 Xiayuan Xu1,2,†Chengcheng Gao3,†Keqin Jin1,2Liping Zhang4Yanfen Yang4Jun Zhang1,2Yun Ye2,5
Xiayuan Xu1,2,†Chengcheng Gao3,†Keqin Jin1,2Liping Zhang4Yanfen Yang4Jun Zhang1,2Yun Ye2,5 Shuangshuang Shen2,5*
Shuangshuang Shen2,5*
- 1Department of Genetic Laboratory, Jinhua Maternity and Child Health Care Hospital, Jinhua, China
- 2Jinhua’s Key Laboratory of Birth Defects Prevention and Treatment, Jinhua Maternity and Child Health Care Hospital, Jinhua, China
- 3Key Laboratory of Digital Technology in Medical Diagnostics of Zhejiang Province, Dian Diagnostics Group Co., Ltd., Hangzhou, China
- 4Department of Ultrasonography, Jinhua Maternity and Child Health Care Hospital, Jinhua, China
- 5Department of Prenatal Diagnostic, Jinhua Maternity and Child Health Care Hospital, Jinhua, China
Background: Congenital heart defects (CHDs) represent the leading cause of neonatal mortality among congenital abnormalities. Genetic factors, such as EVC2 gene mutations and other genetic alterations, constitute a major cause of CHD. Thus, determining the genetic etiology of fetal CHDs is crucial for optimizing pregnancy management and informing future reproductive decisions.
Case presentation: Here, we describe a male fetus with complex CHD who was diagnosed at 25 weeks of gestation, delivered at full term, and died prematurely within a month due to heart failure. The cardiac abnormalities observed included an atrial septal defect developing from a patent foramen ovale, mitral valve regurgitation, dilated right ventricle and left atrium, aortic stenosis, and aortic arch dysplasia. Novel compound heterozygosity of the EVC2 gene, including a non-sense mutation (p.W828Ter) and two cis missense mutations (p.E87G and p.S217C), was identified by prenatal trio-whole-exome sequencing of amniotic fluid, followed by validation using Sanger sequencing. This novel EVC2 genotype was supposed to potentially affect fetal cardiac development, given the variable clinical heterogeneity of the EVC2 mutation-associated phenotype. This case represents the first identification of the EVC2 p.E87G and p.S217C, and the isolated CHD without visible skeletal dysplasia is an important feature of our case.
Conclusions: Our study expands the genotypic and phenotypic spectra of the EVC2 gene. We recommend including the EVC2 gene in preconception carrier screening and prenatal diagnosis for CHDs.
1 Introduction
Congenital heart defects (CHDs) represent a large and rapidly emerging health challenge for neonates worldwide (1). Genetic alterations, including aneuploidy, copy number variation, and inherited sequence variation, can lead to the development of CHD (2, 3). It is essential to determine the cause of CHD in the proband based on the current medical progress as thoroughly as possible, which will help prevent mothers from giving birth to another child with CHD.
Ellis–van Creveld syndrome (EVCS) (MIM: #225500), also called chondroectodermal dysplasia, is a rare autosomal recessive disorder mainly caused by mutations in EVC or EVC2 genes and one of the most severe CHDs in neonates (4, 5). EVCS affects 1 in 60,000–200,000 neonates worldwide. Its characteristic features include short limbs and ribs, postaxial polydactyly, dysplastic nails and teeth, and congenital heart defects (6). Congenital heart defects, most commonly an atrial septal defect, are observed in 60% of affected individuals and contribute to the majority of deaths in EVCS infants (7, 8). Other cardiovascular malformations include persistent left superior vena cava (LSVC), unroofed coronary sinus, and anomalous pulmonary veins (9). The prenatal anomalies of EVCS could be identified as early as 18 weeks of pregnancy (10). However, the phenotypes of EVCS vary among individuals, ranging from lethal to mild clinical presentations. Formal evidence-based diagnostic criteria for EVCS have not been established, and the clinical diagnosis mainly relies on genetic sequencing (molecular diagnosis), clinical characteristics, and radiographic features. Thus, more research is needed to establish the association between EVC2 mutations and their phenotypes.
In the present study, we describe a male fetus diagnosed with complex CHD at 25 weeks of gestation. Biallelic EVC2 mutations (p.W828Ter, p.E87G, and p.S217C) were identified by prenatal trio-based whole-exome sequencing (trio-WES) of amniotic fluid.
2 Case presentation
2.1 Case description
In this family, the father is of Han Chinese descent, while the mother belongs to the Miao ethnic minority group in China. The parents denied a consanguineous marriage and have a healthy daughter. No family history of genetic disorders was reported. The proband was a male fetus, the second child of the 26-year-old mother (gravida 5, para 2). He was diagnosed with CHD in utero at 25 weeks of gestation. Fetal echocardiography revealed several cardiac abnormalities, with a normal cardiac rate (144 bpm); these abnormalities included aortic stenosis with coarctation of the aortic arch (from aortic root diameter measuring 2.3 mm to an isthmus of 1.7 mm), mitral valve regurgitation with reduced mitral valve flow, a patent foramen ovale (left-to-right shunt), an enlarged left atrium (left 13 mm × 13 mm vs. right 11 mm × 11 mm), and an enlarged right ventricle (right 14 mm × 11 mm vs. left 15 mm × 8 mm) (Figures 1A,B). The mother did not terminate the pregnancy despite the obstetrician informing her of the risks. At 39 weeks of gestation, the obstetrician reported no other abnormalities except the CHD.
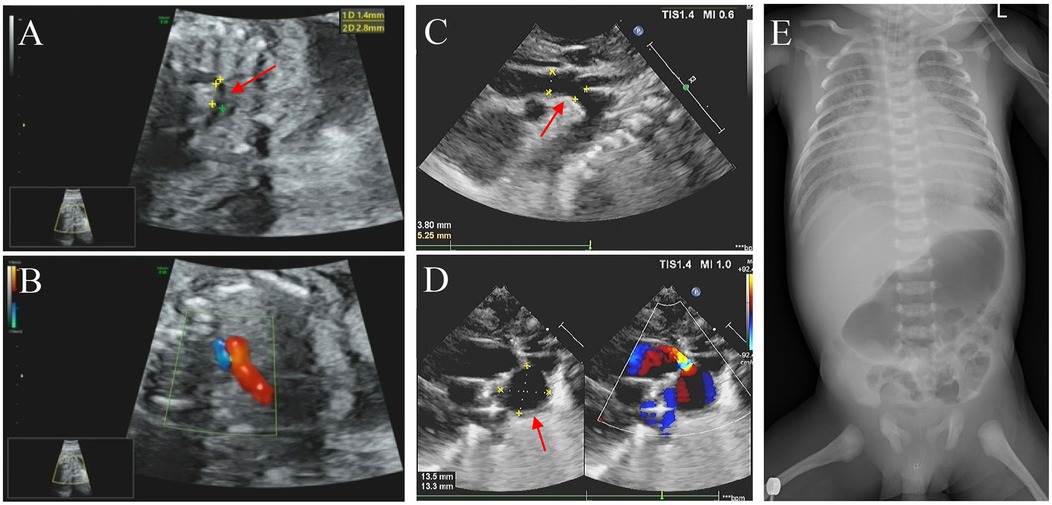
Figure 1. Imaging examination results. (A,C) Echocardiography performed during the fetal period (25 weeks pregnant) and neonatal period, respectively, showed the coarctation of the aortic arch (marked with an arrowhead). (B) The fetus exhibited pulmonary and aortic blood flow incongruity with an increased vessel ratio. (D) Neonatal imaging showing a patent ductus arteriosus aneurysm (marked with an arrowhead) and a bidirectional arterial shunt. (E) Digital x-ray imaging did n't show any obvious developmental abnormalities of the sternum, ribs, or spine.
The proband was delivered with normal growth parameters (birth weight = 3,130 g, length = 50 cm) by cesarean section at 40 weeks of gestation due to severe preeclampsia and then transferred to neonatology for respiratory distress. Imaging examinations did n't reveal any significant developmental anomalies of the sternum, ribs, or spine, but they did reveal more severe CHD symptoms, including an atrial septal defect (3.9 mm), a patent ductus arteriosus aneurysm (DAA, 14 mm × 13 mm aneurysm), aortic stenosis, aortic arch dysplasia (3.8 mm), moderate mitral regurgitation, an enlarged right ventricle (right 30 mm × 15 mm vs. left 27 mm × 16 mm), and an enlarged left atrium (left 26 mm × 23 mm vs. right 23 mm × 19 mm) with reduced left ventricular systolic function (Figures 1C–E). On the ninth day of life, the proband was transferred to the neonatal intensive care unit (NICU) due to heart failure (N-terminal BNP >5,000 pg/ml, troponin = 2.450 μg/L), respiratory failure, gastrointestinal bleeding, and cardiogenic shock accompanied by metabolic disorders (hypoglycemia, hyperlactatemia, and hyperkalemia). Life support measures were initiated, including ventilator-assisted ventilation, milrinone injection, alprostadil injection, and nutritional support therapy. Although medical treatment improved his metabolic acidosis and internal hemorrhage, the proband unfortunately died prematurely within a month due to heart failure.
Karyotyping and chromosomal microarray analysis (CMA) did n't reveal any abnormalities during pregnancy. However, prenatal trio-WES of amniotic fluid identified compound heterozygosity in EVC2 gene, consisting of a paternal non-sense mutation (NM_147127.5: c. 2484G>A, p.W828Ter) and two maternal missense mutations (c.260A>G, p.E87G; c.650C>G, p.S217C) (Table 1, Figure 2A). These three EVC2 mutations were verified by Sanger sequencing (Figures 2B–D). The EVC2 p.W828Ter mutation has been indexed as pathogenic or likely pathogenic for EVCS (ClinVar: #553315), while the other two EVC2 mutations have n't been previously reported or indexed in the database. The EVC2 protein contains a glycosylation site at amino acid position 220 and an EVC2-like domain spanning amino acids 237–660. None of the three detected mutations are located within the known EVC2 protein functional domains. The non-sense mutation, EVC2 p.W828Ter, is located in exon 14 and is predicted to lead to non-sense-mediated mRNA decay (PVS1 criterion met). According to GnomAD, the allele frequency (AF) for p.W828Ter is 0.0003013 in the East Asian population (PM2_supporting criterion met), while the other two missense mutations have not been indexed (PM2_supporting). The non-sense mutation has been reported in trans with pathogenic variants (EVC2 p.R399X and c.871-2_894del) in two patients with EVCS (PM3 criterion met) (11, 12). Integrated analysis—including rare exome variant ensemble learner (REVEL), combined annotation dependent depletion (CADD), ClinPred, and Computational_evidence_ratio—suggests that p.E87G is more likely to be tolerable, whereas most pathogenicity predictions for p.S217C are detrimental (Table 1) (meeting the PP3 criterion). Thus, EVC2 p.W828Ter is classified as pathogenic (PVS1 + PM2_supporting + PM3), while EVC2 p.S217C (PM2_supporting + PM3 + PP3) and p.E87G (PM2_supporting + PM3) are classified as variants of uncertain significance (VUSs). Unfortunately, samples from the last three miscarriages were not preserved for further verification. Thus, it is supposed that the novel identified EVC2 genotype may have affected the cardiac development of the proband.
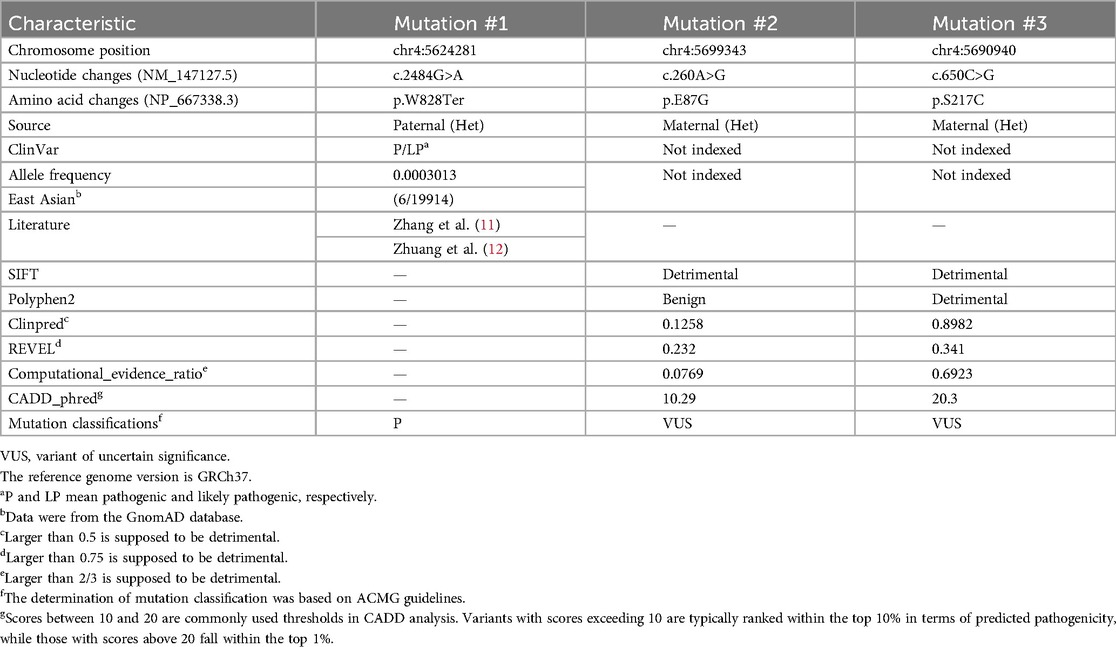
Table 1. Database information and pathogenicity predictions of EVC2 mutations.
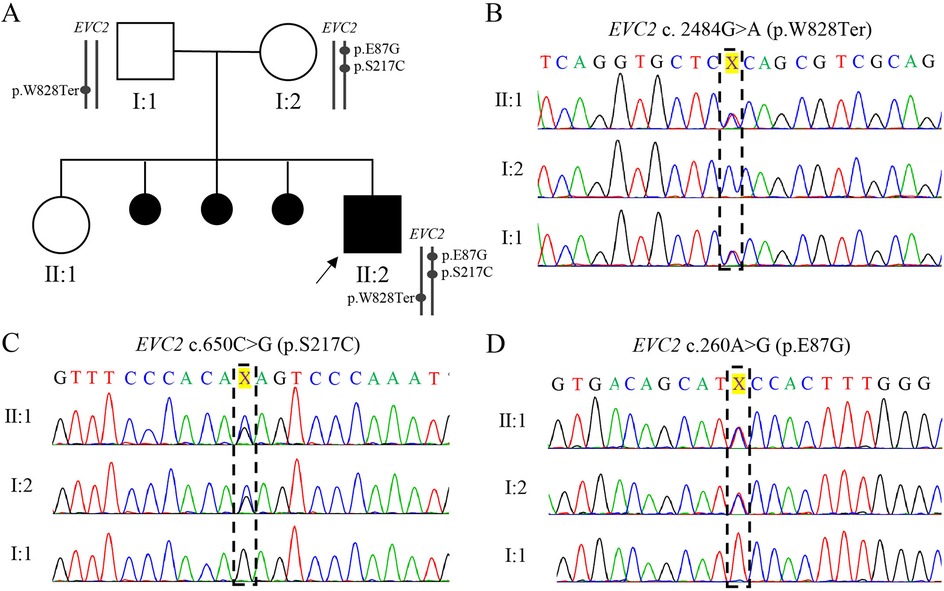
Figure 2. Family pedigree and Sanger sequencing validation of three EVC2 mutations. (A) The family pedigree. The cause of the last three miscarriages remains unknown due to the absence of preserved samples for further detection. (B–D) Chromas showing the mutation in the family. The position of EVC2 p.W828*, p.S217C, and p. E87G is marked with a black box.
2.2 Genetic testing methodology
Trio-WES was performed using peripheral blood samples from the parents and amniotic fluid from the fetus. Genomic DNA was extracted from parental blood samples using the DNeasy Blood & Tissue Kit (Qiagen, Valencia, CA, USA), while fetal DNA was extracted from the amniotic fluid. The sequencing libraries were prepared using the Agilent SureSelect Human All Exon v.6. After PCR amplification, the sequencing libraries were sequenced on the HiSeq X Ten platform (Illumina Inc., San Diego, CA, USA). For all three individuals, the mean sequencing depth exceeded 175×, with more than 99.4% of variants covered at a depth greater than 20×.
Adapter sequences and low-quality reads were filtered out. The clean reads were then aligned to the human reference genome (GRCh37/hg19) using the Burrows–Wheeler Aligner (BWA). Variant calling was performed using the Genome Analysis Toolkit (GATK), followed by variant annotation using ANNOVAR. Candidate mutations were filtered based on AF (<1%), clinical phenotype, inheritance patterns, and pathogenicity classification.
Sanger sequencing was conducted to validate the candidate variants. The primer sequences used for amplification are the following: for EVC2 c. 2484G>A: forward 5′-TGCAGCAGGAGTGTTAGGTG-3′ and reverse 5′- CCTGCAGAACTCAGCCATGA-3′; for EVC2: c.260A>G, forward 5′-ACTGTGCACTAACGCTTCGG-3′ and reverse 5′-TTCACACTTCTGGATGAAAGTGC-3′; for EVC2: c.650C>G, forward 5′-ACATGCCTGACCCAGAACAC-3′ and reverse 5′-CGTGCTACCCTCCTTCTTCC-3′. Sequencing was performed using an ABI 3130 Genetic Analyzer (Applied Biosystems, CA, USA).
3 Discussion
A rising number of disease-causing genes and mutations have been identified in patients with CHD (13). In our CHD case, trio-WES identified novel biallelic mutations in the EVC2 gene. The EVC2 p.W828Ter mutation was reported as a pathogenic variant in two patients with EVCS (11). The other two missense variants, p.E87G and p.S217C, have n't been identified previously.
Notably, our male proband did n't exhibit visible skeletal dysplasia, which differs from the classic features of EVCS. This may be attributable to the high degree of variability in clinical expressivity. In a Pakistani family, three EVCS siblings carried a homozygous deletion mutation: EVC p.Leu244_Ser253delinsPro (14). Of these, two affected males only exhibited polydactyly, along with nail and dental abnormalities, while their sister presented with more severe symptoms, including short stature and CHD. Further study is warranted to determine whether there are differences in disease severity between female and male patients with EVCS. In another family, both compound heterozygous genotypes (p.Arg662Ter and p.Arg663Pro, p.Arg662Ter and c.1316-7A>G) were identified in individuals with EVCS (15). However, the individual carrying a compound heterozygosity genotype, p.Arg663Pro and c.1316-7A>G, only exhibited mild mitral regurgitation, indicating that some hypomorphic mutations could still produce a sufficient amount of functional EVC2 protein. Thus, it is hypothesized that the combination of EVC2 p.W828Ter, p.E87G, and p.S217C identified in our proband may induce CHD but have little effect on fetal skeletal development.
The EVC and EVC2 proteins constitute the EVC–EVC2 complex, located within the primary cilium. This complex functions by transducing extracellular signals to the nucleus via the Hedgehog (HH) signaling pathway (16). The pathogenesis of EVCS phenotypes caused by EVC2 mutations has been partially elucidated. EVC2 loss-of-function results in delayed ameloblast differentiation by disrupting HH signaling in the dental epithelium, clarifying the pathogenesis of enamel hypoplasia in patients with EVCS (17). Apart from the HH signaling pathway, EVC2 mutations could induce dwarfism by elevating fibroblast growth factor (FGF) signaling (18). However, the pathogenesis of other phenotypes, especially the pathogenic mechanism of CHD, needs further research for clarification.
CHD related to EVC2 or EVC mutations can also be repaired surgically. Delayed surgery for CHD among children (1.3 vs. 50.1 months) was associated with fewer postoperative complications and a higher survival rate (19). Respiratory morbidity was identified as the main postoperative complication, while respiratory failure was the leading cause of mortality (20). However, our proband was born with respiratory distress and rapidly progressed to respiratory failure, accompanied by heart failure and cardiogenic shock, within 9 days. Consequently, surgical intervention was no longer appropriate. Thus, it is crucial to establish a clinical management process for infants with CHD carrying EVC2 mutations to get through early life until they are suitable for surgery.
Here, we report on the entire life course of a CHD fetus carrying compound heterozygous mutations in the EVC2 gene. The two novel EVC2 missense variants (p.E87G and p.S217C) were identified for the first time. It is hypothesized that this novel EVC2 genotype might affect heart development but have little effect on skeletal development. Our study provides new insights into prenatal diagnosis and genetic counseling for patients with CHD carrying EVC2 mutations. Meanwhile, we suggest establishing a clinical management process for CHD neonates carrying EVC2 mutations to prevent premature death.
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Ethics statement
The studies involving humans were approved by the Ethics Committee of Jinhua Maternity and Child Health Care Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants' legal guardians/next of kin. Written informed consent was obtained from the individual(s) and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
XX: Data curation, Formal analysis, Funding acquisition, Investigation, Project administration, Resources, Supervision, Writing – original draft, Writing – review & editing. CG: Conceptualization, Investigation, Software, Writing – original draft, Writing – review & editing. KJ: Methodology, Validation, Writing – original draft. LZ: Investigation, Writing – original draft. YYa: Visualization, Writing – original draft. JZ: Validation, Writing – original draft. YYe: Investigation, Writing – original draft. SS: Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by grants from the Jinhua Science and Technology Bureau (Nos. 2020-4-068 and 2024-3-089).
Acknowledgments
The authors thank all staff involved in this study and also express their gratitude to the family who made this study possible.
Conflict of interest
CG was employed by Dian Diagnostics Group Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. GBDCHD Collaborators. Global, regional, and national burden of congenital heart disease, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Child Adolesc Health. (2020) 4(3):185–200. doi: 10.1016/S2352-4642(19)30402-X
2. Williams K, Carson J, Lo C. Genetics of congenital heart disease. Biomolecules. (2019) 9(12):879. doi: 10.3390/biom9120879
3. Zaidi S, Brueckner M. Genetics and genomics of congenital heart disease. Circ Res. (2017) 120(6):923–40. doi: 10.1161/CIRCRESAHA.116.309140
4. Galdzicka M, Patnala S, Hirshman MG, Cai JF, Nitowsky H, Egeland JA, et al. A new gene, EVC2, is mutated in Ellis-van Creveld syndrome. Mol Genet Metab. (2002) 77(4):291–5. doi: 10.1016/S1096-7192(02)00178-6
5. Ruiz-Perez VL, Tompson SW, Blair HJ, Espinoza-Valdez C, Lapunzina P, Silva EO, et al. Mutations in two nonhomologous genes in a head-to-head configuration cause Ellis-van Creveld syndrome. Am J Hum Genet. (2003) 72(3):728–32. doi: 10.1086/368063
6. Ruiz-Perez VL, Ide SE, Strom TM, Lorenz B, Wilson D, Woods K, et al. Mutations in a new gene in Ellis-van Creveld syndrome and Weyers acrodental dysostosis. Nat Genet. (2000) 24(3):283–6. doi: 10.1038/73508
7. Baujat G, Le Merrer M. Ellis-van Creveld syndrome. Orphanet J Rare Dis. (2007) 2:27. doi: 10.1186/1750-1172-2-27
8. Nguyen TQ, Saitoh M, Trinh HT, Doan NM, Mizuno Y, Seki M, et al. Truncation and microdeletion of EVC/EVC2 with missense mutation of EFCAB7 in Ellis-van Creveld syndrome. Congenit Anom (Kyoto). (2016) 56(5):209–16. doi: 10.1111/cga.12155
9. Hills CB, Kochilas L, Schimmenti LA, Moller JH. Ellis-van Creveld syndrome and congenital heart defects: presentation of an additional 32 cases. Pediatr Cardiol. (2011) 32(7):977–82. doi: 10.1007/s00246-011-0006-9
10. Peraita-Ezcurra M, Martinez-Garcia M, Ruiz-Perez VL, Sanchez-Gutierrez ME, Fenollar-Cortes M, Velez-Monsalve C, et al. Ellis-van Creveld syndrome in a fetus with rhizomelia and polydactyly. Report of a case diagnosed by genetic analysis, and correlation with pathological andradiologic findings. Gene. (2012) 499(1):223–5. doi: 10.1016/j.gene.2012.02.030
11. Zhang Z, Bao K, He JW, Fu WZ, Zhang CQ, Zhang ZL. Identification of one novel mutation in the EVC2 gene in a Chinese family with Ellis-van Creveld syndrome. Gene. (2012) 511(2):380–2. doi: 10.1016/j.gene.2012.09.071
12. Zhuang J, Liu S, Wang J, Chen Y, Zhang H, Jiang Y, et al. Prenatal whole exome sequencing identified two rare compound heterozygous variants in EVC2 causing Ellis-van Creveld syndrome. Mol Genet Genomic Med. (2023) 11(10):e2242. doi: 10.1002/mgg3.2242
13. Morton SU, Quiat D, Seidman JG, Seidman CE. Genomic frontiers in congenital heart disease. Nat Rev Cardiol. (2022) 19(1):26–42. doi: 10.1038/s41569-021-00587-4
14. Zaka A, Shahzad S, Rao HZ, Kanwal S, Gul A, Basit S. An intrafamilial phenotypic variability in Ellis-Van Creveld syndrome due to a novel 27 bps deletion mutation. Am J Med Genet A. (2021) 185(10):2888–94. doi: 10.1002/ajmg.a.62360
15. Piceci-Sparascio F, Palencia-Campos A, Soto-Bielicka P, D'Anzi A, Guida V, Rosati J, et al. Common atrium/atrioventricular canal defect and postaxial polydactyly: a mild clinical subtype of Ellis-van Creveld syndrome caused by hypomorphic mutations in the EVC gene. Hum Mutat. (2020) 41(12):2087–93. doi: 10.1002/humu.24112
16. Dorn KV, Hughes CE, Rohatgi R. A smoothened-EVC2 complex transduces the Hedgehog signal at primary cilia. Dev Cell. (2012) 23(4):823–35. doi: 10.1016/j.devcel.2012.07.004
17. Zhang H, Takeda H, Tsuji T, Kamiya N, Kunieda T, Mochida Y, et al. Loss of function of EVC2 in dental mesenchyme leads to hypomorphic enamel. J Dent Res. (2017) 96(4):421–9. doi: 10.1177/0022034516683674
18. Zhang H, Kamiya N, Tsuji T, Takeda H, Scott G, Rajderkar S, et al. Elevated fibroblast growth factor signaling is critical for the pathogenesis of the dwarfism in EVC2/limbin mutant mice. PLoS Genet. (2016) 12(12):e1006510. doi: 10.1371/journal.pgen.1006510
19. Chowdhury D, Williams KB, Chidekel A, Pizarro C, Preedy C, Young M, et al. Management of congenital heart disease associated with Ellis-van Creveld short-rib thoracic dysplasia. J Pediatr. (2017) 191:145–51. doi: 10.1016/j.jpeds.2017.08.073
Keywords: congenital heart defect, fetus, neonate, EVC2, Ellis–van Creveld syndrome, case report
Citation: Xu X, Gao C, Jin K, Zhang L, Yang Y, Zhang J, Ye Y and Shen S (2025) Case Report: A novel compound heterozygosity of the EVC2 gene identified in a Chinese pedigree with congenital heart defect. Front. Pediatr. 13:1352571. doi: 10.3389/fped.2025.1352571
Received: 8 December 2023; Accepted: 18 June 2025;
Published: 14 July 2025.
Edited by:
Andrew S. Day, University of Otago, New ZealandReviewed by:
Emanuele Micaglio, IRCCS San Donato Polyclinic, ItalyAsmat Ullah, Broad Institute of MIT and Harvard, United States
Dina Yagel, Clalit Health Services, Israel
Copyright: © 2025 Xu, Gao, Jin, Zhang, Yang, Zhang, Ye and Shen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shuangshuang Shen, c3NzaGVuX2poZmJAaG90bWFpbC5jb20=
†These authors have contributed equally to this work