 Fang-Mei Luo
Fang-Mei Luo Liang-Liang Fan
Liang-Liang Fan Yue Sheng2
Yue Sheng2 Lv Liu
Lv Liu- 1Department of Respiratory Medicine, The Second Xiangya Hospital of Central South University, Changsha, China
- 2Department of Cell Biology, The School of Life Sciences, Central South University, Changsha, China
- 3Hunan Key Laboratory of Animal Models for Human Disease, School of Life Sciences, Central South University, Changsha, China
Inherited macrothrombocytopenia (IMTP) is a rare disorder characterized by a reduced platelet count and abnormally large platelets. The main clinical symptom of IMTP is mild bleeding in some patients. At present, more than 30 genes have been identified in patients with syndromic and non-syndromic IMTP. In this study, a 3-year-old boy and his mother who presented with mild epistaxis and/or gingival bleeding were diagnosed as having IMTP. Wen then selected whole sequencing to explore the genetic lesion of the patients. After data filtering and mutation validation, a novel frameshift mutation (NM_001130004: c.398_399insTGCG, p.F134AfsX60) of α-actin 1 (ACTN1) was identified in the proband and his mother but absent in other unaffected individuals. Previous studies have proven that mutations in ACTN1 may lead to IMTP with mild to absent bleeding phenotype. The novel mutation, resulting in a truncated protein in exon 4 of the ACTN1 gene, was absent in the public database, such as 1000G and genomAD. Further Western blot revealed that the expression of α-actin 1 in the proband was decreased overtly, which indicated that the novel frameshift mutation may induce non-sense-mediated mRNA decay. In summary, this study not only broadened the variants spectrum of ACTN1 gene, which may contribute to the genetic counseling of IMTP, but also confirmed the diagnosis of IMTP, which may help the management and prognosis for the family members.
Introduction
Inherited macrothrombocytopenia (IMTP) is an important cause of thrombocytopenia, which is defined as a platelet count of <150 × 109/L (1–3). Besides the reduced platelet count, a significant increase in platelet size (>12 fL) is another feature of IMTP (1, 3). As a rare clinical condition, IMTP affects at least 2.7 per 100,000 individuals with mild to absent bleeding phenotype (2, 4). At present, mutations in more than 10 genes including α-actin 1(ACTN1), myosin heavy chain 9 (MYH9), tubulin beta class I (TUBB), etc. have been identified in non-syndromic IMTP with autosomal dominant, recessive, and sex-linked patterns (1, 3). In addition, some syndromes are also typically characterized by low platelet counts and severe bleeding tendency, such as Wiskott-Aldrich syndrome, Bernard-Soulier syndrome, Di George syndrome, and so on (5–7). However, because of varying mutations and clinical manifestations, the IMTP shows obvious heterogeneity, which challenges the clear diagnosis of IMTP and leads to the misdiagnosis as immune thrombocytopenic purpura (8).
In this study, we enrolled a 3-year-old boy and his mother with IMTP and mild epistaxis and/or gingival bleeding. The aim of this study was to explore the genetic lesion of the patients with the help of whole-exome sequencing.
Case Presentation
A family from central south region of China (Hunan province) including seven persons was investigated in this study (Figure 1A). The proband (III-2), a 3-year-old boy, was admitted to our hospital because of mild epistaxis. Blood routine examination of the proband was shown as follows: hemoglobin, 13.0 g/dL; platelet count, 102 × 109/L; mean platelet volume, 12.9 fL; and platelet distribution width, 18.1 fL. In vitro platelet aggregation in response to collagen and ristocetin was normal, but adenosine diphosphate (ADP) was slightly reduced (35%). Medical history survey found that the boy (III-2) has suffered from epistaxis several times with unexplained reason. Further family history investigation revealed that the proband's mother (II-1) and grandfather (I-1) have a history of mild epistaxis and gingival bleeding. Blood routine examination of the proband's mother (II-1) also found the reduced platelets (111 × 109/L) count and increased mean platelet volume (12.7 fL) and platelet distribution width (17.7 fL). Peripheral blood smears May-Grünwald Giemsa staining revealed macrothrombocytopenia in the proband and his mother (Figure 1B). No other family members showed abnormal blood routine examination and bleeding diathesis. In addition, 200 unrelated, ethnically matched healthy controls were used as internal controls to exclude single-nucleotide polymorphisms (SNPs) in local individuals. These healthy controls (male/female: 100/100, aged 36.7 ± 8.6 years) lacked IMTP diagnostic features. Each participant underwent thorough examination for clinical diagnosis or exclusion, including general examination such as blood routine examination and peripheral blood smear May-Grünwald Giemsa staining.
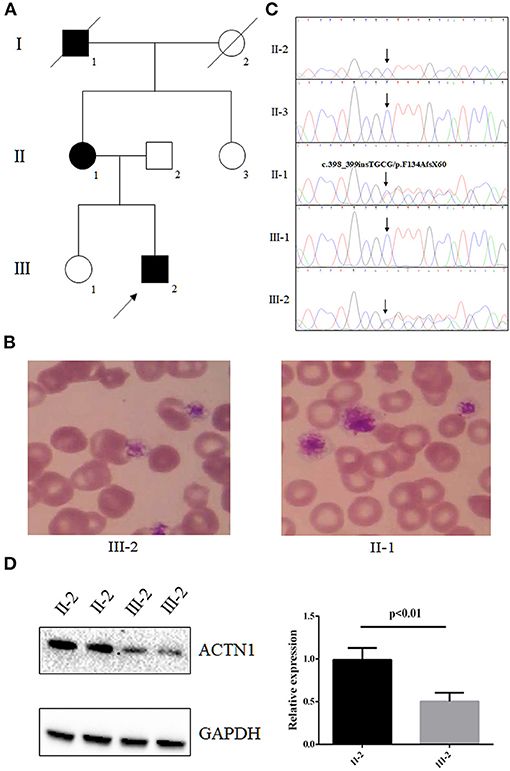
Figure 1. The clinical data and genetic analysis of the family with IMTP and mild bleeding. (A) Pedigree of the family. Black circles/squares are affected; white circles/squares are unaffected; slashed symbol is deceased family member. Arrow indicates the proband. (B) Blood smears (May-Grünwald Giemsa staining) revealed macrothrombocytopenia in the proband and his mother. (C) Sanger DNA sequencing chromatogram demonstrates the heterozygosity for an ACTN1 frameshift mutation (NM_001130004: c.398_399insTGCG, p.F134AfsX60) in affected members. (D) Western blot analysis of the expression of ACTN1 in the proband's platelets.
We then employed whole-exome sequencing to explore the candidate gene mutation in the proband (III-2). Whole-exome sequencing was conducted at BerryGenomic Institute (Beijing, China) (9). Exomes were captured by Agilent SureSelect Human All Exon V6 kits, and high-throughput sequencing was conducted with an Illumina HiSeq 4000 system. The strategies of data filtering were as follows (9, 10): (a) non-synonymous SNPs or frameshift-causing INDELs with an alternative allele frequency >0.01 in the NHLBI Exome Sequencing Project Exome Variant Server (ESP6500), dbSNP155, the 1000 Genomes Project, the genomAD database, or in-house exome databases of BerryGenomic (2,000 exomes) were excluded; (b) the filtered SNVs and INDELs, predicted by SIFT, Polyphen2, and MutationTaster to be damaging, were remained; (c) the variants belong to pathogenic and likely pathogenic according to American College of Medical Genetics (ACMG) guideline remained (11); (d) cosegregation analysis was conducted in the family.
Whole-exome sequencing yielded 9.98-Gb data. After alignment and single-nucleotide variant calling, 70,145 variants were identified in the proband. Via the aforementioned filtering method and Sanger sequencing validation, a novel frameshift mutation (NM_001130004: c.398_399insTGCG, p.F134AfsX60) of ACTN1 was identified in the proband and his mother but absent in unaffected individuals (Figure 1C). No other potential pathogenic mutation for macrothrombocytopenia-related phenotype was found (Table 1). Previous studies have proven that mutations in ACTN1 may lead to IMTP with mild to absent bleeding phenotype (12, 13). The novel mutation, resulting in a truncated protein in exon 4 of the ACTN1 gene, was absent in the public database such as 1000G and genomAD, as well as our 200 healthy controls. Bioinformatics programs predicted that this mutation (NM_001130004: c.398_399insTGCG, p.F134AfsX60) was a pathogenic mutation and located in an evolutionarily conserved site of the α-actin 1 protein. According to ACMG guidelines (11), this mutation was pathogenic (PVS1 + PM2 + PM3). We then further extracted total protein from the platelet of the proband and healthy control (II-2). Western Blot indicated that, compared with the healthy controls, the expression of ACTN1 was decreased by ~49% in the heterozygous carrier (Figure 1D). These data suggested that the variant (NM_001130004: c.398_399insTGCG, p.F134AfsX60) of ACTN1 was a loss-of-function mutation and can lead to non–sense-mediated mRNA decay.
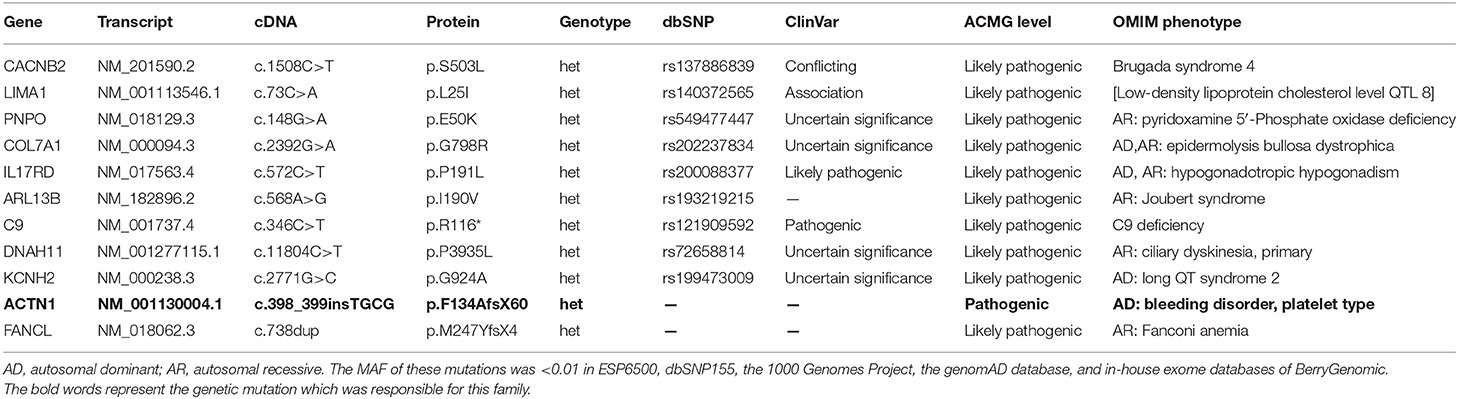
Table 1. Characteristics of the genetic variants identified for the proband via whole-exome sequencing.
Discussion
The human ACTN1 gene encoding a member of the actin-crosslinking protein named α-actinin is located on chromosome 14q24.1, and it consists of 21 exons spanning ~3.78 kilobases. α-Actin 1 participates in the organization of the cytoskeleton, thought to be an anchor actin to a variety of intracellular structures and mainly expressed in platelets and megakaryocytes (14). In 2013, six different mutations of ACTN1 were identified in 13 unrelated families with IMTP, which indicated that ACTN1 was one of the genetic lesions in IMTP (12). At present, thrombocytopenia caused by pathogenic variants in ACTN1 gene has been classified to ACTN1-related thrombocytopenia (15). To date, approximately 44 mutations of ACTN1 have been detected in IMTP patients. Here, we identified a frameshift mutation (NM_001130004: c.398_399insTGCG, p.F134AfsX60) of ACTN1 in a Chinese family with IMTP. As far as we know, this mutation may be first reported in IMTP patients; our study expanded the variant spectrum of ACTN1 gene.
The α-actin superfamily consists of four members including ACTN1, ACTN2, ACTN3, and ACTN4, which are responsible for the organization of the cytoskeleton (3, 16). Previous studies have found that ACTN1 was mainly expressed in platelets and megakaryocytes; the mutated ACTN1 may lead to a decrease of 50% platelet counts and an increase of 30% in platelet size (12, 17). In our study, the proband and his mother with the novel mutation of ACTN1 were also presented with macrothrombocytopenia. The novel frameshift mutation (NM_001130004: c.398_399insTGCG, p.F134AfsX60) can lead to the truncation mutation in the N-terminal of ACTN1. According to non–sense-mediated mRNA decay theory (18), the novel mutation may result in the decreased mRNA levels of ACTN1. Functional studies further confirmed that the novel mutation may lead to the reduction of ACTN1 expression, which may affect the organization of the cytoskeleton in platelets and megakaryocytes, finally resulting in macrothrombocytopenia.
Previous studies in Chinese hamster ovary cells revealed that the mutated ACTN1 may disrupt the normal actin-based cytoskeletal structure (12). The mice with ACTN1 mutation may present with disorganized actin-based cytoskeleton in megakaryocytes, which may further result in abnormal number and size of platelets (12). In this study, the phenotypes of the novel mutation (NM_001130004: c.398_399insTGCG, p.F134AfsX60) carriers (II-1 and III-2) were consistent with mice model and other reported patients, which further confirmed the pathogenicity of this novel mutation (19, 20).
IMTP can easily be misdiagnosed as immune thrombocytopenic purpura, which may further make problems in therapy and management of patients (8). Hence, precise diagnosis is necessary for IMTP patients, especially for the IMTP patients caused by ACTN1 mutations. A recent study that involved ~50 ACTN1 mutation carriers indicated that ACTN1 mutations lead to a benign form of platelet macrocytosis not always associated with thrombocytopenia (21). Precise diagnosis of ACTN1-caused IMTP can provide affected patients and their family members with a good prognosis. In our study, we confirmed the diagnosis of the affected members by whole-exome sequencing and Sanger sequencing, which may aid in the further management and prognosis of the family members. Meanwhile, our study also indicated that whole-exome sequencing combined with Sanger sequencing was an effective method in diagnosis of IMTP.
In summary, by employing whole-exome sequencing, we identified a novel frameshift mutation (NM_001130004: c.398_399insTGCG, p.F134AfsX60) of ACTN1 in a Chinese family with IMTP and mild epistaxis and/or gingival bleeding. Hence, this study not only broadened the variants spectrum of ACTN1 gene, which may contribute to the genetic counseling of IMTP, but also confirmed the diagnosis of IMTP, which may help in the management and prognosis of the family members.
Data Availability Statement
The data presented in the study are deposited in the (BioSample) repository, accession number: SAMN18953804, https://www.ncbi.nlm.nih.gov/biosample/18953804.
Ethics Statement
The studies involving human participants were reviewed and approved by the Second Xiangya Hospital of Central South University, Changsha, China. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author Contributions
F-ML and LL enrolled the samples and clinical data. YS and YD performed DNA isolation and PCR. F-ML and L-LF wrote the manuscript and revised it. L-LF and LL supported the project. All authors reviewed the manuscript.
Funding
This study was supported by National Natural Science Foundation of China (81800220, 82000079, and 82000427) and National Natural Science Foundation of Hunan province (2020JJ5785).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank all subjects for participating in this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2021.679279/full#supplementary-material
References
2. Favier R, Raslova H. Progress in understanding the diagnosis and molecular genetics of macrothrombocytopenias. Br J Haematol. (2015) 170:626–39. doi: 10.1111/bjh.13478
3. Ghosh K, Bhattacharya M, Chowdhury R, Mishra K, Ghosh M. Inherited macrothrombocytopenia: correlating morphology, epidemiology, molecular pathology and clinical features. Indian J Hematol Blood Transfus. (2018) 34:387–97. doi: 10.1007/s12288-018-0950-0
4. Savoia A. Molecular basis of inherited thrombocytopenias: an update. Curr Opin Hematol. (2016) 23:486–92. doi: 10.1097/MOH.0000000000000269
5. Monteferrario D, Bolar NA, Marneth AE, Hebeda KM, Bergevoet SM, Veenstra H, et al. A dominant-negative GFI1B mutation in the gray platelet syndrome. N Engl J Med. (2014) 370:245–53. doi: 10.1056/NEJMoa1308130
6. Grainger JD, Thachil J, Will AM. How we treat the platelet glycoprotein defects; Glanzmann thrombasthenia and Bernard Soulier syndrome in children and adults. Br J Haematol. (2018) 182:621–32. doi: 10.1111/bjh.15409
7. Ferrua F, Cicalese MP, Galimberti S, Giannelli S, Dionisio F, Barzaghi F, et al. Lentiviral haemopoietic stem/progenitor cell gene therapy for treatment of Wiskott-Aldrich syndrome: interim results of a non-randomised, open-label, phase 1/2 clinical study. Lancet Haematol. (2019) 6:e239–53. doi: 10.1016/S2352-3026(19)30021-3
8. Kanhai D, Mulder R, Ploos van Amstel HK, Schutgens R, Lukens M, Tamminga RYJ. Familial macrothrombocytopenia due to a double mutation in cis in the alpha-actinin 1 gene (ACTN1), previously considered to be chronic immune thrombocytopenic purpura. Pediatr Blood Cancer. (2018) 65:e27418. doi: 10.1002/pbc.27418
9. Luo FM, Deng MX, Yu R, Liu L, Fan LL. Case report: chorea-acanthocytosis presents as epilepsy in a consanguineous family with a nonsense mutation of in VPS13A. Front Neurosci. (2021) 15:604715. doi: 10.3389/fnins.2021.687435
10. Yu R, Liu L, Li YL, Fan LL. MITF p.Arg217Thr variant identified in a Han Chinese family with Tietz/Waardenburg syndrome. Biomed Res Int. (2021) 2021:4381272. doi: 10.1155/2021/4381272
11. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
12. Kunishima S, Okuno Y, Yoshida K, Shiraishi Y, Sanada M, Muramatsu H, et al. ACTN1 mutations cause congenital macrothrombocytopenia. Am J Hum Genet. (2013) 92:431–8. doi: 10.1016/j.ajhg.2013.01.015
13. Bottega R, Marconi C, Faleschini M, Baj G, Cagioni C, Pecci A, et al. ACTN1-related thrombocytopenia: identification of novel families for phenotypic characterization. Blood. (2015) 125:869–72. doi: 10.1182/blood-2014-08-594531
14. Haudek VJ, Slany A, Gundacker NC, Wimmer H, Drach J, Gerner C. Proteome maps of the main human peripheral blood constituents. J Proteome Res. (2009) 8:3834–43. doi: 10.1021/pr801085g
15. Boutroux H, David B, Gueguen P, Frange P, Vincenot A, Leverger G, et al. ACTN1-related macrothrombocytopenia: a novel entity in the progressing field of pediatric thrombocytopenia. J Pediatr Hematol Oncol. (2017) 39:e515–8. doi: 10.1097/MPH.0000000000000885
16. Murphy AC, Young PW. The actinin family of actin cross-linking proteins - a genetic perspective. Cell Biosci. (2015) 5:49. doi: 10.1186/s13578-015-0029-7
17. Yasutomi M, Kunishima S, Okazaki S, Tanizawa A, Tsuchida S, Ohshima Y. ACTN1 rod domain mutation associated with congenital macrothrombocytopenia. Ann Hematol. (2016) 95:141–4. doi: 10.1007/s00277-015-2517-6
18. Baker KE, Parker R. Nonsense-mediated mRNA decay: terminating erroneous gene expression. Curr Opin Cell Biol. (2004) 16:293–9. doi: 10.1016/j.ceb.2004.03.003
19. Gueguen P, Rouault K, Chen JM, Raguenes O, Fichou Y, Hardy E, et al. A missense mutation in the alpha-actinin 1 gene. (ACTN1) is the cause of autosomal dominant macrothrombocytopenia in a large French family. PLoS ONE. (2013) 8:e74728. doi: 10.1371/journal.pone.0074728
20. Vincenot A, Saultier P, Kunishima S, Poggi M, Hurtaud-Roux MF, Roussel A, et al. Novel ACTN1 variants in cases of thrombocytopenia. Hum Mutat. (2019) 40:2258–69. doi: 10.1002/humu.23840
Keywords: macrothrombocytopenia, mild bleeding, ACTN1 mutation, non-sense-mediated mRNA decay, whole-exome sequencing
Citation: Luo F-M, Fan L-L, Sheng Y, Dong Y and Liu L (2021) Case Report: Exome Sequencing Identified a Novel Frameshift Mutation of α-Actin 1 in a Chinese Family With Macrothrombocytopenia and Mild Bleeding. Front. Pediatr. 9:679279. doi: 10.3389/fped.2021.679279
Received: 11 March 2021; Accepted: 17 May 2021;
Published: 18 June 2021.
Edited by:
Emiliano González Vioque, University Clinical Hospital of Santiago, SpainReviewed by:
Benedetta Izzi, Istituto Neurologico Mediterraneo Neuromed (IRCCS), ItalyAniefiok John Udoakang, University of Ghana, Ghana
Copyright © 2021 Luo, Fan, Sheng, Dong and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lv Liu, ZG9jbGl1bHZAY3N1LmVkdS5jbg==
†These authors have contributed equally to this work