 Jing Qu
Jing Qu Zhihong Hao2
Zhihong Hao2 Wang Junli
Wang Junli- 1Department of Pediatrics, Guangzhou First People’s Hospital, Guangdong Medical University, Zhanjiang, Guangdong, China
- 2Department of Pediatrics, Guangzhou First People’s Hospital, School of Medicine, South China University of Technology, Guangzhou, Guangdong, China
- 3Department of Pediatrics, The Second Affiliated Hospital, South China University of Technology, Guangzhou, China
Background: Livedoid vasculopathy (LV) is a rare, non-inflammatory, intradermal vascular obstructive skin disorder characterized by purpuric papules and plaques with capillary dilation. These lesions typically progress to crusted ulcers and ultimately result in fixed, white, atrophic stellate scars. The condition is marked by painful ulcers that heal slowly and have a tendency to recur.
Case presentation: We report a case of a pediatric patient presenting with recurrent rashes and pain in both lower extremities. Physical examination revealed purpuric plaques with ulceration, scarring, and white atrophic healing features. Histopathological examination demonstrated intradermal thrombosis, vessel wall necrosis, and surrounding inflammatory cell infiltration with erythrocyte extravasation. Periodic acid-Schiff (PAS) staining was positive. The clinical and pathological findings were consistent with a diagnosis of LV. The patient was treated with oral rivaroxaban.
Conclusion: This case highlights the critical importance of early recognition and intervention in the management of LV. Clinicians should consider LV in the differential diagnosis when encountering patients with painful purpuric rashes. Improvement in pain following treatment with anticoagulants, such as rivaroxaban, may indirectly support the diagnosis. A skin biopsy is essential for definitive diagnosis.
Introduction
Livedoid vasculopathy (LV) is a rare, non-inflammatory, intradermal vascular obstructive skin disease characterized by painful ulcers, reticular cyanotic macules, and porcelain-white atrophic scarring, predominantly affecting the distal portions of the lower extremities (1–3). Although LV was first described in the early 20th century, it was not until the revision of the International Classification of Diseases (ICD-10) in 2004 that the condition was accurately defined. LV is a chronic, relapsing disorder with a distinct seasonal pattern, typically exacerbating in the summer and remitting in the winter (4). The annual incidence of LV is approximately one case per 100,000 individuals, with a predilection for females (5). Despite its clinical significance, the international diagnostic criteria for LV remain poorly defined, with only an expert consensus published in 2013 providing guidance (6). The non-specific clinical manifestations of LV often necessitate histopathological confirmation via skin biopsy for accurate diagnosis. The most typical pathological feature of LV is focal thrombosis within the dermal capillaries (3).
Given the unique clinical and pathological characteristics of LV, differentiating it from other vasculitic diseases is crucial, making clinical diagnosis particularly challenging. Case reports of LV play a vital role in guiding clinical practice, providing valuable insights for the diagnosis and treatment of this condition. They also contribute to the development of more rational diagnostic and therapeutic strategies for similar patients.
Case presentation
Description of patient
A 13-year-old male patient from Guangdong, China, was admitted to our hospital on 23 September 2023, with a 15-day history of recurrent rash and pain in both lower extremities. The patient had been previously diagnosed with “Henoch–Schönlein purpura (HSP)” in January 2021 due to a generalized red rash. The rash began from the lower limbs and spread to the whole body, which resolved with oral cimetidine and cetirizine. He was discharged for regular checkups, during which he received methotrexate 7.5 mg once a week without any recurrence of the disease. In June 2023, the rash reappeared. The local hospital diagnosed the recurrence of allergic purpura, and the symptoms were relieved after 2 weeks of symptomatic drugs—cimetidine and methylprednisolone.
On admission, scattered old rashes were noted throughout the body, with new red eruptions on both lower limbs that did not fade with pressure, accompanied by breakouts and crusts, without pruritus (Figure 1). Hypopigmented spots with a white atrophic appearance were also observed. Ultrasound examination of the extremities performed at an outside hospital did not reveal any abnormalities in the arteries and veins of both lower extremities.
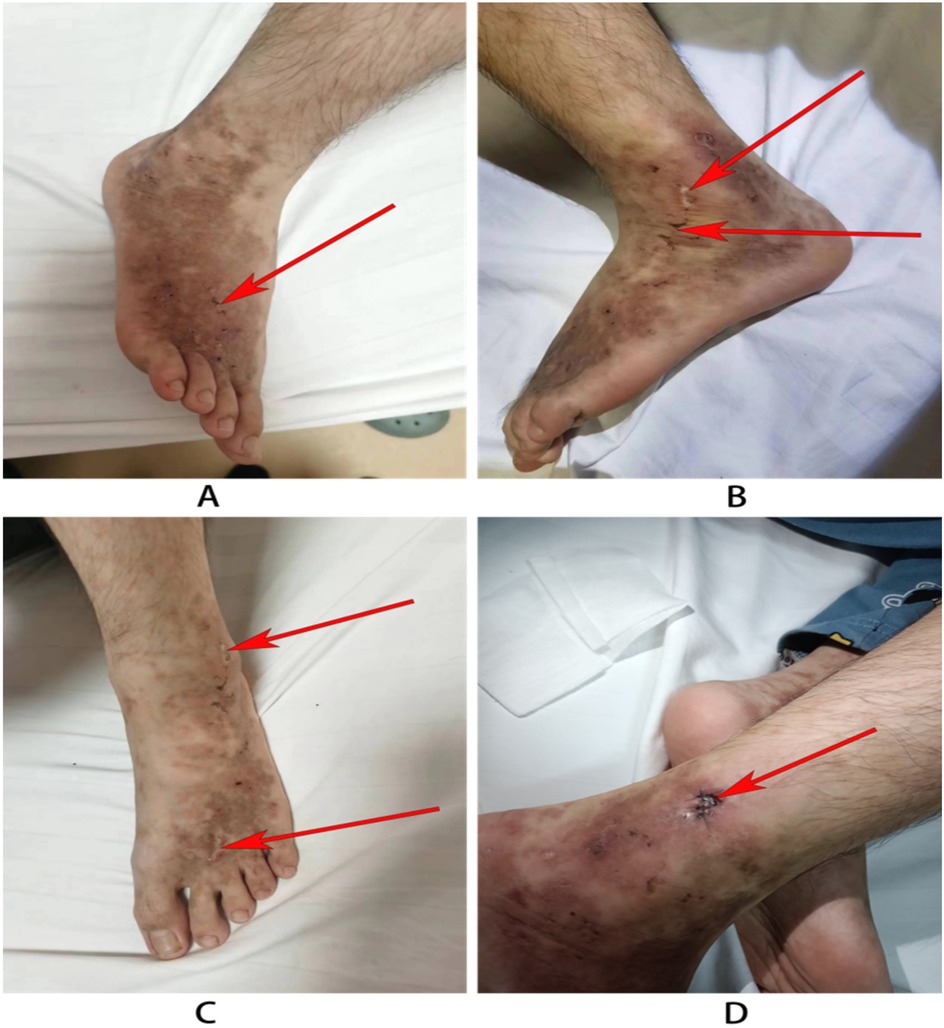
Figure 1. Skin rash in the child. (A) The left leg of the child: atrophic scarring around lateral malleoli (the red arrow). (B,C) The right legs: The arrow indicates porcelain-white atrophic scarring and stellate purpuric macules with telangiectasias. (D) Wound recovery after 3 days after dermatological pathology.
Clinical findings and diagnostic assessment
Upon admission to our hospital, a comprehensive set of laboratory tests (Table 1) was performed, including assessments of coagulation factors, D-dimer levels, vasculitis markers, humoral immunity, pre-transfusion screening, autoantibody profiles, and others. These tests did not reveal any significant abnormalities, effectively ruling out the possibility of HSP recurrence.
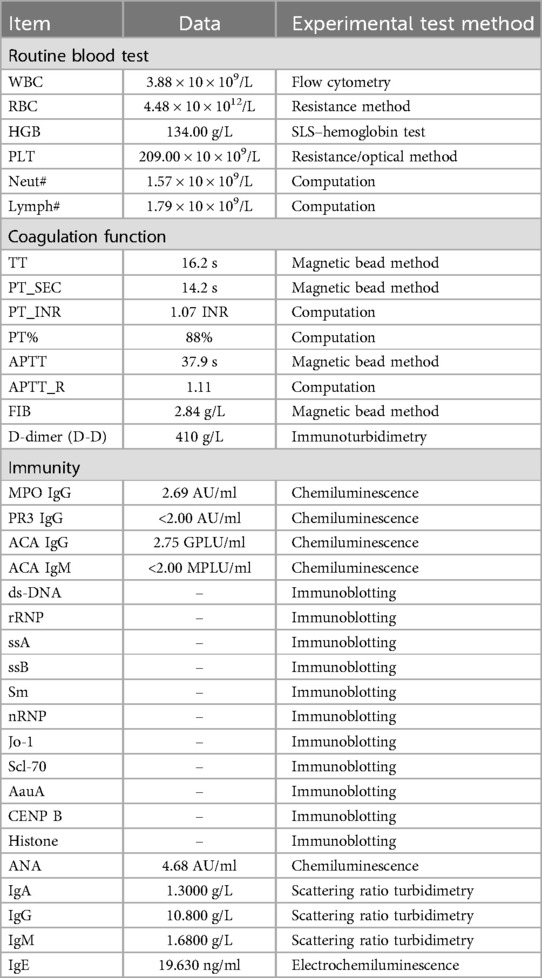
Table 1. Laboratory data at the admission of the patient.
The skin biopsy was requested on September 26. A skin specimen measuring approximately 1 cm × 0.6 cm × 0.5 cm was taken from the right ankle for pathological examination. Microscopic examination of the specimen revealed normal-appearing squamous epithelium on the skin surface, with hyperplasia of small blood vessels in both the superficial and deep dermis, accompanied by a small number of inflammatory cells. No tumor cells were identified. Direct immunofluorescence testing showed negative results for IgG, C3, IgM, and IgA (Figures 2, 3).
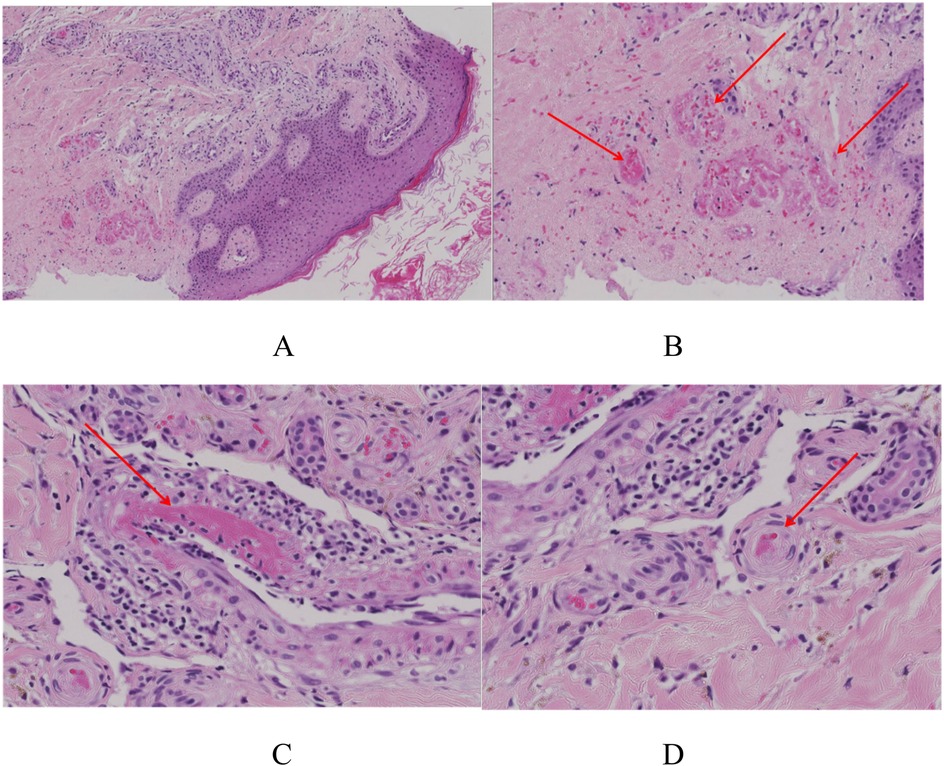
Figure 2. The skin biopsy of this child. (A) The overall picture of skin HE staining. H&E 8×. (B) Capillary telangiectasia, extravasation of red blood cells, and intraluminal capillary thrombi (the red arrows). H&E 10×. (C) Livedoid vasculopathy in a skin biopsy with an occlusive, intraluminal thrombus (the red arrows) surrounding hemorrhage. H&E 40×. (D) Chronic change: thickening of capillary walls with pink, glassy (hyalinized) collagen (the red arrows). H&E 40×.
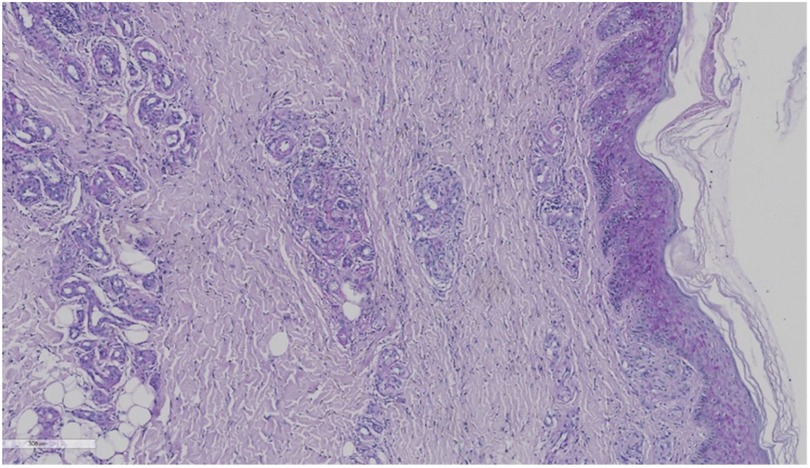
Figure 3. Immunofluorescence examination showing that IgG, C3, IgM, and IgA are negative.
After a thorough evaluation of the patient's condition, the skin specimen was reviewed twice in consultation with dermatology. Histopathological examination revealed epidermal hyperkeratosis, intravascular thrombosis, and necrosis of the vessel wall in the dermis, along with perivascular inflammatory cell infiltration and erythrocyte extravasation. Special staining techniques, such as periodic acid-Schiff (PAS) staining, were positive, further confirming the diagnosis of livedoid vasculopathy (LV) (Figure 3).
The patient, a male adolescent, presented with “rash and pain” as the primary symptoms. He had previously been diagnosed with HSP, and his symptoms had improved with treatment. On admission, physical examination revealed new erythematous eruptions on both lower limbs that did not blanch with pressure, accompanied by breakouts and crusts but no pruritus. Laboratory tests evaluated immune antibodies and coagulation function, ruling out HSP and other immune-mediated diseases. Skin biopsy findings suggested non-inflammatory vascular wall damage and fibrinoid necrosis of the vessel wall. Based on these findings, the patient was ultimately diagnosed with LV.
Therapeutic intervention
Following the diagnosis of LV, the patient was initiated on anticoagulation therapy with intravenous heparin and oral rivaroxaban (10 mg once daily). One week later, the patient's pain was significantly relieved, and he was discharged without recurrence of the rash.
Follow-up and outcomes
The patient continued to take rivaroxaban regularly for 2 months. Follow-up visits revealed a significant reduction in the rash compared with the initial presentation, with no new eruptions observed. The patient was discharged with regular follow-up appointments, and his condition showed marked improvement compared with the initial presentation.
Discussion
LV has a strong gender characteristic; the incidence of women is three times that of men, especially in the age group of 15–50 years old (7). As a disease related to coagulation disorders, it is divided into secondary and primary according to the presence or absence of primary disease. It is often associated with multiple stasis, autoimmune connective tissue diseases, tumors, and immune-related diseases (6). The disease is often exacerbated by temperature changes and a hypercoagulable state of the blood (2, 8). Therefore, clinicians must conduct a comprehensive assessment to diagnose LV accurately and manage its underlying causes. In our case, the patient had a history of Henoch–Schönlein purpura (HSP), which significantly increased the risk of vascular fragility and endothelial damage due to an autoimmune attack. This predisposition to vascular endothelial damage further heightened the likelihood of thrombosis. Upon admission, we first evaluated and ruled out the recurrence of HSP based on clinical and laboratory findings.
The pathogenesis of LV remains elusive, with abnormal coagulation function being the most likely underlying mechanism. Current research primarily focuses on three aspects: flow disruption, endothelial injury, and coagulation disorders (3). This patient had suffered from the basic disease of allergic purpura, which greatly increased the possibility of vascular fragility and endothelial damage after his own immune attack. It has a high-risk factor for vascular endothelial damage, making thrombosis even easier. In pathology, the three major manifestations of LV disease are segmental hyalinization or fibrinoid degeneration of dermal vessels, proliferation of the endothelium, and intraluminal thrombosis (3, 7). The skin biopsy of this patient showed capillary telangiectasia, extravasation of red blood cells, and intraluminal capillary thrombi. Clinically, the rash presented as erythematous macules that were not significantly elevated above the skin surface and were devoid of itching or pain. The lesions progressed through various stages, including ulceration, white atrophic-like changes, and crusting. LV predominantly affects the lower limbs, with rare involvement of the upper limbs (7). Capillary embolism leads to the formation of small ulcers, which typically develop into characteristic “atrophie blanche” 3–4 months later. This condition is often accompanied by burning pain, significantly impacting the patient's quality of life, particularly during active disease phases, affecting psychological, physical, and social aspects (9).
In terms of clinical laboratory findings, most cases of LV lack significant diagnostic markers. However, some studies have suggested an association with hypercoagulable states. For instance, a prospective study found elevated levels of hypercoagulability markers, such as fibrinopeptide A, lipoprotein (a), and plasminogen activator inhibitor-1 (10). In our case, only routine laboratory tests were performed, and more specialized studies were lacking. Currently, various therapeutic regimens are employed for LV, yet standardized treatment protocols remain limited. Among monotherapy options, anticoagulants are the most commonly used, followed by systemic glucocorticoids, antiplatelet agents, and immunomodulatory drugs (2, 11). Given the low incidence of LV, research on its treatment is relatively sparse. First-line treatments typically include antiplatelet drugs, such as aspirin and clopidogrel. Rivaroxaban has emerged as a promising therapeutic option due to its safety and ease of use. A comparative study analyzed 20 articles and 138 patients, finding that the average treatment response time was 2.4 months for rivaroxaban vs. 2.3 months for intravenous immunoglobulin (IVIG). This study further validated the efficacy of rivaroxaban (12).
Rivaroxaban is a direct Xa factor inhibitor that has been widely used to treat and prevent major thromboembolic diseases. Compared with other anticoagulants such as low molecular weight heparin and warfarin, rivaroxaban can be taken orally and does not require international normalized monitoring, making it the preferred drug. A study by Kerk et al. (13) first reported a successful case of treating LV with rivaroxaban. Rivaroxaban was gradually becoming familiar to people. In a recent review, approximately 73 patients were counted, and the therapeutic dose of lifasab was 10–20 mg/day. Approximately 82.2% (60/73) responded to the treatment, and their pain and ulcers were relieved (14). A cross-sectional study by Zhao et al. (15) found that it can monitor the active phase through coagulation factor X (IQR: 102.3–132.5 vs. IQR: 92.9–118.8, P = 0.04). Moreover, 73% of patients achieved complete remission within 12 weeks of rivaroxaban treatment, with low side effects (25%). It is clear from these studies that rivaroxaban is effective for LV patients. In our case, it also demonstrated that appropriate rivaroxaban use in children can effectively control the progression of LV disease. Adverse reactions were observed rarely, with heavy menstrual bleeding being the most common (13, 14).
Rivaroxaban is a direct factor Xa inhibitor widely used for treating and preventing major thromboembolic diseases. Compared to other anticoagulants, such as low molecular weight heparin and warfarin, rivaroxaban offers the advantage of oral administration without the need for international normalized ratio (INR) monitoring, making it a preferred choice (13). A recent review summarized data from 73 patients, with a therapeutic dose of rivaroxaban ranging from 10 to 20 mg/day. Approximately 82.2% (60/73) of patients responded to treatment, experiencing relief from pain and ulceration (14). A cross-sectional study demonstrated that coagulation factor X levels could be used to monitor disease activity. Moreover, 73% of patients achieved complete remission within 12 weeks of rivaroxaban treatment, with a low incidence of adverse effects (14). These studies collectively highlight the efficacy of rivaroxaban in treating LV. In our case, rivaroxaban effectively controlled the progression of LV in the pediatric patient, with minimal adverse reactions, the most common being heavy menstrual bleeding (13, 14). In addition to rivaroxaban, emerging targeted therapies show promise in treating refractory LV. For instance, tofacitinib, a pan-Janus kinase (JAK) inhibitor, has been reported to be effective in LV treatment (16). Another study demonstrated that anti-interleukin 17 A biologics can also control disease progression (17). In conclusion, the application of targeted therapies has expanded our understanding of the therapeutic mechanisms of LV, bringing the possibility of clinical cure closer to reality.
Conclusion
When patients have a purple rash with pain, clinicians should consider the possibility of LV. It is important to first assess whether the child has a hypercoagulable state before analyzing the test results. There is no specific clinical test for this disease, and its diagnosis mainly depends on skin biopsy. However, invasive clinical examinations are difficult for family members to accept and increase the complexity of diagnosing and treating the disease. When diagnosis is challenging, anticoagulants such as rivaroxaban for treatment may be considered. If pain improves, it can indirectly confirm the condition. A skin biopsy must focus on dermal vascular occlusion, which is crucial for diagnosis.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by the Ethics Committee of Guangzhou First People's Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants' legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
JQ: Investigation, Methodology, Writing – original draft. ZH: Methodology, Software, Writing – original draft. WJ: Data curation, Writing – review & editing. SY: Project administration, Resources, Writing – original draft.
Funding
The authors declare that financial support was received for the research and/or publication of this article. This work was supported by grants from the Guangzhou Planned Project of Science and Technology (Nos. 2024A03J1018 and 2024A03J1094) and National Key Research and Development Program of China (Nos. 2022YFC2705100, 2022YFC2705103, and 2022YFC2705104).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Beato Merino MJ, Diago A, Fernandez-Flores A, Fraga J, García Herrera A, Garrido M, et al. Clinical and histopathologic characteristics of the main causes of vascular occusion—part II: coagulation disorders, emboli, and other. Actas Dermosifiliogr (Engl Ed). (2021) 112(2):103–17. doi: 10.1016/j.ad.2020.10.004
2. Bilgic A, Ozcobanoglu S, Bozca BC, Alpsoy E. Livedoid vasculopathy: a multidisciplinary clinical approach to diagnosis and management. Int J Womens Dermatol. (2021) 7(5Part A):588–99. doi: 10.1016/j.ijwd.2021.08.013
3. Eswaran H, Googe P, Vedak P, Marston WA, Moll S. Livedoid vasculopathy: a review with focus on terminology and pathogenesis. Vasc Med. (2022) 27(6):593–603. doi: 10.1177/1358863X221130380
4. Gao Y, Jin H. Livedoid vasculopathy and its association with genetic variants: a systematic review. Int Wound J. (2021) 18(5):616–25. doi: 10.1111/iwj.13563
5. Vasudevan B, Neema S, Verma R. Livedoid vasculopathy: a review of pathogenesis and principles of management. Indian J Dermatol Venereol Leprol. (2016) 82(5):478–88. doi: 10.4103/0378-6323.183635
6. Alavi A, Hafner J, Dutz JP, Mayer D, Sibbald RG, Criado PR, et al. Livedoid vasculopathy: an in-depth analysis using a modified Delphi approach. J Am Acad Dermatol. (2013) 69(6):1033–1042.e1. doi: 10.1016/j.jaad.2013.07.019
7. Rujitharanawong C, Chularojanamontri L, Trakanwittayarak S, Pinkaew S, Nuttawong S, et al. Livedoid vasculopathy: clinical course and long-term outcome in Asian patients with a review of the literature. Dermatol. Ther. (2021) 34(1):e14569. doi: 10.1111/dth.14569
8. Lipsker D. Livedoid vasculopathy: how to diagnose and how to treat? J Eur Acad Dermatol Venereol. (2019) 33(9):1627–8. doi: 10.1111/jdv.15830
9. Polo Gascón MR, de Carvalho JF, de Souza Espinel DP, Barros AM, Alavi A, Criado PR. Quality-of-life impairment in patients with livedoid vasculopathy. J Am Acad Dermatol. (2014) 71(5):1024–6. doi: 10.1016/j.jaad.2014.06.030
10. Gao Y, Jin H. Plasminogen activator inhibitor-1: a potential etiological role in livedoid vasculopathy. Int Wound J. (2020) 17(6):1902–8. doi: 10.1111/iwj.13480
11. Wen J, Li XH, Yang QP, Yu JB. Clinical and dermoscopic features of livedoid vasculopathy. Chin Med J (Engl). (2020) 133(17):2137–8. doi: 10.1097/CM9.0000000000001048
12. Ramphall S, Rijal S, Prakash V, Ekladios H, Mulayamkuzhiyil Saju J, Mandal N, et al. Comparative efficacy of rivaroxaban and immunoglobulin therapy in the treatment of livedoid vasculopathy: a systematic review. Cureus. (2022) 14(8):e28485. doi: 10.7759/cureus.28485
13. Kerk N, Drabik A, Luger TA, Schneider SW, Goerge T. Rivaroxaban prevents painful cutaneous infarctions in livedoid vasculopathy. Br J Dermatol. (2013) 168(4):898–9. doi: 10.1111/bjd.12100
14. Gao Y, Jin H. Rivaroxaban for treatment of livedoid vasculopathy: a systematic review. Dermatol Ther. (2021) 34(5):e15051. doi: 10.1111/dth.15051
15. Zhao C, Yu M, Cheng X, Li S, Feng S. Associations between coagulation factors and livedoid vasculopathy and the efficacy and safety of rivaroxaban in patients with livedoid vasculopathy: a retrospective study. Clin Exp Dermatol. (2024) 49(8):817–24. doi: 10.1093/ced/llae023
16. Jia E, Yan G, Xiao M, Geng H, Wei J, Zhang J. Refractory ulcerations associated with livedoid vasculopathy successfully treated with tofacitinib. Dermatol Ther. (2020) 33(6):e14470. doi: 10.1111/dth.14470
Keywords: livedoid vasculopathy (LV), white atrophy, rivaroxaban, capillaries, children
Citation: Qu J, Hao Z, Junli W and Yu S (2025) A case report and literature review of livedoid vasculopathy in children. Front. Pediatr. 13:1537133. doi: 10.3389/fped.2025.1537133
Received: 30 November 2024; Accepted: 21 April 2025;
Published: 14 May 2025.
Edited by:
Panagiota Anyfanti, Aristotle University of Thessaloniki, GreeceReviewed by:
Dragana Lazarevic, University Clinical Center Nis, SerbiaPaulo Ricardo Criado, Faculdade de Medicina do ABC, Brazil
Copyright: © 2025 Qu, Hao, Junli and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shengyou Yu, ZXl5dXNoZW5neW91QHNjdXQuZWR1LmNu