 Lars Lindberg
Lars Lindberg- Institution of Clinical Sciences, PICU, Children’s Hospital in Lund, Skane University Hospital, Lund University, Lund, Sweden
This case report highlights the challenges in treating bronchopulmonary dysplasia (BPD) in a premature infant with severe pulmonary hypertension, recurrent pulmonary hypertensive crises, and the need of 100% oxygen to achieve acceptable arterial oxygen saturations. Key factors in the infant's improvement involved switching from pulmonary vasodilation to systemic afterload reduction using losartan, an angiotensin II type 1 receptor blocker. This alteration in treatment strategy led to a pronounced and prompt decrease in pulmonary arterial pressure, reduced oxygen dependency and resolution of pulmonary hypertensive crises. The infant's remarkable clinical response suggests that the pulmonary hypertension in BPD may have a pulmonary post-capillary cause, possibly driven by angiotensin II. A literature review corroborates this revision of the current understanding of the pathophysiologic mechanism involved in BPD and suggests that therapies targeting the renin-angiotensin-aldosterone system rather than pulmonary vasodilation may be an effective treatment strategy.
1 Introduction
Bronchopulmonary dysplasia (BPD) is a chronic neonatal lung disease commonly seen in extremely premature infants and along with increasing survival rates a growing burden in neonatal intensive care units (NICU) (1–3). BPD is characterized by injury to the lung parenchyma and vascular compartments (4–6) with histological evidence of alveolar destruction, epithelial injury, smooth muscle hyperplasia, diffuse fibrosis, inflammatory cell infiltration, and pulmonary venous congestion (7–11). Commonly used therapeutic measures in the NICU, such as long term oxygen supplementation and mechanical ventilator support, can themselves exacerbate lung damage via activation of inflammation and oxidative stress pathways (12, 13).
A frequent complication of BPD is the development of pulmonary hypertension (PH) (14–18). PH in BPD is most often interpreted as having a pre-capillary origin, and thus treated with pulmonary vasodilators, such as phosphodiesterase inhibitors, inhaled nitric oxide (iNO), inhaled iloprost, and endothelin receptor antagonists (15, 19). These drugs can temporarily improve oxygenation and reduce PH, but pulmonary vasodilators neither prevent the development of BPD nor lower mortality rates (20–27). This indicates that PH in severe BPD is associated with an increase in morbidity and mortality (18), but not the cause and accordingly a potential confounder. The findings by Cohen et al. (28) that a cohort with BPD and PH treated with sildenafil had the highest overall mortality (26%) of all their cohorts compare with the findings by Yung et al. (29) that a limited use of sildenafil had an extraordinarily low mortality rate of only 5% in their children with severe BPD and PH, indirectly indicate that the use of pulmonary vasodilators may not be appropriate. The understanding of PH in BPD as a pre-capillary phenomenon that should be treated with pulmonary vasodilators warrants a reconsideration.
Moreover, research indicates that left sided cardiac strain caused by systemic hypertension, likely due to increased systemic vascular resistance, is common in cohorts of children with BPD (30–32). This contributes to a subtle left ventricular diastolic dysfunction, increased left atrial pressure, pulmonary venous congestion and ultimately the development of an insidious pulmonary post-capillary PH (24, 33). Elevated left atrial pressure is associated with higher mortality rate in children with BPD (33). In addition, the effectiveness to reduce PH by systemic afterload reduction via renin-angiotensin-aldosteron-system (RAAS) inhibition has been documented and supports the assumption that PH in BPD has a post-capillary rather than pre-capillary origin (34).
The presented case reports a child with severe BPD and PH, who showed a remarkable improvement after switching the treatment strategy from pulmonary vasodilators to the angiotensin receptor blocker (ARB), losartan. The pathophysiological mechanisms and rationale behind using RAAS inhbition instead of pulmonary vasodilators in BPD are discussed.
2 Case description
This case presents a female infant born vaginally at a gestational age of 25 + 1 weeks, with a birth weight of 820 g [appropriate for gestational age (AGA)], which speaks against intrauterine growth restriction (35). She received “less invasive surfactant administration” (LISA) with Curosurf® 240 mg at 3 hours and 120 mg at 2 days of age. The infant developed severe respiratory distress syndrome (RDS) and required endotracheal intubation at 5 days of age due to hypercapnia, hypoxia and fatigue (Figure 1). Duration of invasive mechanical ventilation and non-invasive ventilation are shown in Table 1. An intraventricular hemorrhage (IVH grade 1—subependymal bleeding) was diagnosed on the right side at 3 days of age. Several follow-ups during the course of the disease did not detect any new bleedings or sign of perventricular leukomalacia (PVL).

Figure 1. Representative chest radiographs. (A) PMA (post-menstrual age) 26w, (B) PMA 35w, (C) PMA 41w (before closure of the shunts), (D) PMA 50w (before tracheostomy), (E) PMA 55w (after tracheostomy) and (F) PMA 56w. The child developed early bilateral fine granular opacities in the pulmonary parenchyma and air bronchograms as seen in respiratory distress syndrome (RDS). Pulmonary edema could not be excluded. The granular opacities remained and became coarse. Hyperinflation and cystic changes developed and the cystic structure in left lower lobe was defined as pneumatocele. Transient apical atelectasis and non-specific parenchymal opacities occurred mainly on the right side.
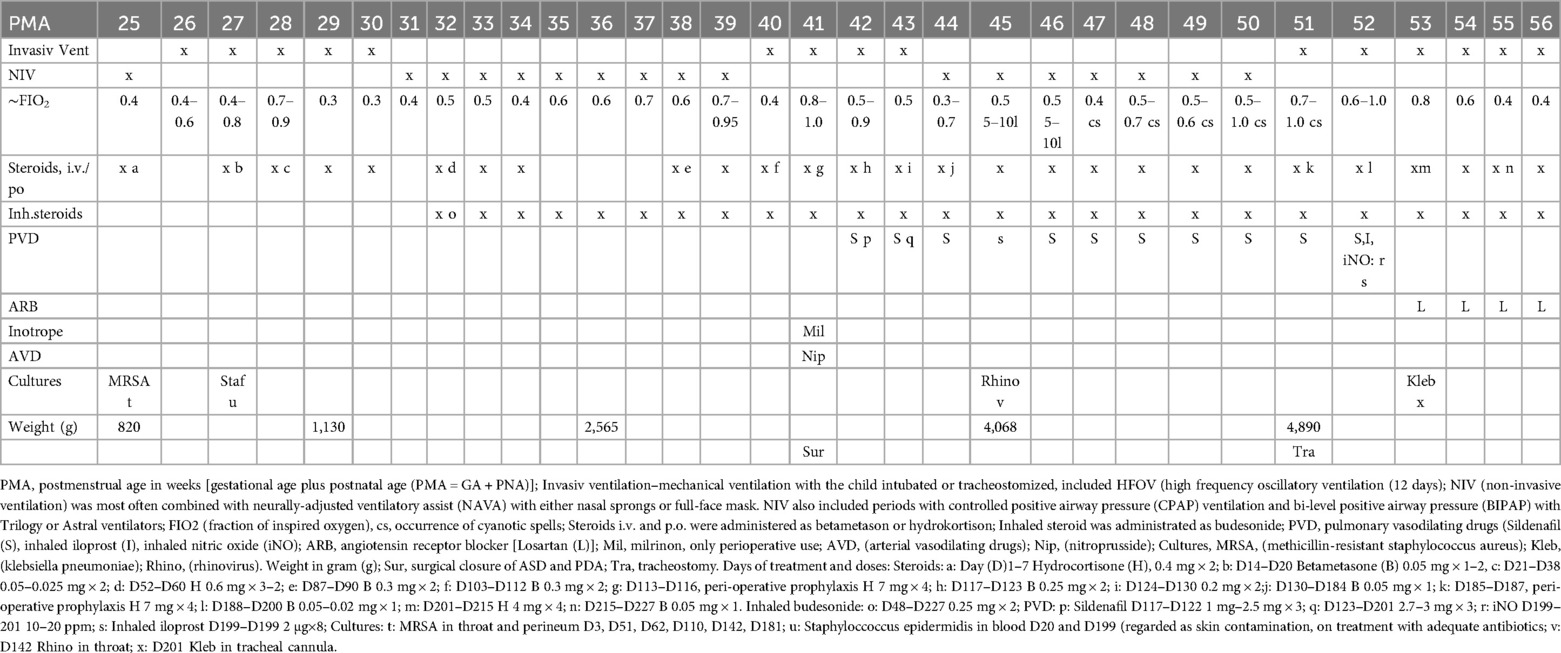
Table 1. A clinical timeline.
The first cardiac ultrasound was performed at 6 days of age and revealed an atrial septal defect (ASD) and a patent ductus arteriosus (PDA, Vmax 1,8 m/s), both with significant left-to-right shunting. A reversible ante-to-retrograde blood flow was noticed in mesenteric blood vessels, which indicated a significant run-off phenomenon of blood flow from the gut into the lung with diastolic left-to-right shunt in the PDA, but no NEC developed. Ibuprofen (PEDEA®) was administered intravenously with five doses with no effect on the PDA. The right ventricle was dilated due to pulmonary overcirculation and on some cardiac ultrasounds judge to be slightly hypertrophied. Left ventricle had normal systolic contractility. A CT scan (Figure 2) and cardiac ultrasounds clearly showed that all four pulmonary veins enter the left atrium without obstruction. The ASD was not suitable for device closure due to its size and location. A cardiac catheterization was discussed, but the cardiologists refrain from it, since pulmonary vein stenosis had been excluded, device closure was not needed and the PH was regarded to be caused by the left-to-right shunts. A surgical closure of the ASD and PDA was advocated and successfully done at 4 months of age.

Figure 2. Ct scans with contrast of the chest. (A) CT (computed tomography) scan was performed at PMA (post-menstrual age) 41w before closure of the shunts. Frontal view on left, anterior to dorsal (A,C,E) and transverse view on right, apical to basal (B,D,F). Pulmonary venous obstruction was excluded and the PDA and ASD could be verified. Pronounced bilateral lung parenchymal abnormalities with crazy paving, ground glass, intralobular lines, thickening of interlobular septa, and dorsal consolidation were noticed in keeping with BPD. Consolidation of the middle lobe with no suspicion of intralobular sequestration was seen. Ventrobasal in the left lower lobe an 18 × 20 × 16 mm air filled cystic structure, probable a pneumatocele was diagnosed. Several small cystic changes were seen bilateral, especially in basal parts of the lung. A mild pulmonary edema could not be excluded.
A significant rise in the systolic and diastolic arterial blood pressure [mean arterial pressure (MAP) ≥90–105 mm Hg] occurred postoperatively. Cardiac ultrasounds showed sign of a remaining sub-systemic PH after the correction and a decision was made to start sildenafil treatment on the third postoperative day aiming at a target dose of 2 mg/kg/day. Repeated cardiac ultrasounds during the treatment with sildenafil demonstrated a persistent PH at sub-systemic levels evaluated by the tricuspid regurgitant pressure gradient (75 mm Hg, 4 m/s). There was no sign of right ventricular failure. The contractility and the systolic function of the left ventricle were described as good. MAP remained high (> 70–80 mm Hg), when the child was awaken for several weeks after the correction. A diagnosis of severe BPD was confirmed based on chest x-ray (Figure 1) and the definition by the National Institute of Child Health and Human Development program (36, 37). A cystic structure in left lower base was seen on the chest x-ray already the day after birth and it was later defined as a congenital pulmonary adenomatoid malformation (CPAM). Emphysematous changes developed after 35 weeks of postmenstrual age (PMA) (Figure 1). The infant required continuous non-invasive ventilator support using a full face mask and oxygen supplementation with FiO2 of 0.5–0.75 to maintain oxygen arterial saturations between 65% and 85%. Despite receiving a sildenafil dose of 1.9 mg/kg/day, the baby developed cyanotic spells with dangerously low to undetectable levels of oxygen saturation and was claimed to have untreatable PH with repeated pulmonary hypertensive crises. It was difficult to increase her feeding due to gastric retentions and what was judge as abdominal spasm or pain. Broad-spectrum antibiotic treatments were given due to the growth of MRSA, and Klebsiella pneumoniae in the upper airways, and rhinovirus was also detected (Table 1).
The infant was considered stable enough to be transferred from the NICU to a general pediatric ward at six months of age. However, her condition deteriorated rapidly after transfer, with severe intercostal respiratory retractions and increased restlessness. Her cyanotic spells became more frequent, despite FiO2 of 1.0, and a decision was made to transfer the child to the pediatric intensive care unit (PICU) for optimization of her treatment.
The infant was intubated and underwent tracheostomy, requiring FiO2 1.0 to maintain arterial oxygen saturations of 65%–75%. Despite two weeks of ventilator optimization, pulmonary vasodilators, targeted antibiotics therapy based on culture results, restricted crystalloid infusion, and diuretic treatment, the PH persisted. iNO and iloprost inhalation were administrated as a complement to sildenafil, but only led to transient improvements in oxygenation. Given the lack of response and the opinion that a post-capillary PH was the reason for the clinical deterioration, it was decided to change the treatment strategy from pulmonary vasodilation to systemic afterload reduction. Sildenafil, iNO, and inhaled iloprost were discontinued, and the treatment was switched to the angiotensin II type-1 receptor (AT1R) blocker, losartan. The starting dose was 0.25 mg/kg/dose, increasing to 1 mg/kg/dose twice daily over five days. Anti-inflammatory treatment with melatonin (0.5 mg/kg twice daily) was introduced based on clinical and experimental data supporting its anti-inflammatory properties in BPD (38–40). A low physiological steroid substitution was continued due to iatrogenic adrenal insufficiency, alongside inhaled budesonide (Pulmicort®) (Table 1). Blood volume was optimized with blood products and 5% albumin to stabilized blood pressure alongside the start of losartan, which unmasked a compensated hypovolemia.
Within two to three days after starting losartan, the infant's oxygenation improved, her circulation stabilized, and the repeated cyanotic spells disappeared. Inspired oxygen levels were reduced, sedation was gradually weaned, and enteral feeding was resumed and progressively increased without gastric retention or sign of abdominal pain.
The sign of pulmonary edema on chest x-ray, which occurred during the treatment with pulmonary vasodilators, disappeared within a few days. The discontinuation of the pulmonary vasodilators may partly have attributed to the improvement in oxygenation by decreasing capillary filtration pressure. Her blood pressure normalized and stabilized at a systolic arterial pressure of 60–70 mm Hg (MAP∼50 mm Hg). Cardiac ultrasounds after the alteration of treatment regimen showed normal bilateral ventricular size and function with no remaining direct or indirect indications of pulmonary hypertension.
After 33 days of intensive care, the infant was transferred back to the pediatric ward without clinical or echocardiographic signs of PH. She still required a high-flow nasal-mask during the day and bi-level positive airway pressure at night (FIO2 0.3–0,35) to maintain oxygen saturation >90%. Arrested alveolar development, pruning of vascular bed and cystic changes might contribute to the lasting V/Q mismatch and need of extra oxygen. Over the following year, she gained weight and showed good psychomotor development and no signs of retinopathy. No genetic testing was performed to exclude all possible underlying chromosomal diagnosis contributing to PH, since it was obviously reversible to the treatment.
3 Discussion
3.1 How RAAS activity influences pro-inflammatory responses and causes lung damage in BPD.
Children with severe BPD and PH often require high oxygen concentrations and mechanical ventilation. Both induce inflammatory responses through oxidative stress and barotrauma (41). Several authors have stressed that this inflammation significantly contributes to the pulmonary damage in BPD (9, 10, 13, 42). Inflammatory responses increase pulmonary and systemic endothelial permeability with subsequent extravascular fluid accumulation and loss of circulatory blood volume (7). Losses of circulatory blood volume in very early born premature infants are also common due to early cord-clamping and the following need of frequent blood sampling. A reduction of intravascular volume decreases systemic cardiac output and serves as an important stimulus for RAAS activation and subsequent increase in systemic vascular resistance. The RAAS cascade is intitated by the cleavage of angiotensinogen to angiotensin I (ANG I) by renin in the kidney. ANG I is subsequently converted to angiotensin II (ANG II) by the enzyme—angiotensin converting enzyme 1 (ACE1) located primarily in pulmonary microvascular endothelium (43). ANG II amplifies several pro-inflammatory responses by activating the angiotensin II type 1 receptor AT1R, leading to immune cell infiltration, augmentation of oxidative stress, increased vascular permeability, and development of pulmonary fibrosis (44, 45). See Table 2 for biological effects of ANG II.
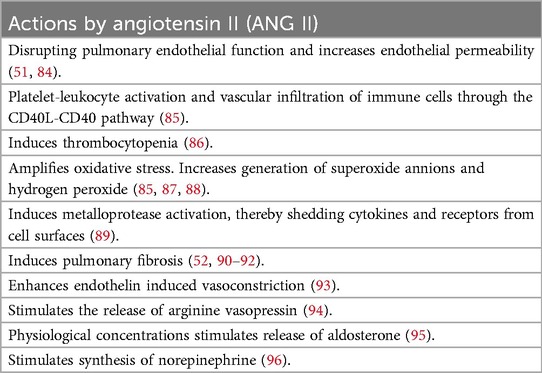
Table 2. Pro-inflammatory and hypertensive actions of angiotensin II (ANG II).
The damaging properties of ANG II have been implicated in various lung injuries, including adult respiratory distress syndrome (ARDS), respiratory syncytial virus infection (RSV), and COVID-19 (46–53). ANG II is normally inactivated by angiotensin converting enzyme 2 (ACE2) and a reduction in ACE2 activity has been shown to result in exacerbation of lung injury. Downregulation of ACE2 has been demonstrated during hyperoxia and may be an important contributing factor to inhibit the inactivation of ANG II in BPD (54). Moreover, mitigating the harmful effects of ANG II, by administration of recombinant ACE2, is protective, as is the administration of ARBs, both in clinical studies and in experimental models of lung injury (47, 48, 50, 55).
Elevated levels of ANG II correlates directly with the severity of pulmonary damage and mortality supporting its role in exacerbating lung pathology (51, 53, 56). Increased circulating concentration of ANG II has been found in prematurely born children with very low birth weight (57). Further support för the critical role of ANG II in neonatal pulmonary pathology comes from the observation of the increased incidence of BPD and PH in premature infants exposed to high placental concentrations of ANG II in preeclamptic mothers (58–60). In addition, several reports have noticed an association between early blood losses and the development of BPD and morbidity, which may be explained by the activation of RAAS (61–63). An initial activation of RAAS in extremely premature born children may continue during the following intensive care period due to need of frequent blood sampling, a continuous inflammatory response caused by use of high levels of inspired oxygen, and a high incidence of left-to-right shunts. All these factors contribute to a low systemic cardiac output and consequently RAAS activation, ultimately contributing to lung injury. The activation of ANG II contributes to arterial vasoconstriction and increases afterload of the left ventricle. This may initital cause a subtle left ventricular diastolic dysfunction that can be difficult to detect by the cardiac ultrasound. With time it will increase left atrial pressure, lead to an increased backward pressure in the pulmonary veins and contribute to post-capillary PH (64). PH in infants with BPD usually occurs insidiously and should be followed by repeat cardiac ultrasounds to be detected. In summary, the damage-inducing properties of ANG II aligns well with the key pathophysiological findings in BPD. Data from experimental models and clinical studies provide a strong rationale for using ARBs to block AT1R and thereby potentially mitigate the harmful effects of ANG II in the development of BPD, especially in infants with concurrent PH (34, 54). The amplifying effect of ANG II on oxidative stress, argues for a continuous use of ARB in children with BPD at least until the oxygen demand has been normalized.
3.2 How RAAS and its metabolites cause systemic vasoconstriction and pulmonary post-capillary pulmonary hypertension
Angiotensin II is a potent systemic vasoconstrictor resulting in increased arterial blood pressure, which has been observed in children with BPD (30–32, 65, 66). An increase in the afterload of left ventricle increases left ventricular strain, left ventricular end-diastolic pressure, impairs left ventricular diastolic function and elevates left atrial pressure, which in turn is transmitted backward into the pulmonary veins, leading to variable degrees of pulmonary venous congestion (33, 64, 67, 68). This congestive state, albeit not pronounced favors the development of post-capillary PH (68). Elevated left atrial pressure is difficult to diagnose in premature infants given the difficulties to obtain a reliable pulmonary capillary wedge pressure (PCWP) measurement and a significant risk of being underestimated (69). Additionally, typical histological findings in children with BPD confirm that pulmonary venous congestion precedes the diagnosis of PH, indicating that left heart impairment plays a critical role in the pathogenesis of BPD-associated PH (11). The proposed mechanism emphasizes the role of RAAS-mediated systemic vasoconstriction as a key contributor to the pathophysiology of PH in BPD and highlights the potential for targeted therapies directed against AT1R. It may explain why PH in BPD responded promptly to afterload reduction with ARB in the presented case.
3.3 The impact of RAAS activation on capillary filtration pressure and the use of pulmonary vasodilators
Pulmonary venous congestion leads to an increase in pulmonary capillary hydrostatic pressure. This raises capillary filtration pressure pushing fluid into the interstitial space of the lung parenchyma. The excess of interstitial fluid increases the diffusion distance for oxygen across the alveolar-capillary membrane, leading to impaired oxygen exchange, oxygen desaturation, and need of oxygen supplementation.
The use of pulmonary vasodilators dilates pulmonary pre-capillary arterioles and exposes the congested pulmonary capillary bed to the pulmonary arterial blood pressure, which increase the capillary filtration pressure further. This may lead to a potential progression of the interstitial fluid leak to a point where the fluid enters the alveolar space. Interstitial lung fluid is difficult to diagnose on a plain chest x-ray, but when the fluid enters the alveolar space it can easily be seen as a pulmonary edema. This was observed in the presented case when the child developed sign of pulmonary edema during the treatment with pulmonary vasodilators. This has led some authors to regard pulmonary vasodilation as contraindicated in BPD (34).
3.4 The impact of a limited circulatory blood volume in children with BPD
Clinically, sign of pulmonary edema may potentially lead to the false assessment that the infants are fluid overloaded and need diuresis (70). However, indiscriminate use of diuretics causes a non-compensated loss of intravascular volume and result in further stimulation of the already activated RAAS and, thus a sustained activation of the pathogenic pathway. It may further reduce systemic blood flow, contribute to worsening the pulmonary venous congestion and the development of PH. This scenario explains the importance of maintaining the intravascular volume and circulatory blood volume at the same time as a controlled and restricted crystalloid fluid intake can prevent accumulation of interstitial fluids (71–74).
Infants with BPD and its concurrent inflammatory condition can be characterized to have a compensated hypovolemia, which implies the presence of a reduced circulatory blood volume without obvious signs of hypovolemia. The infants can easily maintain or increase their systemic blood pressure by vasoconstriction at the cost of reduced systemic blood flow. Venoconstriction increases mean systemic fillling pressure, which maintains right atrium pressure and central venous pressure, making these parameters inadequate to be used in the clinical assessment of the circulatory blood volume (75, 76). However, a typical finding in these infants, as in our child, is striking blood pressure fluctuation during sedation and/or feeding, when compensatory protective vasoconstriction is inhibited.
After the introduction of losartan, which potentially inhibits systemic vasoconstriction and thereby mitigates pulmonary venous congestion, the chest x-ray improved immediately and the blood pressure became stable at a normal level for age, simultaneously as the circulatory blood volume was increased with blood products and albumin.
Overall, capillary filtration pressure is a key factor in the respiratory and cardiovascular complications of BPD. Effective management requires careful balancing of fluid intake, circulatory blood volume and pharmacological interventions.
3.5 The impact of left to right shunts in BPD
Left-to-right shunts such as PDA, ASD, or patent foramen ovale (PFO) have been noticed in up to 85% of infants with BPD and 97% in severe BPD cases (33). They contribute significantly to the pathophysiology of PH and the development of BPD (77, 78). The shunts allow blood to flow from the systemic to the pulmonary circulation, causing pulmonary overcirculation and systemic hypoperfusion (65). This hypoperfusion is probably the reason why children with left-to-right shunts have very high activity in the RAAS and sympathetic nervous system (79, 80). High activity in RAAS results in the production of ANG II in the lung, which amplify established inflammatory activity (Table 2). Activity in RAAS contributes to arterial vasoconstriction and high sympathetic activity to venoconstriction. Pulmonary overcirculation increases the tone in the pulmonary pre-capillary arterioles by myogenic reflexes, which increases the pulmonary arterial pressure (81, 82). In combination with impaired left ventricular diastolic function and pulmonary venous congestion, it may explain the high incidence of BPD with severe PH in this group of patients (33). It seems, therefore crucial to close left-to-right shunts as soon as possible, especially in children with BPD to normalize systemic blood flow and thereby decrease the activation of RAAS. Normalization of systemic blood flow in a child with a long-standing vasoconstriction and a limited vascular growth will result in a transient high systemic blood pressure, which also was noticed in this case.
Long lasting pulmonary overcirculation remodels pre-capillary arterioles in the lungs to protect against an excessive high capillary filtration pressure. Closing a left-to-right shunt may not immediately normalize pulmonary arterial pressure due to this remodeling. The postoperatively remodeling of smooth muscle in the pulmonary arterioles takes time and requires patience until the pulmonary pressure has normalized. After surgical correction of left-to-right shunt lesions, all children normalize their pulmonary arterial pressure without pulmonary vasodilatory treatment, even if the PH has super-systemic levels postoperative, unless the severe PH was caused by an untreatable severe mitral regurgitation (83). Children with severe PH caused by irreversible lung vascular hypoplasia or a genetic etiology conformable with severe BPD and PH are also irreversible condition without cure by surgical corrections or medical treatments.
Children with PH associated with both a remodeling of the pre-capillary arterioles and underlying pulmonary venous congestion, such as those with BPD, may be particular susceptible to pulmonary vasodilators. Vasodilation of the pulmonary arterioles may easily increase pulmonary capillary filtration pressure and exacerbate the accumulation of interstitial lung fluid. Pulmonary vasodilator treatment was, despite this aspect, started in the NICU in the reported infant, after it was noticed that a significant PH remained postoperatively. This may have contributed to the deterioration in the clinical condition, with the occurrence of interstitial lung fluid and pulmonary edema.
ARB was used instead of an ACE inhibitor, since ARBs directly inhibit the effect of ANG II on the AT1R and therefore have the potential to reduce inflammation, oxidative stress and prevent lung fibrosis in BPD, alongside with its effect to reduce afterload and the pulmonary venous congestion.
In conclusion, our child with BPD and severe PH showed prompt improvement after transitioning the treatment regimen from pulmonary vasodilation to afterload reduction using an ARB. A review of the literature highlights the scientific rationale for employing ARBs in BPD, given their ability to ameliorate pulmonary venous congestion by reducing systemic afterload, lower arterial hypertension, and protect against lung-damaging pro-inflammatory responses mediated by ANG II.
In line with these pathophysiological highlights, it is recommended that ARBs, combined with blood volume optimization, should be considered a first-line treatment for BPD children with PH, particularly in cases associated with high systemic arterial pressure and/or left ventricular diastolic dysfunction. However, despite the pathophysiological benefits favoring the use of ARBs in BPD with PH, the widespread reliance on pulmonary vasodilators in this population underscores the need for a randomized, blinded, controlled trial to compare the efficacy of ARBs against pulmonary vasodilators.
Data availability statement
The data analyzed in this study is subject to the following licenses/restrictions: the data presented in the case was collected from medical records. Requests to access these datasets should be directed tobGFycy5saW5kYmVyZ0BtZWQubHUuc2U=.
Ethics statement
Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
LL: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Resources, Software, Validation, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Tracy MK, Berkelhamer SK. Bronchopulmonary dysplasia and pulmonary outcomes of prematurity. Pediatr Ann. (2019) 48(4):e148–53. doi: 10.3928/19382359-20190325-03
2. Nakashima T, Inoue H, Sakemi Y, Ochiai M, Yamashita H, Ohga S, et al. Trends in bronchopulmonary dysplasia among extremely preterm infants in Japan, 2003–2016. J Pediatr. (2021) 230:119–125.e7. doi: 10.1016/j.jpeds.2020.11.041
3. Zhu R, Xu Y, Qin Y, Xu J, Wang R, Wu S, et al. In-hospital mortality and length of hospital stay in infants requiring tracheostomy with bronchopulmonary dysplasia. J Perinatol. (2024) 44(7):957–62. doi: 10.1038/s41372-023-01840-z
4. Higano NS, Bates AJ, Gunatilaka CC, Hysinger EB, Critser PJ, Hirsch R, et al. Bronchopulmonary dysplasia from chest radiographs to magnetic resonance imaging and computed tomography: adding value. Pediatr Radiol. (2022) 52(4):643–60. doi: 10.1007/s00247-021-05250-1
5. Sun YH, Yuan L, Du Y, Zhou JG, Lin SB, Zhang R, et al. Characterization of lung ultrasound imaging in preterm infants with bronchopulmonary dysplasia. Clin Hemorheol Microcirc. (2022) 80(2):83–95. doi: 10.3233/CH-211132
6. van Mastrigt E, Logie K, Ciet P, Reiss IK, Duijts L, Pijnenburg MW, et al. Lung CT imaging in patients with bronchopulmonary dysplasia: a systematic review. Pediatr Pulmonol. (2016) 51(9):975–86. doi: 10.1002/ppul.23446
7. Groneck P, Gotze-Speer B, Oppermann M, Eiffert H, Speer CP. Association of pulmonary inflammation and increased microvascular permeability during the development of bronchopulmonary dysplasia: a sequential analysis of inflammatory mediators in respiratory fluids of high-risk preterm neonates. Pediatrics. (1994) 93(5):712–8. doi: 10.1542/peds.93.5.712
8. Groneck P, Speer CP. Inflammatory mediators and bronchopulmonary dysplasia. Arch Dis Child Fetal Neonatal Ed. (1995) 73(1):F1–3. doi: 10.1136/fn.73.1.F1
9. Heydarian M, Schulz C, Stoeger T, Hilgendorff A. Association of immune cell recruitment and BPD development. Mol Cell Pediatr. (2022) 9(1):16. doi: 10.1186/s40348-022-00148-w
10. Sun Y, Chen C, Zhang X, Weng X, Sheng A, Zhu Y, et al. High neutrophil-to-lymphocyte ratio is an early predictor of bronchopulmonary dysplasia. Front Pediatr. (2019) 7:464. doi: 10.3389/fped.2019.00464
11. Stone A, Poulik J, Koussa S, Xin Y, Sharma A, Sood BG. Early histological changes of bronchopulmonary dysplasia and pulmonary hypertension may precede clinical diagnosis in preterm infants. Early Hum Dev. (2022) 171:105612. doi: 10.1016/j.earlhumdev.2022.105612
12. Groneck P, Speer CP. Pathogenesis of bronchopulmonary dysplasia. Z Geburtshilfe Neonatol. (1995) 199(5):181–9.8528953
13. Speer CP. Pulmonary inflammation and bronchopulmonary dysplasia. J Perinatol. (2006) 26(Suppl 1):S57–62. discussion S63-54. doi: 10.1038/sj.jp.7211476
14. Ambalavanan N, Mourani P. Pulmonary hypertension in bronchopulmonary dysplasia. Birth Defects Res A Clin Mol Teratol. (2014) 100(3):240–6. doi: 10.1002/bdra.23241
15. Hansmann G, Sallmon H, Roehr CC, Kourembanas S, Austin ED, Koestenberger M, et al. Pulmonary hypertension in bronchopulmonary dysplasia. Pediatr Res. (2021) 89(3):446–55. doi: 10.1038/s41390-020-0993-4
16. Altit G, Bhombal S, Feinstein J, Hopper RK, Tacy TA. Diminished right ventricular function at diagnosis of pulmonary hypertension is associated with mortality in bronchopulmonary dysplasia. Pulm Circ. (2019) 9(3):2045894019878598. doi: 10.1177/2045894019878598
17. Hafner F, Johansson C, Schwarzkopf L, Forster K, Kraus Y, Flemmer AW, et al. Current diagnosis and treatment practice for pulmonary hypertension in bronchopulmonary dysplasia-a survey study in Germany (PUsH BPD). Pulm Circ. (2023) 13(4):e12320. doi: 10.1002/pul2.12320
18. Khemani E, McElhinney DB, Rhein L, Andrade O, Lacro RV, Thomas KC, et al. Pulmonary artery hypertension in formerly premature infants with bronchopulmonary dysplasia: clinical features and outcomes in the surfactant era. Pediatrics. (2007) 120(6):1260–9. doi: 10.1542/peds.2007-0971
19. Yang EL, Levy PT, Critser PJ, Dukhovny D, Evers PD. The clinical and cost utility of cardiac catheterizations in infants with bronchopulmonary dysplasia. J Pediatr. (2022) 246:56–63.e53. doi: 10.1016/j.jpeds.2022.04.009
20. Abounahia FF, Abu-Jarir R, Abounahia MF, Al-Badriyeh D, Abushanab D, Abu-Ghalwa M, et al. Prophylactic sildenafil in preterm infants at risk of bronchopulmonary dysplasia: a pilot randomized, double-blinded, placebo-controlled trial. Clin Drug Investig. (2019) 39(11):1093–107. doi: 10.1007/s40261-019-00834-0
21. Barrington KJ, Finer N. Inhaled nitric oxide for respiratory failure in preterm infants. Cochrane Database Syst Rev. (2010) (12):CD000509. doi: 10.1002/14651858.CD000509.pub4
22. Barst RJ, Ivy DD, Gaitan G, Szatmari A, Rudzinski A, Garcia AE, et al. A randomized, double-blind, placebo-controlled, dose-ranging study of oral sildenafil citrate in treatment-naive children with pulmonary arterial hypertension. Circulation. (2012) 125(2):324–34. doi: 10.1161/CIRCULATIONAHA.110.016667
23. Branescu I, Vladareanu S, Shetty S, Alexandru DO, Kulkarni A. Sildenafil in pulmonary hypertension associated with bronchopulmonary dysplasia: friend or foe? Curr Health Sci J. (2023) 49(3):319–24. doi: 10.12865/CHSJ.49.03.03
24. Mourani PM, Sontag MK, Ivy DD, Abman SH. Effects of long-term sildenafil treatment for pulmonary hypertension in infants with chronic lung disease. J Pediatr. (2009) 154(3):379–384.2. doi: 10.1016/j.jpeds.2008.09.021
25. Nyp M, Sandritter T, Poppinga N, Simon C, Truog WE. Sildenafil citrate, bronchopulmonary dysplasia and disordered pulmonary gas exchange: any benefits? J Perinatol. (2012) 32(1):64–9. doi: 10.1038/jp.2011.131
26. Dillon K, Lamba V, Philip RR, Weems MF, Talati AJ. Efficacy of sildenafil in infants with bronchopulmonary dysplasia-associated pulmonary hypertension. Children (Basel). (2023) 10(8):1397. doi: 10.3390/children10081397
27. van der Graaf M, Rojer LA, Helbing W, Reiss I, Etnel JRG, Bartelds B. EXPRESS: sildenafil for bronchopulmonary dysplasia and pulmonary hypertension: a meta-analysis. Pulm Circ. (2019) 9(3):2045894019837875. doi: 10.1177/2045894019837875
28. Cohen JL, Nees SN, Valencia GA, Rosenzweig EB, Krishnan US. Sildenafil use in children with pulmonary hypertension. J Pediatr. (2019) 205:29–34.e21. doi: 10.1016/j.jpeds.2018.09.067
29. Yung D, Jackson EO, Blumenfeld A, Redding G, DiGeronimo R, McGuire JK, et al. A multidisciplinary approach to severe bronchopulmonary dysplasia is associated with resolution of pulmonary hypertension. Front Pediatr. (2023) 11:1077422. doi: 10.3389/fped.2023.1077422
30. Abman SH, Warady BA, Lum GM, Koops BL. Systemic hypertension in infants with bronchopulmonary dysplasia. J Pediatr. (1984) 104(6):928–31. doi: 10.1016/S0022-3476(84)80501-6
31. Kiss JK, Gajda A, Mari J, Bereczki C. Blood pressure in preterm infants with bronchopulmonary dysplasia in the first three months of life. Pediatr Nephrol. (2024) 39(8):2475–81. doi: 10.1007/s00467-024-06354-0
32. Sehgal A, Elsayed K, Nugent M, Varma S. Sequelae associated with systemic hypertension in infants with severe bronchopulmonary dysplasia. J Perinatol. (2022) 42(6):775–80. doi: 10.1038/s41372-022-01372-y
33. Sullivan RT, Tandel MD, Bhombal S, Adamson GT, Boothroyd DB, Tracy M, et al. Role of left atrial hypertension in pulmonary hypertension associated with bronchopulmonary dysplasia. Front Pediatr. (2022) 10:1012136. doi: 10.3389/fped.2022.1012136
34. Sehgal A, Krishnamurthy MB, Clark M, Menahem S. ACE inhibition for severe bronchopulmonary dysplasia - an approach based on physiology. Physiol Rep. (2018) 6(17):e13821. doi: 10.14814/phy2.13821
35. Mestan KK, Check J, Minturn L, Yallapragada S, Farrow KN, Liu X, et al. Placental pathologic changes of maternal vascular underperfusion in bronchopulmonary dysplasia and pulmonary hypertension. Placenta. (2014) 35(8):570–4. doi: 10.1016/j.placenta.2014.05.003
37. Jobe AH, Bancalari E. Bronchopulmonary dysplasia. Am J Respir Crit Care Med. (2001) 163(7):1723–9. doi: 10.1164/ajrccm.163.7.2011060
38. Gharehbaghi MM, Yeganedoust S, Shaseb E, Fekri M. Evaluation of melatonin efficacy in prevention of bronchopulmonary dysplasia in preterm newborn infants. Turk J Pediatr. (2022) 64(1):79–84. doi: 10.24953/turkjped.2021.1334
39. Pan L, Fu JH, Xue XD, Xu W, Zhou P, Wei B. Melatonin protects against oxidative damage in a neonatal rat model of bronchopulmonary dysplasia. World J Pediatr. (2009) 5(3):216–21. doi: 10.1007/s12519-009-0041-2
40. Carloni S, Proietti F, Rocchi M, Longini M, Marseglia L, D'Angelo G, et al. Melatonin pharmacokinetics following oral administration in preterm neonates. Molecules. (2017) 22(12):2115. doi: 10.3390/molecules22122115
41. Van Marter LJ, Allred EN, Pagano M, Sanocka U, Parad R, Moore M, et al. Do clinical markers of barotrauma and oxygen toxicity explain interhospital variation in rates of chronic lung disease? The neonatology committee for the developmental network. Pediatrics. (2000) 105(6):1194–201. doi: 10.1542/peds.105.6.1194
42. Groneck P, Speer CP. Pulmonary inflammation in the pathogenesis of bronchopulmonary dysplasia. Pediatr Pulmonol Suppl. (1997) 16:29–30. doi: 10.1002/ppul.1950230816
43. Satou R, Penrose H, Navar LG. Inflammation as a regulator of the renin-angiotensin system and blood pressure. Curr Hypertens Rep. (2018) 20(12):100. doi: 10.1007/s11906-018-0900-0
44. Suzuki Y, Ruiz-Ortega M, Egido J. Angiotensin II: a double-edged sword in inflammation. J Nephrol. (2000) 13(Suppl 3):S101–110.11132026
45. Suzuki Y, Ruiz-Ortega M, Lorenzo O, Ruperez M, Esteban V, Egido J. Inflammation and angiotensin II. Int J Biochem Cell Biol. (2003) 35(6):881–900. doi: 10.1016/S1357-2725(02)00271-6
46. Arjaans S, Wagner BD, Mourani PM, Mandell EW, Poindexter BB, Berger RMF, et al. Early angiogenic proteins associated with high risk for bronchopulmonary dysplasia and pulmonary hypertension in preterm infants. Am J Physiol Lung Cell Mol Physiol. (2020) 318(4):L644–54. doi: 10.1152/ajplung.00131.2019
47. Gu H, Xie Z, Li T, Zhang S, Lai C, Zhu P, et al. Angiotensin-converting enzyme 2 inhibits lung injury induced by respiratory syncytial virus. Sci Rep. (2016) 6:19840. doi: 10.1038/srep19840
48. Hrenak J, Simko F. Renin-angiotensin system: an important player in the pathogenesis of acute respiratory distress syndrome. Int J Mol Sci. (2020) 21(21):8038. doi: 10.3390/ijms21218038
49. Huang F, Guo J, Zou Z, Liu J, Cao B, Zhang S, et al. Angiotensin II plasma levels are linked to disease severity and predict fatal outcomes in H7N9-infected patients. Nat Commun. (2014) 5:3595. doi: 10.1038/ncomms4595
50. Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med. (2005) 11(8):875–9. doi: 10.1038/nm1267
51. Parikh SM, Mammoto T, Schultz A, Yuan HT, Christiani D, Karumanchi SA, et al. Excess circulating angiopoietin-2 may contribute to pulmonary vascular leak in sepsis in humans. PLoS Med. (2006) 3(3):e46. doi: 10.1371/journal.pmed.0030046
52. Wang J, Chen L, Chen B, Meliton A, Liu SQ, Shi Y, et al. Chronic activation of the renin-angiotensin system induces lung fibrosis. Sci Rep. (2015) 5:15561. doi: 10.1038/srep15561
53. Wu Z, Hu R, Zhang C, Ren W, Yu A, Zhou X. Elevation of plasma angiotensin II level is a potential pathogenesis for the critically ill COVID-19 patients. Crit Care. (2020) 24(1):290. doi: 10.1186/s13054-020-03015-0
54. Gandhi C, Uhal BD. Roles of the angiotensin system in neonatal lung injury and disease. JSM Atheroscler. (2016) 1(3):1014.29806050
55. Duarte M, Pelorosso F, Nicolosi LN, Victoria Salgado M, Vetulli H, Aquieri A, et al. Telmisartan for treatment of COVID-19 patients: an open multicenter randomized clinical trial. EClinicalMedicine. (2021) 37:100962. doi: 10.1016/j.eclinm.2021.100962
56. Liu N, Hong Y, Chen RG, Zhu HM. High rate of increased level of plasma angiotensin II and its gender difference in ccovid-19: an analysis o f55 hospitalized patients with COVID-19 in a single hospital, Wuhan, China. J Clin Toxicol. (2021) 11(S16):1–6. doi: 10.1101/2020.04.27.20080432
57. Miyawaki M, Okutani T, Higuchi R, Yoshikawa N. The plasma angiotensin II level increases in very low-birth weight infants with neonatal chronic lung disease. Early Hum Dev. (2008) 84(6):375–9. doi: 10.1016/j.earlhumdev.2007.10.005
58. Check J, Gotteiner N, Liu X, Su E, Porta N, Steinhorn R, et al. Fetal growth restriction and pulmonary hypertension in premature infants with bronchopulmonary dysplasia. J Perinatol. (2013) 33(7):553–7. doi: 10.1038/jp.2012.164
59. Dravet-Gounot P, Torchin H, Goffinet F, Aubelle MS, El Ayoubi M, Lefevre C, et al. Bronchopulmonary dysplasia in neonates born to mothers with preeclampsia: impact of small for gestational age. PLoS One. (2018) 13(9):e0204498. doi: 10.1371/journal.pone.0204498
60. Spaan JJ, Brown MA. Renin–angiotensin system in pre-eclampsia. Obstet Med. (2012) 5:7. doi: 10.1258/om.2012.120007
61. Gire C, Fournier N, Pirrello J, Marret S, Patural H, Flamant C, et al. Impact of early hemoglobin levels on neurodevelopment outcomes of two-year-olds in very preterm children. Children (Basel). (2023) 10(2):209. doi: 10.3390/children10020209
62. Hellstrom W, Forssell L, Morsing E, Savman K, Ley D. Neonatal clinical blood sampling led to major blood loss and was associated with bronchopulmonary dysplasia. Acta Paediatr. (2020) 109(4):679–87. doi: 10.1111/apa.15003
63. Hellstrom W, Martinsson T, Hellstrom A, Morsing E, Ley D. Fetal haemoglobin and bronchopulmonary dysplasia in neonates: an observational study. Arch Dis Child Fetal Neonatal Ed. (2021) 106(1):88–92. doi: 10.1136/archdischild-2020-319181
64. Itelman E, Segel MJ, Kuperstein R, Feinberg M, Segev A, Segal G, et al. Pulmonary hypertension is associated with systemic arterial hypertension among patients with normal left ventricular diastolic function. J Am Heart Assoc. (2021) 10(24):e023603. doi: 10.1161/JAHA.121.023603
65. Pharande P, Sehgal A, Menahem S. Cardiovascular sequelae of bronchopulmonary dysplasia in preterm neonates born before 32 weeks of gestational age: impact of associated pulmonary and systemic hypertension. J Cardiovasc Dev Dis. (2024) 11(8):233. doi: 10.3390/jcdd11080233
66. Sehgal A, Malikiwi A, Paul E, Tan K, Menahem S. Systemic arterial stiffness in infants with bronchopulmonary dysplasia: potential cause of systemic hypertension. J Perinatol. (2016) 36(7):564–9. doi: 10.1038/jp.2016.10
67. Mourani PM, Ivy DD, Rosenberg AA, Fagan TE, Abman SH. Left ventricular diastolic dysfunction in bronchopulmonary dysplasia. J Pediatr. (2008) 152(2):291–3. doi: 10.1016/j.jpeds.2007.11.006
68. Sehgal A, Malikiwi A, Paul E, Tan K, Menahem S. A new look at bronchopulmonary dysplasia: postcapillary pathophysiology and cardiac dysfunction. Pulm Circ. (2016) 6(4):508–15. doi: 10.1086/688641
69. Gartenberg AJ, O'Byrne ML, Leeth EB, Tang J, Gupta M, Rome JJ, et al. Pulmonary artery wedge pressure can underestimate direct pulmonary vein pressure in pediatric pulmonary vein stenosis. J Soc Cardiovasc Angiogr Interv. (2024) 3(4):101350. doi: 10.1016/j.jscai.2024.101350
70. Oh W, Poindexter BB, Perritt R, Lemons JA, Bauer CR, Ehrenkranz RA, et al. Association between fluid intake and weight loss during the first ten days of life and risk of bronchopulmonary dysplasia in extremely low birth weight infants. J Pediatr. (2005) 147(6):786–90. doi: 10.1016/j.jpeds.2005.06.039
71. Bollaboina SKY, Urakurva AK, Kamsetti S, Kotha R. A systematic review: is early fluid restriction in preterm neonates going to prevent bronchopulmonary dysplasia? Cureus. (2023) 15(12):e50805. doi: 10.7759/cureus.50805
72. Li YJ, Zhu XF, Liu JH, Yi XQ, He H. Influence of early fluid overload on bronchopulmonary dysplasia in very low-birth-weight infants. Front Pediatr. (2022) 10:980179. doi: 10.3389/fped.2022.980179
73. Barrington KJ, Fortin-Pellerin E, Pennaforte T. Fluid restriction for treatment of preterm infants with chronic lung disease. Cochrane Database Syst Rev. (2017) 2(2):CD005389. doi: 10.1002/14651858.CD005389.pub2
74. Diderholm B, Normann E, Ahlsson F, Sindelar R, Agren J. The impact of restricted versus liberal early fluid volumes on plasma sodium, weight change, and short-term outcomes in extremely preterm infants. Nutrients. (2022) 14(4):795. doi: 10.3390/nu14040795
75. Magder S. Volume and its relationship to cardiac output and venous return. Crit Care. (2016) 20(1):271. doi: 10.1186/s13054-016-1438-7
76. Malbrain M. Do we still need central venous pressure monitoring in the ICU? No!. Intensive Crit Care Nurs. (2025) 88:103991. doi: 10.1016/j.iccn.2025.103991
77. Gopagondanahalli KR, Abdul Haium AA, Vora SJ, Sundararaghavan S, Ng WD, Choo TLJ, et al. Serial tissue Doppler imaging in the evaluation of bronchopulmonary dysplasia-associated pulmonary hypertension among extremely preterm infants: a prospective observational study. Front Pediatr. (2024) 12:1349175. doi: 10.3389/fped.2024.1349175
78. Ito M, Kato S, Saito M, Miyahara N, Arai H, Namba F, et al. Bronchopulmonary dysplasia in extremely premature infants: a scoping review for identifying risk factors. Biomedicines. (2023) 11(2):533. doi: 10.3390/biomedicines11020553
79. Buchhorn R, Ross RD, Bartmus D, Wessel A, Hulpke-Wette M, Bursch J. Activity of the renin-angiotensin-aldosterone and sympathetic nervous system and their relation to hemodynamic and clinical abnormalities in infants with left-to-right shunts. Int J Cardiol. (2001) 78(3):225–30. discussion 230-221. doi: 10.1016/S0167-5273(01)00398-9
80. Scammell AM, Diver MJ. Plasma renin activity in infants with congenital heart disease. Arch Dis Child. (1987) 62(11):1136–8. doi: 10.1136/adc.62.11.1136
81. McNamara PJ, Lakshminrusimha S. Maldevelopment of the immature pulmonary vasculature and prolonged patency of the ductus arteriosus: association or cause? Am J Respir Crit Care Med. (2023) 207(7):814–6. doi: 10.1164/rccm.202211-2146ED
82. Storme L, Rairigh RL, Parker TA, Kinsella JP, Abman SH. In vivo evidence for a myogenic response in the fetal pulmonary circulation. Pediatr Res. (1999) 45(3):425–31. doi: 10.1203/00006450-199903000-00022
83. Lindberg L. Long-term follow-up of pediatric patients with severe postoperative pulmonary hypertension after correction of congenital heart defects. Pediatr Cardiol. (2022) 43(4):827–36. doi: 10.1007/s00246-021-02794-9
84. Bodor C, Nagy JP, Vegh B, Nemeth A, Jenei A, MirzaHosseini S, et al. Angiotensin II increases the permeability and PV-1 expression of endothelial cells. Am J Physiol Cell Physiol. (2012) 302(1):C267–276. doi: 10.1152/ajpcell.00138.2011
85. Hausding M, Jurk K, Daub S, Kroller-Schon S, Stein J, Schwenk M, et al. CD40l contributes to angiotensin II-induced pro-thrombotic state, vascular inflammation, oxidative stress and endothelial dysfunction. Basic Res Cardiol. (2013) 108(6):386. doi: 10.1007/s00395-013-0386-5
86. Xu DF, Liu YJ, Mao YF, Wang Y, Xu CF, Zhu XY, et al. Elevated angiotensin II induces platelet apoptosis through promoting oxidative stress in an AT1R-dependent manner during sepsis. J Cell Mol Med. (2021) 25(8):4124–35. doi: 10.1111/jcmm.16382
87. Griendling KK, Ushio-Fukai M. Reactive oxygen species as mediators of angiotensin II signaling. Regul Pept. (2000) 91(1-3):21–7. doi: 10.1016/S0167-0115(00)00136-1
88. Zhang H, Schmeisser A, Garlichs CD, Plotze K, Damme U, Mugge A, et al. Angiotensin II-induced superoxide anion generation in human vascular endothelial cells: role of membrane-bound NADH-/NADPH-oxidases. Cardiovasc Res. (1999) 44(1):215–22. doi: 10.1016/S0008-6363(99)00183-2
89. Obama T, Takayanagi T, Kobayashi T, Bourne AM, Elliott KJ, Charbonneau M, et al. Vascular induction of a disintegrin and metalloprotease 17 by angiotensin II through hypoxia inducible factor 1alpha. Am J Hypertens. (2015) 28(1):10–4. doi: 10.1093/ajh/hpu094
90. Marshall RP, Gohlke P, Chambers RC, Howell DC, Bottoms SE, Unger T, et al. Angiotensin II and the fibroproliferative response to acute lung injury. Am J Physiol Lung Cell Mol Physiol. (2004) 286(1):L156–164. doi: 10.1152/ajplung.00313.2002
91. Marshall RP, McAnulty RJ, Laurent GJ. Angiotensin II is mitogenic for human lung fibroblasts via activation of the type 1 receptor. Am J Respir Crit Care Med. (2000) 161(6):1999–2004. doi: 10.1164/ajrccm.161.6.9907004
92. Uhal BD, Li X, Piasecki CC, Molina-Molina M. Angiotensin signalling in pulmonary fibrosis. Int J Biochem Cell Biol. (2012) 44(3):465–8. doi: 10.1016/j.biocel.2011.11.019
93. Lin YJ, Kwok CF, Juan CC, Hsu YP, Shih KC, Chen CC, et al. Angiotensin II enhances endothelin-1-induced vasoconstriction through upregulating endothelin type A receptor. Biochem Biophys Res Commun. (2014) 451(2):263–9. doi: 10.1016/j.bbrc.2014.07.119
94. Sandgren JA, Linggonegoro DW, Zhang SY, Sapouckey SA, Claflin KE, Pearson NA, et al. Angiotensin AT(1A) receptors expressed in vasopressin-producing cells of the supraoptic nucleus contribute to osmotic control of vasopressin. Am J Physiol Regul Integr Comp Physiol. (2018) 314(6):R770–80. doi: 10.1152/ajpregu.00435.2017
95. Fredlund P, Saltman S, Catt KJ. Aldosterone production by isolated adrenal glomerulosa cells: stimulation by physiological concentrations of angiotensin II. Endocrinology. (1975) 97(6):1577–86. doi: 10.1210/endo-97-6-1577
96. Maruyama R, Hatta E, Yasuda K, Smith NC, Levi R. Angiotensin-converting enzyme-independent angiotensin formation in a human model of myocardial ischemia: modulation of norepinephrine release by angiotensin type 1 and angiotensin type 2 receptors. J Pharmacol Exp Ther. (2000) 294(1):248–54. doi: 10.1016/S0022-3565(24)39063-9
Keywords: bronchopulmonary dysplasia, pulmonary hypertension, angiotensin receptor blocker, phosphodiesterase type 5 inhibitor, prematurity
Citation: Lindberg L (2025) The rationale of using angiotensin receptor blocker instead of pulmonary vasodilators to treat pulmonary hypertension in bronchopulmonary dysplasia: a case report and literature review. Front. Pediatr. 13:1504180. doi: 10.3389/fped.2025.1504180
Received: 30 September 2024; Accepted: 30 April 2025;
Published: 19 May 2025.
Edited by:
Shahana Perveen, Cohen Children’s Medical Center, United StatesReviewed by:
Sara Berkelhamer, University of Washington, United StatesJoanna Beachy, Baystate Medical Center, United States
Copyright: © 2025 Lindberg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lars Lindberg, bGFycy5saW5kYmVyZ0BtZWQubHUuc2U=