 Bunty Sharma1†
Bunty Sharma1† Himanshu Shekhar2†
Himanshu Shekhar2† Anidrisha Sahu2
Anidrisha Sahu2 Damandeep Kaur3
Damandeep Kaur3 Shafiul Haque4,5
Shafiul Haque4,5 Hardeep Singh Tuli6
Hardeep Singh Tuli6 Himanshu Sharma7
Himanshu Sharma7 Ujjawal Sharma2*
Ujjawal Sharma2*- 1Department of Biotechnology, Graphic Era (Deemed to be University), Dehradun, Uttarakhand, India
- 2Department of Human Genetics and Molecular Medicine, Central University of Punjab, Bhatinda, India
- 3University Center for Research and Development (UCRD), Chandigarh University, Mohali, Punjab, India
- 4College of Nursing and Health Sciences, Jazan University, Jazan, Saudi Arabia
- 5School of Medicine, Universidad Espiritu Santo, Samborondon, Ecuador
- 6Department of Bio-Sciences and Technology, Maharishi Markandeshwar Engineering College, Maharishi Markandeshwar (Deemed to be University), Mullana, India
- 7Department of Biochemistry, All India Institute of Medical Sciences, Bathinda, India
Prostate cancer is one of the most prevalent malignancies globally. This cancerous condition originates within the prostate gland, an integral part of the male reproductive system. The molecular mechanism underlying cancer is among the key areas of research in the scientific community. Cancer, being a multifactorial disease, is controlled by many factors ranging from environmental to genetic to epigenetic factors. Epigenetic regulation holds a crucial role in tumorigenesis and its progression. Epigenetics refers to alterations in the genome that happen without any changes to the DNA sequence itself; they may be triggered by multiple factors ranging from environmental to dietary factors. It includes methylation of DNA and histone modifications. Histone modifications, including histone methylation, histone acetylation, and histone ubiquitination, play a crucial role in the pathogenesis and progression of prostate cancer. These epigenetic modifications via transcriptional regulation affect key cellular processes and are thus implicated in prostate cancer and other cancers. These epigenetic markers could be used as both diagnostic and prognostic markers and also could be used as novel therapeutic targets against prostate cancer and other malignancies. Here in this review article, we have summarized different histone modifications and their mechanistic and therapeutic implications in prostate cancer.

Graphical Abstract.
1 Introduction
Prostate tumor stands as the second most prevalent diagnosed malignancy and the fifth most common cause of mortality due to cancer globally, with an approximation of new cases reaching 1.4 million and a death toll reaching 3.75 lakhs in 2020 alone (1). The incidence of prostate cancer is rising among older men. Prostate cancer is associated with many risk factors including age, race, ethnicity, family history, and other factors such as occupational, environmental, and dietary factors (2, 3). In the early stages, prostate cancer may be undetected due to its asymptomatic nature. Delayed diagnosis is a major reason for the high mortality of prostate cancer. Presently, prostate cancer diagnosis mostly relies on prostate-specific antigen (PSA) screening and biopsy techniques, which are not very efficient and may lead to under- or over-diagnosis of the disease. Also, current treatment strategies such as chemotherapy and hormone therapy are associated with risk and side effects. Therefore, researchers are looking for a more precise novel diagnostic and prognostic biomarker and novel therapeutic target for prostate cancer. Epigenetic markers are among such novel markers that are extensively explored nowadays and are reported to be implicated in multiple cancers. Epigenetics plays a key role in tumorigenesis and the progression of tumors, and epigenetic markers are emerging as a novel hallmark of cancer (4). Epigenetic modifications such as methylation of DNA as well as modifications in histone bring out transcriptional activation or suppression and thus modulate key cellular processes. Methylation of DNA is directly involved in gene expression regulation and could be used as both diagnostic and prognostic biomarkers of prostate cancer (5). However, histone modifications are also an important epigenetic marker, which are found to be associated with many cancers including prostate cancer (6). Among major histone modifications are histone methylation and acetylation. Histone acetylation entails attaching an acetyl group to lysine residues located on histone tails (7). This process is facilitated by two key enzyme groups: histone acetyltransferases (HATs) and histone deacetylases (HDACs). A distinct acetylation pattern brings transcriptional changes, thus affecting key pathophysiological pathways of prostate cancer. However, histone methylation refers to methyl group addition at the basic amino acid residue of the histone chain, and it is mediated by two enzymes: histone methyltransferases (HMTs) and histone demethylases (HDMs). Differential methylation patterns as well as differential expression patterns of HMTs and HDMs are reported in prostate cancer, and these could be used as novel biomarkers as well as therapeutic targets (8). In addition to acetylation and methylation, other histone modifications like phosphorylation and ubiquitination have also been reported to be involved in prostate cancer (9, 10). In this article, we have thoroughly reviewed different histone modifications involved in prostate cancer, giving a holistic view of current scientific knowledge of the molecular mechanism of epigenetic regulation by histone modification.
2 Prostate cancer
Prostate cancer is a type of malignancy that arises in the glandular tissues of the prostate, a small, walnut-shaped organ that contributes significantly to male reproductive functions by producing enzymes, lipids, amines, and metal ions critical for the normal functioning of spermatozoa (11). Prostatic enlargement can be due to benign prostatic hyperplasia (BPH) or prostate cancer, two distinct conditions with different pathological and clinical implications. BPH is a non-cancerous enlargement of the prostate, whereas prostate cancer involves uncontrolled malignant cell growth. BPH typically affects the transitional zone, while prostate cancer often arises in the peripheral zone of the prostate. The progression of prostate cancer involves several stages, beginning with prostatic intraepithelial neoplasia, advancing to localized prostate adenocarcinoma, which may invade nearby tissues, and then ultimately leading to metastatic prostate cancer (12). The predominant type of prostate cancer is adenocarcinoma, although there are other variations also. At the early stage, it remains asymptotic, but as it progresses, symptoms like difficulty in starting urination, interrupted or weak urine flow, frequent urination, mainly during the night, inability to complete emptying of the urinary bladder, burning sensation and cramps during urination, blood in semen or urine, and painful ejaculation may occur. Risk elements associated with it may include age, race and ethnicity, genetics and family history, physical activity and sleep, and other dietary and environmental factors (2) (Figure 1).
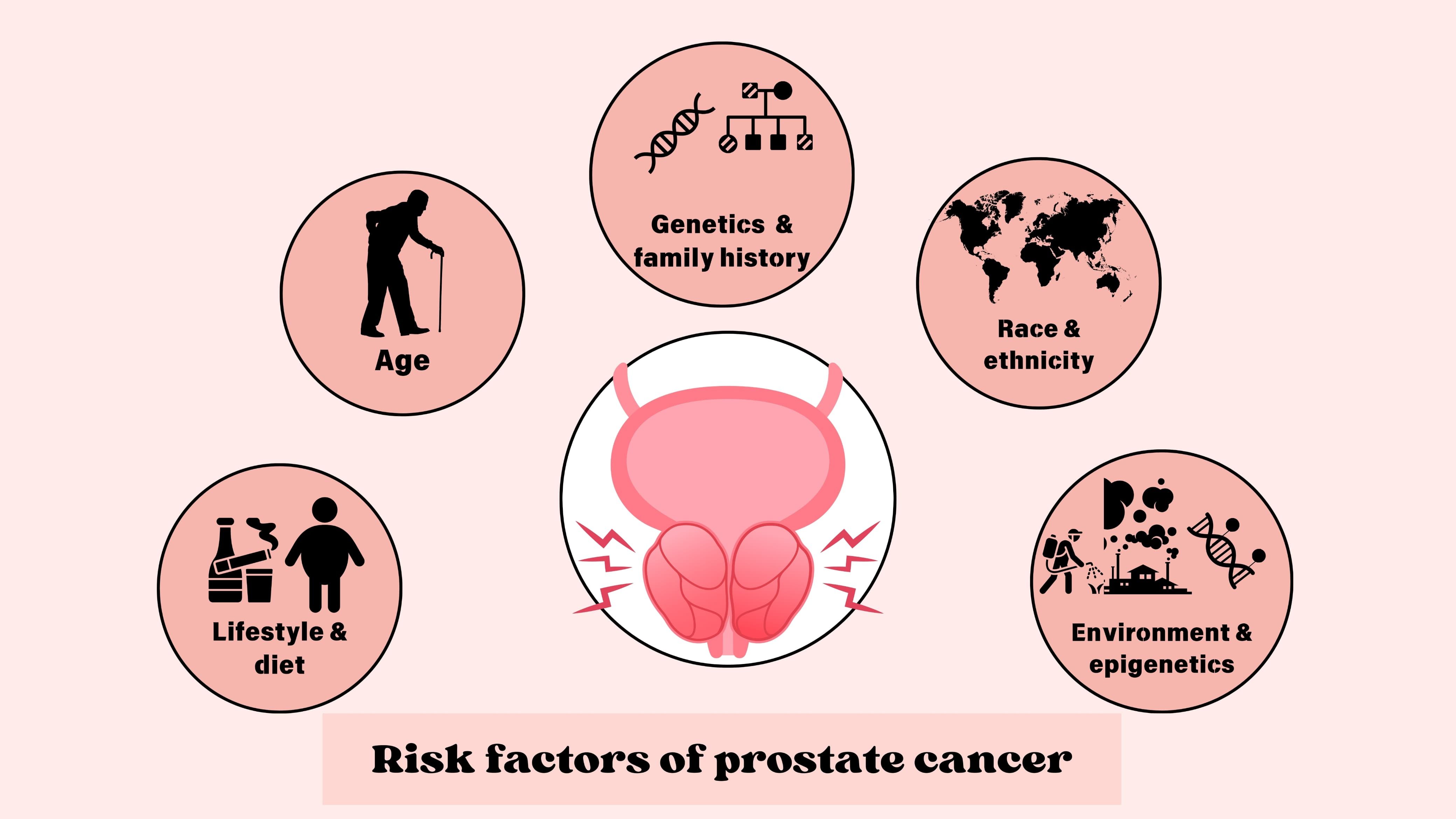
Figure 1. Risk factors of prostate cancer. Major risk factors involved with prostate cancer include age, race and ethnicity, genetics and family history, lifestyle and dietary factors, and epigenetic changes.
3 Molecular pathophysiology of prostate cancer
The multifaceted nature of prostate cancer arises from the complex interplay of various molecular pathways that govern its initiation and progression. Its complex pathophysiology involves various signaling pathways, genetic alterations, and epigenetic changes such as histone modifications and DNA methylation.
3.1 Androgen receptor signaling pathway
The androgen receptor (AR), a transcription factor activated by androgens, belongs to the nuclear receptor family. AR plays a significant role in the progression and development of prostate cancer (13). AR is expressed in almost all primary as well as metastatic prostate cancers, irrespective of their grade and stage. This expression is maintained in most androgen-independent and castration-resistant prostate cancer (CRPC). AR signaling continues to be active, promoting the survival and growth of prostate cancer cells (14). The interaction between androgen receptor signaling and the tumor microenvironment is intricate, exhibiting both tumor-promoting and tumor-suppressing effects (15). The role of AR signaling and androgens in metastatic prostate cancer is well understood, demonstrating that prostate cancer cells are highly adaptive at sustaining functional AR signaling to promote cancer growth (16). In prostate cancer, AR signaling in stromal cells, initially high during prostate development, progressively decreases as cancer advances, correlating with worse clinical outcomes and a shift from androgen-dependent paracrine to autocrine pathways in cancer cells. This reduction in stromal AR expression is linked to disease progression, metastasis, and CRPC progression (14). This direct link of AR signaling to prostate cancer makes it a suitable therapeutic target, and androgen deprivation therapy remains the primary treatment option for advanced prostate cancer. These therapies alleviate symptoms, decrease tumor size, and extend patient survival. However, they seldom achieve a cure for the cancer. Prostate cancer frequently returns, leading to lethal CRPC (Figure 2). Mechanisms enabling this resistance include upregulation of the androgen receptor gene or enhancer, androgen receptor mutation, variants of the androgen receptor, overexpression of coactivators, and intra-tumoral synthesis of androgens (17). While different mutations in the androgen receptor have been documented in prostate cancer, particular mutations (such as L702H, W742L/C, H875Y, F877L, and T878A/S) are commonly detected following the development of treatment resistance (18). While CRPC progression is generally associated with a gain in AR signaling, a different type of CRPC relies on the loss of AR dependency leading to AR-indifferent conditions such as neuroendocrine prostate cancer (NEPC). NEPC differentiation is primarily associated with a lack of expression of RB1, TP53, and PTEN and upregulation of MYCN and AURKA expression. This transition of PC to NEPC is associated with a key cellular feature called lineage plasticity. It refers to the ability of cancer cells to adapt and change their cellular identity in response to therapeutic pressures such as AR signaling inhibitors (19).
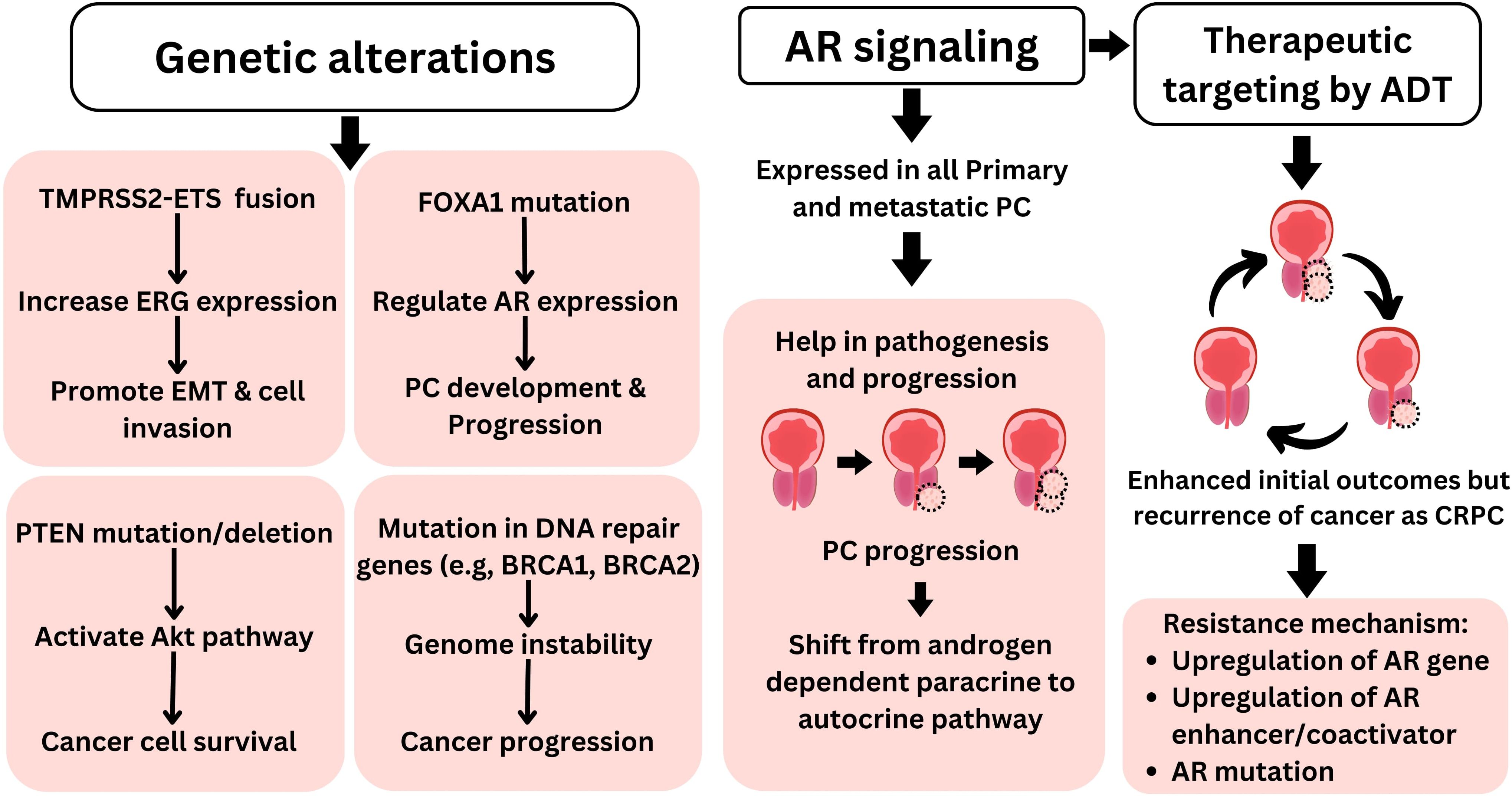
Figure 2. Molecular pathophysiology of prostate cancer. Genetic alterations such as TMPRSS2–ETS fusion, FOXA1 mutation, PTEN deletion/mutation, and defect in DNA repair genes play key roles in prostate cancer pathophysiology. Furthermore, AR signaling plays a central role in prostate cancer pathophysiology, making it suitable therapeutic target for PC. Furthermore, targeting AR with androgen deprivation therapy is among primary therapeutic approaches for PC; however, despite initial favorable outcomes such as reduced tumor size, PC often reoccurs as more lethal CRPC. AR, androgen receptor; PC, prostate cancer; CRPC, castration-resistant prostate cancer.
3.2 Genetic alterations
Apart from AR mutations, other genetic alterations are also observed in prostate cancer. The most common of these are TMPRSS2–ETS gene fusion, FOXA1 mutation, loss of the PTEN gene, and defects in DNA repair genes (Figure 2) (20, 21). In over 50% of prostate tumor cases, TMPRSS2–ETS gene fusion has been documented (20). This fusion involves the androgen-sensitive TMPRSS2 promoter joining with the ERG coding region. In the presence of androgens, this fusion drives increased expression of ERG, significantly influencing cell invasion and the process of epithelial–mesenchymal transition (EMT) (22). FOXA1 is another important transcription factor that is often mutated in multiple types of malignancies including prostate cancer (23). Mutations in FOXA1 are observed in nearly half of primary prostate cancer tumors in Asian men and 20% of tumors in men of other ethnicities (21). FOXA1 is crucial for regulating the expression of numerous genes, particularly AR, during prostate cancer development and progression (24). Moreover, FOXA1 has been identified as an important regulator of alternative splicing in prostate cancer (25). Deletion or mutation of the PTEN gene is found in approximately 20% of the primary prostate cancer samples during radical prostatectomy and up to 50% of CRPC cases (26). PTEN loss leads to the activation of the Akt pathway, enhancing cancer cell survival (20). Furthermore, mutations in genes that code for DNA damage response components, such as BRCA1 and BRCA2, are prevalent in prostate cancer. These mutations diminish the capacity to repair both single- and double-strand DNA damage, thereby compromising the integrity of the genome (27).
3.3 Other contributing factors
In addition to AR signaling and genetic alterations, other factors such as inflammation and epigenetics play important roles in the pathophysiology of prostate cancer. Chronic inflammation creates a pro-tumorigenic environment by producing inflammatory cytokines and reactive oxygen species that lead to DNA damage (28). Epigenetic changes, including methylation at DNA and histone modifications, also contribute to the progression of cancer by altering gene expression. The role of epigenetic modifications, in particular histone modifications in prostate cancer, will be discussed in detail in the upcoming section.
4 Epigenetic dynamics of prostate cancer
Epigenetic modifications are alterations in the expression of genes that occur independently of any changes to the DNA sequence itself. These epigenetic modifications are heritable and reversible. These modifications commonly include methylation of DNA and the modifications of histone, and these are important for the maintenance of gene expression (29). Epigenetic changes can arise from external environmental factors or exposure to elements such as diet, drugs, and food (30). Epigenetic modifiers are one of the key players in cancer development and progression, making them attractive candidates as prognostic markers and therapeutic targets. These non-mutational epigenetic regulations of gene expression are emerging hallmarks of cancer and are subject to extensive study nowadays (4).
Epigenetic abnormalities, such as DNA methylation, histone modifications, and remodeling of nucleosomes, happen at all stages of the development and advancement of prostate cancer (31) (Figure 3). These epigenetic markers can be used as biomarkers for both the diagnosis and prognosis of prostate cancer (32). The methylation of DNA is a fundamental epigenetic mechanism that regulates gene expression and various cell processes. It refers to the addition of methyl group to DNA molecules, specifically at cytosine bases, typically occurring at CpG dinucleotides, and this modification is catalyzed by enzyme group DNA methyltransferases (33). Abnormal DNA methylation patterns, especially CpG island hypermethylator phenotype, are associated with specific clinical characteristics and outcomes in prostate cancer. These include tumor aggressiveness, high Gleason scores, elevated PSA level, advanced stages, poorer prognosis, and reduced survival rates. Commonly hypermethylated genes in prostate cancer are involved in functions such as apoptosis, DNA damage repair, cell cycle regulation, cell adhesion, hormonal responses, and signal transduction. Examples include GSTP1, RARβ, RASSF1A, CDH13, APC, DAPK, p16, FHIT, CDH1, and MGMT (5). Another epigenetic phenomenon related to prostate cancer is DNA hypomethylation, which involves the removal of methyl groups from CpG sites that are typically methylated, leading to increased expression of genes. In prostate cancer, hypomethylation influences genes encoding urokinase-type PLAU (PLAU and its receptor are crucial for tumor invasion and metastasis development by breaking down the extracellular matrix), HPSE (a versatile protein involved in extracellular matrix degradation and heparan sulfate chain breakdown of proteoglycans), and CYP1B1 (essential for estrogen metabolism and activation of procarcinogens) (34).
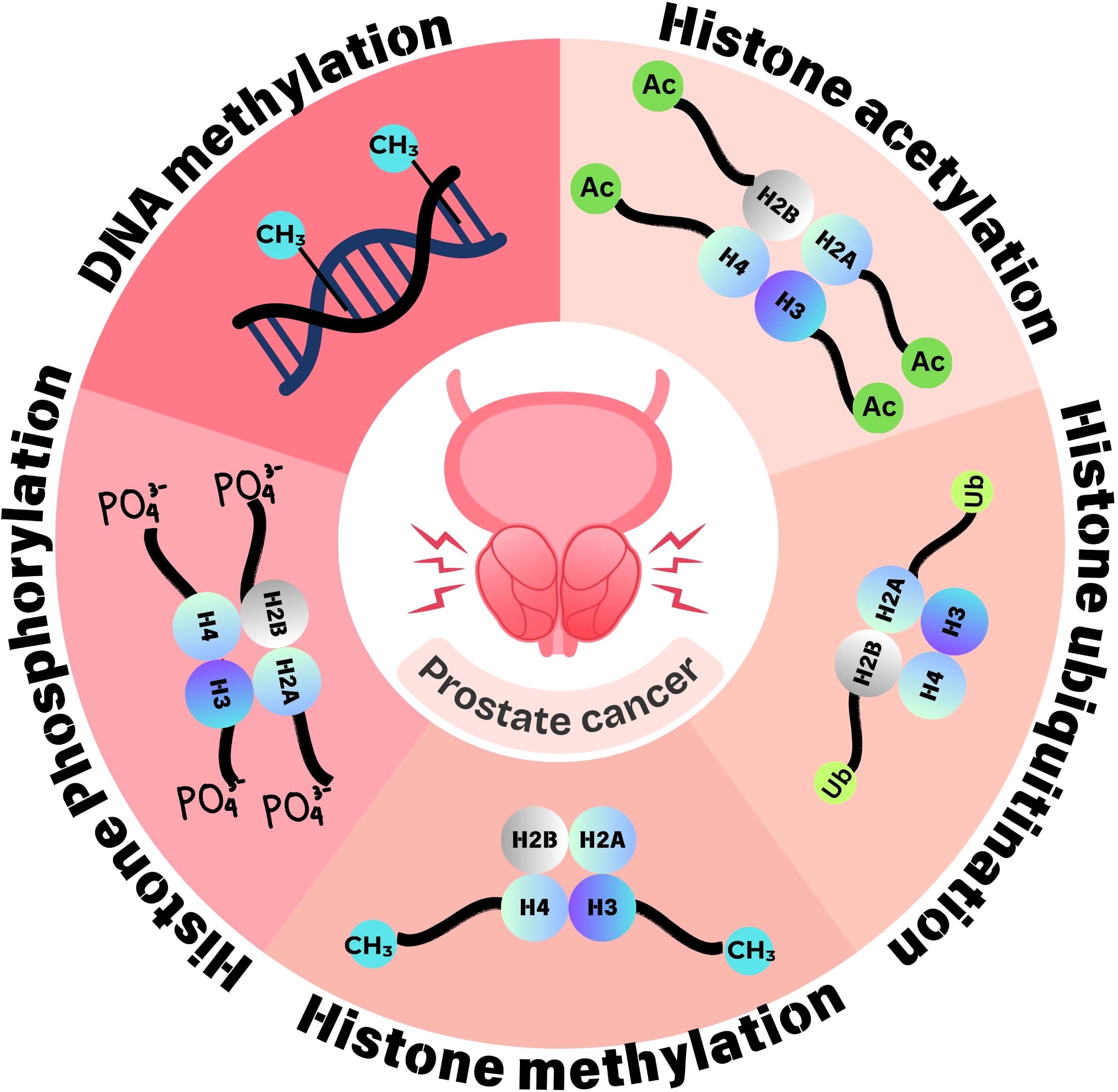
Figure 3. Epigenetic dynamics of prostate cancer. Epigenetic changes implicated in prostate cancer involve aberrant methylation at DNA and histone modifications such as acetylation, methylation, phosphorylation, and ubiquitination. These epigenetic changes regulate gene expression and are thus involved in tumorigenesis and tumor progression.
In addition to DNA methylation, histone modifications like histone acetylation and histone methylation are pivotal in regulating gene expression and influencing cancer development and progression. These histone modifications are briefly discussed in the proceeding sections.
5 Histone modifications in prostate cancer
Among epigenetic changes, histone modifications (histone acetylation, histone methylation, etc.) play a crucial role in gene expression regulation, and their dysregulation has been implicated in cancer development and progression (35). Post-translational modifications, including methylation, phosphorylation, acetylation, sumoylation, deamination, ubiquitylation, proline isomerization, ADP-ribosylation, and β-N-acetylglucosamine, can affect the projecting tail of histones. Histone modification influences DNA replication, repair, and recombination processes as well as chromatin structure regulation and remodeling (36). Histone modifications and differential expression of histone modifiers are reported in different cancers including prostate cancer (Figure 4). The process of addition of an acetyl group to lysine amino acid residue in the projecting tail of histone is known as histone acetylation. Usually linked to transcriptional activity, it is regulated by two distinct sets of enzymes: HDACs, which eliminate acetyl groups, and HATs, which add them (36). Histone methylation is another post-translational modification process that refers to the methyl group addition to the histone proteins. Histone methylation occurs on all basic residues: arginine, lysine, and histidine. Among these, lysine may undergo mono-methylation, di-methylation, or tri-methylation, whereas arginine may undergo mono-methylation, symmetrical di-methylation, or asymmetrical di-methylation, and histidine could be mono-methylated, although this is relatively rare (37). S-Adenosyl methionine, the methyl-donating substrate of histone methyltransferases, mediates histone methylation (38). Apart from acetylation and methylation, histone ubiquitination (removal or addition of ubiquitin at the C-terminal end of histone H2A and H2B) and ubiquitin-activating enzymes are also associated with prostate cancer (34). Other histone modifications implicated in cancer include phosphorylation and ADP-ribosylation (35).
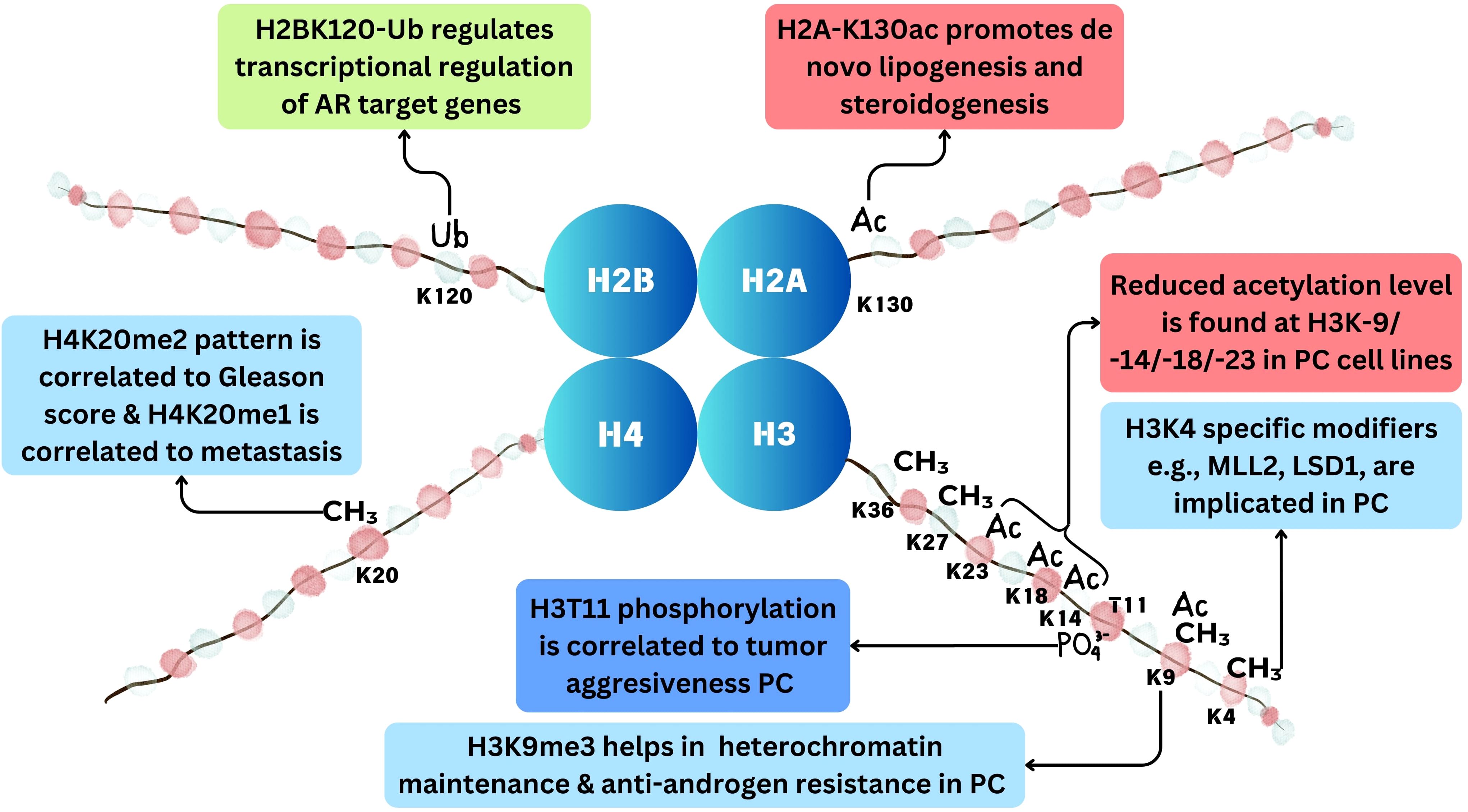
Figure 4. Histone modifications in prostate cancer. Methylation at histone occurs at H3K4, H3K9, H3K27, H3K36, and H4K20 positions. Acetylation occurs at H3K9, H3K14, H3K18, H3K23, and H2AK130 positions. Ubiquitination occurs at H2BK120 position and phosphorylation at H3T11 position in prostate cancer.
5.1 Histone acetylation in prostate cancer
As discussed earlier, histone acetylation refers to the acetyl group addition to lysine amino acid residues at histone tails, and it is regulated by two opposing enzyme groups: HATs and HDACs. HATs (often referred to as writers), using acetyl CoA as a cofactor, add acetyl moiety at the ϵ-amino group of lysine residue; this modification weakens the interaction between histone and DNA, leading to transcriptional activation. However, HDACs, also known as erasers, are enzymes that remove the acetyl moiety from the lysine amino acid residues; this interaction leads to the condensation of chromatin and finally transcriptional repression (36). Bromodomains, also known as “readers”, are tiny protein modules that read the acetylation on lysine amino acid residues (39).
The acetylation level of histone H3 at the N-terminal end at different positions such as lysine 9, lysine 14, lysine 18, and lysine 23 is reduced in cancer cell lines such as PC-3, LNCaP, DU-145, and clinical prostatic adenocarcinomas, in comparison to that in the non-cancerous prostate cell lines such as RC170N/h and RC165N/h. This reduced acetylation is associated with significantly elevated HDAC activity in cancer cells. Inhibiting HDACs restores the acetylation of histone and increases the expression of the p21 gene, highlighting that increased HDAC activity underlies the deficient histone acetylation seen in prostate cancer (40). Furthermore, a recent study by Nguyen et al. revealed a novel mechanism including histone acetylation by which prostate cancer cells adapt to androgen deficiency through dual phosphorylation of sterol regulatory element-binding protein 1 (SREBF1) at Y673 and Y951. Dual-phosphorylated SREBF1 senses low androgen levels and dissociates from AR, enabling its nuclear translocation. In the nucleus, SREBF1 recruits KAT2A/GCN5, leading to the acetylation of H2A at lysine 130. This epigenetic marker promotes the transcription of genes necessary for de novo lipogenesis and steroidogenesis (41).
Furthermore, dysregulation of HAT and HDAC expression is often observed in prostate cancer. Histone acetyltransferases such as p300 and CBP are prominent coactivators of androgen receptors and have a pro-tumor role in prostate cancer (34). A recent study reported that histone acetyltransferase 1 (HAT1) is upregulated in prostate cancer cells and correlated to disease progression to CRPC. The same study further reported that HAT1 knockdown re-sensitizes the drug response to CRPC cells (42). Similarly, histone deacetylase is also found to be implicated in prostate cancer, and the use of histone deacetylase inhibitors has shown enhanced therapeutic outcomes (43, 44).
Different histone acetyltransferases and histone deacetylases implicated in prostate cancer are summarized in Table 1.
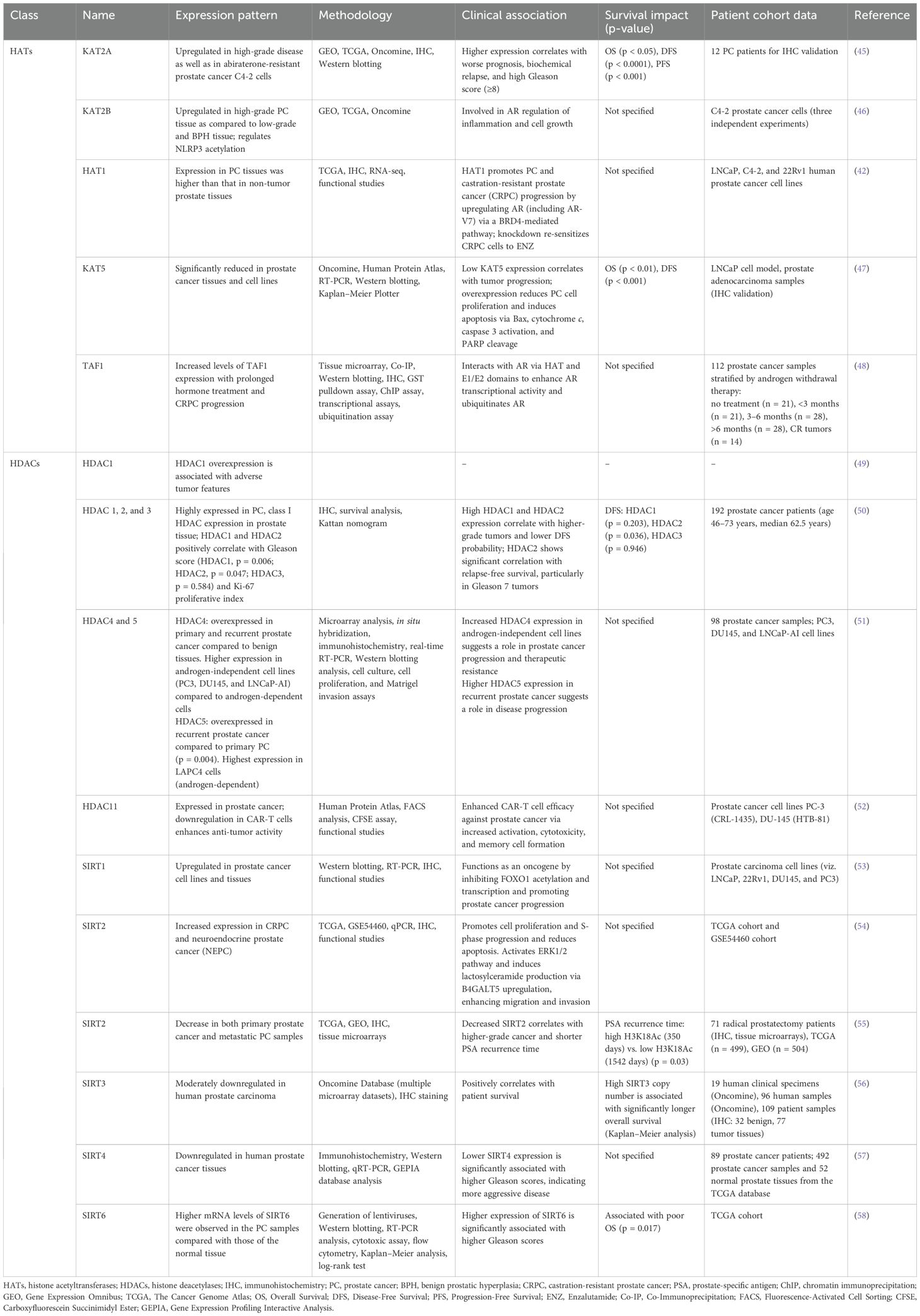
Table 1. Different HATs and HDACs implicated in prostate cancer.
5.2 Histone methylation in prostate cancer
Histone methylation is another epigenetic marker associated with prostate cancer, and distinct patterns of gene expression of HMTs and HDMs have been reported in different studies. Histone methylation refers to the addition of methyl group to basic amino acid residue (arginine, lysine, and histidine) of the histone chain. It is mediated by S-adenosyl methionine, which acts as a methyl-donating substrate (59). Some of the most studied histone methylation patterns include H3K4 methylation (i.e., methylation at lysine 4 residue of histone H3), H3K9 methylation (i.e., methylation at lysine 9 of histone H3), H3K27 methylation (histone H3 lysine 27 methylation), H3K36 methylation (histone H3 lysine 36 methylation), and H4K20 methylation (histone H4 lysine 20 methylation) (60, 61). These methylation patterns can be associated with either transcription activation (H3K4me3) or repression (H3K9me2/3), thus modulating key cellular processes (61). Methylation, in contrast to acetylation, does not change the histone protein’s net charge. Instead, methylation recruits different effector proteins, referred to as histone methylation readers, to cause the transcriptional alteration (60).
A number of studies have reported differential methylation patterns and dysregulated expression of HDMs and HMTs in prostate cancer. Pang et al. reported that androgen-stimulated H3K4me2 methylation in prostate cancer is mediated by PI3 K/p110beta-dependent signaling, which may be exploited as a new biomarker for disease prognosis and targeted therapy (62). MLL2 is an important H3K4 methyltransferase that is involved in the activation of the PI3K/EMT process and also induces DNA damage in prostate cancer (61). SMYD3 is another HMT that is found to be implicated in prostate tumors. Its downregulation in prostate cancer inhibits cancer development by suppressing the transcription activation of cyclin D2 or AR (63). Among H3K4 demethylases, LSD1 directly suppresses androgen receptor transcriptional activity through H3K4 demethylation (64). Additionally, LSD1 can enhance CRPC by controlling mitotic kinesin and the centromere-binding protein CENPE (65). Another H3K4 HDM, KDM5D, is also implicated in androgen receptor signaling and thus prostate cancer (66). Furthermore, H3K9me3 is critical for the maintenance of heterochromatin and the development of anti-androgen resistance in prostate cancer, and the methylation of H3K9 by EHMT1 has been linked to poor patient outcomes from hormone therapy. Also, H3K9me3 writers’ inhibition suppresses anti-androgen resistance (67). A 2017 study highlighted that H3K27me3 is a crucial epigenetic modification in the progression of prostate cancer (68). Furthermore, H3K27- and H3K36-specific HMTs and HDMs are also implicated in prostate cancer (69). Among H4 methylation sites, methylation at H4K20me2 has shown a significant difference in tumor vs. normal tissues. H4K20me1 pattern is found to significantly differentiate CRPC tissues from other kinds of prostate tissues. Additionally, a significant correlation between H4K20me1 with lymph node metastases and H4K20me2 Gleason score was found (70). Different HMTs and HDMs implicated in prostate cancer are summarized in Table 2.
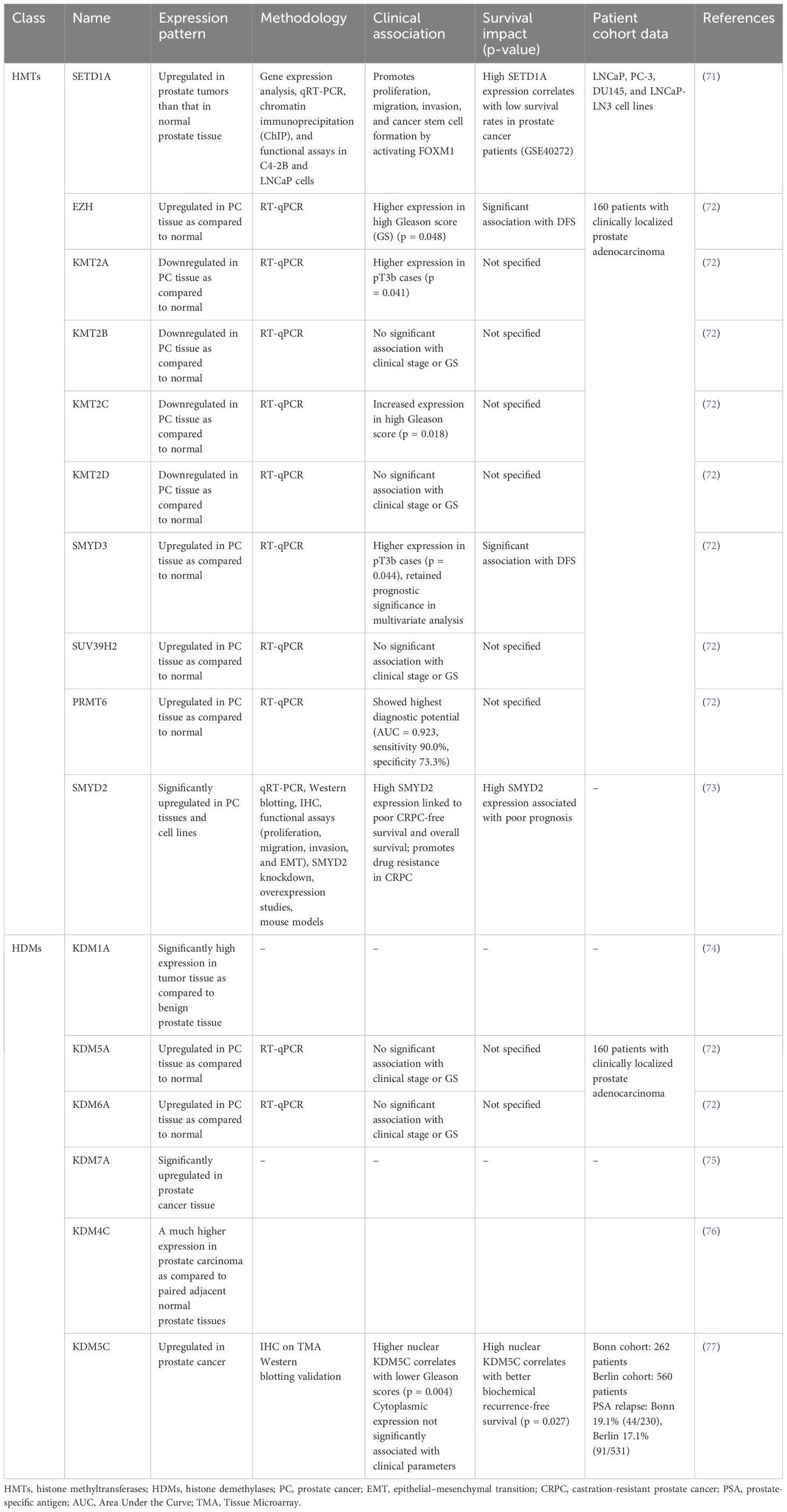
Table 2. Different HMTs and HDMs implicated in prostate cancer.
5.3 Other histone modifications in prostate cancer
Other important histone modifications implicated in cancer include phosphorylation and ubiquitination. Histone phosphorylation happens at threonine and serine amino acid residues. This process is modulated by kinase and phosphatase. Phosphorylation, like acetylation, leads to the opening of the chromatin structure by negative charge addition to the histone and also leads to the recruitment of effector proteins (69). In prostate cancer, PRK1 phosphorylates histone H3 at threonine 11 (H3T11) upon AR activation, facilitating AR-dependent transcription by promoting histone demethylation and RNA polymerase II recruitment. Elevated PRK1 levels and phosphorylated H3T11 correlate with prostate cancer aggressiveness, suggesting PRK1 inhibition as a potential therapeutic strategy for blocking AR-induced tumor proliferation (78).
Histone ubiquitination is another post-translational modification where ubiquitin molecules are covalently attached to histone proteins within chromatin (79). This modification primarily occurs on histone H2A and histone H2B. Histone ubiquitination regulates various chromatin-related processes, including transcriptional regulation, DNA repair, and chromatin compaction (80). Histone ubiquitination, specifically at H2BK120 regulated by RNF20 and RNF40, plays a crucial role in the transcriptional regulation of AR target genes in prostate cancer. The dynamic nature of H2BK120-ub and its interplay with other histone modifications are important for gene expression regulation. Variations in RNF20 expression and its interaction with AR play a complex role in prostate cancer progression, offering potential targets for therapeutic intervention (10).
6 Therapeutic implications of histone modifications in prostate cancer
As we have discussed in previous sections, epigenetic modifications play an important role in prostate cancer pathogenesis and progression. In this section, we will discuss the therapeutic implications of targeting epigenetic modifications in prostate cancer, exploring various strategies and their potential benefits. Epigenetic modifiers such as HATs, HDACs, HMTs, and HDMs play key roles in prostate cancer progression through various mechanisms, and targeting these modulators has shown significant therapeutic outcomes (81).
HAT inhibitors are being explored as potential therapeutic agents in prostate cancer, particularly in the context of more lethal CRPC. Presently, several inhibitors targeting HATs have been explored and shown therapeutic effects against different cancers such as breast cancer, Renal Cell Carcinoma (RCC), bladder cancer, and prostate cancer (82). CCS1477, a novel small-molecule inhibitor targeting the bromodomain of histone acetyltransferases p300 and CBP, demonstrates antitumor activity by inhibiting AR and C-MYC signaling in CRPC. It shows potential in reducing AR signaling and affecting metastatic CRPC target expression in clinical settings (83). Furthermore, various HDAC inhibitors are also in clinical trials for different cancers including prostate cancer. Several HDAC inhibitors, such as vorinostat (SAHA), belinostat (PXD-101), panobinostat (LBH589), chidamide (CS055, HBI-8000), and romidepsin (FK228), have been approved by the U.S. Food and Drug Administration (FDA) as medicines for the treatment of skin T-cell lymphoma (TCL) and peripheral TCL (79). Hematological malignancies have demonstrated encouraging responses to HDAC inhibitors. However, HDAC inhibitors have not successfully passed clinical trials for solid tumors, even though there have been promising results in biological studies, preclinical research, and early clinical trials. For instance, clinical trials involving vorinostat (SAHA) have shown drug toxicity and no promising results in clinical outcomes (84).
Furthermore, inhibitors of HMTs and HDMs are also explored for their possible therapeutic benefits in different cancers including prostate cancer. One of the most thoroughly researched histone methylation inhibitors is the class of EZH2 inhibitors. Blocking EZH2 has shown promising outcomes, especially in lymphomas; however, targeting EZH2 has not yet demonstrated significant clinical activity in solid tumors (85). Among SMYD family HMTs, SMYD3 inhibition by inhibitor molecule BCI-121 has shown decreased proliferation in colon cancer cell lines (86). Among inhibitors of HDM, LSD1 inhibitors are the most extensively studied, and many LSD2 inhibitors such as tranylcypromine (TCP), ORY-1001 iadademstat, and bomedemstat are already in phase 1/2 of clinical trials, but mostly in breast cancer and leukemia (87). In prostate cancer, pargyline (an inhibitor of LSD1) has also shown promising therapeutic potential. Pargyline decreased the migration and invasion capabilities of LNCap cells and hindered the EMT process by upregulating E-cadherin expression while downregulating N-cadherin and vimentin expressions both in vitro and in vivo. Additionally, pargyline delayed the transition of prostate cancer from an androgen-dependent state to an androgen-independent one (88).
In conclusion, targeting histone modifications offers promising therapeutic potential for prostate cancer, particularly in overcoming treatment-resistant forms like CRPC. Ongoing research and clinical trials are crucial to optimize these strategies.
7 Conclusion and future prospects
Histone modifications are pivotal in the regulation of gene expression in prostate cancer, influencing various aspects of the disease’s progression and resistance to therapy. The dysregulation of histone acetylation and methylation, along with other modifications, has been implicated in critical processes such as DNA damage repair, cell cycle regulation, and hormone response. These modifications provide valuable biomarkers for the diagnosis and prognosis of prostate cancer, highlighting the potential for personalized treatment approaches. Although significant advancements have been made in epigenetic biomarkers and their association with prostate cancer, several challenges remain in their clinical transition. These include evaluating the role of epigenetic alterations in normal biological processes and disease progression and interpreting large-scale epigenetic data to ensure robust validation of biomarkers.
Current research emphasizes the importance of developing therapies that target specific histone modifications and the enzymes that regulate them, such as HDACs, HATs, HMTs, and HDMs. The therapeutic potential of these targets is particularly promising in addressing treatment-resistant forms of prostate cancer, such as CRPC. Future studies should focus on further elucidating the role of these modifications in cancer biology and exploring novel inhibitors that can selectively modulate these epigenetic changes. By advancing our understanding of histone modifications in prostate cancer, we can pave the way for more effective, targeted therapeutic strategies that improve patient outcomes and address the complexities of this disease.
Author contributions
BS: Data curation, Formal Analysis, Writing – original draft, Writing – review & editing. HShe: Data curation, Writing – original draft. AS: Data curation, Writing – original draft. DK: Data curation, Writing – review & editing. SH: Data curation, Resources, Writing – review & editing. HT: Supervision, Writing – review & editing. HSha: Methodology, Supervision, Writing – review & editing. US: Conceptualization, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2021) 71(3):209–49. doi: 10.3322/caac.21660
2. Bergengren O, Pekala KR, Matsoukas K, Fainberg J, Mungovan SF, Bratt O, et al. 2022 Update on prostate cancer epidemiology and risk factors-A systematic review. Eur Urol. (2023) 84:191–206. doi: 10.1016/j.eururo.2023.04.021
3. Cui H, Zhang W, Zhang L, Qu Y, Xu Z, Tan Z, et al. Risk factors for prostate cancer: An umbrella review of prospective observational studies and mendelian randomization analyses. PLoS Med. (2024) 21:e1004362. doi: 10.1371/journal.pmed.1004362
4. Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discovery. (2022) 12:31–46. doi: 10.1158/2159-8290.CD-21-1059
5. Shin HJ, Hua JT, Li H. Recent advances in understanding DNA methylation of prostate cancer. Front Oncol. (2023) 13:1182727. doi: 10.3389/fonc.2023.1182727
6. Chinaranagari S, Sharma P, Bowen NJ, Chaudhary J. Prostate cancer epigenome. Methods Mol Biol (Clifton N.J.). (2015) 1238:125–40. doi: 10.1007/978-1-4939-1804-1_7
7. Martin BJE, Brind’Amour J, Kuzmin A, Jensen KN, Liu ZC, Lorincz M, et al. Transcription shapes genome-wide histone acetylation patterns. Nat Commun. (2021) 12:210. doi: 10.1038/s41467-020-20543-z
8. Liao Y, Xu K. Epigenetic regulation of prostate cancer: the theories and the clinical implications. Asian J androl. (2019) 21:279–90. doi: 10.4103/aja.aja_53_18
9. Cang S, Xu X, Ma Y, Liu D, Chiao JW. Hypoacetylation, hypomethylation, and dephosphorylation of H2B histones and excessive histone deacetylase activity in DU-145 prostate cancer cells. J Hematol Oncol. (2016) 9:3. doi: 10.1186/s13045-016-0233-x
10. Jääskeläinen T, Makkonen H, Visakorpi T, Kim J, Roeder RG, Palvimo JJ. Histone H2B ubiquitin ligases RNF20 and RNF40 in androgen signaling and prostate cancer cell growth. Mol Cell Endocrinol. (2012) 350:87–98. doi: 10.1016/j.mce.2011.11.025
11. Kumar VL, Majumder PK. Prostate gland: structure, functions and regulation. Int Urol Nephrol. (1995) 27:231–43. doi: 10.1007/BF02564756
12. Wang G, Zhao D, Spring DJ, DePinho RA. Genetics and biology of prostate cancer. Genes Dev. (2018) 32:1105–40. doi: 10.1101/gad.315739.118
13. Lin CY, Jan YJ, Kuo LK, Wang BJ, Huo C, Jiang SS, et al. Elevation of androgen receptor promotes prostate cancer metastasis by induction of epithelial-mesenchymal transition and reduction of KAT5. Cancer Sci. (2018) 109:3564–74. doi: 10.1111/cas.13776
14. Aurilio G, Cimadamore A, Mazzucchelli R, Lopez-Beltran A, Verri E, Scarpelli M, et al. Androgen receptor signaling pathway in prostate cancer: from genetics to clinical applications. Cells. (2020) 9:2653. doi: 10.3390/cells9122653
15. McAllister MJ, Underwood MA, Leung HY, Edwards J. A review on the interactions between the tumor microenvironment and androgen receptor signaling in prostate cancer. Transl Res. (2019) 206:91–106. doi: 10.1016/j.trsl.2018.11.004
16. Zhou Y, Bolton EC, Jones JO. Androgens and androgen receptor signaling in prostate tumorigenesis. J Mol Endocrinol. (2015) 54:R15–29. doi: 10.1530/JME-14-0203
17. Feng Q, He B. Androgen receptor signaling in the development of castration-resistant prostate cancer. Front Oncol. (2019) 9:858. doi: 10.3389/fonc.2019.00858
18. Shiota M, Akamatsu S, Tsukahara S, Nagakawa S, Matsumoto T, Eto M. Androgen receptor mutations for precision medicine in prostate cancer. Endocr Relat Cancer. (2022) 29:R143–55. doi: 10.1530/ERC-22-0140
19. Imamura J, Ganguly S, Muskara A, Liao RS, Nguyen JK, Weight C, et al. Lineage plasticity and treatment resistance in prostate cancer: the intersection of genetics, epigenetics, and evolution. Front Endocrinol (Lausanne). (2023) 14:1191311. doi: 10.3389/fendo.2023.1191311
20. Masud N. Symphony in the crowd: Key genetic alterations in prostate cancer. Cancer Innov. (2023) 2:203–9. doi: 10.1002/cai2.52
21. Schumacher FR, Basourakos SP, Lewicki PJ, Vince R, Spratt DE, Barbieri CE, et al. Race and genetic alterations in prostate cancer. JCO Precis Oncol. (2021) 5:PO.21.00324. doi: 10.1200/PO.21.00324
22. Adamo P, Ladomery MR. The oncogene ERG: a key factor in prostate cancer. Oncogene. (2016) 35:403–14. doi: 10.1038/onc.2015.109
23. Tam KJ, Liu L, Hsing M, Dalal K, Thaper D, McConeghy B, et al. Clinically-observed FOXA1 mutations upregulate SEMA3C through transcriptional derepression in prostate cancer. Sci Rep. (2024) 14(1):7082. doi: 10.1038/s41598-024-57854-w
24. Dong HY, Ding L, Zhou TR, Yan T, Li J, Liang C. FOXA1 in prostate cancer. Asian J androl. (2023) 25:287–95. doi: 10.4103/aja202259
25. Del Giudice M, Foster JG, Peirone S, Rissone A, Caizzi L, Gaudino F, et al. FOXA1 regulates alternative splicing in prostate cancer. Cell Rep. (2022) 40:111404. doi: 10.1016/j.celrep.2022.111404
26. Jamaspishvili T, Berman DM, Ross AE, Scher HI, De Marzo AM, Squire JA, et al. Clinical implications of PTEN loss in prostate cancer. Nat Rev Urol. (2018) 15:222–34. doi: 10.1038/nrurol.2018.9
27. Lozano R, Castro E, Aragón IM, Cendón Y, Cattrini C, López-Casas PP, et al. Genetic aberrations in DNA repair pathways: a cornerstone of precision oncology in prostate cancer. Br J Cancer. (2021) 124:552–63. doi: 10.1038/s41416-020-01114-x
28. Kay J, Thadhani E, Samson L, Engelward B. Inflammation-induced DNA damage, mutations and cancer. DNA Repair (Amst). (2019) 83:102673. doi: 10.1016/j.dnarep.2019.102673
29. Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. (2010) 31:27–36. doi: 10.1093/carcin/bgp220
30. Metere A, Graves CE. Factors influencing epigenetic mechanisms: is there A role for bariatric surgery? High-throughput. (2020) 9:6. doi: 10.3390/ht9010006
31. Conteduca V, Hess J, Yamada Y, Ku SY, Beltran H. Epigenetics in prostate cancer: clinical implications. Transl Androl Urol. (2021) 10:3104–16. doi: 10.21037/tau-20-1339
32. Wu Y, Sarkissyan M, Vadgama JV. Epigenetics in breast and prostate cancer. Methods Mol Biol. (2015) 1238:425–66. doi: 10.1007/978-1-4939-1804-1_23
33. Dhar GA, Saha S, Mitra P, Nag Chaudhuri R. DNA methylation and regulation of gene expression: Guardian of our health. Nucleus. (2021) 64:259–70. doi: 10.1007/s13237-021-00367-y
34. Sugiura M, Sato H, Kanesaka M, Imamura Y, Sakamoto S, Ichikawa T, et al. Epigenetic modifications in prostate cancer. Int J Urol. (2021) 28:140–9. doi: 10.1111/iju.14406
35. Audia JE, Campbell RM. Histone modifications and cancer. Cold Spring Harbor Perspect Biol. (2016) 8:a019521. doi: 10.1101/cshperspect.a019521
36. Gujral P, Mahajan V, Lissaman AC, Ponnampalam AP. Histone acetylation and the role of histone deacetylases in normal cyclic endometrium. Reprod Biol Endocrinol. (2020) 18:84. doi: 10.1186/s12958-020-00637-5
37. Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. (2012) 13:343–57. doi: 10.1038/nrg3173
38. Serefidou M, Venkatasubramani AV, Imhof A. The impact of one carbon metabolism on histone methylation. Front Genet. (2019) 10:764. doi: 10.3389/fgene.2019.00764
39. Marmorstein R, Zhou MM. Writers and readers of histone acetylation: structure, mechanism, and inhibition. Cold Spring Harbor Perspect Biol. (2014) 6:a018762. doi: 10.1101/cshperspect.a018762
40. Cang S, Feng J, Konno S, Han L, Liu K, Sharma SC, et al. Deficient histone acetylation and excessive deacetylase activity as epigenomic marks of prostate cancer cells. Int J Oncol. (2009) 35:1417–22. doi: 10.3892/ijo_00000459
41. Nguyen T, Sridaran D, Chouhan S, Weimholt C, Wilson A, Luo J, et al. Histone H2A Lys130 acetylation epigenetically regulates androgen production in prostate cancer. Nat Commun. (2023) 14:3357. doi: 10.1038/s41467-023-38887-7
42. Hong Z, Xiang Z, Zhang P, Wu Q, Xu C, Wang X, et al. Histone acetyltransferase 1 upregulates androgen receptor expression to modulate CRPC cell resistance to enzalutamide. Clin Trans Med. (2021) 11:e495. doi: 10.1002/ctm2.495
43. Pramanik SD, Kumar Halder A, Mukherjee U, Kumar D, Dey YN, R M. Potential of histone deacetylase inhibitors in the control and regulation of prostate, breast and ovarian cancer. Front Chem. (2022) 10:948217. doi: 10.3389/fchem.2022.948217
44. Kaushik D, Vashistha V, Isharwal S, Sediqe SA, Lin MF. Histone deacetylase inhibitors in castration-resistant prostate cancer: molecular mechanism of action and recent clinical trials. Ther Adv Urol. (2015) 7:388–95. doi: 10.1177/1756287215597637
45. Lu D, Song Y, Yu Y, Wang D, Liu B, Chen L, et al. KAT2A-mediated AR translocation into nucleus promotes abiraterone-resistance in castration-resistant prostate cancer. Cell Death Dis. (2021) 12:787. doi: 10.1038/s41419-021-04077-w
46. Zhao AN, Yang Z, Wang DD, Shi B, Zhang H, Bai Y, et al. Disturbing NLRP3 acetylation and inflammasome assembly inhibits androgen receptor-promoted inflammatory responses and prostate cancer progression. FASEB J. (2022) 36:e22602. doi: 10.1096/fj.202200673RRR
47. Kim CH, Lee DH. KAT5 Negatively regulates the proliferation of prostate cancer LNCaP cells via the caspase 3-dependent apoptosis pathway. Anim Cells Syst (Seoul). (2019) 23:253–9. doi: 10.1080/19768354.2019.1644372
48. Tavassoli P, Wafa LA, Cheng H, Zoubeidi A, Fazli L, Gleave M, et al. TAF1 differentially enhances androgen receptor transcriptional activity via its N-terminal kinase and ubiquitin-activating and -conjugating domains. Mol Endocrinol. (2010) 24:696–708. doi: 10.1210/me.2009-0229
49. Burdelski C, Ruge OM, Melling N, Koop C, Simon R, Steurer S, et al. HDAC1 overexpression independently predicts biochemical recurrence and is associated with rapid tumor cell proliferation and genomic instability in prostate cancer. Exp Mol Pathol. (2015) 98:419–26. doi: 10.1016/j.yexmp.2015.03.024
50. Weichert W, Röske A, Gekeler V, Beckers T, Stephan C, Jung K, et al. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br J Cancer. (2008) 98:604–10. doi: 10.1038/sj.bjc.6604199
51. Wang L, Zou X, Berger AD, Twiss C, Peng Y, Li Y, et al. Increased expression of histone deacetylaces (HDACs) and inhibition of prostate cancer growth and invasion by HDAC inhibitor SAHA. Am J Trans Res. (2009) 1:62–71.
52. Zhang H, Yao J, Ajmal I, Farooq MA, Jiang W. shRNA-mediated gene silencing of HDAC11 empowers CAR-T cells against prostate cancer. Front Immunol. (2024) 15:1369406. doi: 10.3389/fimmu.2024.1369406
53. Jung-Hynes B, Nihal M, Zhong W, Ahmad N. Role of sirtuin histone deacetylase SIRT1 in prostate cancer. A target for prostate cancer management via its inhibition? J Biol Chem. (2009) 284:3823–32. doi: 10.1074/jbc.M807869200. Lin R, Yang Y, Wu E, Zhou M, Wang S, Zhang Q. SIRT2 promotes cell proliferation and migration through mediating ERK1/2 activation and lactosylceramide accumulation in prostate cancer. Prostate. 2023 Jan;83(1):71-81. doi: 10.1002/pros.24437. Epub 2022 Sep 8. PMID: 36082450.
54. Damodaran S, Damaschke N, Gawdzik J, Yang B, Shi C, Allen GO, et al. Dysregulation of Sirtuin 2 (SIRT2) and histone H3K18 acetylation pathways associates with adverse prostate cancer outcomes. BMC Cancer. (2017) 17:874. doi: 10.1186/s12885-017-3853-9
55. Quan Y, Wang N, Chen Q, Xu J, Cheng W, Di M, et al. SIRT3 inhibits prostate cancer by destabilizing oncoprotein c-MYC through regulation of the PI3K/Akt pathway. Oncotarget. (2015) 6:26494–507. doi: 10.18632/oncotarget.4764
56. Cai G, Ge Z, Xu Y, Cai L, Sun P, Huang G. SIRT4 functions as a tumor suppressor during prostate cancer by inducing apoptosis and inhibiting glutamine metabolism. Sci Rep. (2022) 12:12208. doi: 10.1038/s41598-022-16610-8
57. Fu W, Li H, Fu H, Zhao S, Shi W, Sun M, et al. The SIRT3 and SIRT6 promote prostate cancer progression by inhibiting necroptosis-mediated innate immune response. J Immunol Res. (2020) 2020:8820355. doi: 10.1155/2020/8820355
58. Yang L, Jin M, Park SJ, Seo SY, Jeong KW. SETD1A promotes proliferation of castration-resistant prostate cancer cells via FOXM1 transcription. Cancers. (2020) 12:1736. doi: 10.3390/cancers12071736
59. Mentch SJ, Mehrmohamadi M, Huang L, Liu X, Gupta D, Mattocks D, et al. Histone methylation dynamics and gene regulation occur through the sensing of one-carbon metabolism. Cell Metab. (2015) 22:861–73. doi: 10.1016/j.cmet.2015.08.024
60. Kumar A, Kumari N, Nallabelli N, Prasad R. Pathogenic and therapeutic role of H3K4 family of methylases and demethylases in cancers. Indian J Clin biochem: IJCB. (2019) 34:123–32. doi: 10.1007/s12291-019-00828-x
61. Yang L, Jin M, Jeong KW. Histone H3K4 methyltransferases as targets for drug-resistant cancers. Biol (Basel). (2021) 10:581. doi: 10.3390/biology10070581
62. Pang J, Yang YW, Huang Y, Yang J, Zhang H, Chen R, et al. P110β Inhibition reduces histone H3K4 di-methylation in prostate cancer. Prostate. (2017) 77:299–308. doi: 10.1002/pros.23271
63. Vieira FQ, Costa-Pinheiro P, Almeida-Rios D, Graça I, Monteiro-Reis S, Simões-Sousa S, et al. SMYD3 contributes to a more aggressive phenotype of prostate cancer and targets Cyclin D2 through H4K20me3. Oncotarget. (2015) 6:13644–57. doi: 10.18632/oncotarget.3767
64. Cai C, He HH, Chen S, Coleman I, Wang H, Fang Z, et al. Androgen receptor gene expression in prostate cancer is directly suppressed by the androgen receptor through recruitment of lysine-specific demethylase 1. Cancer Cell. (2011) 20:457–71. doi: 10.1016/j.ccr.2011.09.001
65. Liang Y, Ahmed M, Guo H, Soares F, Hua JT, Gao S, et al. LSD1-mediated epigenetic reprogramming drives CENPE expression and prostate cancer progression. Cancer Res. (2017) 77:5479–90. doi: 10.1158/0008-5472.CAN-17-0496
66. Komura K, Jeong SH, Hinohara K, Qu F, Wang X, Hiraki M, et al. Resistance to docetaxel in prostate cancer is associated with androgen receptor activation and loss of KDM5D expression. Proc Natl Acad Sci U.S.A. (2016) 113:6259–64. doi: 10.1073/pnas.1600420113
67. Baratchian M, Tiwari R, Khalighi S, Chakravarthy A, Yuan W, Berk M, et al. H3K9 methylation drives resistance to androgen receptor-antagonist therapy in prostate cancer. Proc Natl Acad Sci U.S.A. (2022) 119(21):e2114324119. doi: 10.1073/pnas.2114324119
68. Ngollo M, Lebert A, Daures M, Judes G, Rifai K, Dubois L, et al. Global analysis of H3K27me3 as an epigenetic marker in prostate cancer progression. BMC Cancer. (2017) 17(1):261. doi: 10.1186/s12885-017-3256-y
69. Rossetto D, Avvakumov N, Côté J. Histone phosphorylation: a chromatin modification involved in diverse nuclear events. Epigenetics. (2012) 7:1098–108. doi: 10.4161/epi.21975
70. Behbahani TE, Kahl P, von der Gathen J, Heukamp LC, Baumann C, Gütgemann I, et al. Alterations of global histone H4K20 methylation during prostate carcinogenesis. BMC Urol. (2012) 12:5. doi: 10.1186/1471-2490-12-5
71. Vieira FQ, Costa-Pinheiro P, Ramalho-Carvalho J, Pereira A, Menezes FD, Antunes L, et al. Deregulated expression of selected histone methylases and demethylases in prostate carcinoma. Endocr Relat Cancer. (2013) 21:51–61. doi: 10.1530/ERC-13-0375
72. Li J, Hong Z, Zhang J, Zheng S, Wan F, Liu Z, et al. Lysine methyltransferase SMYD2 enhances androgen receptor signaling to modulate CRPC cell resistance to enzalutamide. Oncogene. (2024) 43(10):744–57. doi: 10.1038/s41388-024-02945-1
73. Kashyap V, Ahmad S, Nilsson EM, Helczynski L, Kenna S, Persson JL, et al. The lysine specific demethylase-1 (LSD1/KDM1A) regulates VEGF-A expression in prostate cancer. Mol Oncol. (2013) 7:555–66. doi: 10.1016/j.molonc.2013.01.003
74. Lee KH, Kim BC, Jeong SH, Jeong CW, Ku JH, Kim HH, et al. Histone demethylase KDM7A regulates androgen receptor activity, and its chemical inhibitor TC-E 5002 overcomes cisplatin-resistance in bladder cancer cells. Int J Mol Sci. (2020) 21:5658. doi: 10.3390/ijms21165658
75. Lin CY, Wang BJ, Chen BC, Tseng JC, Jiang SS, Tsai KK, et al. Histone Demethylase KDM4C Stimulates the Proliferation of Prostate Cancer Cells via Activation of AKT and c-Myc. Cancers. (2019) 11:1785. doi: 10.3390/cancers11111785
76. Stein J, Majores M, Rohde M, Lim S, Schneider S, Krappe E, et al. KDM5C is overexpressed in prostate cancer and is a prognostic marker for prostate-specific antigen-relapse following radical prostatectomy. Am J Pathol. (2014) 184:2430–7. doi: 10.1016/j.ajpath.2014.05.022
77. Du C, Lv C, Feng Y, Yu S. Activation of the KDM5A/miRNA-495/YTHDF2/m6A-MOB3B axis facilitates prostate cancer progression. J Exp Clin Cancer Res. (2020) 39:223. doi: 10.1186/s13046-020-01735-3
78. Metzger E, Yin N, Wissmann M, Kunowska N, Fischer K, Friedrichs N, et al. Phosphorylation of histone H3 at threonine 11 establishes a novel chromatin mark for transcriptional regulation. Nat Cell Biol. (2008) 10:53–60. doi: 10.1038/ncb1668
79. Mishra T, Singh S, Singh TG. Therapeutic implications and regulations of protein post-translational modifications in Parkinsons disease. Cell Mol Neurobiol. (2024) 44:53. doi: 10.1007/s10571-024-01471-8
80. Chen JJ, Stermer D, Tanny JC. Decoding histone ubiquitylation. Front Cell Dev Biol. (2022) 10:968398. doi: 10.3389/fcell.2022.968398
81. Thapa K, Kumar S, Sharma A, Arora S, Grewal AK, SINGH TG. Histone deacetylase inhibitors as potential therapeutic agents for various disorders. J Pharm Technology Res Manage. (2017) 5:235–25. doi: 10.15415/jptrm.2017.52014
82. Wu D, Qiu Y, Jiao Y, Qiu Z, Liu D. Small molecules targeting HATs, HDACs, and BRDs in cancer therapy. Front Oncol. (2020) 10:560487. doi: 10.3389/fonc.2020.560487
83. Welti J, Sharp A, Brooks N, Yuan W, McNair C, Chand SN, et al. Targeting the p300/CBP axis in lethal prostate cancer. Cancer Discovery. (2021) 11:1118–37. doi: 10.1158/2159-8290.CD-20-0751
84. Rana Z, Diermeier S, Hanif M, Rosengren RJ. Understanding failure and improving treatment using HDAC inhibitors for prostate cancer. Biomedicines. (2020) 8:22. doi: 10.3390/biomedicines8020022
85. Noerenberg D, Damm F. Beyond the code: the role of histone methylation in cancer resistance and therapy. Sig Transduct Target Ther. (2024) 9:161. doi: 10.1038/s41392-024-01878-1
86. Bottino C, Peserico A, Simone C, Caretti G. SMYD3: an oncogenic driver targeting epigenetic regulation and signaling pathways. Cancers. (2020) 12:142. doi: 10.3390/cancers12010142
87. Noce B, Di Bello E, Fioravanti R, Mai A. LSD1 inhibitors for cancer treatment: Focus on multi-target agents and compounds in clinical trials. Front Pharmacol. (2023) 14:1120911. doi: 10.3389/fphar.2023.1120911
Keywords: prostate cancer, epigenetics, histone modification, therapeutics, epigenetic modifications
Citation: Sharma B, Shekhar H, Sahu A, Kaur D, Haque S, Tuli HS, Sharma H and Sharma U (2025) Epigenetic insights into prostate cancer: exploring histone modifications and their therapeutic implications. Front. Oncol. 15:1570193. doi: 10.3389/fonc.2025.1570193
Received: 03 February 2025; Accepted: 27 March 2025;
Published: 28 April 2025.
Edited by:
Daekyu Sun, University of Arizona, United StatesReviewed by:
R. C. Koumar, Yenepoya University, IndiaRitika Tiwari, University of Miami, United States
Copyright © 2025 Sharma, Shekhar, Sahu, Kaur, Haque, Tuli, Sharma and Sharma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ujjawal Sharma, dWpqYXdhbC5zaGFybWFAY3VwLmVkdS5pbg==
†These authors have contributed equally to this work