 Chen Chen1†
Chen Chen1† Qijun Zhang
Qijun Zhang Haixia Shi
Haixia Shi- 1Department Traditional of Chinese Medicine (TCM), Shanghai Pudong New Area Pulmonary Hospital, Shanghai, China
- 2Department of Oncology, Integrated Traditional Chinese and Western Medicine Hospital of Shanghai Minhang District, Shanghai, China
- 3Department of Traditional Chinese Medicine, Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China
Cellular senescence exerts dual roles in lung cancer pathogenesis: initially suppressing tumorigenesis via p53/p21/p16-mediated cell cycle arrest, but promoting malignancy through the senescence-associated secretory phenotype (SASP). SASP secretes cytokines, proteases, and growth factors, reshaping the tumor microenvironment (TME) to drive immune evasion, metastasis, and therapy resistance. NF-κB activation induces APOBEC3B mutagenesis and PD-L1 overexpression, while mTOR signaling enhances glycolysis and OXPHOS to fuel tumor growth. Clinically, telomere attrition, p16/p21 expression, and SASP components serve as prognostic biomarkers. Therapeutic strategies target senescent cells and SASP. Future directions focus on single-cell multi-omics to decode senescence heterogeneity, spatially controlled drug delivery, and therapies targeting senescence-immune-metabolic crosstalk. By unraveling senescence’s dual regulatory mechanisms, this review highlights precision approaches to overcome resistance and improve lung cancer outcomes.
1 Introduction
Lung cancer remains the leading cause of global cancer mortality and the most prevalent malignant tumor, with adenocarcinoma constituting its predominant histological subtype (1). According to GLOBOCAN 2020 statistics, there were over 2.2 million new cases of lung cancer worldwide, accounting for 11.4% of all cancer cases. The number of deaths was approximately 1.79 million, representing 18% of the total cancer mortality. Among them, lung adenocarcinoma is the most common subtype of non-small cell lung cancer, accounting for about 40%–50% of NSCLC cases, with a higher incidence in women and non-smokers (2). As the most common form of non-small cell lung cancer (NSCLC), lung adenocarcinoma (LUAD) arises from malignant transformation of bronchial glandular cells, pathologically defined by glandular differentiation patterns and mucin-producing cellular architecture (3, 4). Distinct from other NSCLC subtypes, LUAD predominantly originates in peripheral lung structures including distal airways and alveoli, exhibiting characteristic histomorphological patterns such as acinar, papillary, micropapillary, and invasive mucinous adenocarcinoma (5). Clinical presentation often involves nonspecific respiratory symptoms—persistent cough, hemoptysis, dyspnea, and chest pain—frequently accompanied by constitutional manifestations like unexplained weight loss and fatigue (6). Its indolent early-stage progression explains why 20–30% of cases are incidentally detected through routine chest imaging (X-ray/CT), while over 60% present with locally advanced or metastatic disease at diagnosis (7). Epidemiologically, LUAD accounts for 40–50% of global lung cancer diagnoses, displaying unique demographic patterns: increased incidence in never-smokers, female predominance, higher prevalence in Asian populations, and elevated urban versus rural rates, potentially reflecting differential air pollution exposure (8). These epidemiological shifts, coupled with rising adenocarcinoma incidence rates, position LUAD as a critical driver of lung cancer’s persistent mortality burden (9). Despite the advancements in targeted therapies and immunotherapies that have significantly improved survival outcomes for some patients, the overall five-year survival rate for lung cancer remains below 20% (10). Particularly in advanced lung adenocarcinoma, issues such as drug resistance, recurrence, and immune evasion continue to pose significant challenges in clinical treatment, underscoring the urgent need to elucidate their molecular mechanisms and explore novel therapeutic strategies.
Molecular pathogenesis of LUAD centers on dysregulation of proliferative signaling cascades mediated by driver mutations (11). Epidermal growth factor receptor (EGFR) mutations represent the most prevalent oncogenic drivers, occurring in 10–50% of cases depending on population ethnicity and smoking status (12). Mutant EGFR acquires ligand-independent tyrosine kinase activity, constitutively activating downstream effectors including mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK, pro-survival signaling), phosphatidylinositol 3-kinase/protein kinase B (PI3K/AKT, anti-apoptotic signaling), and janus kinase/signal transducer and activator of transcription (JAK/STAT, proliferative/invasive regulation) pathways (13). Concurrently, PI3K/AKT/mTOR pathway hyperactivation promotes tumor metabolism and survival through enhanced glucose utilization and protein synthesis (14). Wnt/β-catenin signaling aberrations further contribute by sustaining cancer stem cell populations and apoptotic resistance (15). Moreover, kirsten rat sarcoma viral oncogene (KRAS) mutations, rearranged during transfection gene (RET) fusions, and ROS proto-oncogene 1 (ROS1) rearrangements represent pivotal oncogenic drivers in LUAD. KRAS mutations constitute the most prevalent driver mutation in EGFR-negative LUAD, notably with the smoking-associated KRAS G12C subtype predominating, accounting for around 13% of all NSCLC cases. By activating multiple downstream signaling pathways, including MAPK, PI3K/AKT, and RalGDS, KRAS mutations facilitate tumor proliferation, migration, and immune evasion (16). RET fusions predominantly occur in younger LUAD patients who are either non-smokers or light smokers, with an incidence rate of approximately 1–2%, and they are closely linked with accelerated tumor progression. RET fusions frequently result in fusion proteins with partner genes such as kinesin family member 5B (KIF5B) or coiled-coil domain containing 6 (CCDC6), thereby activating the MAPK and STAT pathways. As highly selective RET inhibitors, Selpercatinib and Pralsetinib have demonstrated remarkable objective response rates and favorable safety profiles in advanced RET fusion-positive LUAD, subsequently being incorporated into the recommended first-line treatment regimens (17). ROS1 fusion proteins propel tumor progression by engaging signaling pathways including PI3K/AKT and JAK/STAT. Crizotinib stands as the first approved targeted therapeutic for ROS1-rearranged NSCLC, demonstrating an efficacy rate exceeding 70% (18).
Similarly, epigenetic mechanisms, including DNA methylation, histone modifications and the dysregulation of non-coding RNAs (such as microRNAs and long non-coding RNAs), play pivotal roles in the initiation and progression of lung adenocarcinoma (19), while epigenetic dysregulation via abnormal DNA methylation, histone modifications, and non-coding RNA expression (miR-21 overexpression and lncRNA HOTAIR dysregulation) drives malignant transformation without altering genomic sequences (20).
The biological continuum of aging intersects critically with LUAD pathogenesis through cellular senescence mechanisms. Aging involves progressive functional decline across organ systems, mediated by hallmarks including genomic instability, telomere attrition, epigenetic drift, and mitochondrial dysfunction (21, 22). Cellular senescence—a permanent cell-cycle arrest triggered by oncogenic stress, DNA damage, or tumor suppressor activation—exerts context-dependent tumor-modulating effects (23). While initially tumor-suppressive by halting malignant transformation, senescent cells develop a senescence-associated secretory phenotype (SASP) (24), releasing inflammatory cytokines (IL-6, IL-8), growth factors (TGF-β), and proteases that remodel the tumor microenvironment (TME) (25). SASP components induce paracrine senescence in adjacent cells, recruit immunosuppressive myeloid cells, and paradoxically promote angiogenesis and metastasis through TME modulation (26, 27).
Emerging evidence positions senescence as a dual-axis regulator in LUAD progression (28). Lin et al. developed a 16-gene senescence-related signature (SRS) demonstrating that SASP-mediated immune microenvironment remodeling predicts immunotherapy response and survival outcomes (29). Complementary transcriptomic analyses of 278 senescence-associated genes revealed distinct senescence subtypes correlated with differential immune infiltration patterns in LUAD (30). These findings illuminate senescence as a dynamic interface between tumor biology and immune regulation, offering novel therapeutic targets—particularly for immunotherapy-resistant LUAD subtypes where SASP factors may mediate immune evasion. This review aims to systematically summarize the current understanding of cellular senescence in lung adenocarcinoma, with an emphasis on its dual roles in both tumor suppression and promotion. We particularly focus on the SASP and how it impacts tumor progression, immune modulation, and therapy resistance. Additionally, we discuss potential therapeutic opportunities and challenges in this context.
2 Association of cellular senescence with lung cancer
2.1 Telomere attrition
Telomere attrition serves as a critical nexus between cellular aging and lung carcinogenesis, driving chromosomal instability while paradoxically influencing tumor-suppressive and oncogenic pathways (31). Telomeres—terminal chromosomal regions composed of repetitive TTAGGG sequences and stabilized by shelterin protein complexes (TRF1/TRF2, TPP1)—prevent aberrant DNA repair by masking chromosomal ends from damage recognition systems (32). In somatic cells, the end-replication problem results in progressive telomere shortening (50–200 bp per division), culminating in replicative senescence when critical length thresholds (Hayflick limit) are breached; This triggers DNA damage response (DDR) activation through ataxia-telangiectasia mutated/ATM and rad3-related (ATM/ATR) kinases, stabilizing p53 to induce p21-mediated cell cycle arrest—a fundamental tumor-suppressive mechanism (33).
Contrasting this protective role, 85–90% of lung cancers exhibit pathological telomerase reactivation via telomerase reverse transcriptase (TERT, catalytic subunit) overexpression and telomerase RNA component (TERC, RNA template) dysregulation, enabling replicative immortality; Reactivation mechanisms include recurrent TERT promoter mutations (C228T/C250T) and epigenetic remodeling of telomere maintenance genes (34). Paradoxically, despite telomerase activity, lung tumors frequently display ongoing telomere attrition, generating chromosomal fusions and breakage-fusion-bridge cycles that amplify oncogenic signaling through PI3K/AKT and RAS-MAPK pathways while enhancing immune evasion via programmed death protein 1 (PD-L1)/PD-1 axis upregulation (35). Telomere attrition can elicit a persistent DNA damage response, thereby activating the ATM/ATR-checkpoint kinase 1 (CHK1) pathway and facilitating immune surveillance evasion by upregulating PD-L1 expression, ultimately promoting tumor cell immune tolerance and progression (36). Telomere disruption and the consequent loss of telomere-binding protein functionality can also activate cell survival signaling via the PI3K/AKT pathway, thereby enhancing tumor cell adaptability to oxidative stress and nutrient-poor conditions (37, 38). Clinically, leukocyte telomere length demonstrates bidirectional associations with lung adenocarcinoma risk: longer telomeres in peripheral blood correlate with heightened susceptibility, potentially reflecting inherited telomere maintenance defects or accelerated age-related shortening (39, 40). Mechanistically, while elongated telomeres can delay replicative senescence and extend the proliferative lifespan of somatic cells, they may also elevate the risk of accumulating genetic and epigenetic alterations under carcinogenic exposures, such as tobacco smoke, thereby increasing the potential for oncogenesis (41). Elongated telomeres are frequently accompanied by increased telomerase activity, which not only maintains chromosomal stability but also interacts with oncogenic pathways, such as MYC and TERT promoter mutations (42). This dual role positions telomere dynamics as both a biomarker and therapeutic target. Emerging strategies include TERT promoter inhibition using oligonucleotide antagonists such as GRN163L, telomerase splicing modulation through NOVA1-dependent alternative splicing blockade, and senescence-targeted therapies that combine senolytics (navitoclax) with SASP pathway inhibitors to mitigate pro-tumorigenic microenvironment effects, all of which represent promising approaches to advance lung cancer treatment (Shown in Figure 1) (43).
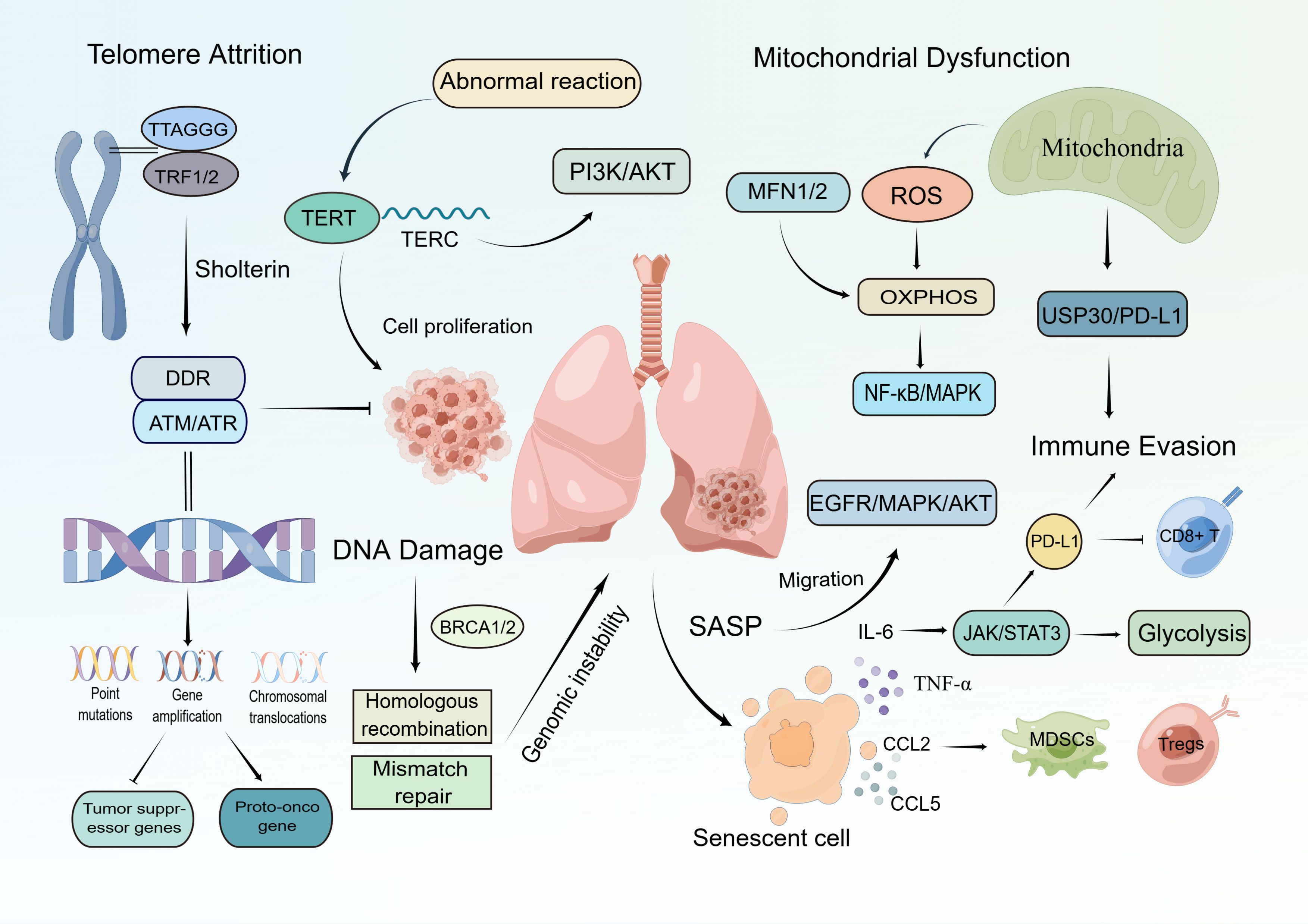
Figure 1. The Role of Cellular Senescence in Lung Cancer Progression. This figure illustrates the biological network through which cellular senescence drives lung cancer progression. (1)Telomere Attrition: Shortening of TTAGGG sequences and TRF1/2 imbalance disrupts Shelterin complex protection, activating ATM/ATR-mediated DDR. This induces genomic instability, leading to oncogene activation (EGFR, KRAS) and tumor suppressor inactivation (BRCA1/2); (2)DNA Damage: Defective homologous recombination repair (BRCA1/2) and mismatch repair (MMR) accelerate malignant clonal evolution; (3)Mitochondrial Dysfunction: MFN1/2 abnormalities elevate ROS, activating NF-κB/MAPK pathways to enhance cancer cell migration. Simultaneously, increased OXPHOS supports cancer stem cell survival; (4)Immune Evasion: ROS and PD-L1 suppress CD8+ T cell function, while USP30-mediated glycolytic reprogramming further weakens antitumor immunity; (5)Senescence-Associated Secretory Phenotype (SASP): Senescent cells secrete IL-6, TNF-α, and CCL2/5, recruiting MDSCs and Tregs to create an immunosuppressive microenvironment. This synergizes with EGFR/MAPK/AKT signaling to promote tumor growth and metastasis.
2.2 Carcinogenic effects of DNA damage and mutation accumulation
The accumulation of DNA damage and mutations constitutes a pivotal carcinogenic mechanism, driven by structural genomic alterations from endogenous sources—including replication errors and oxidative stress—and exogenous environmental carcinogens such as ultraviolet radiation and chemical agents (44).Unrepaired DNA lesions induce oncogenic transformation through point mutations, gene amplifications, or chromosomal translocations, which activate proto-oncogenes via gain-of-function mutations or inactivate tumor suppressors through loss of heterozygosity and epigenetic silencing (45). Compromised DNA repair pathways further amplify genomic instability: for example, breast cancer susceptibility gene 1/2(BRCA1/2) mutations disrupt homologous recombination (HR) repair, forcing reliance on error-prone mechanisms like non-homologous end joining, thereby accelerating oncogenic mutation accrual, while mismatch repair (MMR) deficiencies propagate microsatellite instability (46). Deficiency in HR repair serves as a principal driver of genomic instability across various tumor types. In LUAD, TP53 and KRAS mutations are intricately associated with defects in HR repair mechanisms. The loss of TP53 function compromises DNA damage checkpoint control, thereby synergistically promoting genomic instability and increased reliance on HR pathways. Conversely, KRAS mutations elevate ROS levels, thereby intensifying DNA damage stress and compelling tumor cells to rely more heavily on residual HR mechanisms for survival (47, 48). On the other hand, while MMR deficiencies are relatively uncommon in LUAD, their presence often results in a high tumor mutational burden (TMB), facilitating neoantigen formation and increasing sensitivity to immune checkpoint inhibitors (ICIs). The loss of function in core MMR genes such as muts homolog 2 (MSH2) and muts homolog 1 (MSH1) can lead to the upregulation of PD-L1 expression while activating the cyclic GMP-AMP synthase-stimulator of interferon genes (cGAS-STING) pathway, thereby inducing IFN-γ signaling to enhance tumor immunogenicity (49). PARP inhibitors obstruct the repair of single-strand breaks, thereby compelling cells to rely on the HR pathway to rectify DNA damage. Consequently, in tumor cells harboring BRCA1/2 mutations, where HRR function is compromised, the action of PARP inhibitors leads to the accumulation of DNA damage, triggering apoptosis—a phenomenon known as “synthetic lethality” (50).
Emerging approaches focus on replication stress mitigation using ATR/CHK1 inhibitors to bypass therapy resistance, alongside novel agents disrupting DNA damage tolerance pathways, collectively advancing precision oncology paradigms that capitalize on repair pathway dysregulation (51, 52). ATR and CHK1 are key kinases within the DDR network, primarily responding to replication stress and single-stranded DNA (ssDNA) damage. In LUAD, frequent mutations in genes such as TP53, KRAS, and ATM result in increased reliance of tumor cells on the ATR/CHK1 pathway (53). Ceralasertib (AZD6738), an ATR kinase inhibitor, markedly increases apoptosis, induces G2/M arrest, and enhances p21 expression while reducing CDC2 levels in SNU478 and SNU869 cell lines, demonstrating enhanced antitumor activity when combined with paclitaxel (54, 55). Prexasertib (LY2606368) is a substrate ATP competitive selective inhibitor of CHK1 and checkpoint kinase 2 (CHK2). In a phase I clinical trial involving patients with advanced squamous cell carcinoma, prexasertib monotherapy exhibited notable antitumor activity, with some patients achieving disease control after 3 months (56).
2.3 Mitochondrial dysfunction and cancer cell metabolism
Mitochondrial dysfunction drives cancer metabolic reprogramming by enabling survival advantages through energy metabolism remodeling, oxidative stress modulation, apoptosis evasion, and anabolic precursor synthesis (57). Although the Warburg effect historically dominated cancer metabolism paradigms, recent studies demonstrate that oxidative phosphorylation (OXPHOS) sustains the survival of therapy-resistant tumor subpopulations and metastatic cancer stem cells (58)—a phenomenon exemplified by glioblastoma stem cells that maintain immortality through mitochondrial fusion-mediated OXPHOS enhancement and NAD+ metabolic rewiring (59).This metabolic plasticity underpins therapeutic challenges, as lung cancers with elevated OXPHOS activity exhibit immunotherapy resistance, prompting the development of precision strategies like the OXPHOS inhibitor IACS-010759 to target refractory malignancies (60).
Mitochondrial reactive oxygen species (ROS) exhibit context-dependent oncogenic roles: mtDNA mutations or electron transport chain defects induce ROS overproduction, activating nuclear factor kappa-B (NF-κB) and MAPK pathways to drive lung cancer metastasis (61), while pharmacologic ROS modulation exerts antitumor effects. For instance, metformin suppresses ROS via complex I inhibition to sensitize tumors to chemotherapy, whereas pro-oxidant therapies exploit ROS overload to eliminate cancer stem cells (62). Parallel mechanisms involve mitochondrial regulation of apoptosis—overexpression of anti-apoptotic BCL-2 proteins (e.g., in lymphomas and breast cancers) blocks cytochrome c-mediated apoptosome activation, a vulnerability successfully targeted by the BCL-2 inhibitor venetoclax (63).
Mitochondrial metabolism plays a pivotal regulatory role in shaping the tumor immune microenvironment (TIME), significantly influencing immune evasion and antitumor immune responses (64). Tumor cells, by enhancing oxidative phosphorylation (OXPHOS) and aerobic glycolysis metabolism, accelerate nutrient consumption and produce lactate, leading to glucose and oxygen scarcity in TIME and the formation of an acidic microenvironment. This suppresses CD8+ effector T cell activity and promotes the expansion of regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs), creating an immunosuppressive niche (65). On the other hand, the proliferation and sustained functionality of effector and memory CD8+ T cells depend on mitochondrial OXPHOS and the tricarboxylic acid cycle (TCA). Mitochondrial dysfunction, such as loss of membrane potential and accumulation of reactive oxygen species (ROS), can lead to T cell exhaustion, closely related to the upregulation of immune checkpoint molecules like PD-1 (66). Therefore, targeting mitochondrial metabolism to remodel TIME and enhance T cell-mediated immune responses has become a key research direction in tumor immunotherapy. For instance, inhibiting pyruvate dehydrogenase kinase (PDK) can enhance TCA activity, promote acetyl-CoA production, and lead to increased histone acetylation, thereby boosting PD-L1 expression on tumor cells (67). In a triple-negative breast cancer (TNBC) mouse model, combined treatment with metformin and PD-1 antibodies significantly inhibited tumor growth and metastasis, increased CD8+ T cell infiltration, and reduced PD-L1 expression, indicating synergistic antitumor effects of the combination (68).
2.4 Promoting role of SASP in the carcinogenic microenvironment
The senescence-associated secretory phenotype (SASP) drives tumor progression through a multifaceted molecular network—comprising cytokines (IL-6, IL-8), chemokines (CXCL1, CCL2), proteases (MMPs), and growth factors (VEGF, TGF-β)—that remodels the tumor microenvironment (TME) into a pro-carcinogenic niche (69). SASP components directly amplify tumor proliferation and invasion: EREG/EGFR signaling activation via the MAPK/AKT axis mediates chemotherapy-induced progression in prostate cancer (70), while MMP1 and MMP3 degrade extracellular matrix (ECM) components to facilitate glioblastoma and lung cancer metastasis (71). Concurrently, SASP reprograms cancer metabolism; IL-6-induced STAT3 activation shifts energy production toward glycolysis while suppressing oxidative phosphorylation (OXPHOS), thereby fueling rapid tumor growth (72).
immune evasion (73). In KRAS-mutant lung cancer, senescent macrophages secrete CCL2 to recruit MDSCs, while IL-10 and TGF-β polarize tumor-associated macrophages (TAMs) toward an immunosuppressive M2 phenotype, crippling cytotoxic T cell activity (74). This immunosuppressive axis is reinforced by PD-L1 upregulation: IL-6 and VEGF activate PD-L1 expression on tumor cells, impairing NK cell function and CD8+ T cell-mediated cytotoxicity (75). Beyond immune modulation, SASP reshapes the stromal architecture by inducing fibrotic barriers. Cancer-associated fibroblasts (CAFs) secrete collagen and fibronectin under SASP influence, while lysyl oxidase (LOX)-mediated ECM crosslinking increases tissue stiffness, activating integrin-FAK signaling to accelerate metastasis and confer therapy resistance (76).
2.5 Regulation of immune evasion and inflammatory response by senescence
Cellular senescence orchestrates immune evasion and chronic inflammation in lung cancer through SASP-mediated immunosuppression and mitochondrial dysfunction (28).SASP-derived pro-inflammatory cytokines (IL-6, IL-8, TNF-α) and chemokines (CCL2, CXCL1) directly suppress antitumor immunity: IL-6 activates JAK/STAT3 signaling to upregulate PD-L1 expression on tumor cells and dendritic cells, inducing CD8+ T cell exhaustion (77). This mechanism corroborated by the 30% higher PD-L1 expression prevalence in elderly lung cancer patients compared to younger counterparts (78). Chemokine-driven immunosuppression is exemplified in KRAS-mutant lung cancer, where senescent cells recruit MDSCs and regulatory Tregs via CCL2 secretion, establishing an immune-privileged niche (79).
Senescent lung cancer cells further sabotage immune function through mitochondrial hijacking. Mutant mitochondria are transferred to T cells via tunneling nanotubes (TNTs), reducing T cell oxidative phosphorylation activity by 60% while activating the USP30-PD-L1 axis to amplify immune evasion—a process demonstrating direct crosstalk between metabolic dysfunction and checkpoint signaling (80). Concurrently, senescence-associated chronic inflammation fuels tumor progression through genomic destabilization: apolipoprotein B mRNA editing enzyme catalytic subunit 3B (APOBEC3B)-mediated cytosine deamination elevates mutation burden (2.5-fold higher in elderly patients) while NF-κB activation sustains pro-tumorigenic cytokine release (81). Therapeutic inhibition of NF-κB in aged preclinical models reduces lung tumor volume by 70%, underscoring the pathway’s centrality in senescence-driven malignancy (82). Collectively, these mechanisms drive immune surveillance failure and aggressive progression within the senescent lung cancer microenvironment (Shown in Table 1).
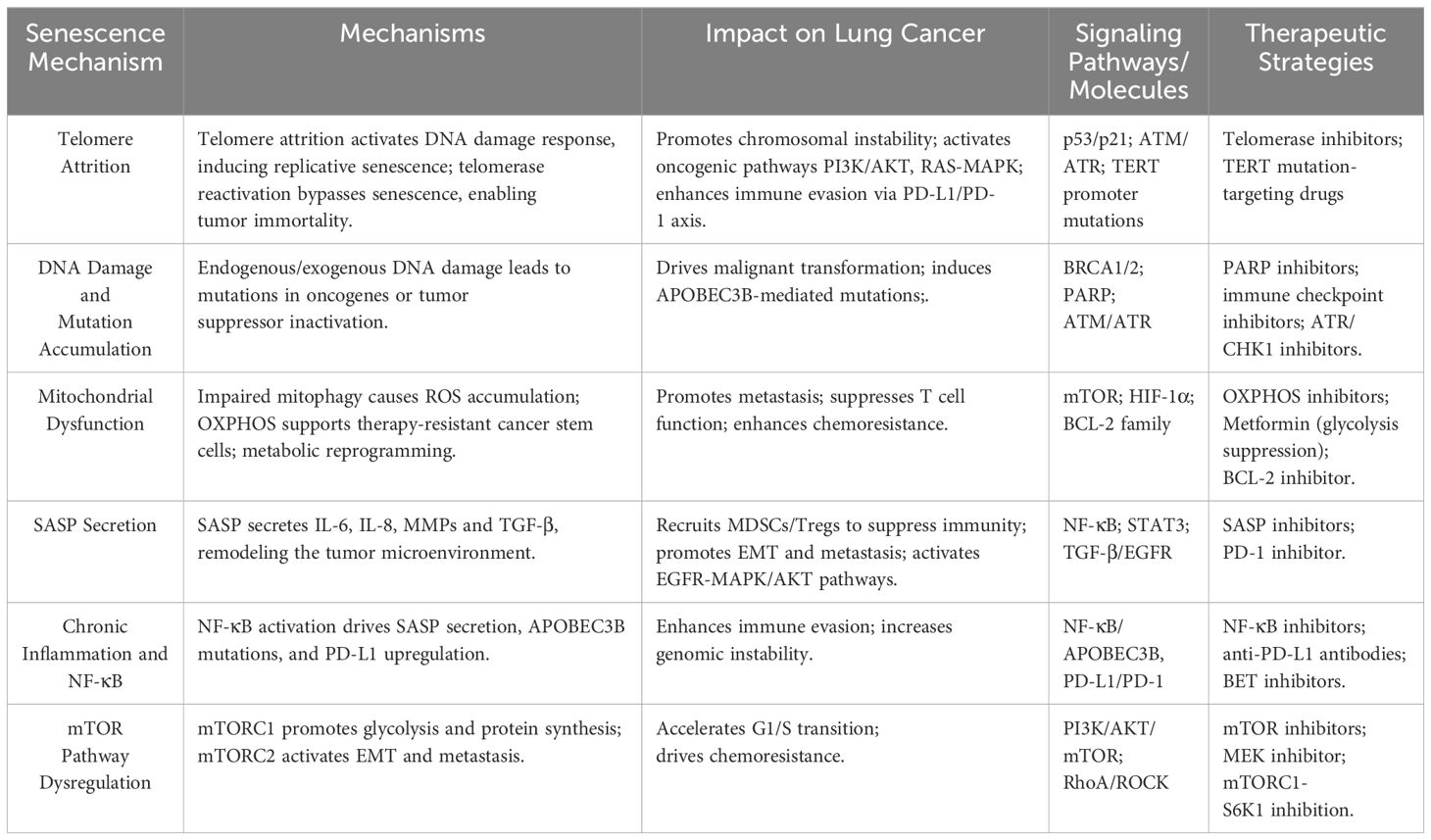
Table 1. Mechanisms of the impact of cellular senescence on lung cancer.
3 Cellular senescence and lung cancer-related signaling pathways
3.1 Dual Roles of the p53/p21/p16 Signaling Pathway
The p53/p21/p16 axis exerts context-dependent tumor-suppressive and oncogenic effects in lung cancer through dynamic molecular crosstalk. Canonically, wild-type p53 activates p21 (cyclin dependent kinase Inhibitor 1A, CDKN1A) to enforce G1/S cell cycle arrest, enabling DNA repair or apoptosis initiation (83). This mechanism impaired in ~50% of lung cancers harboring TP53 mutations (84). Mutant p53 acquires oncogenic functions via epigenetic remodeling, including senescence-associated heterochromatin foci (SAHF) formation, which derepresses MYC transcription while silencing the cyclin dependent kinase Inhibitor 2A (CDKN2A) locus encoding p16 (85).
The p21 demonstrates paradoxical roles contingent on p53 status. In p53-wildtype tumors, p21-mediated CDK2/4 inhibition enhances chemotherapy response (86). Conversely, mutant p53 redirects p21 to upregulate RAD21, promoting homologous recombination repair and cisplatin resistance (87). Microenvironmental cues further modulate p21 activity: EGFR-STAT3 signaling phosphorylates p21 to induce its cytoplasmic translocation, where the p21/STAT3 complex activates AKT-mTOR signaling to drive metastasis (88).
The p16 (CDKN2A) inactivation—primarily through promoter hypermethylation—represents a hallmark of lung cancer progression, enabling cell cycle dysregulation via CDK4/6-RB pathway activation (89). Paradoxically, p16 loss upregulates telomerase (hTERT) to immortalize tumor cells, whereas its overexpression induces senescence, highlighting its dual regulatory capacity (90). Clinically, p16/p21 co-expression patterns predict immunotherapy efficacy: NSCLC patients with low p16 expression exhibit elevated PD-L1 levels but paradoxically inferior responses to PD-1 inhibitors, suggesting p16 loss primes an immune-evasive phenotype resistant to checkpoint blockade (91). These findings underscore the pathway’s complexity as a therapeutic determinant in lung cancer.
3.2 Cross-talk of NF-κB in lung cancer and cellular senescence
The NF-κB pathway functions as a molecular nexus linking cellular senescence to lung cancer progression through dual pro-survival and pro-inflammatory mechanisms. In senescent cells, DNA damage triggers ATM/ATR kinase activation, which phosphorylates the IKK complex to degrade IκBα, enabling nuclear translocation of the p65/p50 heterodimer—a prerequisite for SASP factor transcription (IL-6, IL-8, MMP9) that fosters a tumor-promoting inflammatory niche (92). Lung cancer cells amplify this cascade autonomously via TNF-α and HMGB1 secretion, creating a self-reinforcing NF-κB activation loop (93).
NF-κB-driven immune evasion operates through PD-L1 upregulation on tumor cells and stromal elements, suppressing CD8+ T cell cytotoxicity while recruiting myeloid-derived suppressor cells (MDSCs) and Tregs to establish an immunosuppressive barrier—a mechanism validated in therapy-resistant NSCLC subtypes (94). Concurrently, NF-κB exacerbates genomic instability by elevating APOBEC3B deaminase activity, inducing mutagenic C-to-T transitions in driver genes (EGFR, KRAS) that accelerate clonal evolution (95). Pro-inflammatory stimuli, such as TNF-α or IL-1β, activate NF-κB, which then binds to the promoter region of the APOBEC3B gene, resulting in increased transcriptional output. Elevated levels of APOBEC3B lead to widespread cytosine deamination in single-stranded DNA, contributing to a hypermutator phenotype and increased intratumoral heterogeneity. Recent studies have confirmed this axis in various cancers, including LUAD, linking chronic inflammation to tumor evolution through NF-κB-APOBEC3B-driven mutagenesis (96). Radiotherapy can induce nuclear translocation of NF-κB transcription factors, such as p65, by activating the ATM/IKK axis, thereby upregulating stemness genes like SOX2, NANOG, and ALDH1, and promoting self-renewal and survival of cancer stem cells (CSCs). Studies indicate that NF-κB activation is closely related to the enrichment of CSCs following radiotherapy, and its inhibition can significantly reverse CSC-associated phenotypes (97, 98). Furthermore, NF-κB drives the expression of pro-inflammatory factors such as IL-6, TNF-α, and CXCL1/2, promoting the infiltration of immunosuppressive cells like Tregs and MDSCs, and inducing upregulation of PD-L1. This culminates in the formation of TIME, weakening CD8+ T cell functionality and responsiveness to immunotherapy (99).
3.3 Regulation of the mTOR signaling pathway in cellular senescence and tumor development
The mTOR pathway functions as a metabolic integrator with dichotomous roles in cellular senescence and lung cancer progression, governed by its distinct complexes mTORC1 and mTORC2 (100). mTORC1 hyperactivation impairs mitophagy, causing accumulation of dysfunctional mitochondria and ROS overproduction, which drives p21-dependent senescence (101). Pharmacologic mTORC1 inhibition (rapamycin) reduces senescence-associated β-galactosidase (SA-β-Gal)-positive cells by 60% and rescues mitophagy, as demonstrated in in vitro senescence models (102). In contrast, mTORC2 exacerbates oxidative stress by suppressing SOD2 and catalase expression via AKT-mediated FOXO inactivation (103). Preclinical studies in lung preneoplasia show that mTORC2-selective inhibitor PP242 restores SOD2 levels to 80% of baseline and attenuates senescence-associated fibrosis by blocking FOXO3a phosphorylation (104). However, during lung cancer progression, aberrant activation of the mTOR pathway strongly promotes tumor malignancy (105). In established lung cancers, mTORC1 phosphorylates 4E-BP1/S6K1 to enhance ribosome biogenesis and oncoprotein synthesis (cyclin D1, c-MYC), accelerating G1/S progression (106). Clinically, 65% of lung tumors exhibit elevated p-S6K1 (indicating mTORC1 hyperactivity), correlating with poor prognosis (107). Studies show that mTORC2 is significantly activated in EGFR-mutant non-small cell lung cancer (NSCLC), and its functional upregulation is closely associated with tumor invasiveness, epithelial-mesenchymal transition (EMT), and TKI resistance (108). Furthermore, mTORC2 can enhance tumor cell metabolic adaptability by upregulating c-Myc and HIF-1α, thereby further promoting survival advantage under hypoxic conditions or treatment pressure (109). mTORC1 upregulates HIF-1α-dependent GLUT1 and LDHA to potentiate glycolysis, while mTORC2 enhances lipid biosynthesis via ACC activation, fulfilling anabolic demands of proliferating tumors (110, 111). These dual roles position mTOR as a context-dependent regulator: constraining senescence via metabolic homeostasis in pre-malignant states, yet driving malignancy through proliferative, invasive, and metabolic rewiring in advanced disease. In terms of treatment, although mTOR inhibitors such as rapamycin and its derivatives have entered clinical trials, they are mostly selective for mTORC1. Long-term use often induces feedback activation of the mTORC2-AKT pathway, limiting efficacy and potentially promoting resistance (112). Currently, rational combination strategies, such as pairing mTOR inhibitors with EGFR-TKI, PD-1/PD-L1 antibodies, or metabolic inhibitors, are considered key directions in enhancing efficacy and overcoming resistance (113).
4 Cellular senescence and lung cancer treatment
In lung cancer therapy, chemotherapy- or radiotherapy-induced senescent cells drive treatment resistance through senescence-associated secretory phenotype (SASP) activation. Senescent cells within the tumor microenvironment secrete pro-inflammatory cytokines, chemokines, and matrix remodeling enzymes, fostering chronic inflammation and immunosuppressive signaling (114). A key mechanism involves post-chemotherapy fibroblasts transferring zinc ions to cancer cells via the ZRT/IRT-like protein 1-connexin 43 (ZIP1-CX43) axis, which upregulates ABCB1-mediated drug efflux pumps to confer platinum resistance. ZIP1, as a zinc ion transporter, is extensively involved in maintaining intracellular zinc homeostasis, oxidative stress response, and metabolic regulation. Studies have shown that ZIP1 is underexpressed in various tumors, including prostate cancer and lung cancer, and is closely associated with metabolic reprogramming and apoptosis inhibition of tumor cells (115). Recently, studies focusing on the cooperative regulation between ZIP1 and gap junction protein CX43 have gained attention. ZIP1’s role in upregulating CX43 to form gap junctions between fibroblasts and lung cancer cells, facilitating zinc transfer and leading to chemotherapy resistance, has been highlighted (116). SASP factors further promote immune evasion: IL-6 enhances PD-L1 expression through STAT3 signaling to suppress CD8+ T cell activity, while CCL2 recruits MDSCs and regulatory Tregs, amplifying immunosuppression (116, 117). Senescent stromal cells exacerbate resistance by secreting serine peptidase inhibitor kazal type 1 (SPINK1), which activates the EGFR/STAT3 axis to inhibit apoptosis and stimulate metastasis (118). SPINK1 is a serine protease inhibitor that primarily inhibits trypsin activity under normal physiological conditions. Recent studies have shown that SPINK1 is abnormally overexpressed in prostate, pancreatic, and lung cancers, and is involved in regulating EGFR pathway activity, anti-apoptosis, and tumor stemness maintenance (119). In NSCLC, high SPINK1 expression is associated with poor prognosis in patients. SPINK1 promotes tumor cell growth and inhibits apoptosis by maintaining cellular redox homeostasis through activation of the nuclear factor erythroid 2-related factor 2 (NRF2) pathway; SPINK1 can also enhance migration and invasion capabilities of lung adenocarcinoma cells by upregulating the expression of matrix metalloproteinase 12 (120, 121). This interplay between SASP secretion, immune modulation, and metabolic remodeling underscores the critical role of senescent cells in driving therapeutic resistance and tumor progression in lung cancer.
Therapeutic elimination of senescent cells using senolytics has emerged as a strategy to overcome resistance (122). The BCL-2 inhibitor Navitoclax selectively targets chemotherapy-induced senescent lung cancer cells, demonstrating efficacy in preclinical models (123). In KRAS-mutant tumors, combining Navitoclax with PD-1 inhibitors elevates complete remission rates from 15% to 60% (124). Navitoclax has demonstrated the capability to eliminate senescent cells in clinical trials targeting idiopathic pulmonary fibrosis and myelofibrosis, such as NCT03289771 and NCT04592885 (125, 126). However, in oncological applications, the utility of Navitoclax is markedly constrained by dose-limiting thrombocytopenia (127). Similarly, Dasatinib-Quercetin co-treatment clears senescent fibroblasts by inhibiting SRC kinase and PI3K/AKT signaling, restoring T cell-mediated antitumor responses (128). The combined treatment strategy of Dasatinib-Quercetin has been validated in managing non-cancerous age-related conditions such as chronic kidney disease and osteoarthritis (NCT02848131) (129). In oncological models, Dasatinib-Quercetin has shown efficacy in eliminating chemotherapy-induced senescent cells and in retarding disease progression (130) However, in the domain of solid tumors, this approach remains in the early clinical stages, with long-term safety and efficacy requiring further evaluation.
SPINK1-neutralizing monoclonal antibodies block SASP-induced EGFR activation, synergizing with carboplatin to enhance cytotoxicity (118). In studies of hepatocellular carcinoma, SPINK1 neutralizing antibodies significantly downregulate VEGF and phosphorylated EGFR levels, thereby inhibiting tumor angiogenesis and the EMT process, subsequently delaying tumor progression (127). In a murine model of castration-resistant prostate cancer (CRPC), SPINK1 monoclonal antibodies markedly reduce the expression of neuroendocrine markers such as SYN and CHGA within tumors (131). CDK4/6 inhibitors, such as Palbociclib, Ribociclib, and Abemaciclib, have achieved significant advancements in the treatment of HR+/HER2- breast cancer by blocking the cell cycle transition from G1 to S phase (132). However, tumor cells may circumvent CDK4/6 inhibition by upregulating the expression of CDK2, CCNE1, or E2F target genes, leading to treatment failure (133). Additionally, CDK4/6 inhibitors are metabolized via CYP3A4 and share metabolic pathways with various chemotherapeutic agents, which may result in abnormal drug plasma concentrations and increase the risk of adverse effects (134). Future directions include integrating single-cell metabolomics and spatial transcriptomics to map SASP regulatory networks (135), enabling precision strategies such as dual PD-1/SPINK1 checkpoint blockade or metabolic reprogramming with agents like metformin (136). Nevertheless, the field remains constrained by several technical challenges. Current mass spectrometry platforms often struggle with low metabolite abundance and limited dynamic range at the single-cell level, potentially compromising quantification accuracy (137). Moreover, the spatial and temporal resolution of metabolomic analysis remains insufficient, particularly in tissue contexts with complex microenvironments, such as lung tumors (137). Bioinformatics pipelines for integrating single-cell metabolomics data with transcriptomics or proteomics datasets are still under development, limiting interpretability (138). Addressing these bottlenecks is vital for realizing the full potential of SASP network analysis in mechanistic and clinical research.
5 Discussion
The dual roles of cellular senescence in lung cancer—acting as a tumor-suppressive mechanism via p53/p21/p16-mediated cell cycle arrest while driving malignancy through SASP-mediated inflammation—underscore its context-dependent impact on disease progression (139). SASP factors such as IL-6, IL-8, and MMPs activate oncogenic NF-κB and STAT3 signaling, with NF-κB upregulating APOBEC3B to induce EGFR/KRAS mutagenesis (140, 141), and STAT3 enhancing PD-L1 expression to suppress T cell cytotoxicity (142, 143). In NSCLC, SASP exhibits typical pro-inflammatory characteristics, primarily including factors such as IL-6, IL-8, CXCL1, and MMPs. These secretions can significantly enhance tumor invasiveness and heterogeneity by inducing EMT, promoting angiogenesis, and activating proliferative cancer-adjacent cells (144). Additionally, SASP can attract MDSCs and Tregs, shaping an immunosuppressive tumor microenvironment, thereby weakening the efficacy of immune checkpoint inhibitors (145). These mechanisms offer a therapeutic window for targeting SASP, especially in patients undergoing chemotherapy or radiotherapy that induces senescence, where senolytic drugs may help reduce recurrence and increase responses to immunotherapy (146). In contrast, SCLC is usually accompanied by the loss of p53 and Rb pathways, making it difficult for cells to enter the classic senescence program and hence lacking the typical SASP phenotype (147). Nevertheless, some studies indicate that SCLC can still exhibit atypical SASP-like phenotype post-treatment, with the released signaling factors potentially affecting tumor plasticity and cellular state transitions, such as neuroendocrine transdifferentiation (148). The high mutational burden of SCLC does not correlate with immunogenicity, possibly due in part to evasion of immune recognition by mechanisms such as downregulation of MHC-I, rather than relying on SASP’s constructed immunosuppressive network (149). Therefore, in SCLC, strategies targeting SASP have yet to show distinct clinical advantages, but the concept of inducing senescence or mimicking SASP to inhibit tumor activity still holds research potential.
Concurrently, mitochondrial dysfunction in senescent cells promotes metabolic reprogramming and ROS accumulation, fostering cancer stem cell survival and chemotherapy resistance. This is exacerbated by immunosenescence, exemplified by mitochondrial transfer to T cells via tunneling nanotubes, which cripples antitumor immunity and establishes a “metabolic-immune” barrier (150, 151). Senescence-associated biomarkers provide critical prognostic and therapeutic insights. Telomere length and TERT activity stratify immunotherapy responsiveness, with longer telomeres paradoxically correlating with poorer outcomes (152). Combined p16/p21 expression analysis predicts efficacy of immune checkpoint inhibitors (153), while dynamic monitoring of SASP factors (IL-6, CCL2) and APOBEC3B mutation burden—2.5-fold higher in elderly patients—guides synthetic lethality strategies like PARP inhibition (154). Single-cell sequencing has identified senescence-related gene signatures (senescence risk score, SRS) that enable molecular subtyping for precision therapy (155).
Therapeutic strategies targeting senescence focus on three pillars: senolytic elimination, SASP inhibition, and metabolic normalization. Navitoclax, a BCL-2 inhibitor, clears chemotherapy-induced senescent cells and synergizes with PD-1 inhibitors to boost tumor remission rates (156). JQ1, a BET inhibitor, epigenetically suppresses the IL-6/STAT3 axis to overcome EGFR-TKI resistance (157). Metformin reverses SASP-driven glycolysis and enhances T cell function, improving 5-year survival by 35% in diabetic lung cancer cohorts (158). Emerging approaches include DR5 agonist/cFLIP inhibitor combinations identified through multi-omics analysis and chronotherapy-optimized mTOR inhibitors to enhance CD8+ T cell activity (159). Despite significant progress in elucidating the role of senescence in lung adenocarcinoma, several challenges persist. Firstly, the heterogeneity and dynamic nature of senescent cells complicate the identification of universal markers or therapeutic targets. Secondly, the SASP demonstrates environment-dependent dual roles in tumor suppression and promotion, thereby complicating therapeutic modulation. Thirdly, the absence of reliable and specific senescence biomarkers in clinical lung cancer samples impedes effective patient stratification and comprehensive treatment monitoring. Finally, although senolytics and SASP inhibitors offer promising therapeutic avenues, their safety profiles, efficacy, and delivery mechanisms pose challenges, especially in combination therapies. Addressing these gaps remains critical for the successful translation of senescence-targeting strategies into effective clinical practice. Future research must address senescence heterogeneity and spatiotemporal dynamics. Spatial transcriptomics and metabolic flux analysis can map senescent cell niches, while AI-driven models integrating epigenetic, microbiome, and immune datasets may predict optimal therapeutic targets. Multidisciplinary innovations targeting the senescence-immune-metabolic axis will be pivotal in overcoming resistance and improving lung cancer survival.
Author contributions
CC: Writing – original draft, Writing – review & editing. JC: Writing – original draft, Writing – review & editing. YZ: Conceptualization, Writing – original draft. QZ: Writing – original draft, Writing – review & editing. HS: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was funded by the Pudong New Area Science and Technology Development Fund Public Welfare Research Project (PKJ2023-Y64); Shanghai Pudong New Area Health System Discipline Leader Training Program (PWRd2023-16); Shanghai Hongkou District Health Commission Traditional Chinese Medicine Research Project (HKQ-ZYY-2021-03).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Jiang C, Zhang N, Hu X, and Wang HTumor-associated exosomes promote lung cancer metastasis through multiple mechanisms. Mol Cancer. (2021) 20:117. doi: 10.1186/s12943-021-01411-w
2. Li C, Lei S, Ding L, Xu Y, Wu X, Wang H, et al. Global burden and trends of lung cancer incidence and mortality. Chin Med J. (2023) 136:1583–90. doi: 10.1097/cm9.0000000000002529
3. Jiang H and Bu L. Progress in the treatment of lung adenocarcinoma by integrated traditional Chinese and Western medicine. Front Med. (2023) 10:1323344. doi: 10.3389/fmed.2023.1323344
4. Liu B, Wang C, Fang Z, Bai J, Qian Y, Ma Y, et al. Single-cell RNA sequencing reveals the cellular and molecular changes that contribute to the progression of lung adenocarcinoma. Front Cell Dev Biol. (2022) 10:927300. doi: 10.3389/fcell.2022.927300
5. Shi L, Zhao J, Wei Z, Wu H, and Sheng M. Radiomics in distinguishing between lung adenocarcinoma and lung squamous cell carcinoma: a systematic review and meta-analysis. Front Oncol. (2024) 14:1381217. doi: 10.3389/fonc.2024.1381217
6. Pannu BS and Iyer VN. Lung adenocarcinoma presenting with isolated ‘chronic cough’ of 3 years duration-a cautionary tale. Respir Med Case Rep. (2015) 16:157–9. doi: 10.1016/j.rmcr.2015.10.005
7. Wang C, Li J, Chen J, Wang Z, Zhu G, Song L, et al. Multi-omics analyses reveal biological and clinical insights in recurrent stage I non-small cell lung cancer. Nat Commun. (2025) 16:1477. doi: 10.1038/s41467-024-55068-2
8. Chen P, Quan Z, Song X, Gao Z, and Yuan K. MDFI is a novel biomarker for poor prognosis in LUAD. Front Oncol. (2022) 12:1005962. doi: 10.3389/fonc.2022.1005962
9. Li Y, Tao L, and Cai W. Profiles of immune infiltration and prognostic immunoscore in lung adenocarcinoma. BioMed Res Int. (2020) 2020:5858092. doi: 10.1155/2020/5858092
10. Jeon DS, Kim HC, Kim SH, Kim TJ, Kim HK, Moon MH, et al. Five-year overall survival and prognostic factors in patients with lung cancer: results from the korean association of lung cancer registry (KALC-R) 2015. Cancer Res Treat. (2023) 55:103–11. doi: 10.4143/crt.2022.264
11. Seguin L, Durandy M, and Feral CC. Lung adenocarcinoma tumor origin: A guide for personalized medicine. Cancers. (2022) 14(7):1759. doi: 10.3390/cancers14071759
12. Li AR, Chitale D, Riely GJ, Pao W, Miller VA, Zakowski MF, et al. EGFR mutations in lung adenocarcinomas: clinical testing experience and relationship to EGFR gene copy number and immunohistochemical expression. J Mol Diagn: JMD. (2008) 10:242–8. doi: 10.2353/jmoldx.2008.070178
13. Griesing S, Liao BC, and Yang JC. CD73 is regulated by the EGFR-ERK signaling pathway in non-small cell lung cancer. Anticancer Res. (2021) 41:1231–42. doi: 10.21873/anticanres.14880
14. Li Q, Li Z, Luo T, and Shi H. Targeting the PI3K/AKT/mTOR and RAF/MEK/ERK pathways for cancer therapy. Mol Biomed. (2022) 3:47. doi: 10.1186/s43556-022-00110-2
15. Song P, Gao Z, Bao Y, Chen L, Huang Y, Liu Y, et al. Wnt/β-catenin signaling pathway in carcinogenesis and cancer therapy. J Hematol Oncol. (2024) 17:46. doi: 10.1186/s13045-024-01563-4
16. Skoulidis F, Li BT, Dy GK, Price TJ, Falchook GS, Wolf J, et al. Sotorasib for lung cancers with KRAS p. G12C Mutation New Engl J Med. (2021) 384:2371–81. doi: 10.1056/NEJMoa2103695
17. Subbiah V, Wolf J, Konda B, Kang H, Spira A, Weiss J, et al. Tumour-agnostic efficacy and safety of selpercatinib in patients with RET fusion-positive solid tumours other than lung or thyroid tumours (LIBRETTO-001): a phase 1/2, open-label, basket trial. Lancet Oncol. (2022) 23:1261–73. doi: 10.1016/s1470-2045(22)00541-1
18. Shaw AT, Riely GJ, Bang YJ, Kim DW, Camidge DR, Solomon BJ, et al. Crizotinib in ROS1-rearranged advanced non-small-cell lung cancer (NSCLC): updated results, including overall survival, from PROFILE 1001. Ann Oncol: Off J Eur Soc Med Oncol. (2019) 30:1121–6. doi: 10.1093/annonc/mdz131
19. Langevin SM, Kratzke RA, and Kelsey KT. Epigenetics of lung cancer. Trans Res: J Lab Clin Med. (2015) 165:74–90. doi: 10.1016/j.trsl.2014.03.001
20. Kanwal R and Gupta S. Epigenetic modifications in cancer. Clin Genet. (2012) 81:303–11. doi: 10.1111/j.1399-0004.2011.01809.x
21. López-Otín C, Blasco MA, Partridge L, Serrano M, and Kroemer G. Hallmarks of aging: An expanding universe. Cell. (2023) 186:243–78. doi: 10.1016/j.cell.2022.11.001
22. López-Otín C, Blasco MA, Partridge L, Serrano M, and Kroemer G. The hallmarks of aging. Cell. (2013) 153:1194–217. doi: 10.1016/j.cell.2013.05.039
23. Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. (2013) 75:685–705. doi: 10.1146/annurev-physiol-030212-183653
24. Yamauchi S and Takahashi A. Cellular senescence: mechanisms and relevance to cancer and aging. J Biochem. (2025) 177:163–9. doi: 10.1093/jb/mvae079
25. Gonzalez-Meljem JM, Apps JR, Fraser HC, and Martinez-Barbera JP. Paracrine roles of cellular senescence in promoting tumourigenesis. Br J Cancer. (2018) 118:1283–8. doi: 10.1038/s41416-018-0066-1
26. Ortiz-Montero P, Londoño-Vallejo A, and Vernot JP. Senescence-associated IL-6 and IL-8 cytokines induce a self- and cross-reinforced senescence/inflammatory milieu strengthening tumorigenic capabilities in the MCF-7 breast cancer cell line. Cell Communication Signaling: CCS. (2017) 15:17. doi: 10.1186/s12964-017-0172-3
27. Liu H, Zhao H, and Sun Y. Tumor microenvironment and cellular senescence: Understanding therapeutic resistance and harnessing strategies. Semin Cancer Biol. (2022) 86:769–81. doi: 10.1016/j.semcancer.2021.11.004
28. Jha SK, De Rubis G, Devkota SR, Zhang Y, Adhikari R, Jha LA, et al. Cellular senescence in lung cancer: Molecular mechanisms and therapeutic interventions. Ageing Res Rev. (2024) 97:102315. doi: 10.1016/j.arr.2024.102315
29. Lin W, Wang X, Wang Z, Shao F, Yang Y, Cao Z, et al. Comprehensive analysis uncovers prognostic and immunogenic characteristics of cellular senescence for lung adenocarcinoma. Front Cell Dev Biol. (2021) 9:780461. doi: 10.3389/fcell.2021.780461
30. Lin W, Wang X, Xu Z, Wang Z, Liu T, Cao Z, et al. Identification and validation of cellular senescence patterns to predict clinical outcomes and immunotherapeutic responses in lung adenocarcinoma. Cancer Cell Int. (2021) 21:652. doi: 10.1186/s12935-021-02358-0
31. Ruiz A, Flores-Gonzalez J, Buendia-Roldan I, and Chavez-Galan L. Telomere shortening and its association with cell dysfunction in lung diseases. Int J Mol Sci. (2021) 23(1):425. doi: 10.3390/ijms23010425
32. de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. (2005) 19:2100–10. doi: 10.1101/gad.1346005
33. Herbig U, Jobling WA, Chen BP, Chen DJ, and Sedivy JM. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Mol Cell. (2004) 14:501–13. doi: 10.1016/s1097-2765(04)00256-4
34. Yang L, Wang M, Li N, Yan LD, Zhou W, Yu ZQ, et al. TERT mutations in non-small cell lung cancer: clinicopathologic features and prognostic implications. Clin Med Insights Oncol. (2023) 17:11795549221140781. doi: 10.1177/11795549221140781
35. Zhang H, Dai Z, Wu W, Wang Z, Zhang N, Zhang L, et al. Regulatory mechanisms of immune checkpoints PD-L1 and CTLA-4 in cancer. J Exp Clin Cancer Res: CR. (2021) 40:184. doi: 10.1186/s13046-021-01987-7
36. Mouw KW and Konstantinopoulos PA. From checkpoint to checkpoint: DNA damage ATR/Chk1 checkpoint signalling elicits PD-L1 immune checkpoint activation. Br J Cancer. (2018) 118:933–5. doi: 10.1038/s41416-018-0017-x
37. Méndez-Pertuz M, Martínez P, Blanco-Aparicio C, Gómez-Casero E, Belen García A, Martínez-Torrecuadrada J, et al. Modulation of telomere protection by the PI3K/AKT pathway. Nat Commun. (2017) 8:1278. doi: 10.1038/s41467-017-01329-2
38. Tang Y, Zhang Z, Chen Y, Qin S, Zhou L, Gao W, et al. Metabolic adaptation-mediated cancer survival and progression in oxidative stress. Antioxid (Basel Switzerland). (2022) 11(7):1324. doi: 10.3390/antiox11071324
39. Cortez Cardoso Penha R, Smith-Byrne K, Atkins JR, Haycock PC, Kar S, Codd V, et al. Common genetic variations in telomere length genes and lung cancer: a Mendelian randomisation study and its novel application in lung tumour transcriptome. eLife. (2023) 12. doi: 10.7554/eLife.83118
40. Fabiani R, Chiavarini M, Rosignoli P, and Giacchetta I. Leucocyte telomere length and lung cancer risk: A systematic review and meta-analysis of prospective studies. Cancers. (2024) 16. doi: 10.3390/cancers16183218
41. Deb S, Berei J, Miliavski E, Khan MJ, Broder TJ, Akurugo TA, et al. The effects of smoking on telomere length, induction of oncogenic stress, and chronic inflammatory responses leading to aging. Cells. (2024) 13(11):884. doi: 10.3390/cells13110884
42. Borah S, Xi L, Zaug AJ, Powell NM, Dancik GM, Cohen SB, et al. Cancer. TERT promoter mutations and telomerase reactivation in urothelial cancer. Sci (New York NY). (2015) 347:1006–10. doi: 10.1126/science.1260200
43. Jung SJ, Kim DS, Park WJ, Lee H, Choi IJ, Park JY, et al. Mutation of the TERT promoter leads to poor prognosis of patients with non-small cell lung cancer. Oncol Lett. (2017) 14:1609–14. doi: 10.3892/ol.2017.6284
44. Yu Q, Ge X, Wang Z, Ding S, Jin Y, and Chen L. Novel DNA damage-related subtypes characterization identifies uterine corpus endometrial carcinoma (UCEC) based on machine learning. J Oncol. (2022) 2022:3588117. doi: 10.1155/2022/3588117
45. Zhang L, Meng F, and Zhong D. DNA damage repair system and antineoplastic agents in lung cancer. Chin J Lung Cancer. (2022) 25:434–42. doi: 10.3779/j.issn.1009-3419.2022.101.24
46. Royfman R, Whiteley E, Noe O, Morand S, Creeden J, Stanbery L, et al. BRCA1/2 signaling and homologous recombination deficiency in breast and ovarian cancer. Future Oncol (London England). (2021) 17:2817–30. doi: 10.2217/fon-2021-0072
47. Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, et al. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov. (2018) 8:822–35. doi: 10.1158/2159-8290.Cd-18-0099
48. Dong ZY, Zhong WZ, Zhang XC, Su J, Xie Z, Liu SY, et al. Potential predictive value of TP53 and KRAS mutation status for response to PD-1 blockade immunotherapy in lung adenocarcinoma. Clin Cancer Res: An Off J Am Assoc Cancer Res. (2017) 23:3012–24. doi: 10.1158/1078-0432.Ccr-16-2554
49. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. New Engl J Med. (2015) 372:2509–20. doi: 10.1056/NEJMoa1500596
50. Muvaffak A and Coleman KG. PARP inhibitor synthetic lethality in ATM biallelic mutant cancer cell lines is associated with BRCA1/2 and RAD51 downregulation. Front Oncol. (2024) 14:1380633. doi: 10.3389/fonc.2024.1380633
51. Cui Y, Palii SS, Innes CL, and Paules RS. Depletion of ATR selectively sensitizes ATM-deficient human mammary epithelial cells to ionizing radiation and DNA-damaging agents. Cell Cycle (Georgetown Tex). (2014) 13:3541–50. doi: 10.4161/15384101.2014.960729
52. Bonagas N, Gustafsson NMS, Henriksson M, Marttila P, Gustafsson R, Wiita E, et al. Pharmacological targeting of MTHFD2 suppresses acute myeloid leukemia by inducing thymidine depletion and replication stress. Nat Cancer. (2022) 3:156–72. doi: 10.1038/s43018-022-00331-y
53. Schmitt A, Knittel G, Welcker D, Yang TP, George J, Nowak M, et al. ATM deficiency is associated with sensitivity to PARP1- and ATR inhibitors in lung adenocarcinoma. Cancer Res. (2017) 77:3040–56. doi: 10.1158/0008-5472.Can-16-3398
54. Nam AR, Jin MH, Bang JH, Oh KS, Seo HR, Oh DY, et al. Inhibition of ATR increases the sensitivity to WEE1 inhibitor in biliary tract cancer. Cancer Res Treat. (2020) 52:945–56. doi: 10.4143/crt.2020.080
55. Kim ST, Smith SA, Mortimer P, Loembé AB, Cho H, Kim KM, et al. Phase I study of ceralasertib (AZD6738), a novel DNA damage repair agent, in combination with weekly paclitaxel in refractory cancer. Clin Cancer Res: An Off J Am Assoc Cancer Res. (2021) 27:4700–9. doi: 10.1158/1078-0432.Ccr-21-0251
56. Hong DS, Moore K, Patel M, Grant SC, Burris HA 3rd, William WN Jr., et al. Evaluation of prexasertib, a checkpoint kinase 1 inhibitor, in a phase ib study of patients with squamous cell carcinoma. Clin Cancer Res: An Off J Am Assoc Cancer Res. (2018) 24:3263–72. doi: 10.1158/1078-0432.Ccr-17-3347
57. Singh S, Bhat MY, Sathe G, Gopal C, Sharma J, Madugundu AK, et al. Proteomic signatures of diffuse and intestinal subtypes of gastric cancer. Cancers. (2021) 13(23):5930. doi: 10.3390/cancers13235930
58. Huang Y, Du Y, Zheng Y, Wen C, Zou H, Huang J, et al. Ct-OATP1B3 promotes high-grade serous ovarian cancer metastasis by regulation of fatty acid beta-oxidation and oxidative phosphorylation. Cell Death Dis. (2022) 13:556. doi: 10.1038/s41419-022-05014-1
59. Iranmanesh Y, Jiang B, Favour OC, Dou Z, Wu J, Li J, et al. Mitochondria’s role in the maintenance of cancer stem cells in glioblastoma. Front Oncol. (2021) 11:582694. doi: 10.3389/fonc.2021.582694
60. Boreel DF, Beerkens APM, Heskamp S, Boswinkel M, Peters JPW, Adema GJ, et al. Inhibition of OXPHOS induces metabolic rewiring and reduces hypoxia in murine tumor models. Clin Trans Radiat Oncol. (2024) 49:100875. doi: 10.1016/j.ctro.2024.100875
61. Chen LY, Wang Y, Terkeltaub R, and Liu-Bryan R. Activation of AMPK-SIRT3 signaling is chondroprotective by preserving mitochondrial DNA integrity and function. Osteoarthritis Cartilage. (2018) 26:1539–50. doi: 10.1016/j.joca.2018.07.004
62. An X, Yu W, Liu J, Tang D, Yang L, and Chen X. Oxidative cell death in cancer: mechanisms and therapeutic opportunities. Cell Death Dis. (2024) 15:556. doi: 10.1038/s41419-024-06939-5
63. Roberts AW, Davids MS, Pagel JM, Kahl BS, Puvvada SD, Gerecitano JF, et al. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. New Engl J Med. (2016) 374:311–22. doi: 10.1056/NEJMoa1513257
64. Bai R and Cui J. Mitochondrial immune regulation and anti-tumor immunotherapy strategies targeting mitochondria. Cancer Lett. (2023) 564:216223. doi: 10.1016/j.canlet.2023.216223
65. Ganjoo S, Gupta P, Corbali HI, Nanez S, Riad TS, Duong LK, et al. The role of tumor metabolism in modulating T-Cell activity and in optimizing immunotherapy. Front Immunol. (2023) 14:1172931. doi: 10.3389/fimmu.2023.1172931
66. Bengsch B, Johnson AL, Kurachi M, Odorizzi PM, Pauken KE, Attanasio J, et al. Bioenergetic insufficiencies due to metabolic alterations regulated by the inhibitory receptor PD-1 are an early driver of CD8(+) T cell exhaustion. Immunity. (2016) 45:358–73. doi: 10.1016/j.immuni.2016.07.008
67. Zhang R, Mao G, Tang Y, Li C, Gao Y, Nie W, et al. Inhibition of glycolysis enhances the efficacy of immunotherapy via PDK-mediated upregulation of PD-L1. Cancer Immunol Immunother: CII. (2024) 73:151. doi: 10.1007/s00262-024-03735-0
68. Tan X, Li Y, Hou Z, Zhang M, Li L, and Wei J. Combination therapy with PD-1 inhibition plus rapamycin and metformin enhances anti-tumor efficacy in triple negative breast cancer. Exp Cell Res. (2023) 429:113647. doi: 10.1016/j.yexcr.2023.113647
69. Jiang YH, Jiang LY, Wang YC, Ma DF, and Li X. Quercetin attenuates atherosclerosis via modulating oxidized LDL-induced endothelial cellular senescence. Front Pharmacol. (2020) 11:512. doi: 10.3389/fphar.2020.00512
70. Zhang JJ, Cao CX, Wan LL, Zhang W, Liu ZJ, Wang JL, et al. Forkhead Box q1 promotes invasion and metastasis in colorectal cancer by activating the epidermal growth factor receptor pathway. World J Gastroenterol. (2022) 28:1781–97. doi: 10.3748/wjg.v28.i17.1781
71. Park SY, Cui Z, Kim B, Park G, and Choi YW. Treatment with gold nanoparticles using cudrania tricuspidata root extract induced downregulation of MMP-2/-9 and PLD1 and inhibited the invasiveness of human U87 glioblastoma cells. Int J Mol Sci. (2020) 21(4):1282. doi: 10.3390/ijms21041282
72. Cantanhede IG, Liu H, Liu H, Balbuena Rodriguez V, Shiwen X, Ong VH, et al. Exploring metabolism in scleroderma reveals opportunities for pharmacological intervention for therapy in fibrosis. Front Immunol. (2022) 13:1004949. doi: 10.3389/fimmu.2022.1004949
73. Geng F, Dong L, Bao X, Guo Q, Guo J, Zhou Y, et al. CAFs/tumor cells co-targeting DNA vaccine in combination with low-dose gemcitabine for the treatment of Panc02 murine pancreatic cancer. Mol Ther Oncolytics. (2022) 26:304–13. doi: 10.1016/j.omto.2022.07.008
74. He M, Yu W, Chang C, Miyamoto H, Liu X, Jiang K, et al. Estrogen receptor α promotes lung cancer cell invasion via increase of and cross-talk with infiltrated macrophages through the CCL2/CCR2/MMP9 and CXCL12/CXCR4 signaling pathways. Mol Oncol. (2020) 14:1779–99. doi: 10.1002/1878-0261.12701
75. Shayan G, Kansy BA, Gibson SP, Srivastava RM, Bryan JK, Bauman JE, et al. Phase ib study of immune biomarker modulation with neoadjuvant cetuximab and TLR8 stimulation in head and neck cancer to overcome suppressive myeloid signals. Clin Cancer Res: An Off J Am Assoc Cancer Res. (2018) 24:62–72. doi: 10.1158/1078-0432.Ccr-17-0357
76. Zeltz C, Alam J, Liu H, Erusappan PM, Hoschuetzky H, Molven A, et al. α11β1 integrin is induced in a subset of cancer-associated fibroblasts in desmoplastic tumor stroma and mediates in vitro cell migration. Cancers. (2019) 11(6):765. doi: 10.3390/cancers11060765
77. Coppé JP, Desprez PY, Krtolica A, and Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. (2010) 5:99–118. doi: 10.1146/annurev-pathol-121808-102144
78. Palicelli A, Bonacini M, Croci S, Magi-Galluzzi C, Cañete-Portillo S, Chaux A, et al. What do we have to know about PD-L1 expression in prostate cancer? A systematic literature review. Part 1: focus on immunohistochemical results with discussion of pre-analytical and interpretation variables. Cells. (2021) 10(11):3166. doi: 10.3390/cells10113166
79. Tanita K, Fujimura T, Sato Y, Lyu C, Kambayashi Y, Ogata D, et al. Bexarotene reduces production of CCL22 from tumor-associated macrophages in cutaneous T-cell lymphoma. Front Oncol. (2019) 9:907. doi: 10.3389/fonc.2019.00907
80. Guan F, Wu X, Zhou J, Lin Y, He Y, Fan C, et al. Mitochondrial transfer in tunneling nanotubes-a new target for cancer therapy. J Exp Clin Cancer Res: CR. (2024) 43:147. doi: 10.1186/s13046-024-03069-w
81. Zhang M, Qi Y, Li H, Cui J, Dai L, Frank JA, et al. AIM2 inflammasome mediates Arsenic-induced secretion of IL-1 β and IL-18. Oncoimmunology. (2016) 5:e1160182. doi: 10.1080/2162402x.2016.1160182
82. Wang S, Jia M, He Z, and Liu XS. APOBEC3B and APOBEC mutational signature as potential predictive markers for immunotherapy response in non-small cell lung cancer. Oncogene. (2018) 37:3924–36. doi: 10.1038/s41388-018-0245-9
83. Venit T, Semesta K, Farrukh S, Endara-Coll M, Havalda R, Hozak P, et al. Nuclear myosin 1 activates p21 gene transcription in response to DNA damage through a chromatin-based mechanism. Commun Biol. (2020) 3:115. doi: 10.1038/s42003-020-0836-1
84. Nguyen TA, Bieging-Rolett KT, Putoczki TL, Wicks IP, Attardi LD, and Pang KC. SIDT2 RNA transporter promotes lung and gastrointestinal tumor development. iScience. (2019) 20:14–24. doi: 10.1016/j.isci.2019.09.009
85. Ishiguro N and Yoshida H. ASPL-TFE3 oncoprotein regulates cell cycle progression and induces cellular senescence by up-regulating p21. Neoplasia (New York NY). (2016) 18:626–35. doi: 10.1016/j.neo.2016.08.001
86. Kim Y, Park JB, Fukuda J, Watanabe M, and Chun YS. The effect of neddylation blockade on slug-dependent cancer cell migration is regulated by p53 mutation status. Cancers. (2021) 13(3):531. doi: 10.3390/cancers13030531
87. Gou R, Li X, Dong H, Hu Y, Liu O, Liu J, et al. RAD21 confers poor prognosis and affects ovarian cancer sensitivity to poly(ADP-ribose)Polymerase inhibitors through DNA damage repair. Front Oncol. (2022) 12:936550. doi: 10.3389/fonc.2022.936550
88. Lu L, Zhang S, Li C, Zhou C, Li D, Liu P, et al. Cryptotanshinone inhibits human glioma cell proliferation in vitro and in vivo through SHP-2-dependent inhibition of STAT3 activation. Cell Death Dis. (2017) 8:e2767. doi: 10.1038/cddis.2017.174
89. Dhir T, Schultz CW, Jain A, Brown SZ, Haber A, Goetz A, et al. Abemaciclib is effective against pancreatic cancer cells and synergizes with huR and YAP1 inhibition. Mol Cancer Res: MCR. (2019) 17:2029–41. doi: 10.1158/1541-7786.Mcr-19-0589
90. Tsang CM, Yip YL, Lo KW, Deng W, To KF, Hau PM, et al. Cyclin D1 overexpression supports stable EBV infection in nasopharyngeal epithelial cells. Proc Natl Acad Sci United States America. (2012) 109:E3473–82. doi: 10.1073/pnas.1202637109
91. Lee SE, Kim YJ, Sung M, Lee MS, Han J, Kim HK, et al. Association with PD-L1 expression and clinicopathological features in 1000 lung cancers: A large single-institution study of surgically resected lung cancers with a high prevalence of EGFR mutation. Int J Mol Sci. (2019) 20(19):4794. doi: 10.3390/ijms20194794
92. Zhang Y, Lapidus RG, Liu P, Choi EY, Adediran S, Hussain A, et al. Targeting IκB kinase β/NF-κB signaling in human prostate cancer by a novel IκB kinase β Inhibitor cmpdA. Mol Cancer Ther. (2016) 15:1504–14. doi: 10.1158/1535-7163.Mct-15-0999
93. Zhao R, Kaakati R, Liu X, Xu L, Lee AK, Bachelder R, et al. CRISPR/cas9-mediated BRCA1 knockdown adipose stem cells promote breast cancer progression. Plast Reconstructive Surg. (2019) 143:747–56. doi: 10.1097/prs.0000000000005316
94. He Y, Qu Y, Meng B, Huang W, Tang J, Wang R, et al. Mesenchymal stem cells empower T cells in the lymph nodes via MCP-1/PD-L1 axis. Cell Death Dis. (2022) 13:365. doi: 10.1038/s41419-022-04822-9
95. Faure-Dupuy S, Riedl T, Rolland M, Hizir Z, Reisinger F, Neuhaus K, et al. Control of APOBEC3B induction and cccDNA decay by NF-κB and miR-138-5p. JHEP Reports: Innovation Hepatol. (2021) 3:100354. doi: 10.1016/j.jhepr.2021.100354
96. Butler K and Banday AR. APOBEC3-mediated mutagenesis in cancer: causes, clinical significance and therapeutic potential. J Hematol Oncol. (2023) 16:31. doi: 10.1186/s13045-023-01425-5
97. Wang Y, Li W, Patel SS, Cong J, Zhang N, Sabbatino F, et al. Blocking the formation of radiation-induced breast cancer stem cells. Oncotarget. (2014) 5:3743–55. doi: 10.18632/oncotarget.1992
98. Zakaria N, Mohd Yusoff N, Zakaria Z, Widera D, and Yahaya BH. Inhibition of NF-κB signaling reduces the stemness characteristics of lung cancer stem cells. Front Oncol. (2018) 8:166. doi: 10.3389/fonc.2018.00166
99. Antonangeli F, Natalini A, Garassino MC, Sica A, Santoni A, and Di Rosa F. Regulation of PD-L1 expression by NF-κB in cancer. Front Immunol. (2020) 11:584626. doi: 10.3389/fimmu.2020.584626
100. Dong Y, Gong Y, Kuo F, Makarov V, Reznik E, Nanjangud GJ, et al. Targeting the mTOR pathway in hurthle cell carcinoma results in potent antitumor activity. Mol Cancer Ther. (2022) 21:382–94. doi: 10.1158/1535-7163.Mct-21-0224
101. Park JH, Park JW, Lee JH, Kim DY, Hahm JH, and Bae YS. Role of phospholipase D in the lifespan of Caenorhabditis elegans. Exp Mol Med. (2018) 50:1–10. doi: 10.1038/s12276-017-0015-8
102. Sagini K, Urbanelli L, Costanzi E, Mitro N, Caruso D, Emiliani C, et al. Oncogenic H-ras expression induces fatty acid profile changes in human fibroblasts and extracellular vesicles. Int J Mol Sci. (2018) 19(11):3515. doi: 10.3390/ijms19113515
103. Chen J, Cui Z, Wang Y, Lyu L, Feng C, Feng D, et al. Cyclic Polypeptide D7 Protects Bone Marrow Mesenchymal Cells and Promotes Chondrogenesis during Osteonecrosis of the Femoral Head via Growth Differentiation Factor 15-Mediated Redox Signaling. Oxid Med Cell Longev. (2022) 2022:3182368. doi: 10.1155/2022/3182368
104. Lou JS, Xia YT, Wang HY, Kong XP, Yao P, Dong TTX, et al. The WT1/MVP-mediated stabilization on mTOR/AKT axis enhances the effects of cisplatin in non-small cell lung cancer by a reformulated yu ping feng san herbal preparation. Front Pharmacol. (2018) 9:853. doi: 10.3389/fphar.2018.00853
105. Liu J, Yang J, Hou Y, Zhu Z, He J, Zhao H, et al. Casticin inhibits nasopharyngeal carcinoma growth by targeting phosphoinositide 3-kinase. Cancer Cell Int. (2019) 19:348. doi: 10.1186/s12935-019-1069-6
106. Jeon SH and Choung SY. Oyster hydrolysates attenuate muscle atrophy via regulating protein turnover and mitochondria biogenesis in C2C12 cell and immobilized mice. Nutrients. (2021) 13(12):4385. doi: 10.3390/nu13124385
107. Su W, Feng S, Chen X, Yang X, Mao R, Guo C, et al. Silencing of Long Noncoding RNA MIR22HG Triggers Cell Survival/Death Signaling via Oncogenes YBX1, MET, and p21 in Lung Cancer. Cancer Res. (2018) 78:3207–19. doi: 10.1158/0008-5472.Can-18-0222
108. Chiang CT, Demetriou AN, Ung N, Choudhury N, Ghaffarian K, Ruderman DL, et al. mTORC2 contributes to the metabolic reprogramming in EGFR tyrosine-kinase inhibitor resistant cells in non-small cell lung cancer. Cancer Lett. (2018) 434:152–9. doi: 10.1016/j.canlet.2018.07.025
109. Mu H, Yu G, Li H, Wang M, Cui Y, Zhang T, et al. Mild chronic hypoxia-induced HIF-2α interacts with c-MYC through competition with HIF-1α to induce hepatocellular carcinoma cell proliferation. Cell Oncol (Dordrecht Netherlands). (2021) 44:1151–66. doi: 10.1007/s13402-021-00625-w
110. Kim YJ, Lee S, Jin J, Woo H, Choi YK, and Park KG. Cassiaside C inhibits M1 polarization of macrophages by downregulating glycolysis. Int J Mol Sci. (2022) 23(3):1696. doi: 10.3390/ijms23031696
111. Merino-Ramos T, Vázquez-Calvo Á, Casas J, Sobrino F, Saiz JC, and Martín-Acebes MA. Modification of the host cell lipid metabolism induced by hypolipidemic drugs targeting the acetyl coenzyme A carboxylase impairs west nile virus replication. Antimicrobial Agents Chemother. (2016) 60:307–15. doi: 10.1128/aac.01578-15
112. Rozengurt E, Soares HP, and Sinnet-Smith J. Suppression of feedback loops mediated by PI3K/mTOR induces multiple overactivation of compensatory pathways: an unintended consequence leading to drug resistance. Mol Cancer Ther. (2014) 13:2477–88. doi: 10.1158/1535-7163.Mct-14-0330
113. Middleton G, Robbins HL, Fletcher P, Savage J, Mehmi M, Summers Y, et al. A phase II trial of mTORC1/2 inhibition in STK11 deficient non small cell lung cancer. NPJ Precis Oncol. (2025) 9:67. doi: 10.1038/s41698-025-00838-4
114. Wang B, Han J, Elisseeff JH, and Demaria M. The senescence-associated secretory phenotype and its physiological and pathological implications. Nat Rev Mol Cell Biol. (2024) 25:958–78. doi: 10.1038/s41580-024-00727-x
115. Furuta T, Ohshima C, Matsumura M, Takebayashi N, Hirota E, Mawaribuchi T, et al. Oxidative stress upregulates zinc uptake activity via Zrt/Irt-like protein 1 (ZIP1) in cultured mouse astrocytes. Life Sci. (2016) 151:305–12. doi: 10.1016/j.lfs.2016.03.025
116. Ni C, Lou X, Yao X, Wang L, Wan J, Duan X, et al. ZIP1(+) fibroblasts protect lung cancer against chemotherapy via connexin-43 mediated intercellular Zn(2+) transfer. Nat Commun. (2022) 13:5919. doi: 10.1038/s41467-022-33521-4
117. Chibaya L, Snyder J, and Ruscetti M. Senescence and the tumor-immune landscape: Implications for cancer immunotherapy. Semin Cancer Biol. (2022) 86:827–45. doi: 10.1016/j.semcancer.2022.02.005
118. Chen F, Long Q, Fu D, Zhu D, Ji Y, Han L, et al. Targeting SPINK1 in the damaged tumour microenvironment alleviates therapeutic resistance. Nat Commun. (2018) 9:4315. doi: 10.1038/s41467-018-06860-4
119. Ozaki N, Ohmuraya M, Hirota M, Ida S, Wang J, Takamori H, et al. Serine protease inhibitor Kazal type 1 promotes proliferation of pancreatic cancer cells through the epidermal growth factor receptor. Mol Cancer Res: MCR. (2009) 7:1572–81. doi: 10.1158/1541-7786.Mcr-08-0567
120. Guo M, Zhou X, Han X, Zhang Y, and Jiang L. SPINK1 is a prognosis predicting factor of non-small cell lung cancer and regulates redox homeostasis. Oncol Lett. (2019) 18:6899–908. doi: 10.3892/ol.2019.11005
121. Xu L, Lu C, Huang Y, Zhou J, Wang X, Liu C, et al. SPINK1 promotes cell growth and metastasis of lung adenocarcinoma and acts as a novel prognostic biomarker. BMB Rep. (2018) 51:648–53. doi: 10.5483/BMBRep.2018.51.12.205
122. Dong Z, Luo Y, Yuan Z, Tian Y, Jin T, and Xu F. Cellular senescence and SASP in tumor progression and therapeutic opportunities. Mol Cancer. (2024) 23:181. doi: 10.1186/s12943-024-02096-7
123. Pan J, Li D, Xu Y, Zhang J, Wang Y, Chen M, et al. Inhibition of bcl-2/xl with ABT-263 selectively kills senescent type II pneumocytes and reverses persistent pulmonary fibrosis induced by ionizing radiation in mice. Int J Radiat Oncol Biol Physics. (2017) 99:353–61. doi: 10.1016/j.ijrobp.2017.02.216
124. Takasaka N, Seed RI, Cormier A, Bondesson AJ, Lou J, Elattma A, et al. Integrin αvβ8-expressing tumor cells evade host immunity by regulating TGF-β activation in immune cells. JCI Insight. (2018) 3(20):e122591. doi: 10.1172/jci.insight.122591
125. Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, et al. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell. (2015) 14:644–58. doi: 10.1111/acel.12344
126. Kirkland JL and Tchkonia T. Clinical strategies and animal models for developing senolytic agents. Exp Gerontol. (2015) 68:19–25. doi: 10.1016/j.exger.2014.10.012
127. Roberts AW, Seymour JF, Brown JR, Wierda WG, Kipps TJ, Khaw SL, et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. J Clin Oncol: Off J Am Soc Clin Oncol. (2012) 30:488–96. doi: 10.1200/jco.2011.34.7898
128. Zhang C, Quan Y, Yang L, Bai Y, and Yang Y. 6-Methoxyflavone induces S-phase arrest through the CCNA2/CDK2/p21CIP1 signaling pathway in HeLa cells. Bioengineered. (2022) 13:7277–92. doi: 10.1080/21655979.2022.2047496
129. Justice JN, Nambiar AM, Tchkonia T, LeBrasseur NK, Pascual R, Hashmi SK, et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine. (2019) 40:554–63. doi: 10.1016/j.ebiom.2018.12.052
130. Zhu Y, Doornebal EJ, Pirtskhalava T, Giorgadze N, Wentworth M, Fuhrmann-Stroissnigg H, et al. New agents that target senescent cells: the flavone, fisetin, and the BCL-X(L) inhibitors, A1331852 and A1155463. Aging. (2017) 9:955–63. doi: 10.18632/aging.101202
131. Ateeq B, Tomlins SA, Laxman B, Asangani IA, Cao Q, Cao X, et al. Therapeutic targeting of SPINK1-positive prostate cancer. Sci Trans Med. (2011) 3:72ra17. doi: 10.1126/scitranslmed.3001498
132. O’Sullivan CC, Clarke R, Goetz MP, and Robertson J. Cyclin-dependent kinase 4/6 inhibitors for treatment of hormone receptor-positive, ERBB2-negative breast cancer: A review. JAMA Oncol. (2023) 9:1273–82. doi: 10.1001/jamaoncol.2023.2000
133. Li Z, Zou W, Zhang J, Zhang Y, Xu Q, Li S, et al. Mechanisms of CDK4/6 inhibitor resistance in luminal breast cancer. Front Pharmacol. (2020) 11:580251. doi: 10.3389/fphar.2020.580251
134. Scagnoli S, Pisegna S, Toss A, Caputo R, De Laurentiis M, Palleschi M, et al. Clinical impact of drug-drug interactions on abemaciclib in the real-world experience of AB-ITALY study. NPJ Breast Cancer. (2024) 10:58. doi: 10.1038/s41523-024-00657-z
135. Lin JJ, Cardarella S, Lydon CA, Dahlberg SE, Jackman DM, Jänne PA, et al. Five-year survival in EGFR-mutant metastatic lung adenocarcinoma treated with EGFR-TKIs. J Thorac Oncol: Off Publ Int Assoc Study Lung Cancer. (2016) 11:556–65. doi: 10.1016/j.jtho.2015.12.103
136. Mall C, Sckisel GD, Proia DA, Mirsoian A, Grossenbacher SK, Pai CS, et al. Repeated PD-1/PD-L1 monoclonal antibody administration induces fatal xenogeneic hypersensitivity reactions in a murine model of breast cancer. Oncoimmunology. (2016) 5:e1075114. doi: 10.1080/2162402x.2015.1075114
137. Schirmer M and Dusny C. Microbial single-cell mass spectrometry: status, challenges, and prospects. Curr Opin Biotechnol. (2023) 83:102977. doi: 10.1016/j.copbio.2023.102977
138. Zhu X, Xu T, Peng C, and Wu S. Advances in MALDI mass spectrometry imaging single cell and tissues. Front Chem. (2021) 9:782432. doi: 10.3389/fchem.2021.782432
139. Mijit M, Caracciolo V, Melillo A, Amicarelli F, and Giordano A. Role of p53 in the regulation of cellular senescence. Biomolecules. (2020) 10(3):420. doi: 10.3390/biom10030420
140. Davalos AR, Coppe JP, Campisi J, and Desprez PY. Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis Rev. (2010) 29:273–83. doi: 10.1007/s10555-010-9220-9
141. Wang L, Lankhorst L, and Bernards R. Exploiting senescence for the treatment of cancer. Nat Rev Cancer. (2022) 22:340–55. doi: 10.1038/s41568-022-00450-9
142. Boumelha J, de Carné Trécesson S, Law EK, Romero-Clavijo P, Coelho MA, Ng KW, et al. An immunogenic model of KRAS-mutant lung cancer enables evaluation of targeted therapy and immunotherapy combinations. Cancer Res. (2022) 82:3435–48. doi: 10.1158/0008-5472.Can-22-0325
143. Watterson A and Coelho MA. Cancer immune evasion through KRAS and PD-L1 and potential therapeutic interventions. Cell Communication Signaling: CCS. (2023) 21:45. doi: 10.1186/s12964-023-01063-x
144. Lei Y, Zhong C, Zhang J, Zheng Q, Xu Y, Li Z, et al. Senescent lung fibroblasts in idiopathic pulmonary fibrosis facilitate non-small cell lung cancer progression by secreting exosomal MMP1. Oncogene. (2025) 44:769–81. doi: 10.1038/s41388-024-03236-5
145. Liu X, Hoft DF, and Peng G. Senescent T cells within suppressive tumor microenvironments: emerging target for tumor immunotherapy. J Clin Invest. (2020) 130:1073–83. doi: 10.1172/jci133679
146. Wang Z, Chen Y, Fang H, Xiao K, Wu Z, Xie X, et al. Reprogramming cellular senescence in the tumor microenvironment augments cancer immunotherapy through multifunctional nanocrystals. Sci Advances. (2024) 10:eadp7022. doi: 10.1126/sciadv.adp7022
147. Papavassiliou KA, Sofianidi AA, Gogou VA, Anagnostopoulos N, and Papavassiliou AG. P53 and rb aberrations in small cell lung cancer (SCLC): from molecular mechanisms to therapeutic modulation. Int J Mol Sci. (2024) 25(5):2479. doi: 10.3390/ijms25052479
148. Kong SH, Ma L, Yuan Q, Liu X, Han Y, Xiang W, et al. Inhibition of EZH2 alleviates SAHA-induced senescence-associated secretion phenotype in small cell lung cancer cells. Cell Death Discov. (2023) 9:289. doi: 10.1038/s41420-023-01591-y
149. Tian Y, Zhai X, Han A, Zhu H, and Yu J. Potential immune escape mechanisms underlying the distinct clinical outcome of immune checkpoint blockades in small cell lung cancer. J Hematol Oncol. (2019) 12:67. doi: 10.1186/s13045-019-0753-2
150. Wang Q, Yuan Y, Liu J, Li C, and Jiang X. The role of mitochondria in aging, cell death, and tumor immunity. Front Immunol. (2024) 15:1520072. doi: 10.3389/fimmu.2024.1520072
151. Wang Z, Chen C, Ai J, Gao Y, Wang L, Xia S, et al. The crosstalk between senescence, tumor, and immunity: molecular mechanism and therapeutic opportunities. MedComm. (2025) 6:e70048. doi: 10.1002/mco2.70048
152. Kuo CL, Pilling LC, Kuchel GA, Ferrucci L, and Melzer D. Telomere length and aging-related outcomes in humans: A Mendelian randomization study in 261,000 older participants. Aging Cell. (2019) 18:e13017. doi: 10.1111/acel.13017
153. Qin R, Peng W, Wang X, Li C, Xi Y, Zhong Z, et al. Identification of genes related to immune infiltration in the tumor microenvironment of cutaneous melanoma. Front Oncol. (2021) 11:615963. doi: 10.3389/fonc.2021.615963
154. Suzuki K, Tange M, Yamagishi R, Hanada H, Mukai S, Sato T, et al. SMG6 regulates DNA damage and cell survival in Hippo pathway kinase LATS2-inactivated Malignant mesothelioma. Cell Death Discov. (2022) 8:446. doi: 10.1038/s41420-022-01232-w
155. Yang C, Pang Y, Huang Y, Ye F, Chen X, Gao Y, et al. Single-cell transcriptomics identifies premature aging features of TERC-deficient mouse brain and bone marrow. GeroScience. (2022) 44:2139–55. doi: 10.1007/s11357-022-00578-4
156. González-Gualda E, Pàez-Ribes M, Lozano-Torres B, Macias D, Wilson JR 3rd, González-López C, et al. Galacto-conjugation of Navitoclax as an efficient strategy to increase senolytic specificity and reduce platelet toxicity. Aging Cell. (2020) 19:e13142. doi: 10.1111/acel.13142
157. Yeo MK, Kim Y, Lee DH, Chung C, and Bae GE. Cosuppression of NF-κB and AICDA overcomes acquired EGFR-TKI resistance in non-small cell lung cancer. Cancers. (2022) 14(12):2940. doi: 10.3390/cancers14122940
158. An T, Zhang Z, Li Y, Yi J, Zhang W, Chen D, et al. Integrin β1-mediated cell- Cell adhesion augments metformin-induced anoikis. Int J Mol Sci. (2019) 20(5):1161. doi: 10.3390/ijms20051161
Keywords: cellular senescence, lung cancer, SASP, therapy resistance, precision medicine
Citation: Chen C, Chen J, Zhang Y, Zhang Q and Shi H (2025) Senescence-associated secretory phenotype in lung cancer: remodeling the tumor microenvironment for metastasis and immune suppression. Front. Oncol. 15:1605085. doi: 10.3389/fonc.2025.1605085
Received: 02 April 2025; Accepted: 12 May 2025;
Published: 29 May 2025.
Edited by:
Xiaosheng Tan, Rutgers, United StatesReviewed by:
Hao Zhang, Chongqing Medical University, ChinaZheng Liu, Xiangtan Central Hospital, China
Yanjun Gao, George Washington University, United States
Yaping Li, Yale University, United States
Copyright © 2025 Chen, Chen, Zhang, Zhang and Shi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qijun Zhang, emhhbmdxaWp1bjgzNTRAMTYzLmNvbQ==; Haixia Shi, aGFpeGlhLjAxMDFAMTYzLmNvbQ==
†These authors have contributed equally to this work