces of CD4 T cells: immunological and structural characteristics. Int J Mol Sci. (2020) 22:73. doi: 10.3390/ijms22010073
74. Kostic M, Zivkovic N, Cvetanovic A, and Marjanović G. CD4+ T cell phenotypes in the pathogenesis of immune thrombocytopenia. Cell Immunol. (2020) 351:104096. doi: 10.1016/j.cellimm.2020.104096
75. Wang ZN, Xu T, and Liu KS. Research progress on Th22 cells and related cytokines in tumors: current status and future perspectives. Am J Cancer Res. (2023) 13:3315–23.
76. Ayass MA, Tripathi T, Zhu K, Nair RR, Melendez K, Zhang J, et al. T helper (Th) cell profiles and cytokines/chemokines in characterization, treatment, and monitoring of autoimmune diseases. Methods. (2023) 220:115–25. doi: 10.1016/j.ymeth.2023.11.003
77. Seth P and Dubey S. IL-22 as a target for therapeutic intervention: Current knowledge on its role in various diseases. Cytokine. (2023) 169:156293. doi: 10.1016/j.cyto.2023.156293
78. Zhang K, Chen L, Zhu C, Zhang M, and Liang C. Current knowledge of th22 cell and IL-22 functions in infectious diseases. Pathogens. (2023) 12:176. doi: 10.3390/pathogens12020176
79. Qi J, Liu C, Bai Z, Li X, and Yao G. T follicular helper cells and T follicular regulatory cells in autoimmune diseases. Front Immunol. (2023) 14:1178792. doi: 10.3389/fimmu.2023.1178792
80. Huang X, Hao S, Liu J, Huang Y, Liu M, Xiao C, et al. The ubiquitin ligase Peli1 inhibits ICOS and thereby Tfh-mediated immunity. Cell Mol Immunol. (2021) 18:969–78. doi: 10.1038/s41423-021-00660-5
81. Hart AP and Laufer TM. A review of signaling and transcriptional control in T follicular helper cell differentiation. J Leukoc Biol. (2022) 111:173–95. doi: 10.1002/JLB.1RI0121-066R
82. Chaurio RA, Anadon CM, Lee Costich T, Payne KK, Biswas S, Harro CM, et al. TGF-β-mediated silencing of genomic organizer SATB1 promotes Tfh cell differentiation and formation of intra-tumoral tertiary lymphoid structures. Immunity. (2022) 55:115–128.e9. doi: 10.1016/j.immuni.2021.12.007
83. Ji LS, Sun XH, Zhang X, Zhou ZH, Yu Z, Zhu XJ, et al. Mechanism of follicular helper T cell differentiation regulated by transcription factors. J Immunol Res. (2020) 2020:1826587. doi: 10.1155/2020/1826587
84. Zhu F, McMonigle RJ, Schroeder AR, Xia X, Figge D, Greer BD, et al. Spatiotemporal resolution of germinal center Tfh cell differentiation and divergence from central memory CD4+ T cell fate. Nat Commun. (2023) 14:3611. doi: 10.1038/s41467-023-39299-3
85. Walker LSK. The link between circulating follicular helper T cells and autoimmunity. Nat Rev Immunol. (2022) 22:567–75. doi: 10.1038/s41577-022-00693-5
86. Jogdand GM, Mohanty S, and Devadas S. Regulators of tfh cell differentiation. Front Immunol. (2016) 7:520. doi: 10.3389/fimmu.2016.00520
87. Zander R, Kasmani MY, Chen Y, Topchyan P, Shen J, Zheng S, et al. Tfh-cell-derived interleukin 21 sustains effector CD8 T cell responses during chronic viral infection. Immunity. (2022) 55:475–493.e5. doi: 10.1016/j.immuni.2022.01.018
88. Gutiérrez-Melo N and Baumjohann D. T follicular helper cells in cancer. Trends Cancer. (2023) 9:309–25. doi: 10.1016/j.trecan.2022.12.007
89. Wei X and Niu X. T follicular helper cells in autoimmune diseases. J Autoimmun. (2023) 134:102976. doi: 10.1016/j.jaut.2022.102976
90. Ding T, Su R, Wu R, Xue H, Wang Y, Su R, et al. Frontiers of autoantibodies in autoimmune disorders: crosstalk between tfh/tfr and regulatory B cells. Front Immunol. (2021) 12:641013. doi: 10.3389/fimmu.2021.641013
91. Xia X, Yang J, and Wang S. Follicular regulatory T cells in systemic lupus erythematosus. J Immunol Res. (2021) 2021:9943743. doi: 10.1155/2021/9943743
92. Tan D, Yin W, Guan F, Zeng W, Lee P, Candotti F, et al. B cell-T cell interplay in immune regulation: A focus on follicular regulatory T and regulatory B cell functions. Front Cell Dev Biol. (2022) 10:991840. doi: 10.3389/fcell.2022.991840
93. Koh B, Ulrich BJ, Nelson AS, Panangipalli G, Kharwadkar R, Wu W, et al. Bcl6 and Blimp1 reciprocally regulate ST2 Treg-cell development in the context of allergic airway inflammation. J Allergy Clin Immunol. (2020) 146:1121–1136.e9. doi: 10.1016/j.jaci.2020.03.002
94. Chung Y, Tanaka S, Chu F, Nurieva RI, Martinez GJ, Rawal S, et al. Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat Med. (2011) 17:983–8. doi: 10.1038/nm.2426
95. Leong YA, Chen Y, Ong HS, Wu D, Man K, Deleage C, et al. CXCR5(+) follicular cytotoxic T cells control viral infection in B cell follicles. Nat Immunol. (2016) 17:1187–96. doi: 10.1038/ni.3543
96. Shen E, Rabe H, Luo L, Wang L, Wang Q, Yin J, et al. Control of Germinal Center Localization and Lineage Stability of Follicular Regulatory T Cells by the Blimp1 Transcription Factor [published correction appears in Cell Rep. 2020 Apr 28;31(4):107575. Cell Rep. (2019) 29:1848–1861.e6. doi: 10.1016/j.celrep.2019.10.012
97. Ribeiro F, Perucha E, and Graca L. T follicular cells: The regulators of germinal center homeostasis. Immunol Lett. (2022) 244:1–11. doi: 10.1016/j.imlet.2022.02.008
98. Hao H, Nakayamada S, and Tanaka Y. Differentiation, functions, and roles of T follicular regulatory cells in autoimmune diseases. Inflammation Regen. (2021) 41:14. doi: 10.1186/s41232-021-00164-9
99. Sun M, Wang X, Zhang N, Wang L, Wang X, Fan W, et al. Imbalance of follicular regulatory T (Tfr) cells/follicular helper T (Tfh) cells in adult patients with primary immune thrombocytopenia. Exp Biol Med (Maywood). (2023) 248:959–65. doi: 10.1177/15353702231168142
100. Mohammed MT, Cai S, Hanson BL, Zhang H, Clement RL, Daccache J, et al. Follicular T cells mediate donor-specific antibody and rejection after solid organ transplantation. Am J Transplant. (2021) 21:1893–901. doi: 10.1111/ajt.16484
101. Lu Y, Jiang R, Freyn AW, Wang J, Strohmeier S, Lederer K, et al. CD4+ follicular regulatory T cells optimize the influenza virus-specific B cell response. J Exp Med. (2021) 218:e20200547. doi: 10.1084/jem.20200547
102. Vecchione A, Jofra T, Gerosa J, Shankwitz K, Di Fonte R, Galvani G, et al. Reduced follicular regulatory T cells in spleen and pancreatic lymph nodes of patients with type 1 diabetes. Diabetes. (2021) 70:2892–902. doi: 10.2337/db21-0091
103. Hori S, Nomura T, and Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. (2003) 299:1057–61. doi: 10.1126/science.1079490
104. Murphy TJ, Choileain NN, Zang Y, Zang Y, Mannick JA, and Lederer JA. CD4+ CD25+ regulatory T cells control innate immune reactivity after injury. J Immunol. (2005) 174:2957–63. doi: 10.4049/jimmunol.174.5.2957
105. Wing JB and Sakaguchi S. Multiple treg suppressive modules and their adaptability. Front Immunol. (2012) 3:178. doi: 10.3389/fimmu.2012.00178
106. Feng Y, Arvey A, Chinen T, van der Veeken J, Gasteiger G, and Rudensky AY. Control of the inheritance of regulatory T cell identity by a cis element in the FoxP3locus. Cell. (2014) 158:749–63. doi: 10.1016/j.cell.2014.07.031
107. Klatzmann D and Abbas AK. The promise of low-dose interleukin-2 therapy for autoimmune and inflammatory diseases. Nat Rev Immunol. (2015) 15:283–94. doi: 10.1038/nri3823
108. Sakaguchi S, Ono M, Setoguchi R, Yagi H, Hori S, Fehervari Z, et al. Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol Rev. (2006) 212:8–27. doi: 10.1111/j.0105-2896.2006.00427.x
109. Sakaguchi S, Miyara M, Costantino CM, and Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol. (2010) 10:490–500. doi: 10.1038/nri2785
110. Abbas AK, Benoist C, Bluestone JA, Campbell DJ, Ghosh S, Hori S, et al. Regulatory T cells: recommendations to simplify the nomenclature. Nat Immunol. (2013) 14:307–8. doi: 10.1038/ni.2554
111. Zheng Y, Chaudhry A, Kas A, deRoos P, Kim JM, Chu TT, et al. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control TH2 responses. Nature. (2009) 458:351–6. doi: 10.1038/nature07674
112. Koch MA, Perdue NR, Killebrew JR, Killebrew JR, Urdahl KB, and Campbell DJ. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol. (2009) 10:595–602. doi: 10.1038/ni.1731
113. Joller N, Lozano E, Burkett PR, Patel B, Xiao S, Zhu C, et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity. (2014) 40:569–81. doi: 10.1016/j.immuni.2014.02.012
114. Campbell DJ and Koch MA. Phenotypical and functional specialization of FOXP3+ regulatory T cells. Nat Rev Immunol. (2011) 11:119–30. doi: 10.1038/nri2916
115. Fuhrman CA, Yeh WI, Seay HR, Saikumar Lakshmi P, Chopra G, Zhang L, et al. Divergent phenotypes of human regulatory T cells expressing the receptors TIGIT and CD226. J Immunol. (2015) 195:145–55. doi: 10.4049/jimmunol.1402381
116. Ait-Oufella H, Salomon BL, Potteaux S, Robertson AK, Gourdy P, Zoll J, et al. Natural regulatory T cells control the development of atherosclerosis in mice. Nat Med. (2006) 12:178–80. doi: 10.1038/nm1343
117. Cruz LO, Hashemifar SS, Wu CJ, Cho S, Nguyen DT, Lin LL, et al. Excessive expression of miR-27 impairs Treg-mediated immunological tolerance. J Clin Invest. (2017) 127. doi: 10.1172/JCI88415
118. Caramalho I, Lopes-Carvalho T, Ostler D, Zelenay S, Haury M, and Demengeot J. Regulatory T cells selectively express toll-like receptors and are activated by lipopolysaccharide. J Exp Med. (2003) 197:403–11. doi: 10.1084/jem.20021633
119. Pasare C and Medzhitov R. Toll pathway-dependent blockade of CD4+ CD25+ T cell-mediated suppression by dendritic cells. Science. (2003) 299:1033–6. doi: 10.1126/science.1078231
120. Schwartz RH. Natural regulatory T cells and self-tolerance. Nat Immunol. (2005) 6:327–30. doi: 10.1038/ni1184
121. Körholz D, Banning U, Bönig H, Grewe M, Schneider M, Mauz-Körholz C, et al. The role of interleukin-10 (IL-10) in IL-15–mediated T-cell responses. Blood. (1997) 90:4513–21. doi: 10.1016/s1359-6101(02)00025-4
122. Letterio JJ and Roberts AB. Regulation of immune responses by TGF-β. Annu Rev Immunol. (1998) 16:137–61. doi: 10.1146/annurev.immunol.16.1.137
123. Gagliani N, Vesely MCA, Iseppon A, Brockmann L, Xu H, Palm NW, et al. Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature. (2015) 523:221–5. doi: 10.1038/nature14452
124. Yang R, Qu C, Zhou Y, Konkel JE, Shi S, Liu Y, et al. Hydrogen sulfide promotes Tet1-and Tet2-mediated FoxP3demethylation to drive regulatory T cell differentiation and maintain immune homeostasis. Immunity. (2015) 43:251–63. doi: 10.1016/j.immuni.2015.07.017
125. Hosokawa K, Muranski P, Feng X, Townsley DM, Liu B, Knickelbein J, et al. Memory stem T cells in autoimmune disease: high frequency of circulating CD8+ Memory stem cells in acquired aplastic anemia. J Immunol. (2016) 196:1568–78. doi: 10.4049/jimmunol.1501739
126. Schoettler ML and Nathan DG. The pathophysiology of acquired aplastic anemia: current concepts revisited. Hematol Oncol Clin North Am. (2018) 32:581–94. doi: 10.1016/j.hoc.2018.03.001
127. Zeng Y and Katsanis E. The complex pathophysiology of acquired aplastic anaemia. Clin Exp Immunol. (2015) 180:361–70. doi: 10.1111/cei.12605
128. Baudi I, Kawashima K, and Isogawa M. HBV-specific CD8+ T-cell tolerance in the liver. Front Immunol. (2021) 12:721975. doi: 10.3389/fimmu.2021.721975
129. Zhang Z, Duan Z, and Cui Y. CD8+ T cells in brain injury and neurodegeneration. Front Cell Neurosci. (2023) 17:1281763. doi: 10.3389/fncel.2023.1281763
130. De Rosa SC, Herzenberg LA, Herzenberg LA, and Roederer M. 11-color, 13-parameter flow cytometry: identification of human naïve T cells by phenotype, function, and T-cell receptor diversity. Nat Med. (2001) 7:245–8. doi: 10.1038/84701
131. Seder RA and Ahmed R. Similarities and differences in CD4+ and CD8+ effector and memory T cell generation. Nat Immunol. (2003) 4:835–42. doi: 10.1038/ni969
132. Perfetto SP, Chattopadhyay PK, and Roederer M. Seventeen-colour flow cytometry: unravelling the immune system. Nat Rev Immunol. (2004) 4:648–55. doi: 10.1038/nri1416
133. McBreen S, Imlach S, Shirafuji T, Scott GR, Leen C, Bell JE, et al. Infection of the CD45RA+ (naïve) subset of peripheral CD8+ lymphocytes by human immunodeficiency virus type 1 in vivo. J Virol. (2001) 75:4091–102. doi: 10.1128/JVI.75.9.4091-4102.2001
134. Restifo NP, Dudley ME, and Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol. (2012) 12:269. doi: 10.1038/nri3191
135. Zhou H, Yang J, Tian J, and Wang S. CD8+ T lymphocytes: crucial players in sjögren's syndrome. Front Immunol. (2021) 11:602823. doi: 10.3389/fimmu.2020.602823
136. Casalegno Garduño R and Däbritz J. New insights on CD8+ T cells in inflammatory bowel disease and therapeutic approaches. Front Immunol. (2021) 12:738762. doi: 10.3389/fimmu.2021.738762
137. Pepper M and Jenkins MK. Origins of CD4+ effector and central memory T cells. Nat Immunol. (2011) 12:467. doi: 10.1038/ni.2038
138. Busch DH, Fräßle SP, Sommermeyer D, Buchholz VR, and Riddell SR. Role of memory T cell subsets for adoptive immunotherapy[C]//Seminars in immunology. Acad Press. (2016) 28:28–34. doi: 10.1016/j.smim.2016.02.001
139. Awasthi S, Lubinski JM, Eisenberg RJ, Cohen GH, and Friedman HM. An HSV-1 gD mutant virus as an entry-impaired live virus vaccine. Vaccine. (2008) 26:1195–203. doi: 10.1016/j.vaccine.2007.12.032
140. Chang JT, Wherry EJ, and Goldrath AW. Molecular regulation of effector and memory T cell differentiation. Nat Immunol. (2014) 15:1104–15. doi: 10.1038/ni.3031
141. Caccamo N, Joosten SA, Ottenhoff THM, and Dieli F. Atypical human effector/memory CD4+ T cells with a naive-like phenotype. Front Immunol. (2018) 9:2832. doi: 10.3389/fimmu.2018.02832
142. Fleischer J, Gragegriebenow E, Kasper B, Heine H, Ernst M, Brandt E, et al. Platelet factor 4 inhibits proliferation and cytokine release of activated human T cells. J Immunol. (2002) 169:770–7. doi: 10.4049/jimmunol.169.2.770
143. Hundelshausen PV and Weber C. Platelets as immune cells: bridging inflammation and cardiovascular disease. Circ Res. (2007) 100:27–40. doi: 10.1161/01.RES.0000252802.25497.b7
144. Sallusto F, Geginat J, and Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol. (2004) 22:745–63. doi: 10.1146/annurev.immunol.22.012703.104702
145. Pagès F, Berger A, Camus M, Sanchez-Cabo F, Costes A, Molidor R, et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. New Engl J Med. (2005) 353:2654–66. doi: 10.1056/NEJMoa051424
146. Van Panhuys N, Perret R, Prout M, Ronchese F, and Le Gros G. Effector lymphoid tissue and its crucial role in protective immunity. Trends Immunol. (2005) 26:242–7. doi: 10.1016/j.it.2005.03.005
147. Fritsch RD, Shen X, Sims GP, Hathcock KS, Hodes RJ, and Lipsky PE. Stepwise differentiation of CD4 memory T cells defined by expression of CCR7 and CD27. J Immunol. (2005) 175:6489–97. doi: 10.4049/jimmunol.175.10.6489
148. Soares AP, Scriba TJ, Joseph S, Harbacheuski R, Murray RA, Gelderbloem SJ, et al. Bacillus Calmette-Guerin vaccination of human newborns induces T cells with complex cytokine and phenotypic profiles. J Immunol. (2008) 180:3569–77. doi: 10.4049/jimmunol.180.5.3569
149. Martin MD, Kim MT, Shan Q, Sompallae R, Xue HH, Harty JT, et al. Phenotypic and functional alterations in circulating memory CD8 T cells with time after primary infection. PloS Pathog. (2015) 11:e1005219. doi: 10.1371/journal.ppat.1005219
150. Kaech SM, Wherry EJ, and Ahmed R. Effector and memory T-cell differentiation: implications for vaccine development. Nat Rev Immunol. (2002) 2:251–62. doi: 10.1038/nri778
151. Turner SJ, Bennett TJ, and La Gruta NL. CD8+ T-cell memory: the why, the when, and the how. Cold Spring Harb Perspect Biol. (2021) 13:a038661. doi: 10.1101/cshperspect.a038661
152. Wang YY, Zhen C, Hu W, Huang HH, Li YJ, Zhou MJ, et al. Elevated glutamate impedes anti-HIV-1 CD8 + T cell responses in HIV-1-infected individuals on antiretroviral therapy. Commun Biol. (2023) 6:696. doi: 10.1038/s42003-023-04975-z
153. Seok J, Cho SD, Lee J, Choi Y, Kim SY, Lee SM, et al. A virtual memory CD8+ T cell-originated subset causes alopecia areata through innate-like cytotoxicity. Nat Immunol. (2023) 24:1308–17. doi: 10.1038/s41590-023-01547-5
154. Savid-Frontera C, Viano ME, Baez NS, Lidon NL, Fontaine Q, Young HA, et al. Exploring the immunomodulatory role of virtual memory CD8+ T cells: Role of IFN gamma in tumor growth control. Front Immunol. (2022) 13:971001. doi: 10.3389/fimmu.2022.971001
155. Schultze-Florey CR, Chukhno E, Goudeva L, Blasczyk R, Ganser A, Prinz I, et al. Distribution of major lymphocyte subsets and memory T-cell subpopulations in healthy adults employing GLP-conforming multicolor flow cytometry. Leukemia. (2021) 35:3021–5. doi: 10.1038/s41375-021-01348-5
156. Fazeli P, Kalani M, and Hosseini M. T memory stem cell characteristics in autoimmune diseases and their promising therapeutic values. Front Immunol. (2023) 14:1204231. doi: 10.3389/fimmu.2023.1204231
157. Di Benedetto S, Derhovanessian E, Steinhagen-Thiessen E, Goldeck D, Müller L, and Pawelec G. Impact of age, sex and CMV-infection on peripheral T cell phenotypes: results from the Berlin BASE-II Study. Biogerontology. (2015) 16:631–43. doi: 10.1007/s10522-015-9563-2
158. Fuertes Marraco SA, Soneson C, Cagnon L, Cagnon L, Gannon PO, Allard M, et al. Long-lasting stem cell-like memory CD8+ T cells with a naïve-like profile upon yellow fever vaccination. Sci Transl Med. (2015) 7:282ra48. doi: 10.1126/scitranslmed.aaa3700
159. Verma V, Jafarzadeh N, Boi S, Kundu S, Jiang Z, Fan Y, et al. MEK inhibition reprograms CD8+ T lymphocytes into memory stem cells with potent antitumor effects. Nat Immunol. (2021) 22:53–66. doi: 10.1038/s41590-020-00818-9
160. Hotta K, Sho M, Fujimoto K, Shimada K, Yamato I, Anai S, et al. Prognostic significance of CD45RO+ memory T cells in renal cell carcinoma. Br J Cancer. (2011) 105:1191–6. doi: 10.1038/bjc.2011.368
161. Sallusto F and Lanzavecchia A. Understanding dendritic cell and T-lymphocyte traffic through the analysis of chemokine receptor expression. Immunol Rev. (2000) 177:134–40. doi: 10.1034/j.1600-065X.2000.17717.x
162. Friedrich M, Krammig S, Henze M, Döcke WD, Sterry W, and Asadullah K. Flow cytometric characterization of lesional T cells in psoriasis: intracellular cytokine and surface antigen expression indicates an activated, memory/effector type 1 immunophenotype. Arch Dermatol Res. (2000) 292:519–21. doi: 10.1007/s004030000167
163. Beeton C, Wulff H, Barbaria J, Clot-Faybesse O, Pennington M, Bernard D, et al. Selective blockade of T lymphocyte K(+) channels ameliorates experimental autoimmune encephalomyelitis, a model for multiple sclerosis. Proc Natl Acad Sci. (2001) 98:13942–7. doi: 10.1073/pnas.241497298
164. Gattorno M, Prigione I, Morandi F, Gregorio A, Chiesa S, Ferlito F, et al. Phenotypic and functional characterisation of CCR7+ and CCR7-CD4+ memory T cells homing to the joints in juvenile idiopathic arthritis. Arthritis Res Ther. (2005) 7:1. doi: 10.1186/ar1485
165. Sallusto F, Lenig D, Förster R, Lipp M, and Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. (1999) 401:708–12. doi: 10.1038/44385
166. Farber DL, Yudanin NA, and Restifo NP. Human memory T cells: generation, compartmentalization and homeostasis. Nat Rev Immunol. (2014) 14:24–35. doi: 10.1038/nri3567
167. Ammirati E, Cianflone D, Vecchio V, Banfi M, Vermi AC, De Metrio M, et al. Effector memory T cells are associated with atherosclerosis in humans and animal models. J Am Heart Assoc. (2012) 1:27–41. doi: 10.1161/xJAHA.111.000125
168. Olson NC, Doyle MF, Jenny NS, Huber SA, Psaty BM, Kronmal RA, et al. Decreased naive and increased memory CD4(+) T cells are associated with subclinical atherosclerosis: the multi-ethnic study of atherosclerosis. PloS One. (2013) 8:e71498. doi: 10.1371/journal.pone.0071498
169. Karthikeyan S, Geschwind JF, and Ganapathy-Kanniappan S. Tumor cells and memory T cells converge at glycolysis: Therapeutic implications. Cancer Biol Ther. (2014) 15:483–5. doi: 10.4161/cbt.28160
170. Tabler CO, Lucera MB, Haqqani AA, McDonald DJ, Migueles SA, Connors M, et al. CD4+ memory stem cells are infected by HIV-1 in a manner regulated in part by SAMHD1 expression. J Virol. (2014) 88:4976–86. doi: 10.1128/JVI.00324-14
171. Jiang W, Younes SA, Funderburg NT, Mudd JC, Espinosa E, Davenport MP, et al. Cycling memory CD4+ T cells in HIV disease have a diverse T cell receptor repertoire and a phenotype consistent with bystander activation. J Virol. (2014) 88:5369–80. doi: 10.1128/JVI.00017-14
172. Williams MA and Bevan MJ. Effector and memory CTL differentiation. Annu Rev Immunol. (2007) 25:171–92. doi: 10.1146/annurev.immunol.25.022106.141548
173. Kalia V, Penny LA, Yuzefpolskiy Y, Baumann FM, and Sarkar S. Quiescence of memory CD8+ T cells is mediated by regulatory T cells through inhibitory receptor CTLA-4. Immunity. (2015) 42:1116–29. doi: 10.1016/j.immuni.2015.05.023
174. Bour-Jordan H and Bluestone JA. Regulating the regulators: costimulatory signals control the homeostasis and function of regulatory T cells. Immunol Rev. (2009) 229:41–66. doi: 10.1111/j.1600-065X.2009.00775.x
175. Sprent J and Tough DF. T cell death and memory. Science. (2001) 293:245–8. doi: 10.1126/science.1062416
176. Hu H, Huston G, Duso D, Lepak N, Roman E, and Swain SL. CD4+ T cell effectors can become memory cells with high efficiency and without further division. Nat Immunol. (2001) 2:705–10. doi: 10.1038/90643
177. Masopust D, Vezys V, Marzo AL, and Lefrançois L. Preferential localization of effector memory cells in nonlymphoid tissue. Science. (2001) 291:2413–7. doi: 10.1126/science.1058867
178. Willinger T, Freeman T, Hasegawa H, McMichael AJ, and Callan MF. Molecular signatures distinguish human central memory from effector memory CD8 T cell subsets. J Immunol. (2005) 175:5895–903. doi: 10.4049/jimmunol.175.9.5895
179. Kundig TM, Bachmann MF, Ohashi PS, Pircher H, Hengartner H, Zinkernagel RM, et al. On T cell memory: arguments for antigen dependence. Immunol Rev. (1996) 150:63–90. doi: 10.1111/j.1600-065X.1996.tb00696.x
180. Kündig TM, Bachmann MF, Oehen S, Hoffmann UW, Simard JJ, Kalberer CP, et al. On the role of antigen in maintaining cytotoxic T-cell memory. Proc Natl Acad Sci. (1996) 93:9716–23. doi: 10.1073/pnas.93.18.9716
181. Sprent J and Tough DF. Lymphocyte life-span and memory. Science-New York Then Washington. (1994) 265:1395–5. doi: 10.1126/science.8073282
182. Frostegard J, Ulfgren AK, Nyberg P, Hedin U, Swedenborg J, Andersson U, et al. Cytokine expression in advanced human atherosclerotic plaques: dominance of pro-inflammatory (Th1) and macrophage-stimulating cytokines. Atheroscl. (1999) 145:33–43. doi: 10.1016/S0021-9150(99)00011-8
183. Renno T, Hahne M, and MacDonald HR. Proliferation is a prerequisite for bacterial superantigen-induced T cell apoptosis in vivo. J Exp Med. (1995) 181:2283–7. doi: 10.1084/jem.181.6.2283
184. Swain SL, Hu H, and Huston G. Class II-independent generation of CD4 memory T cells from effectors. Science. (1999) 286:1381–3. doi: 10.1126/science.286.5443.1381
185. Surh CD and Sprent J. Homeostasis of naïve and memory T cells. Immunity. (2008) 29:848–62. doi: 10.1016/j.immuni.2008.11.002
186. Ahmed R and Gray D. Immunological memory and protective immunity: understanding their relation. Science. (1996) 272:54. doi: 10.1126/science.272.5258.54
187. McLean AR. Modelling T cell memory. J Theor Biol. (1994) 170:63–74. doi: 10.1006/jtbi.1994.1168
188. Morrell CN, Aggrey AA, Chapman LM, and Modjeski KL. Emerging roles for platelets as immune and inflammatory cells. Blood. (2014) 123:2759–67. doi: 10.1182/blood-2013-11-462432
189. Erbel C, Tyka M, Helmes CM, Akhavanpoor M, Rupp G, Domschke G, et al. CXCL4-induced plaque macrophages can be specifically identified by co-expression of MMP7+S100A8+ in vitro and in vivo. Innate Immun. (2015) 21:255–65. doi: 10.1177/1753425914526461
190. Cochain C, Vafadarnejad E, Arampatzi P, Pelisek J, Winkels H, Ley K, et al. Single-cell RNA-seq reveals the transcriptional landscape and heterogeneity of aortic macrophages in murine atherosclerosis. Circ Res. (2018) 122:1661–74. doi: 10.1161/CIRCRESAHA.117.312509
191. Yeo L, Adlard N, Biehl M, Juarez M, Smallie T, Snow M, et al. Expression of chemokines CXCL4 and CXCL7 by synovial macrophages defines an early stage of rheumatoid arthritis. Ann Rheumatol Dis. (2016) 75:763–71. doi: 10.1136/annrheumdis-2014-206921
192. Affandi AJ, Carvalheiro T, Ottria A, de Haan JJ, Brans MAD, Brandt MM, et al. CXCL4 drives fibrosis by promoting several key cellular and molecular processes. Cell Rep. (2022) 38:110189. doi: 10.1016/j.celrep.2021.110189
193. Martini E, Kunderfranco P, Peano C, Carullo P, Cremonesi M, Schorn T, et al. Single-cell sequencing of mouse heart immune infiltrate in pressure overloaddriven heart failure reveals extent of immune activation. Circulation. (2019) 140:2089–107. doi: 10.1161/CIRCULATIONAHA.119.041694
194. Hansson GK. The heart of immunology: immune mechanisms in cardiovascular medicine. Cardiovasc Res. (2021) 117:e166–8. doi: 10.1093/cvr/cvab314
195. Meikle CK, Meisler AJ, Bird CM, Jeffries JA, Azeem N, Garg P, et al. Platelet-T cell aggregates in lung cancer patients: Implications for thrombosis. PloS One. (2020) 15:e0236966. doi: 10.1371/journal.pone.0236966
196. Saris A, Steuten J, Schrijver DP, van Schijndel G, Zwaginga JJ, van Ham SM, et al. Inhibition of dendritic cell activation and modulation of T cell polarization by the platelet secretome. Front Immunol. (2021) 12:631285. doi: 10.3389/fimmu.2021.631285
197. Jiang LP, Zhu T, Tang K, Wu Y, Fu M, Ji JZ, et al. Enhanced metabolic activation of and platelet response to clopidogrel in T cell-deficient mice through induction of Cyp2c and Cyp3a and inhibition of Ces1. J Thromb Haemost. (2023) 21:1322–35. doi: 10.1016/j.jtha.2023.01.028
198. Schmied L, Höglund P, and Meinke S. Platelet-mediated protection of cancer cells from immune surveillance - possible implications for cancer immunotherapy. Front Immunol. (2021) 12:640578. doi: 10.3389/fimmu.2021.640578
199. Maouia A, Rebetz J, Kapur R, and Semple JW. The immune nature of platelets revisited. Transfus Med Rev. (2020) 34:209–20. doi: 10.1016/j.tmrv.2020.09.005
200. Cognasse F, Duchez AC, Audoux E, Ebermeyer T, Arthaud CA, Prier A, et al. Platelets as key factors in inflammation: focus on CD40L/CD40. Front Immunol. (2022) 13:825892. doi: 10.3389/fimmu.2022.825892
201. Scopelliti F, Cattani C, Dimartino V, Mirisola C, and Cavani A. Platelet derivatives and the immunomodulation of wound healing. Int J Mol Sci. (2022) 23:8370. doi: 10.3390/ijms23158370
202. Zhang S, Liu Y, Wang X, Yang L, Li H, Wang Y, et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J Hematol Oncol. (2020) 13:120. doi: 10.1186/s13045-020-00954-7
203. Manne BK, Denorme F, Middleton EA, Portier I, Rowley JW, Stubben C, et al. Platelet gene expression and function in patients with COVID-19. Blood. (2020) 136:1317–29. doi: 10.1182/blood.2020007214
204. Zhu L, Huang Z, Stålesen R, and Hansson GK and Li N. Platelets provoke distinct dynamics of immune responses by differentially regulating CD4+ T cell proliferation. J Thromb Haemost. (2014) 12:1156–65. doi: 10.1111/jth.12612
205. Geltink RIK and Kyle RL and Pearce EL. Unraveling the complex interplay between T cell metabolism and function. Annu Rev Immunol. (2018) 36:461–88. doi: 10.1146/annurev-immunol-042617-053019
206. Alaynick WA. Nuclear receptors, mitochondria and lipid metabolism. Mitochondrion. (2008) 8:329–37. doi: 10.1016/j.mito.2008.02.001
207. Lasagni L, Francalanci M, Annunziato F, Lazzeri E, Giannini S, Cosmi L, et al. An alternatively spliced variant of CXCR3 mediates the inhibition of endothelial cell growth induced by IP-10, Mig, and I-TAC, and acts as functional receptor for platelet factor 4. J Exp Med. (2003) 197:1537–49. doi: 10.1084/jem.20021897
208. Guo K, Yombo DJK, Wang Z, Navaeiseddighi Z, Xu J, Schmit T, et al. The chemokine receptor CXCR3 promotes CD8+ T cell-dependent lung pathology during influenza pathogenesis. Sci Adv. (2024) 10:eadj1120. doi: 10.1126/sciadv.adj1120
209. Van der Putten C, Remmerswaal EBM, Terpstra ML, van der Bom ND, Kers J, Ten Berge IJM, et al. CD8 and CD4 T cell populations in human kidneys. Cells. (2021) 10:288. doi: 10.3390/cells10020288
210. Pontes Ferreira C, Moro Cariste L, Henrique Noronha I, Fernandes Durso D, Lannes-Vieira J, Ramalho Bortoluci K, et al. CXCR3 chemokine receptor contributes to specific CD8+ T cell activation by pDC during infection with intracellular pathogens [published correction appears in PLoS Negl Trop Dis. 2021 Apr 7;15(4):e0009326. PloS Negl Trop Dis. (2020) 14:e0008414. doi: 10.1371/journal.pntd.0008414
211. Ravera S, Signorello MG, and Panfoli I. Platelet metabolic flexibility: A matter of substrate and location. Cells. (2023) 12:1802. doi: 10.3390/cells12131802
212. Yu H, Hu W, Song X, and Zhao Y. Immune modulation of platelet-derived mitochondria on memory CD4+ T cells in humans. Int J Mol Sci. (2020) 21:6295. doi: 10.3390/ijms21176295
213. Yap ML, McFadyen JD, Wang X, Ziegler M, Chen YC, Willcox A, et al. Activated platelets in the tumor microenvironment for targeting of antibody-drug conjugates to tumors and metastases. Theranostics. (2019) 9:1154–69. doi: 10.7150/thno.29146
214. Hu Q, Sun W, Wang J, Ruan H, Zhang X, Ye Y, et al. Conjugation of haematopoietic stem cells and platelets decorated with anti-PD-1 antibodies augments anti-leukaemia efficacy. Nat BioMed Eng. (2018) 2:831–40. doi: 10.1038/s41551-018-0310-2
215. Tang S, Zhang F, Gong H, Wei F, Zhuang J, Karshalev E, et al. Enzyme-powered Janus platelet cell robots for active and targeted drug delivery. Sci Robot. (2020) 5:eaba6137. doi: 10.1126/scirobotics.aba6137
216. Ortiz-Otero N, Marshall JR, Lash BW, and King MR. Platelet mediated TRAIL delivery for efficiently targeting circulating tumor cells. Nanoscale Adv. (2020) 2:3942–53. doi: 10.1039/d0na00271b
217. Morris K, Schnoor B, and Papa AL. Platelet cancer cell interplay as a new therapeutic target. Biochim Biophys Acta Rev Cancer. (2022) 1877:188770. doi: 10.1016/j.bbcan.2022.188770
218. Giudice V and Selleri C. Aplastic anemia: pathophysiology. Semin Hematol. (2022) 59:13–20. doi: 10.1053/j.seminhematol.2021.12.002
219. Zhu C, Lian Y, Wang C, Wu P, Li X, Gao Y, et al. Single-cell transcriptomics dissects hematopoietic cell destruction and T-cell engagement in aplastic anemia. Blood. (2021) 138:23–33. doi: 10.1182/blood.2020008966
220. Javan MR, Saki N, and Moghimian-Boroujeni B. Aplastic anemia, cellular and molecular aspects. Cell Biol Int. (2021) 45:2395–402. doi: 10.1002/cbin.11689
221. Alvarado LJ, Huntsman HD, Cheng H, Townsley DM, Winkler T, Feng X, et al. Eltrombopag maintains human hematopoietic stem and progenitor cells under inflammatory conditions mediated by IFN-γ. Blood. (2019) 133:2043–55. doi: 10.1182/blood-2018-11-884486
222. Ivashkiv LB. IFNγ: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat Rev Immunol. (2018) 18:545–58. doi: 10.1038/s41577-018-0029-z
223. Ghanima W, Geyer JT, Lee CS, Boiocchi L, Imahiyerobo AA, Orazi A, et al. Bone marrow fibrosis in 66 patients with immune thrombocytopenia treated with thrombopoietin-receptor agonists: a single-center, long-term follow-up. Haematologica. (2014) 99:937–44. doi: 10.3324/haematol.2013.098921
224. Lin S, Hou L, Liu S, Wang J, Chen Q, Zhang B, et al. Roles of regulatory T cells in the pathogenesis of pediatric aplastic anemia. Pediatr Hematol Oncol. (2019) 36:198–210. doi: 10.1080/08880018.2019.1621968
225. Trivigno SMG, Guidetti GF, Barbieri SS, and Zarà M. Blood platelets in infection: the multiple roles of the platelet signalling machinery. Int J Mol Sci. (2023) 24:7462. doi: 10.3390/ijms24087462
226. Feng X, Scheinberg P, Samsel L, et al. Decreased plasma cytokines are associated with low platelet counts in aplastic anemia and immune thrombocytopenic purpura. J Thromb Haemost. (2012) 10:1616–23. doi: 10.1111/j.1538-7836.2012.04757.x
227. Mulet M, Zamora C, Porcel JM, Nieto JC, Pajares V, Muñoz-Fernandez AM, et al. Platelet factor 4 regulates T cell effector functions in Malignant pleural effusions. Cancer Lett. (2020) 491:78–86. doi: 10.1016/j.canlet.2020.06.014
228. Li C, Li J, and Ni H. Crosstalk between platelets and microbial pathogens. Front Immunol. (2020) 11:1962. doi: 10.3389/fimmu.2020.01962
229. Zhang L, Liu J, Qin X, and Liu W. Platelet-acute leukemia interactions. Clin Chim Acta. (2022) 536:29–38. doi: 10.1016/j.cca.2022.09.015
230. Schuhmann MK, Stoll G, Bieber M, Vögtle T, Hofmann S, Klaus V, et al. CD84 links T cell and platelet activity in cerebral thrombo-inflammation in acute stroke. Circ Res. (2020) 127:1023–35. doi: 10.1161/CIRCRESAHA.120.316655
231. Carnaz Simões AM, Holmström MO, Aehnlich P, Rahbech A, Radziwon-Balicka A, Zamora C, et al. Patients With Myeloproliferative Neoplasms Harbor High Frequencies of CD8 T Cell-Platelet Aggregates Associated With T Cell Suppression [published correction appears in Front Immunol. 2022 Aug 05;13:970322. Front Immunol. (2022) 13:866610. doi: 10.3389/fimmu.2022.866610
232. Zamora C, Cantó E, Nieto JC, Ortiz MA, Diaz-Torné C, Diaz-Lopez C, et al. Functional consequences of platelet binding to T lymphocytes in inflammation. J Leukoc Biol. (2013) 94:521–9. doi: 10.1189/jlb.0213074
233. He H, Wang Y, Zhang X, Li X, Liu C, Yan D, et al. Age-related noncanonical TRMT6-TRMT61A signaling impairs hematopoietic stem cells. Nat Aging. (2024) 4:213–30. doi: 10.1038/s43587-023-00556-1
234. McCabe A, Smith JNP, Costello A, Maloney J, Katikaneni D, and MacNamara KC. Hematopoietic stem cell loss and hematopoietic failure in severe aplastic anemia is driven by macrophages and aberrant podoplanin expression. Haematologica. (2018) 103:1451–61. doi: 10.3324/haematol.2018.189449
235. Li L, Ni R, Li Z, Ming Y, Liu L, Peng D, et al. Insights into regulatory factors in megakaryocyte development and function: basic mechanisms and potential targets. Front Biosci (Landmark Ed). (2022) 27:313. doi: 10.31083/j.fbl2711313
236. Cheng G, Saleh MN, Marcher C, Vasey S, Mayer B, Aivado M, et al. Eltrombopag for management of chronic immune thrombocytopenia (RAISE): a 6-month, randomised, phase 3 study. Lancet. (2011) 377:393–402. doi: 10.1016/S0140-6736(10)60959-2
237. Seyfried AN, McCabe A, Smith JNP, Calvi LM, and MacNamara KC. CCR5 maintains macrophages in the bone marrow and drives hematopoietic failure in a mouse model of severe aplastic anemia. Leukemia. (2021) 35:3139–51. doi: 10.1038/s41375-021-01219-z
238. Giudice V, Feng X, Lin Z, Hu W, Zhang F, Qiao W, et al. Deep sequencing and flow cytometric characterization of expanded effector memory CD8+CD57+ T cells frequently reveals T-cell receptor Vβ oligoclonality and CDR3 homology in acquired aplastic anemia. Haematologica. (2018) 103:759–69. doi: 10.3324/haematol.2017.176701
239. Zhao S, Zhang Y, Huang G, Luo W, Li Y, Xiao Y, et al. Increased CD8+CD27+perforin+ T cells and decreased CD8+CD70+ T cells may be immune biomarkers for aplastic anemia severity. Blood Cells Mol Dis. (2019) 77:34–42. doi: 10.1016/j.bcmd.2019.03.009
240. You X, Yang Q, Yan K, Wang SR, Huang RR, Wang SQ, et al. Multi-omics profiling identifies pathways associated with CD8+ T-cell activation in severe aplastic anemia. Front Genet. (2022) 12:790990. doi: 10.3389/fgene.2021.790990
241. Huang G, Zhang Y, Wei X, Yu Z, Lai J, Shen Q, et al. CD8+GITR+ T cells may negatively regulate T cell overactivation in aplastic anemia. Immunol Invest. (2021) 50:406–15. doi: 10.1080/08820139.2020.1770785
242. Omokaro SO, Desierto MJ, Eckhaus MA, Ellison FM, Chen J, and Young NS. Lymphocytes with aberrant expression of Fas ligand attenuate immune bone marrow failure in a mouse model. J Immunol. (2009) 182:3414–22. doi: 10.4049/jimmunol.0801430
243. Chen J, Feng X, Desierto MJ, Keyvanfar K, and Young NS. IFN-γ-mediated hematopoietic cell destruction in murine models of immune-mediated bone marrow failure. Blood. (2015) 126:2621–31. doi: 10.1182/blood-2015-06-652453
244. Rosselli F, Sanceau J, Gluckman E, Wietzerbin J, and Moustacchi E. Abnormal lymphokine production: a novel feature of the genetic disease Fanconi anemia. II. In vitro and in vivo spontaneous overproduction of tumor necrosis factor alpha. Blood. (1994) 83:1216–25. doi: 10.1182/blood.V83.5.1216.1216
245. Hara T, Ando K, Tsurumi H, and Moriwaki H. Excessive production of tumor necrosis factor-alpha by bone marrow T lymphocytes is essential in causing bone marrow failure in patients with aplastic anemia. Eur J Haematol. (2004) 73:10–16.26. doi: 10.1111/j.1600-0609.2004.00259.x
246. Dufour C, Ferretti E, Bagnasco F, Burlando O, Lanciotti M, Ramenghi U, et al. Marrow Failure Study Group of the AIEOP. Changes in cytokine profile pre- and post- immunosuppression in acquired aplastic anemia. Haematologica. (2009) 94:1743–7. doi: 10.3324/haematol.2009.007815
247. Kamada T, Togashi Y, Tay C, Ha D, Sasaki A, Nakamura Y, et al. PD-1+ regulatory T cells amplified by PD-1 blockade promote hyperprogression of cancer. Proc Natl Acad Sci U S A. (2019) 116:9999–10008. doi: 10.1073/pnas.1822001116
248. Zhang B, Chikuma S, Hori S, Fagarasan S, and Honjo T. Nonoverlapping roles of PD-1 and FoxP3 in maintaining immune tolerance in a novel autoimmune pancreatitis mouse model. Proc Natl Acad Sci U S A. (2016) 113:8490–5. doi: 10.1073/pnas.1608873113
249. Wherry EJ and Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. (2015) 15:486–99. doi: 10.1038/nri3862
250. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. (2012) 366:2455–65. doi: 10.1056/NEJMoa1200694
251. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. (2012) 366:2443–54. doi: 10.1056/NEJMoa1200690
252. Zou W, Wolchok JD, and Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci Transl Med. (2016) 8:328rv4. doi: 10.1126/scitranslmed.aad7118
253. Topalian SL, Drake CG, and Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. (2015) 27:450–61. doi: 10.1016/j.ccell.2015.03.001
254. Nagasaki J and Togashi Y. A variety of 'exhausted' T cells in the tumor microenvironment. Int Immunol. (2022) 34:563–70. doi: 10.1093/intimm/dxac013
255. Chen Y, Yu M, Zheng Y, Fu G, Xin G, Zhu W, et al. CXCR5+PD-1+ follicular helper CD8 T cells control B cell tolerance. Nat Commun. (2019) 10:4415. doi: 10.1038/s41467-019-12446-5
256. Cao G, Chi S, Wang X, Sun J, and Zhang Y. CD4+CXCR5+PD-1+ T follicular helper cells play a pivotal role in the development of rheumatoid arthritis. Med Sci Monit. (2019) 25:3032–40. doi: 10.12659/MSM.914868
257. Li Y, Tang L, Guo L, Chen C, Gu S, Zhou Y, et al. CXCL13-mediated recruitment of intrahepatic CXCR5+CD8+ T cells favors viral control in chronic HBV infection. J Hepatol. (2020) 72:420–30. doi: 10.1016/j.jhep.2019.09.031
258. Valentine KM, Mullins GN, Davalos OA, Seow LW, and Hoyer KK. CD8 follicular T cells localize throughout the follicle during germinal center reactions and maintain cytolytic and helper properties. J Autoimmun. (2021) 123:102690. doi: 10.1016/j.jaut.2021.102690
259. Turner CN, Mullins GN, and Hoyer KK. CXCR5+ CD8 T cells: Potential immunotherapy targets or drivers of immune-mediated adverse events? Front Med (Lausanne). (2022) 9:1034764. doi: 10.3389/fmed.2022.1034764
260. Sharma S, Tyagi T, and Antoniak S. Platelet in thrombo-inflammation: Unraveling new therapeutic targets. Front Immunol. (2022) 13:1039843. doi: 10.3389/fimmu.2022.1039843
261. Xia D, Hang D, Li Y, Jiang W, Zhu J, Ding Y, et al. Au-hemoglobin loaded platelet alleviating tumor hypoxia and enhancing the radiotherapy effect with low-dose X-ray. ACS Nano. (2020) 14:15654–68. doi: 10.1021/acsnano.0c06541
262. Wang Y, Meng X, Wang A, Xie X, Pan Y, Johnston SC, et al. Ticagrelor versus clopidogrel in CYP2C19 loss-of-function carriers with stroke or TIA. N Engl J Med. (2021) 385:2520–30. doi: 10.1056/NEJMoa2111749
Keywords: aplastic anemia (AA), CD8 + T cell/CTL, CD4 + T cell, platelet, mitochondria, immunology
Citation: Tan S, He H, Li Y, Shang M, Cao Y, Zou D, Hu R, Hui W, Chang X, Ni J, Ma Q, Su L, Sun J, He W, Feng X and Sun W (2025) Platelets as a potential new immune coordinator in T cell-mediated aplastic anemia. Front. Oncol. 15:1568169. doi: 10.3389/fonc.2025.1568169
Received: 28 January 2025; Accepted: 12 May 2025;
Published: 09 June 2025.
Edited by:
Carmine Selleri, University of Salerno, ItalyReviewed by:
Valentina Giudice, University of Salerno, ItalyLilian Varricchio, Icahn School of Medicine at Mount Sinai, United States
Copyright © 2025 Tan, He, Li, Shang, Cao, Zou, Hu, Hui, Chang, Ni, Ma, Su, Sun, He, Feng and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shuai Tan, dGFuc2h1YWlAeHdob3NwLm9yZw==; Xingmin Feng, ZmVuZ3gyQG5obGJpLm5paC5nb3Y=; Wanling Sun, d2FubGluZ3N1bkB4d2hvc3Aub3Jn
†These authors have contributed equally to this work