 Min Zhang1,†
Min Zhang1,† Zhaodong Lin2,†
Zhaodong Lin2,† Meihuan Chen1Danhua Guo1Qiaomei Yang3Qianqian He1
Meihuan Chen1Danhua Guo1Qiaomei Yang3Qianqian He1 Bin Mao1
Bin Mao1 Bin Liang1Lingji Chen1
Bin Liang1Lingji Chen1 Meiying Cai1*
Meiying Cai1* Hailong Huang1*Liangpu Xu1*
Hailong Huang1*Liangpu Xu1*
- 1Fujian Key Laboratory of Prenatal Diagnosis and Birth Defect, Medical Genetic Diagnosis and Therapy Center, Fujian Maternity and Child Health Hospital, Fuzhou, Fujian, China
- 2Department of Clinical Laboratory, Fuzhou First General Hospital, Fuzhou, Fujian, China
- 3Department of Gynecology, Fujian Maternity and Child Health Hospital, College of Clinical Medicine for Obstetrics & Gynecology and Pediatrics, Fujian Medical University, Fuzhou, Fujian, China
Background: DMD genetic variants cause a spectrum of phenotypes, from severe progressive proximal muscle weakness and degeneration leading to wheelchair dependence and death from cardiac and/or respiratory failure to very mild muscular phenotypes; very rarely, cases are completely asymptomatic. Few cases have been reported in males carrying DMD deletions who are asymptomatic.
Methods: Family clinical information was collected from the patients. A single nucleotide polymorphism array (SNP-array) was used to detect abnormalities in prenatal diagnosis, and multiplex ligation-dependent probe amplification (MLPA) and long-read sequencing (LRS) were used to confirm the detected variant.
Results: We incidentally identified DMD exons 48–55 deletion using SNP-array in prenatal diagnosis; the variant was confirmed using MLPA and LRS, and the fragment size and precise locations of breakpoints were determined. The variant was precisely located at genomic position chrX:31640088–31945085, spanning from intron 47 to intron 56 in DMD. Serum biochemical indicators were normal in the male with the deletion.
Conclusion: Our study is the first to report a DMD exons 48–55 deletion in prenatal diagnosis. The phenotypes of DMD variants are diverse, and this study suggests that prediction of clinical severity based solely on molecular findings should be interpreted with caution.
1 Introduction
Duchenne muscular dystrophy (DMD) is an X-linked recessive disorder characterized by progressive muscle degeneration and weakness caused by variations in the DMD gene, including deletions (68%), duplications (10%), small variants such as point mutations (22%), and rare complex rearrangements (<0.5%), which may vary slightly in different regions (1–3). It usually occurs in males and affects 1/6,000–1/3,600 live births worldwide (4). The clinical phenotypes of muscular dystrophy associated with mutations in DMD are variable, ranging from severe DMD and mild Becker's muscular dystrophy (BMD) to an extremely rare, completely asymptomatic phenotype (5–7).
DMD encodes dystrophin and includes 79 exons spanning 2.2 Mb on Xp21.1; it is the largest gene in the human genome. Traditional molecular detection methods such as multiplex ligation-dependent probe amplification (MLPA) are widely used for DMD mutation screening in clinical laboratories to rapidly and effectively detect exon copy number variations. Next-generation sequencing (NGS) technology enables the detection of single nucleotide variations among the 79 exons within DMD. MLPA with NGS is commonly used clinically to screen for DMD abnormalities, and more than 90% of the variations can be detected in patients with DMD/BMD (8, 9). However, traditional methods cannot identify complex structural variants or determine whether the variations occur intragenically or extragenically. Long-read sequencing (LRS) can effectively discriminate the precise physical locations of breakpoints and structural features of genomic rearrangement and is increasingly being adopted (10–14). With newly emerging genetic technologies, more DMD variations unrelated to indications are being detected.
Here, we report a DMD exons 48–55 deletion in clinical that was inadvertently detected during prenatal diagnosis using a single nucleotide polymorphism array (SNP-array) and confirmed with MLPA and LRS. Our study reinforces the importance of detailed clinical evaluation, precise molecular testing, and appropriate genetic counseling when clinical phenotypes are inconsistent with genotypes.
2 Materials and methods
2.1 Participants
A 35-year-old pregnant female was admitted to our hospital for prenatal diagnosis at an advanced gestational age of 18 weeks. She had no family history of complications. Her spouse was 35 years old and showed no symptoms related to DMD or BMD (such as myasthenia, amyotrophia, pseudohypertrophy of the gastrocnemius muscle); in addition, he reported no muscle fatigue, or walking discomfort. Although he refused to undergo muscle testing, a physical examination showed that, his upper and lower limbs demonstrated sequential joint extension and flexion movements, and could resist force exerted by the examiner, showing normal muscle strength. He did not have cardiac discomfort and also refused to undergo cardiac-related examinations (echocardiography etc.), and nor a family history of muscle or heart diseases. The biochemical indicators showed normal serum creatine kinase (CK) (198 IU/L, reference range: 20–270 IU/L), creatine kinase isoenzyme MB (CK-MB) (18 IU/L, reference range: 0–32 IU/L), alanine aminotransferase (ALT) (27.0 IU/L, reference range: 7–50 IU/L), aspartate aminotransferase (AST) (22.6 IU/L, reference range: 12–40 IU/L), lactate dehydrogenase (LDH) (166 IU/L, reference range: 90–282 IU/L), and hypersensitive troponin Ⅰ (TNIU) (<0.01 μg/L, reference range: 0–0.01 μg/L) concentrations. Informed consent was obtained for all study subjects prior to the molecular studies. This study was approved by the Ethical Review Committee of Fujian Maternity and Child Health Hospital.
2.2 Sample collection
Amniocentesis was performed in pregnant women under ultrasound guidance, and 25–30 ml of amniotic fluid was sampled for cell culture, SNP-array, MLPA, and LRS. Peripheral blood was drawn by venipuncture with 5% ethylene-diamine tetraacetic acid as anticoagulant for the pregnant female and her spouse, the other pedigree members were not genetically tested. Genomic deoxyribonucleic acid (DNA) was extracted using the QIAGEN DNA Mini Kit (Qiagen, Hilden, Germany) following standard procedures, the extracted DNA was quantified by a Nano Drop 2000 Spectrophotometer (Thermo Fisher Scientifc, MA, USA) to ensure DNA concentration was >50 ng/μl, and the 260/280 nm optical density ratio was 1.8–2.0.
2.3 Single nucleotide polymorphism array
Chromosomal microarray analysis was performed using the CytoScan 750K array with 550,000 nonpolymorphic probes for determination of copy number changes and 200,000 single nucleotide polymorphism probes for allelic confirmation of copy number changes and detection of loss of heterozygosity. Array analyses were performed using the Chromosome Analysis Suite software (ChAS), version 3.3 (Affymetrix, Santa Clara, CA, USA). CNVs analysis was performed using the DGV (http://dgv.tcag.ca/dgv/app/home), DECIPHER (https://decipher.sanger.ac.uk/), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), ClinGen Dosage Sensitivity Map (https://dosage.clinicalgenome.org), OMIM (https://omim.org/) for analysis of genes associated with diseases.
2.4 Multiplex ligation-dependent probe amplification
MLPA was performed to detect the exonic deletions and duplications of DMD using the SALSA P034/P035 DMD Kit (MRC Holland, Amsterdam, the Netherlands) that according to the manufacturer's recommendations. ABI 3500xL (Applied Biosystems, USA) was used to detect the reaction products. Data analysis was performed using Coffalyser (MRC Holland, Amsterdam, the Netherlands) software. The reference samples were tested simultaneously with clinical samples.
2.5 Long-read sequencing
Genomic DNA was sequenced using PacBio SMRT target sequencing of the whole DMD (Grandomics, Beijing, China) according to the standard manufacturer's conditions. The Qubit 3.0 (Thermo Fisher Scientific Inc, Carlsbad, CA, USA) and agarose gel electrophoresis was assessed of DNA quantity and quality, respectively. Purified DNA fragments were amplified by polymerase chain reaction and quantified, then subjected to sequencing on the Pacific Biosciences with Sequel II platform according to the manufacturer standard protocols. The data were read and analyzed by bioinformatics after the sequencing data were assessed as qualified by SMRT Link.
2.6 Follow-up of pregnancy outcomes
The pregnant outcomes and the growth and development of the baby were collected through review of medical records and telephone follow-up.
2.7 Ethics approval and consent to participate
All procedures were performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. Written informed consent to participate was obtained from all the participants, including the minors that were signed by their parent. This research received approval from the Ethical Review Committee of Fujian Maternity and Child Health Hospital (Approval Number: 2023KYLLR01016).
3 Results
3.1 SNP-array
The SNP array on amniocytes showed arr[hg19] Xp21.1(31632790_31944075) x1 in the female fetus, suggesting a deletion spanning approximately 311 kb within exons 48–55 of DMD (NM_004006.3). The results of the SNP-array in the parents confirmed that the deletion was derived from the father. No mutations were detected in the mother (Figure 1).
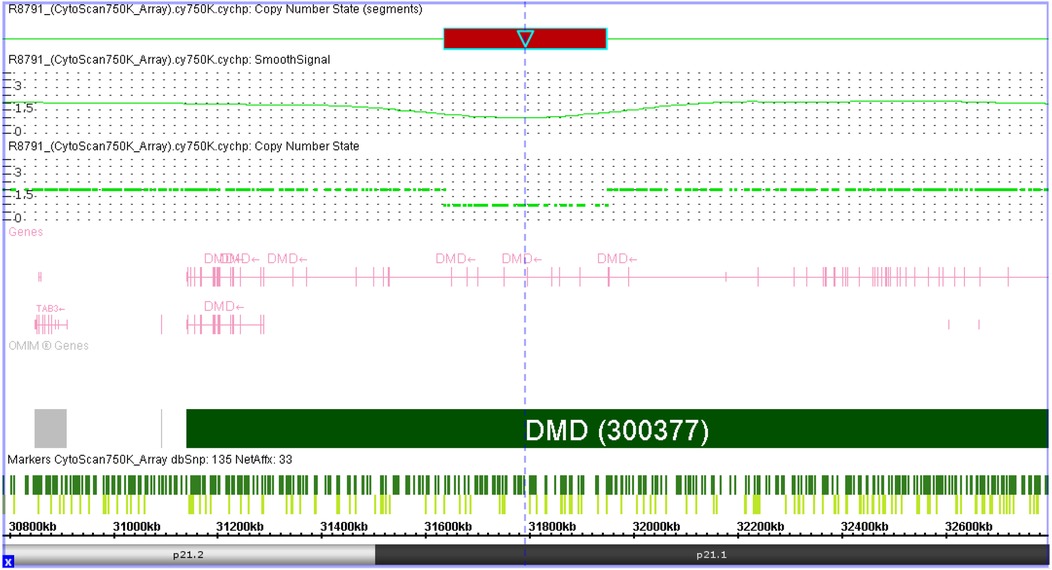
Figure 1. Result of SNP-array detected in female fetus. SNP-array result showed arr[hg19] Xp21.1(31632790_31944075) x1, suggesting a deletion spanning approximately 311 kb within exons 48–55 of DMD.
3.2 MLPA results
In the fetus and her father, the deletion variant of exons 48–55 in DMD was confirmed, and no deletion/duplication variants were detected in the mother (Figure 2).
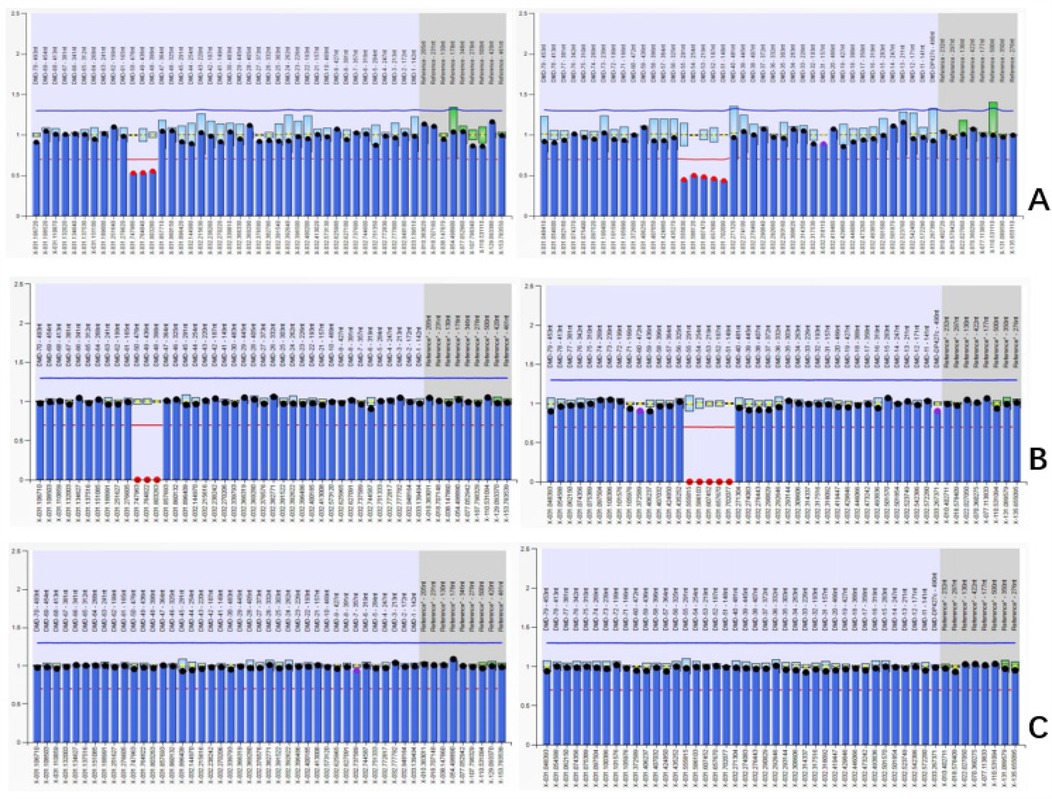
Figure 2. Results of MLPA in the family (A) DMD exon 48–55 heterozygous deletion in female fetus. (B) DMD exon 48–55 hemizygous deletion in father. (C) Normal copy number in mother.
3.3 Breakpoint identification by LRS
A 305 kb deletion spanning chrXp21.1 was identified in the father and fetus, and the breakpoints were precisely located at genomic chrX:31640088–31945085, located within introns 47 and 56 in DMD (Figure 3).

Figure 3. Breakpoint identification by LRS in fetus and her father. A 305 kb deletion spanning chrXp21.1 was identified, and the breakpoints were precisely located at genomic chrX:31640088–31945085, located within introns 47 and 56 in DMD. (A) Diagrammatic of Binary Alignment/Map format. (B) Schematic representation of variation. Red arrows: the breakpoints; Green arrows: the deleted region.
3.4 Follow-up
The female vaginally delivered a full-term female baby, and no abnormalities were observed in the baby at birth. The baby was followed up until 1 year of age, and her growth and development were normal.
4 Discussion
In clinical practice, various methods have been used to diagnose mutations in patients with DMD and BMD. Variations in DMD are mainly caused by deletions or duplications. MLPA is the primary method used to detect copy number variations within the 79 exons of this gene, and approximately 78% of patients can be diagnosed with this test (1). Many laboratories combine MLPA and NGS to screen for DMD abnormalities, allowing more than 90% of variations to be detected (8, 9). However, even after using these techniques, a few patients remain genetically undiagnosed owing to the presence of rare deep intronic and complex structural variants. LRS is increasingly being adopted to solve these difficulties in clinical settings and may provide a more effective method for discriminating the precise locations of breakpoints and structural features of genomic rearrangement through the entire DMD sequence (10–14).
In this study, we identified a deletion using SNP-array in amniocytes that showed arr[hg19] Xp21.1(31632790_31944075) x1 in the female fetus, suggesting a deletion spanning approximately 311 kb within exons 48–55 in DMD (NM_004006.3). The results of the parental SNP-array confirmed that the deletion of DMD was derived from the father; this was subsequently confirmed using MLPA, which identified a DMD exons 48–55 heterozygous deletion in the female fetus and hemizygous deletion in the father. A deletion of DMD exons 48–55 and flanking regions, which were located within intron 47 to intron 55 of DMD spanning 305 kb in chrXp21.1 with a precise position in chrX:31640088–31945085, was identified through LRS.
Due to the X-linked inheritance of dystrophinopathies, the majority of female carriers with pathogenic DMD variants have no clinical manifestations, but 2.5%–25.7% have varying degrees of skeletal muscle and/or myocardial involvement (15, 16). Given the normal values of biochemical indicators and the lack of symptoms related to DMD/BMD in the father, we suspected that the DMD deletions (exons 48–55) in the family were benign variants, and it was recommended that the fetus be preserved. The baby was born without complications and was followed up until 1 year of age, and her growth and development were normal.
Many DMD mutations have been identified that produce non- or partially functional dystrophins, which encompass a broad range of clinical manifestations. Severe cases may manifest as a progressive muscle wasting disease that leads to wheelchair dependence at approximately 10 years of age and premature death with cardiac and/or respiratory failure at approximately 20 years of age without treatment; milder cases can have a later onset and slower progression of independent ambulation or may even be asymptomatic (5–7, 17, 18). Phenotypic differences in DMD and BMD are mainly predicted using the reading frame. Mutations that disrupt the reading frame (out-of-frame) produce no or minimal amounts of dystrophin, which results in a DMD phenotype, whereas mutations that leave the reading frame intact (in-frame) typically result in the production of truncated but partially functional dystrophin that is milder and manifests as BMD. Approximately 90% of cases are in agreement with this rule; however, a fraction is not (19–22). With the development of molecular technologies, more mild and asymptomatic cases have been reported. Some asymptomatic cases with DMD mutations have complex structural variants, comprising a complete copy as well as a recombination fragment of DMD, or are external variations of DMD, thus preserving normal DMD function (10, 23, 24). However, the attenuating factors in many mild and asymptomatic cases of dystrophinopathies have remained unclear. Changes in the intronic architecture at the variant junctions may result in altered splicing signals, that promote aberrant splicing events; despite the relative inefficiency of altered splicing, asymptomatic and mild BMD phenotypes might result from the presence of residual levels of wild type dystrophin transcripts produced by normal splicing, maintaining full or partial dystrophin function (25–27). Some DMD mutations alter splicing and translation sites, resulting in partial exon skipping and bypassing the pathogenic variant; the resulting production of internally shortened but functional proteins may cause a milder phenotype (28–31). Phenotypes might also be influenced by genetic modifiers; for example, LTBP4 has been reported as a modifier of ambulation, and THBS1 plays a role in phenotype mitigating (32). More studies are needed to research other factors that influence clinical phenotypes.
Our results showed the deletion of DMD exons 48–55, which was predicted to be in-frame by the reading frame rule. However, the male had no symptoms related to DMD or BMD, such as myasthenia, amyotrophia, pseudohypertrophy of the gastrocnemius muscle, or cardiac discomfort, the physical examinations of the asymptomatic male showed normal muscle strength, and his biochemical indicators were normal. To the best of our knowledge, this is the first report of a likely asymptomatic male patient with a deletion of DMD exons 48–55. The region between exons 45–55 was a frequent and known “hotspot” for DMD mutations, and the deletion of DMD exons 48–55 has been reported in some studies (33–35). Diegoli et al. (27) reported a male patient diagnosed with DCM with DMD exons 48–55 deletion; his serum creatine phosphokinase level was 456 mU/ml (normal: <200 mU/ml), and an electrocardiogram displayed a left bundle branch block. Baseline echocardiographic results showed that the left ventricular ejection fraction was 20%, and the left ventricular end-diastolic diameter was 78 mm, with a New York Heart Association functional class Functional Class III. The patient eventually died of congestive heart failure. De Palma et al. (28) reported two patients with DMD/BMD due to the deletion of DMD exons 48–55 in a pilot study in a southern Italian cohort, but the related clinical manifestations were not described. In a study by Luce et al. (29), a mother and child were identified as asymptomatic females with DMD exons 48–55 deletion. In our study, the asymptomatic male patient was 35 years old; whether related symptoms would appear with age requires further follow-up.
DMD is one of the largest known genes in humans and encodes a 427 kDa dystrophin protein. Full-length dystrophins are composed of four major domains: an N-terminal actin F-binding domain (encoded by exons 1–8), a large central rod domain (encoded by exons 8–64), a cysteine-rich region (encoded by exons 64–70), and a C-terminal domain (encoded by exons 71–79) (36, 37). Dystrophin is essential as a shock absorber in muscle fiber contraction, physically anchoring cellular skeletal actin fibers and mediating interactions between the cytoskeleton, membrane, and extracellular matrix (38–40). Four dystrophin hinges in the rod domain influence muscle maturation and maintenance. The hinge 3 region is particularly prone to deletion mutations, which are expected to lead to BMD with great heterogeneity in clinical manifestations (18, 41). We report an asymptomatic pedigree of deleted exons 48–55 in DMD, in which the deletion region included hinge 3. The frequency of this variant in the Database of Genomic Variants and gnomADSV general East Asian population databases was zero. It is exceptionally rare for male DMD patients with dystrophin mutations to remain asymptomatic throughout life. Although the male patient in our study was asymptomatic, regular muscle and cardiac evaluations would be recommended to identify the relevant clinical features that may progress with age.
In conclusion, prenatal diagnosis with no family history of DMD/BMD may incidentally detect DMD mutations in fetuses. Our study highlights the importance of detailed clinical evaluations and accurate molecular detection. Precise breakpoint analysis is necessary when clinical manifestations are inconsistent with genotype, and the combined application of multiple techniques can be more comprehensive in evaluating pathogenic differences. When variants are incidental findings in a prenatal diagnosis, caution should be exercised to provide accurate guidance for genetic counseling and prenatal diagnosis.
Data availability statement
The original contributions presented in the study are publicly available. These data can be found here: [ClinVar/accession number SUB15209272 and SUB15243222].
Ethics statement
The studies involving humans were approved by the Ethical Review Committee of Fujian Maternity and Child Health Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants' legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
MZ: Writing – original draft, Writing – review & editing. ZL: Writing – original draft, Writing – review & editing. MCh: Writing – review & editing. DG: Writing – review & editing. QY: Writing – review & editing. QH: Writing – review & editing. BM: Writing – review & editing. BL: Writing – review & editing. LC: Writing – review & editing. MCa: Writing – review & editing. HH: Writing – review & editing. LX: Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by Fujian Maternity and Child Health Hospital Natural Science Foundation (FY YCXY 21-05).
Acknowledgments
The authors thank the family members for their collaboration during this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Fratter C, Dalgleish R, Allen SK, Santos R, Abbs S, Tuffery-Giraud S, et al. EMQN best practice guidelines for genetic testing in dystrophinopathies. Eur J Hum Genet. (2020) 28:1141–59. doi: 10.1038/s41431-020-0643-7
2. Neri M, Rossi R, Trabanelli C, Mauro A, Selvatici R, Falzarano MS, et al. The genetic landscape of dystrophin mutations in Italy: a nationwide study. Front Genet. (2020) 11:131. doi: 10.3389/fgene.2020.00131
3. Duan D, Goemans N, Takeda S, Mercuri E, Aartsma-Rus A. Duchenne muscular dystrophy. Nat Rev Dis Primers. (2021) 7:13. doi: 10.1038/s41572-021-00248-3
4. Birnkrant DJ, Bushby K, Bann CM, Apkon SD, Blackwell A, Brumbaugh D, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. (2018) 17:251–67. doi: 10.1016/S1474-4422(18)30024-3
5. Kekou K, Svingou M, Vogiatzakis N, Nitsa E, Veltra D, Marinakis NM, et al. Retrospective analysis of persistent HyperCKemia with or without muscle weakness in a case series from Greece highlights vast DMD variant heterogeneity. Expert Rev Mol Diagn. (2023) 23:999–1010. doi: 10.1080/14737159.2023.2264181
6. Saengpattrachai M, Ray PN, Hawkins CE, Berzen A, Banwell BL. Grandpa and I have dystrophinopathy? Approach to asymptomatic HyperCKemia. Pediatr Neurol. (2006) 35:145–9. doi: 10.1016/j.pediatrneurol.2006.01.004
7. Wakefield SE, Dimberg EL, Moore SA, Tseng BS. Dystrophinopathy presenting with arrhythmia in an asymptomatic 34-year-old man: a case report. J Med Case Rep. (2009) 3:8625. doi: 10.4076/1752-1947-3-8625
8. Kong X, Zhong X, Liu L, Cui S, Yang Y, Kong L. Genetic analysis of 1051 Chinese families with Duchenne/Becker muscular dystrophy. BMC Med Genet. (2019) 20:139. doi: 10.1186/s12881-019-0873-0
9. Xiao T, Wu B, Cao Y, Liu R, Cheng G, Wang L, et al. Genetic identification of pathogenic variations of the DMD gene: a retrospective study from 10,481 neonatal patients based on next-generation sequencing data. Ann Transl Med. (2021) 9:766. doi: 10.21037/atm-20-7102
10. Bai Y, Liu J, Xu J, Sun Y, Li J, Gao Y, et al. Long-read sequencing revealed extragenic and intragenic duplications of exons 56–61 in DMD in an asymptomatic male and a DMD patient. Front Genet. (2022) 13:878806. doi: 10.3389/fgene.2022.878806
11. Kubota A, Ishiura H, Porto KJL, Tanaka M, Mitsui J, Unuma A, et al. DMD exon 2 duplication due to a complex genomic rearrangement is associated with a somatic mosaicism. Neuromuscul Disord. (2022) 32:263–9. doi: 10.1016/j.nmd.2021.12.004
12. Folland C, Ganesh V, Weisburd B, McLean C, Kornberg AJ, O'Donnell-Luria A, et al. Transcriptome and genome analysis uncovers a DMD structural variant. Neurol Genet. (2023) 9:e200064. doi: 10.1212/NXG.0000000000200064
13. Chen L, Luo X, Wang H, Tian Y, Liu Y. Identifying inversions with breakpoints in the dystrophin gene through long-read sequencing: report of two cases. BMC Med Genomics. (2024) 17:227. doi: 10.1186/s12920-024-01997-2
14. Shen J, Ding T, Sun X, Yang J, Zhang Y, Wang J, et al. Comprehensive analysis of genomic complexity in the 5’ end coding region of the DMD gene in patients of exons 1–2 duplications based on long-read sequencing. BMC Genomics. (2024) 25:292. doi: 10.1186/s12864-024-10224-2
15. Ishizaki M, Kobayashi M, Adachi K, Matsumura T, Kimura E. Female dystrophinopathy: review of current literature. Neuromuscul Disord. (2018) 28:572–81. doi: 10.1016/j.nmd.2018.04.005
16. Liu C, Ma J, Lu Y, Lu Y, Mai J, Bai L, et al. Clinical, pathological, and genetic characterization in a large Chinese cohort with female dystrophinopathy. Neuromuscul Disord. (2023) 33:728–36. doi: 10.1016/j.nmd.2023.08.008
17. Mercuri E, Bönnemann CG, Muntoni F. Muscular dystrophies. Lancet. (2019) 394:2025–38. doi: 10.1016/S0140-6736(19)32910-1
18. Fortunato F, Tonelli L, Farnè M, Selvatici R, Ferlini A. DMD deletions underlining mild dystrophinopathies: literature review highlights phenotype-related mutation clusters and provides insights about genetic mechanisms and prognosis. Front Neurol. (2024) 14:1288721. doi: 10.3389/fneur.2023.1288721
19. Aartsma-Rus A, Van Deutekom JC, Fokkema IF, Van Ommen GJ, Den Dunnen JT. Entries in the Leiden Duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve. (2006) 34:135–44. doi: 10.1002/mus.20586
20. Kesari A, Pirra LN, Bremadesam L, McIntyre O, Gordon E, Dubrovsky AL, et al. Integrated DNA, cDNA, and protein studies in Becker muscular dystrophy show high exception to the reading frame rule. Hum Mutat. (2008) 29:728–37. doi: 10.1002/humu.20722
21. Egorova TV, Galkin II, Velyaev OA, Vassilieva SG, Savchenko IM, Loginov VA, et al. In-frame deletion of dystrophin exons 8–50 results in DMD phenotype. Int J Mol Sci. (2023) 24:9117. doi: 10.3390/ijms24119117
22. Ma YL, Zhang WH, Chen GH, Song LF, Wang Y, Yuan RL, et al. Walking alone milestone combined reading-frame rule improves early prediction of Duchenne muscular dystrophy. Front Pediatr. (2022) 10:985878. doi: 10.3389/fped.2022.985878
23. Zepeda-Mendoza CJ, Bontrager JE, Fisher CF, McDonald A, George-Abraham JK, Hasadsri L. Molecular characterization and reclassification of a 1.18 mbp DMD duplication following positive carrier screening for Duchenne/Becker muscular dystrophy. Clin Case Rep. (2022) 10:e6008. doi: 10.1002/ccr3.6008
24. He W, Meng G, Hu X, Dai J, Liu J, Li X, et al. Reclassification of DMD duplications as benign: recommendations for cautious interpretation of variants identified in prenatal screening. Genes. (2022) 13:1972. doi: 10.3390/genes13111972
25. Torella A, Budillon A, Zanobio M, Del Vecchio Blanco F, Picillo E, Politano L, et al. Alu-mediated insertions in the DMD gene: a difficult puzzle to interpret clinically. Int J Mol Sci. (2023) 24:9241. doi: 10.3390/ijms24119241
26. Gurvich OL, Tuohy TM, Howard MT, Finkel RS, Medne L, Anderson CB, et al. DMD pseudoexon mutations: splicing efficiency, phenotype, and potential therapy. Ann Neurol. (2007) 63:81–9. doi: 10.1002/ana.21290
27. Tuffery-Giraud S, Miro J, Koenig M, Claustres M. Normal and altered pre-mRNA processing in the DMD gene. Hum Genet. (2017) 136:1155–72. doi: 10.1007/s00439-017-1820-9
28. Aartsma-Rus A, van Deutekom JCT, Fokkema IF, den Dunnen JT. G.P.6 07 an overview of mutations reported in the Leiden DMD mutation database that appear not to follow the reading frame rule. Neuromuscul Disord. (2006) 16:700. doi: 10.1016/j.nmd.2006.05.182
29. Waldrop MA, Yaou RB, Lucas KK, Martin AS, O'Rourke E, FILNEMUS, et al. Clinical phenotypes of DMD exon 51 skip equivalent deletions: a systematic review. J Neuromuscul Dis. (2020) 7:217–29. doi: 10.3233/JND-200483
30. Abaji M, Gorokhova S, Da Silva N, Busa T, Grelet M, Missirian C, et al. Novel exon-skipping therapeutic approach for the DMD gene based on asymptomatic deletions of exon 49. Genes. (2022) 13:1277. doi: 10.3390/genes13071277
31. Echigoya Y, Yokota T. Restoring dystrophin expression with exon 44 and 53 skipping in the DMD gene in immortalized myotubes. Methods Mol Biol. (2023) 2587:125–39. doi: 10.1007/978-1-0716-2772-3_7
32. Weiss RB, Vieland VJ, Dunn DM, Kaminoh Y, Flanigan KM. Long-range genomic regulators of THBS1 and LTBP4 modify disease severity in Duchenne muscular dystrophy. Ann Neurol. (2018) 84:234–45. doi: 10.1002/ana.25283
33. Diegoli M, Grasso M, Favalli V, Serio A, Gambarin FI, Klersy C, et al. Diagnostic work-up and risk stratification in X-linked dilated cardiomyopathies caused by dystrophin defects. J Am Coll Cardiol. (2011) 58:925–34. doi: 10.1016/j.jacc.2011.01.072
34. De Palma FDE, Nunziato M, D'Argenio V, Savarese M, Esposito G, Salvatore F. Comprehensive molecular analysis of DMD gene increases the diagnostic value of dystrophinopathies: a pilot study in a southern Italy cohort of patients. Diagnostics. (2021) 11:1910. doi: 10.3390/diagnostics11101910
35. Luce LN, Dalamon V, Ferrer M, Parma D, Szijan I, Giliberto F. MLPA analysis of an Argentine cohort of patients with dystrophinopathy: association of intron breakpoints hot spots with STR abundance in DMD gene. J Neurol Sci. (2016) 365:22–30. doi: 10.1016/j.jns.2016.03.047
36. Davies KE, Nowak KJ. Molecular mechanisms of muscular dystrophies: old and new players. Nat Rev Mol Cell Biol. (2006) 7:762–73. doi: 10.1038/nrm2024
37. Delalande O, Molza AE, Dos Santos Morais R, Chéron A, Pollet É, Raguenes-Nicol C, et al. Dystrophin’s central domain forms a complex filament that becomes disorganized by in-frame deletions. J Biol Chem. (2018) 293:6637–46. doi: 10.1074/jbc.M117.809798
38. Gao QQ, McNally EM. The dystrophin complex: structure, function, and implications for therapy. Compr Physiol. (2015) 5:1223–39. doi: 10.1002/cphy.c140048
39. Dumont NA, Wang YX, von Maltzahn J, Pasut A, Bentzinger CF, Brun CE, et al. Dystrophin expression in muscle stem cells regulates their polarity and asymmetric division. Nat Med. (2015) 21:1455–63. doi: 10.1038/nm.3990
40. Serrano AL, Mann CJ, Vidal B, Ardite E, Perdiguero E, Muñoz-Cánoves P. Cellular and molecular mechanisms regulating fibrosis in skeletal muscle repair and disease. Myogenesis. (2011) 96:167–201. doi: 10.1016/B978-0-12-385940-2.00007-3
Keywords: DMD, asymptomatic, SNP-array, MLPA, LRS
Citation: Zhang M, Lin Z, Chen M, Guo D, Yang Q, He Q, Mao B, Liang B, Chen L, Cai M, Huang H and Xu L (2025) Incidental finding of a DMD exons 48–55 deletion during prenatal diagnosis. Front. Pediatr. 13:1541468. doi: 10.3389/fped.2025.1541468
Received: 7 December 2024; Accepted: 20 March 2025;
Published: 17 April 2025.
Edited by:
Roy Morello, University of Arkansas for Medical Sciences, United StatesReviewed by:
Flavia Balbo Piazzon, AP-HM, FranceAmy Sato, University of Arkansas for Medical Sciences, United States
Copyright: © 2025 Zhang, Lin, Chen, Guo, Yang, He, Mao, Liang, Chen, Cai, Huang and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Meiying Cai, MjIyMzQ1MzRAcXEuY29t; Hailong Huang, aHVhbmdoYWlsb25nQGZqbXUuZWR1LmNu; Liangpu Xu, eGlsaWFuZ3B1QGZqbXUuZWR1LmNu
†These authors share first authorship