 Ting Dong
Ting Dong Jiajia Luo2
Jiajia Luo2 Lina Ma
Lina Ma- 1Department of Lanzhou University, Lanzhou, China
- 2Department of Pediatrics, Lanzhou University Second Hospital, Lanzhou, China
Nephronophthisis is an autosomal recessive disorder associated with the tubular interstitium of the kidney, and can lead to renal failure in children and adolescents. Mutations in the gene encoding nephrocystin-1, NPHP1, are frequently associated with the disease. Here, we describe the case of a child who presented to the clinic with febrile convulsions and who was ultimately diagnosed with nephronophthisis caused by a homozygous deletion of the NPHP1 gene. Alerting pediatricians to the recognition of atypical renal wasting disease and reclarifying the diagnostic value of genetic diagnosis for this disease.
1 Introduction
Nephronophthisis (NPHP) is recognized as the most common inherited cause of renal failure in children (1). According to the age of onset of end-stage renal disease (ESRD), NPHP can be classified as infantile, juvenile and adolescent forms. Among them, juvenile NPHP is the most common. Typical clinical features include polyuria, polydipsia and isosthenuria due to renal concentration function impairment, no hematuria, proteinuria, hypertension and other manifestations. If the disease progresses to a higher stage of chronic kidney disease (stage III and above), fatigue may occur. Fever, fatigue, skin itching, nausea, vomiting, uremic gastritis, anemia, and growth retardation may occur (2). Approximately 10%–20% of cases involve NPHP patients also display additional extrarenal abnormalities, such as Senior-Loken syndrome (SLS), Joubert syndrome, Mainzer-Saldino's syndrome, and liver fibrosis, situs inversus, etc. Among these, the eye is the most commonly affected extrarenal organ in NPHP, with retinitis pigmentosa being the most common (3).
Genes that have been found to be associated with the disease encode proteins involved in the functioning of primary cilia, basal bodies, and centrosomes, and mutations can lead to renal damage, as well as extrarenal manifestations associated with NPHP. Mutations in the nephrocystin-1 gene (NPHP1) on chromosome 2q13, the most common cause of the disease, are either pure or compound heterozygous mutations (4). In this report, we describe the case of a child with renal wasting disease caused by a pure deletion of NPHP1 and analyze and summarize the manifestations of NPHP in this child, to provide a reference for the diagnosis and treatment of similar children in the future and to enhance the understanding and identification of the disease in clinical practice, thus providing timely diagnosis and treatment.
2 Case report (presentation)1
2.1 Medical history
The child was 6 years and 9 months old and presented to the clinic with “one day of fever with two convulsions”. The convulsions were characterized by loss of consciousness, bluish lips, generalized twitching, rigidity of the limbs, no incontinence, and foaming at the mouth; these lasted for two minutes and then subsided before starting again with the same manifestations and lasting between three and four minutes. After an interval of approximately 30 min, the convulsions began again. Cranial nuclear magnetic resonance imaging (MRI), electroencephalography (EEG), laboratory tests (see Table 1), and genetic tests were performed. The diagnosis was renal wasting disease, stage 4 renal impairment, secondary hyperparathyroidism, renal anemia, and renal tubular damage. The child was given human erythropoietin 2,000 IU/session once a week, as well as levetiracetam, uremic granules, capsules of aldoxyl starch, compound a-ketoacid tablets, vitamin D drops, and other medications, and was followed up regularly for three months, with advice to undergo kidney transplantation in case of deterioration of renal function.
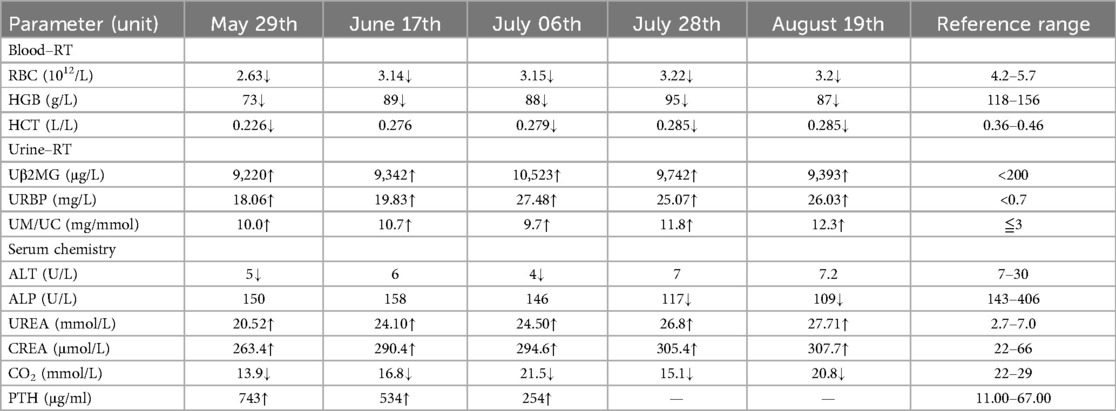
Table 1. Pertinent laboratory results.
2.2 Antecedent history
The patient was in good health, with no symptoms of polyuria, thirst or renal dysfunction. The patient denied history of surgery, trauma or blood transfusion. The patient denied close contact with infectious diseases such as hepatitis and tuberculosis, and denied food and drug allergies.
2.3 Family history
The parents were healthy, the mother had one pregnancy and no abortion, and the marriage was close relatives. The family history of hereditary diseases was denied. The maternal grandmother had a history of nephritis (Figure 1).
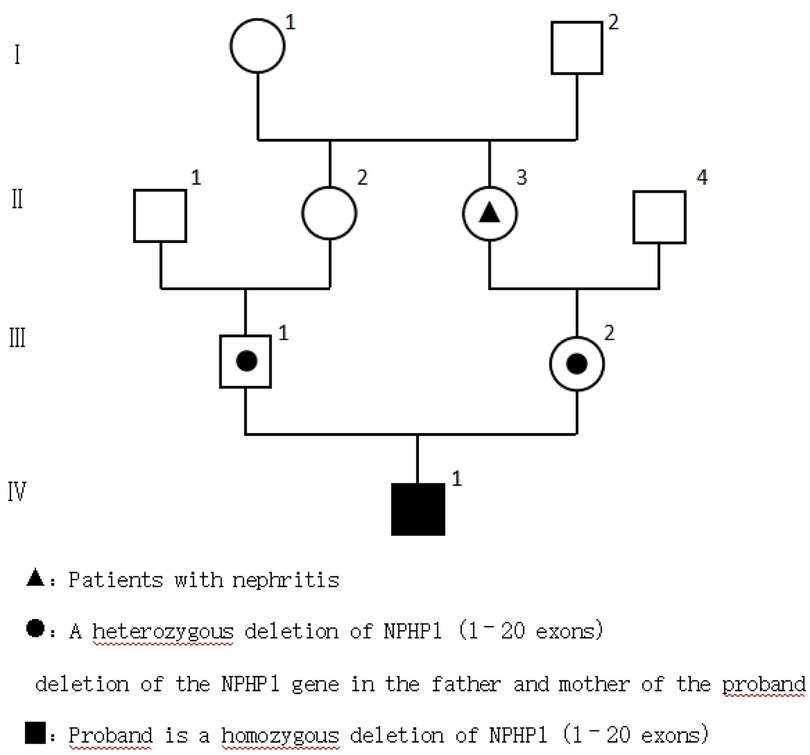
Figure 1. Genealogical chart of the child.
2.4 Personal history
First pregnancy, first delivery, with full-term normal delivery. The birth weight was 3.3 kg and the mother is health during pregnancy.
2.5 Laboratory results
During the hospitalization period, the level of relevant renal function indexes (Urine-RT, UREA, CREA, etc.) was significantly higher than the normal level (Figures 2,3), showing an upward trend, which made it clear that the child's renal function was impaired, and the blood routine was improved, suggesting that: the red blood cell count and hemoglobin were lower than the normal level, which was secondary renal anemia, and the child was given active treatment with human erythropoietin, and the level of the child's parathyroid hormone was significantly elevated. At the same time, the parathyroid hormone level of the child was found to be significantly elevated, which was considered to be secondary hyperparathyroidism, in line with the secondary manifestation of NPHP.
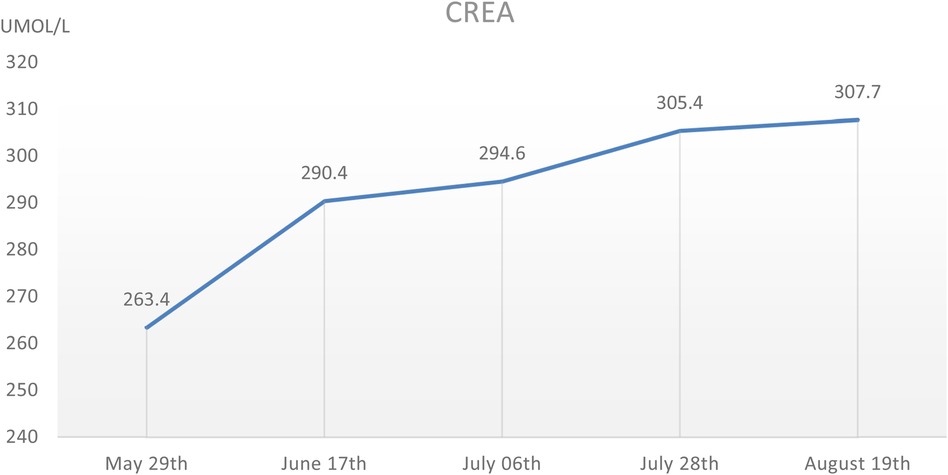
Figure 2. The CREA level of this child during hospitalization, which showed a gradual increase.
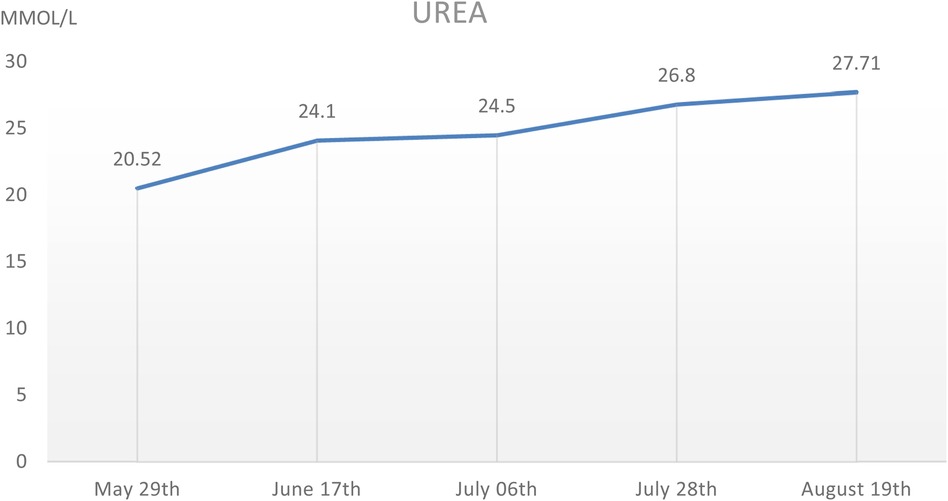
Figure 3. The UREA level of this child during hospitalization, which showed a gradual increase.
2.6 Imaging results
2.61 MR urinary tract plain scan + MRU (including kidney, ureter, and bladder)
Diagnostic opinions: (1) T1WI showed slightly reduced and diffuse bilateral renal signals, with an unclear boundary between the cortex and medulla. (2)Mild expansion of the middle ureter on both sides (Figure 4).
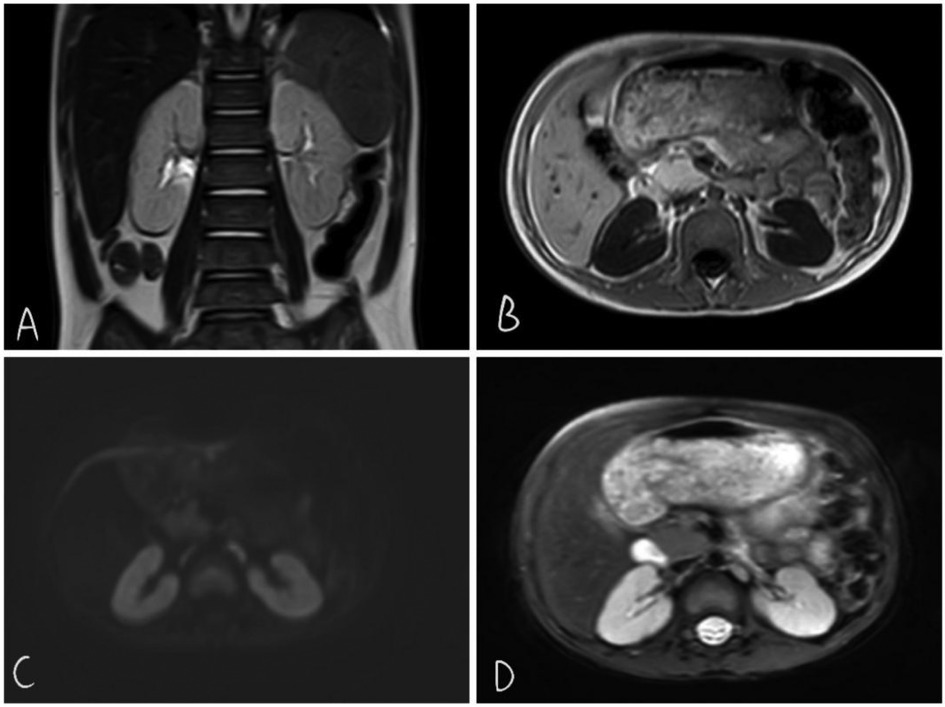
Figure 4. T2wi coronal view shows somewhat poorly defined corticomedullary boundaries in both kidneys (A) T1WI shows long TIWI signals in both kidneys (B) DWI shows no restriction of diffusion (C) T2WI signal shows slightly increased cortical signal in T2WI (D)
2.62 Other examinations
(1) Abdominal color Doppler ultrasound: Diffuse lesions in both kidneys, no abnormality was detected in the liver, gallbladder, pancreas and spleen.
(2) Echocardiography: Patent foramen ovale (occasionally visible).
(3) Fundus color Doppler ultrasound: binocular ametropia, binocular amblyopia.
(4) No abnormality was found on EEG.
(5) MR brain stereotactic + intraoperative navigation + epileptic sequence scan (large-scale fine scan): No obvious abnormality was found in the brain stereotactic scan.
2.67 Genetic testing
A homozygous deletion variant in the NPHP1 gene was detected: [NM_001128178.3(NPHP1):loss2(EXON:1-20)(all)], resulting in homozygous deletion of exon 1 to exon 20 of the NPHP1 gene, and which was suspected to involve the homozygous deletion of the entire gene (Figure 5).
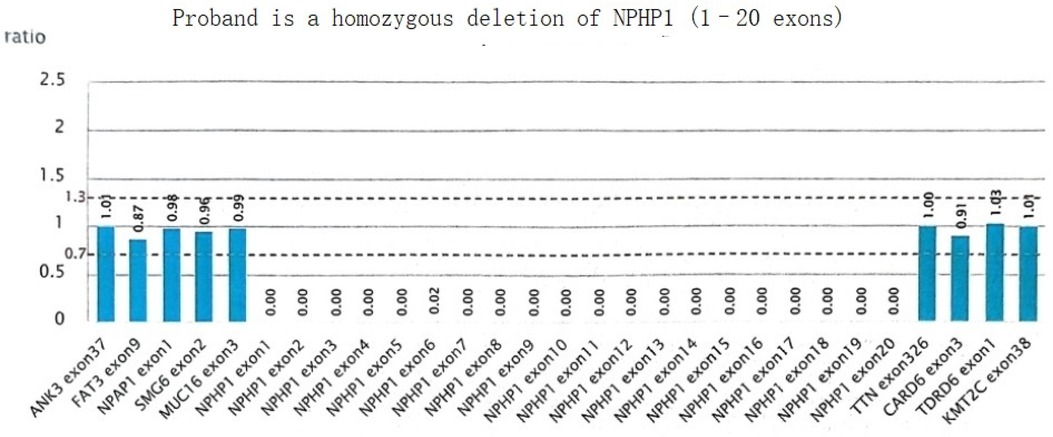
Figure 5. NPHP1 gene sequencing results in subjects.
Both parents of the subject had loss of heterozygosity in NPHP1 (EXON:1–20) (Figure 6).
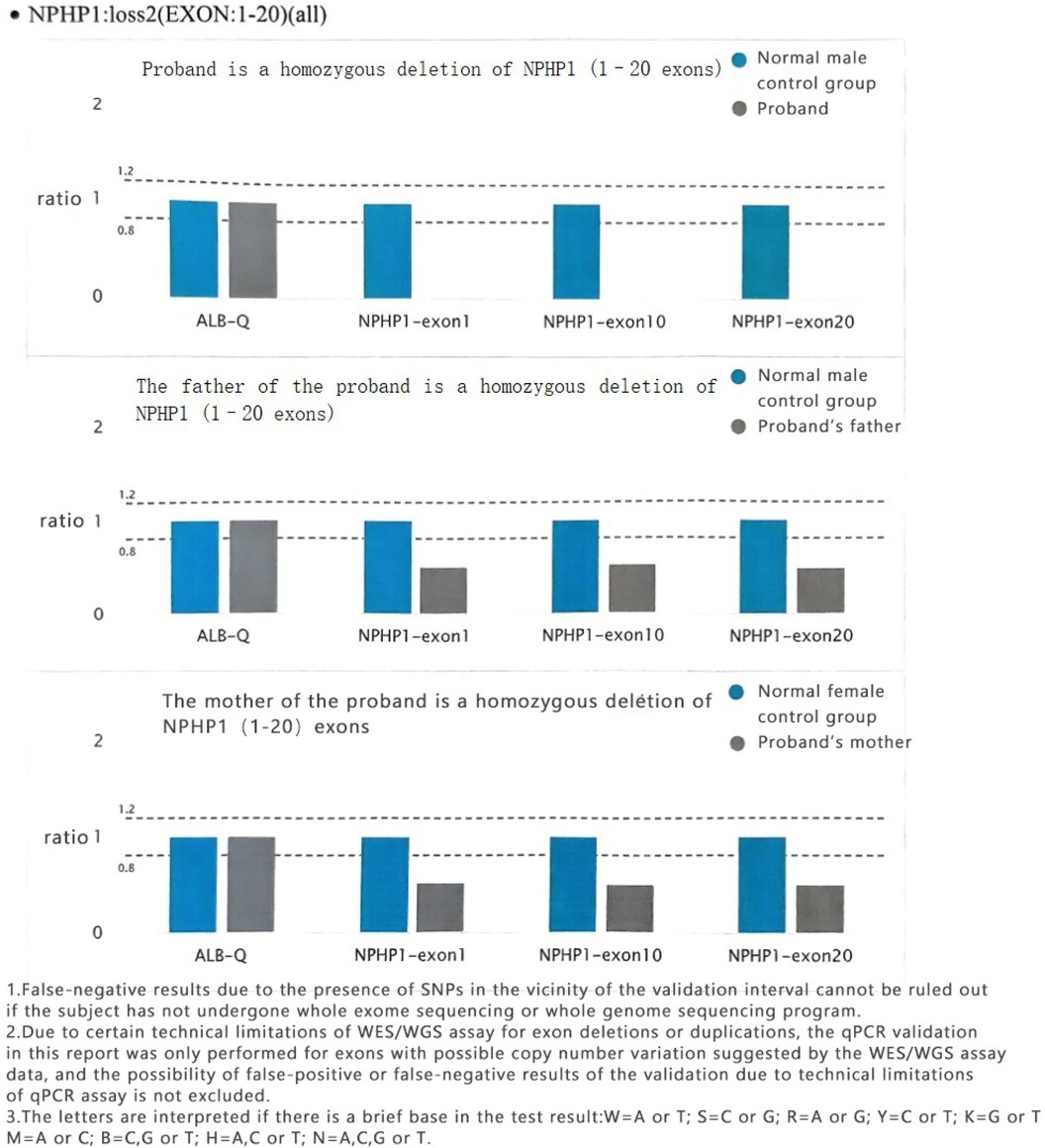
Figure 6. Sequencing results of the NPHP1 gene in the subject's parents.
2.68 Diagnostic
In this case, the child was admitted to hospital due to sudden convulsions. However, no obvious abnormalities were found on neurological examinations. During hospitalization, high levels of serum creatinine and urea nitrogen were found, although the cause of the renal dysfunction was unknown. After evaluation, the child was diagnosed with kidney impairment stage 4. Typical symptoms of renal wasting disease had not yet appeared. And ultrasound examination only indicated diffuse lesions in both kidneys. The parents of the child were consanguineous, and genetic sequencing was performed to identify the cause of the renal impairment. The sequencing results identified a homozygous deletion of the NPHP1 gene. And the diagnosis thus requires differentiation from medullary cystic nephropathy, early autosomal dominant polycystic kidney disease (PKD), autosomal recessive PKD, and acquired tubulointerstitial injury. Although these diseases overlap in some features, they can be distinguished by genetic testing and renal biopsy (5). Infantile NPHP typically presents with ESRD before the age of 4, characterized by NPHP interstitial fibrosis, tubular atrophy, and extensive cyst formation similar to polycystic kidney disease. Due to the widespread formation of renal cysts, the kidneys appear not significantly reduced in size or even enlarged on ultrasound. In this case, the patient developed ESRD at age 6 and is not considered to have infantile NPHP (6).
This, together with the extrarenal manifestations of renal depletion disease (convulsions, decreased visual acuity in both eyes, secondary hyperparathyroidism), results of auxiliary examinations, genetic testing and differential diagnosis, led to the diagnosis of juvenile NPHP.
3 Discussion
Nephronophthisis (NPHP) is one of the most common causes of monogenic single-gene renal failure (CKF), with over 25 genes have been associated with NPHP. These genes encode proteins involved in the functioning of primary cilia, the matrix, and the centrosome, leading to kidney diseases and extrarenal manifestations (7, 8). Among them, the most common cause of juvenile NPHP is a mutation in the NPHP1 gene, which accounts for 20%–25% of reported NPHP cases; 10 cases of renal wasting disease have been found to be caused by homozygous deletion of a large fragment of NPHP1, with the onset of the disease occurring at around 9–13 years old (9). Due to renal concentrating function, most patients exhibit typical symptoms of nephronophthisis, such as polydipsia, polyuria, enuresis, and significantly decreased urine osmolality. Moreover, ultrasound examinations of patients with nephronophthisis caused by NPHP1 gene deletion have consistently shown renal cysts, but the child in this case only suggests that the echo of the parenchyma of both kidneys is enhanced, the boundary between cortex and medulla is blurred, and the blood flow of both kidneys is less.
Renal cortical cysts may appear in the late stage of the disease. These are primarily caused by the NPHP1 protein (also known as renal capsule protein) which is expressed on the surfaces of primary cilia and renal tubular epithelial cells, and can interact with molecules involved in cell adhesion and signal transduction, as well as other capsule proteins. Fibronectin is an important component of the extracellular matrix and is involved in cell proliferation, apoptosis, and polarization (10). Mutations in NPHP1 can prevent its binding to fibronectin, thus restricting its function. Apart from the appearance of a series of extrarenal manifestations, renal cysts develop as the disease progresses. It has been reported that the initial imaging results of patients with NPHP may be non-specific, while the normal shape of the kidney can be assessed by ultrasound, indicating a loss of corticomedullary differentiation. However, with disease progression, ultrasound imaging conducted at later stages may show smaller atrophic kidneys with obvious corticomedullary cysts (11). Therefore, it is thus important to pay active attention to the manifestations associated with renal cysts in such children.
Renal tubulointerstitial damage in renal wasting disease is often characterized by medullary cystic changes. Myelocystic nephrotic include juvenile renal wasting disease (JN), medullary cystic kidney disease (MCKD), and medullary sponge kidney (MSK) (12). MSK is a kind of congenital, benign renal developmental abnormality, primarily characterized by spindle-shaped or small cystic dilation of the collecting ducts within one or more renal pyramids in both kidneys. Clinical manifestations often include kidney stones, renal calcification, tubular concentrating dysfunction, and recurrent urinary tract infections. The prognosis is relatively good, with rare cases causing renal function abnormalities during adolescence. Ultrasound (ultrasound examination) may show small anechoic areas and echogenic foci radiating around the renal pelvis. In this case, no relevant ultrasound findings were observed in the patient's kidneys, so this diagnosis is not considered for now. JN and MCKD share similarities in clinical and pathological manifestations, collectively referred to as the JN-MCKD syndrome. JN-MCKD is a group of cystic kidney diseases characterized by the formation of medullary cysts and occult chronic renal insufficiency, with pathological features consistent with chronic tubulointerstitial nephritis. The two can be distinguished based on inheritance pattern, age of onset, and clinical presentation: renal wasting disease is an autosomal recessive disorder, commonly seen in children and adolescents, and may be accompanied by other extrarenal manifestations. Medullary cystic disease, which is more common in adults, is an autosomal dominant inheritance, mainly with renal lesions and less extrarenal manifestations. Both are genetic diseases, which are often diagnosed by genetic testing.
Feng Chen mentioned that the clinical diagnosis of atypical NPHP is challenging, proposed NPHP1 deletion in Chinese twins with nephronophthisis, and demonstrated in the text the superiority of whole exome sequencing (WES) for diagnosing this type of disease (13). This case patient is also an atypical NPHP patient. Given that the average age of onset of ESRD in juvenile NPHP is 13 years, with only a few cases entering ESRD around age 6, the diagnosis of this patient is even more difficult. This reaffirms the importance of genetic testing for diagnosing atypical NPHP patients.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
TD: Writing – original draft. JL: Writing – review & editing. HW: Writing – review & editing. QZ: Writing – review & editing. TS: Writing – review & editing. LM: Writing – review & editing. JY: Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This article was obtained from a project funded by the Natural Science Foundation of Gansu Province, China (No. 22JR11RA052).
Acknowledgments
The authors would like to thank the family for allowing us to publish this report.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnote
1. ^Project supported by the Nature Science Foundation of Gansu Province, China (No. 22JR11RA052)
References
1. Abdelwahed M, Maaloul I, Benoit V, Hilbert P, Hachicha M, Kamoun H, et al. Copy-number variation of the NPHP1 gene in patients with juvenile nephronophthisis. Acta Clin Belg. (2021) 76:16–24. doi: 10.1080/17843286.2019.1655231
2. Srivastava S, Molinari E, Raman S, Sayer JA. Many genes—one disease? Genetics of nephronophthisis (NPHP) and NPHP-associated disorders. Front Pediatr. (2018) 5:287. doi: 10.3389/fped.2017.00287
3. Doreille A, Raymond L, Lebre A-S, Linster C, Saraeva Lamri R, Karras A, et al. Nephronophthisis in young adults phenocopying thrombotic microangiopathy and severe nephrosclerosis. Clin J Am Soc Nephrol. (2021) 16:615–7. doi: 10.2215/CJN.11890720
4. Gupta S, Ozimek-Kulik JE, Phillips JK. Nephronophthisis-pathobiology and molecular pathogenesis of a rare kidney genetic disease. Genes (Basel). (2021) 12:1762. doi: 10.3390/genes12111762
5. Kramers BJ, Koorevaar IW, van Gastel MDA, van Goor H, Hallows KR, Heerspink HL, et al. Effects of hydrochlorothiazide and metformin on aquaresis and nephroprotection by a vasopressin V2 receptor antagonist in ADPKD: a randomized crossover trial. Clin J Am Soc Nephrol. (2022) 17:507–17. doi: 10.2215/CJN.11260821
6. Huiying S, Yu A. Advances in molecular genetics and pathophysiology of nephronophthisis. J Nephrol Dialy Transplant. (2024) 33:463–8. doi: 10.3969/j.issn.1006-298X.2024.05.012
7. Bhimma R, Jembere E, Hariparshad S. Case report of a child with nephronophthisis from South Africa. BMC Pediatr. (2024) 24:431. doi: 10.1186/s12887-024-04872-2
8. Macia MS, Halbritter J, Delous M, Bredrup C, Gutter A, Filhol E, et al. Mutations in MAPKBP1 cause juvenile or late-onset cilia-independent nephronophthisis. Am J Hum Genet. (2017) 100:323–33. doi: 10.1016/j.ajhg.2016.12.011
9. Lifei Z, Linlu Z, Wenyi W, Jianlei L, Danli Z, Pingping Z, et al. Clinical characterization and genetic analysis of a family line with renal wasting disease caused by a pure deletion of NPHP1 gene. J Shandong Univ (Health Sci). (2024) 62:108–11. doi: 10.6040/j.issn.1671-7554.0.2023.1143
10. McConnachie DJ, Stow JL, Mallett AJ. Ciliopathies and the kidney: a review. Am J Kidney Dis. (2021) 77:410–9. doi: 10.1053/j.ajkd.2020.08.012
11. Chan Q, Zhi-yan M, Shu-fen L, Fei-yan W. Juvenile nephronophthisis type I: a case report and literature review. J Clin Nephrol. (2025) 25:352–4. doi: 10.3969/j.issn.1671-2390.2025.04.012
12. Yuchao Z, Yan Z, Lixuan L, Zhen N, Zhihong L. Hyposthenuria,corticomedullary border cysts and renal dysfunction. J Nephrol Dialy Transplant. (2016) 25:394–8. doi: 10.3969/cndt.j.issn.1006-298X.2016.04.020
Keywords: renal wasting disease, NPHP1 gene, pediatric, case report, sequencing of genome case report (presentation)
Citation: Dong T, Luo J, Sun T, Wu H, Zhao Q, Ma L and Yang J (2025) Case Report: A renal wasting disease caused by a pure deletion of nephrocystin-1. Front. Pediatr. 13:1541411. doi: 10.3389/fped.2025.1541411
Received: 7 December 2024; Accepted: 4 June 2025;
Published: 20 June 2025.
Edited by:
John Andrew Sayer, Newcastle University, United KingdomReviewed by:
Payal Gaggar, Nizam’s Institute of Medical Sciences, IndiaJuliana Estefanía Arcila Galvis, Newcastle University, United Kingdom
Rebecca Walker, The University of Sheffield, United Kingdom
Copyright: © 2025 Dong, Luo, Sun, Wu, Zhao, Ma and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jing Yang, amluZ3lhbmdsZGV5QDE2My5jb20=