 Guo-qin Zhu1,2
Guo-qin Zhu1,2 Ying Hua
Ying Hua Guo-min Li
Guo-min Li- 1Department of Nephrology, Rheumatology and Immunology, Children’s Hospital of Jiangnan University, Wuxi, Jiangsu, China
- 2Department of Nephrology, Rheumatology and Immunology, Wuxi Children's Hospital, Wuxi, Jiangsu, China
- 3Department of Neurology, Children’s Hospital of Jiangnan University, Wuxi, Jiangsu, China
Background Type II DNA topoisomerases (EC5.99.1.3) are enzymes that catalyze topological changes during DNA replication and gene transcription in an ATP-dependent manner. Vertebrates have two isoforms: topoisomerase IIα and β. Type II topoisomerase β is encoded by TOP2B. For TOP2B, a number of germline pathogenic variants have been identified as causative for human diseases, including Hoffman syndrome, ablepharon-macrostomia syndrome with immunodeficiency, B-cell immunodeficiency, distal limb anomalies, and urogenital malformations syndrome. To date, only 14 patients with the above diseases from seven families have been reported in PubMed. Herein, we describe an additional case of a child who presented with “infantile epileptic spasms syndrome” (IESS) as the first symptom, B-cell immunodeficiency, dysmorphic facial features, and a pathogenic variant in TOP2B. The c.1901A > G variant in TOP2B is new to our study, which further enriches the genotype of TOP2B deficiency. Our patient manifested as a typical triad: infantile spasms, hypsarrhythmia on electroencephalogram, and developmental arrest at the age of 7 months. Although epilepsy and neurodevelopmental disorders have been reported in patients with TOP2B deficiency, typical IESS has not been described previously. IESS in our patient further expands the phenotype of TOP2B. The patient was started on monthly intravenous immunoglobulin replacement therapy after being diagnosed with TOP2B deficiency and since then has not suffered from severe infections. TOP2B deficiency is a group of heterogeneous diseases, which is ultrarare. The results from our study extend the phenotype and genotype spectrum of TOP2B deficiency. TOP2B may be a causative gene for IESS.
Introduction
DNA topoisomerases are essential enzymes required for the relaxation of topological stress during DNA replication and gene transcription (1, 2). Mammalian proteins have two isoforms: type II topoisomerase alpha and type II topoisomerase beta. Type II topoisomerase β, encoded by TOP2B, was first reported in 1987 by Fred Drake's group at Smith Kline French (SKF) (3). In mammalian cells, TOP2B is expressed in proliferative and terminally differentiated postmitotic cells (4, 5). A microarray analysis of TOP2B expression in primary human cells (https://BioGPS.org) revealed broad expression and high expression in CD34+ bone marrow cells (6). TOP2B plays a critical role in B-cell development and dominant pathogenic variants in TOP2B have been shown to lead to B-cell deficiency (7, 8). However, a transcriptome analysis of human tissues via the GTEx portal (https://www.gtexportal.org) has also revealed a high expression of TOP2B, with the highest expression in the cerebellum (9). TOP2B essentially affects neuronal differentiation, survival, DNA repair, and neurite outgrowth and is involved in brain development and neural differentiation. Recently, several de novo missense TOP2B variants have been identified in patients with neurodevelopmental disorders (NDDs) (10, 11).
West syndrome (WS) is characterized by infantile spasms, hypsarrhythmia on electroencephalogram (EEG), and developmental arrest or regression (12, 13). WS has recently been reclassified into infantile epileptic spasms syndrome (IESS), a devastating developmental epileptic encephalopathy (DEE) consisting of epileptic spasms, as well as one or both of developmental regression or stagnation and hypsarrhythmia on EEG (14–16). To date, over 28 copy number variants (CNVs) and 70 single-gene pathogenic variants related to IECSS have been discovered (17). However, TOP2B was not included in the list of genes associated with IESS (12, 17). Here, we report a Chinese girl who presented with IESS and B-cell deficiency caused by a de novo TOP2B variant.
Case presentation
The patient was a 2-year-old Chinese girl who was the first child of a non-consanguineous couple. She was born by cesarean section due to prolonged labor and had no asphyxia, with a birth weight of 3.25 kg. At 7 months of age, she could not roll over or reach out to objects. She was admitted to the Department of Neurology at our hospital because of axial spasms in clusters at the age of 9 months. A physical examination revealed that she was hypotonic but had normal muscle power and deep tendon reflexes. Her head circumference, length, and weight were 39 cm (less than the 3rd percentile, 41.7 cm), 68 cm (3rd–10th percentile), and 7.3 kg (75th–90th percentile), respectively. An EEG was performed and it revealed hypsarrhythmia (Figure 1a). MRI scans of the brain revealed marked cerebral atrophy with subsequent enlargement of the subarachnoid spaces (Figures 1b–d). She was diagnosed with IESS based on infantile spasms, hypsarrhythmia, and developmental arrest. Despite the use of different combinations of anticonvulsants (vigabatrin, lamotrigine, and vitamin B6), her epilepsy could not be completely controlled.
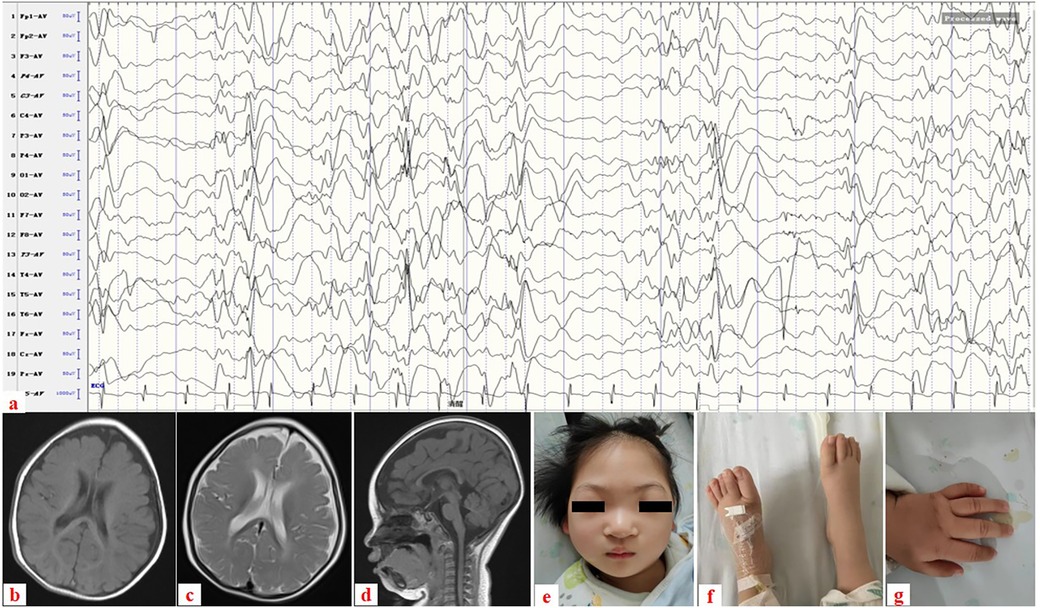
Figure 1. (a) Electroencephalogram showing hypsarrhythmia. (b–d) Brain MRI taken at 7 months’ old: (b) axial image in T1-weighted image, (c) axial image in T2-weighted image, and (d) sagittal image in T1-weighted image reveal cerebral atrophy with subsequent enlargement of the subarachnoid spaces. (e–g) Clinical photograph taken at 1 year and 6 months’ old: (e) Dysmorphic facial features with microcephaly and ocular hypertelorism, (f) normal feet, and (g) normal left hand.
She was referred to our department for evaluation because of recurrent infections and hypogammaglobulinemia at the age of 1 year. A clinical examination revealed dysmorphic facial features with microcephaly and ocular hypertelorism (Figure 1e), global developmental delay with severe muscular hypotonia, significantly decreased head control, and only sparse spontaneous movements. The distal limb was normal (Figures 1f,g). Ocular fixation was not evident. She had poor eye contact and no eye fixation to moving objects. Laboratory findings revealed severe hypogammaglobulinemia: IgG, 1.8 g/L (normal: 5.4–15.1 g/L); IgM, 0.12 g/L (normal: 0.48–2.31 g/L); and IgA, 0.0 g/L (normal: 0.52–2.74 g/L). Total white blood cell count was normal, with normal absolute neutrophil and lymphocyte counts. However, a lymphocyte subset analysis using flow cytometry revealed undetectable CD19+ B lymphocytes (Table 1). Two variants (Table 2) were detected using tri-based whole-exome sequencing (WES): a heterozygous de novo c.1901A > G (p.Y634C) variant in TOP2B (NM_001330700) and a homozygous c.3163G > A (p.V1055I) variant in VARS1 (NM_006295). The c.1901A > G variant in TOP2B in the patient was confirmed using Sanger sequencing and was not detected in her parents (Figure 2a). The c.1901A > G (p.Y634C) variant in TOP2B was evaluated as a pathogenic variant through the American College of Medical Genetics and Genomics/Association for Medical Pathology (ACMG/AMP) variant classification guidelines, whereas the c.3163G > A (p.V1055I) variant is of uncertain significance (VUS). The c.1901A > G variant was not found in ExAC (https://gnomad.broadinstitute.org/) or 1000G (http://browser.1000genomes.org). The c.1901A > G variant is predicted to be “probably damaging” by Polyphen2 with a score of 1.0 (sensitivity: 0.0; specificity: 1.0; Figure 2b), and MutationTaster2021 software predicted this variant to be disease causing. In addition, alignment of the mutated p.Y634C TOP2B protein with different species revealed the complete conservation of the amino acid (Figure 2c). However, both the heterozygous and the homozygous c.3163G > A variants have been found in both ExAC (https://gnomad.broadinstitute.org/) and 1000G (http://browser.1000genomes.org). The c.3163G > A variant was predicted to be “possibly damaging” by Polyphen2 with a score of 0.69 (sensitivity: 0.86; specificity: 0.92), and MutationTaster2021 software predicted this variant to be a polymorphism. The patient was started on monthly intravenous immunoglobulin (IVIG) replacement therapy after being diagnosed with TOP2B deficiency and since then has not suffered from severe infections.
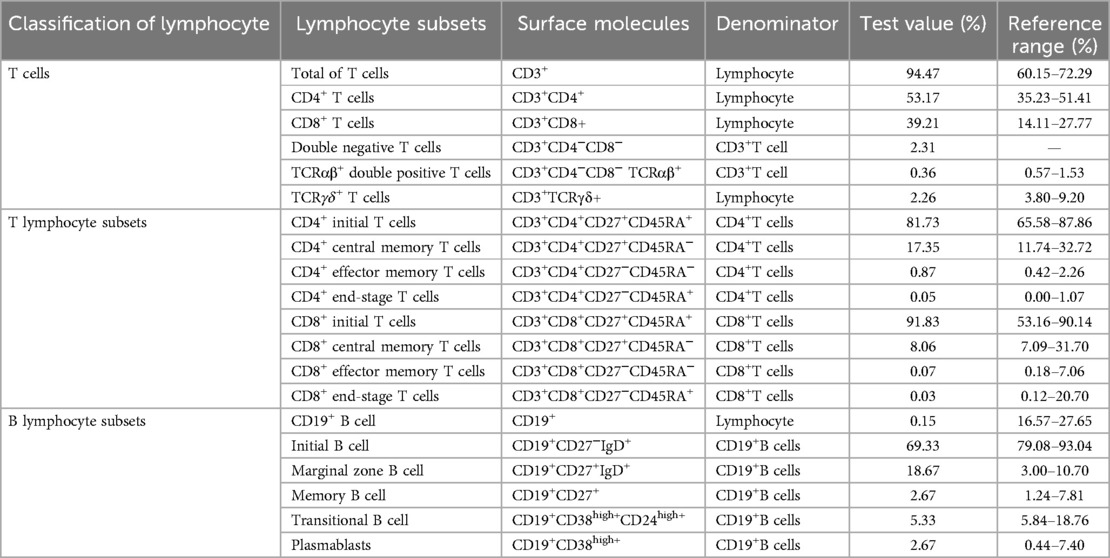
Table 1. Lymphocyte subsets in the patient.
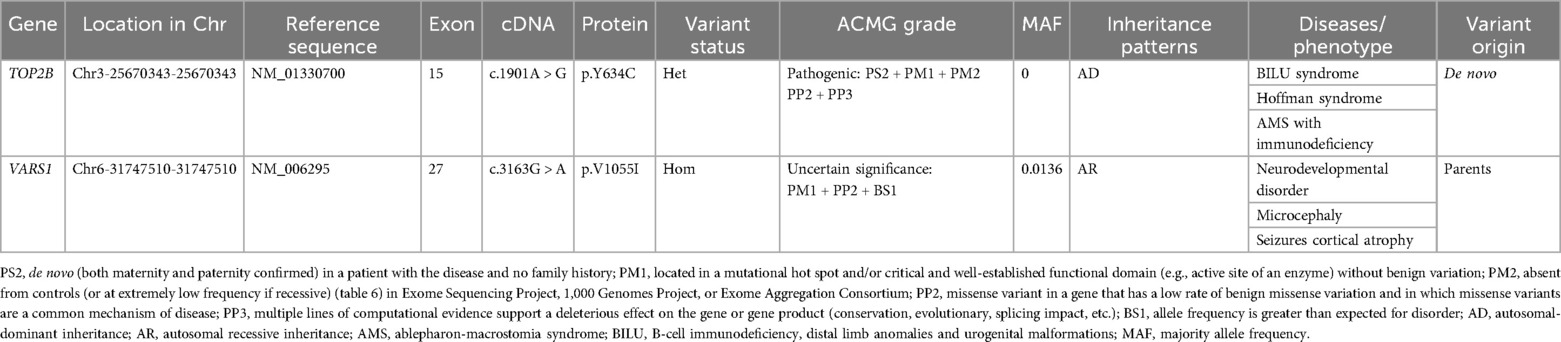
Table 2. Variants in the patients.

Figure 2. (a) familial pedigrees and sanger sequencing of TOP2B. (b) The p.Y634C variant is predicted to be “probably damaging” by Polyphen2. (c) Alignment of the mutated p.Y634C TOP2B protein with different species shows the complete conservation of the amino acid. (d) The TOP2B protein consists of 1,621 amino acids that form an ATPase domain, TOPRIM domain and DNA-binding region, and a C-terminal region, and all variants reported from other studies are located in different structural domains.
This study was approved by the Ethics Committee at the Children's Hospital of Jiangnan University, and written informed consent was obtained from the parents of the patient.
Discussion and conclusions
TOP2B encodes a DNA topoisomerase, which is involved in processes such as chromosome condensation, chromatid separation, and the relief of torsional stress that occurs during DNA transcription and replication (5). For TOP2B, a number of germline pathogenic variants have been identified as causative factors for human diseases. The diseases associated with TOP2B (DAT) include four clinical entities: (1) Hoffman syndrome; (2) B-cell immunodeficiency, distal limb anomalies, and urogenital malformations (BILU) syndrome (MIM #609296); (3) ablepharon-macrostomia syndrome (AMS; MIM #200110) with immunodeficiency; and (4) autosomal-dominant hereditary hearing loss (18–25). To date, only 14 patients with Hoffman syndrome, BILU, and AMS from seven families have been reported in PubMed (10, 11, 18–24). Therefore, this condition is very rare, with a prevalence of <1/1,000,000 (26). Herein, we describe an additional case of a child who presented with WS as the first symptom, B-cell immunodeficiency, and dysmorphic facial features. A c.1901A > G variant in TOP2B was detected by trio-WES in the patient. The c.1901A > G variant was evaluated to be pathogenic through ACMG/AMP variant classification guidelines. The c.1901A > G variant is predicted to be “probably damaging” by Polyphen2, and MutationTaster2021 software was used to predict this variant to be disease causing. In addition, alignment of the mutated p.Y634C TOP2B protein with different species revealed the complete conservation of the amino acid. Although functional studies of the variant were not performed, the above bioinformatics analysis suggested that the variant is pathogenic. A total of 23 variants were reported in TOP2B including 15 missense, 2 synonymous, 1 non-sense, 1 splicing, 3 small deletions, and 1 small insertion mutation in the human gene mutation database (HGMD) professional 2023.4 (Figure 2d) (27). The c.1901A > G variant is new in our study, further enriching the genotype of TOP2B.
A homozygous c.3163G > A variant in VARS1 was found by trio-WES in the patient and evaluated to be VUS through ACMG/AMP variant classification guidelines. The c.3163G > A variant is predicted to be “possibly damaging” by Polyphen2 and MutationTaster2021 software was used to predict this variant to be polymorphic. Thus, the homozygous c.3163G > A variant in VARS1 may not lead to neurological symptoms or signs in the patient.
Eukaryotic TOP2B proteins have evolutionarily conserved domain structures: they consist of 1621 amino acids that form an ATPase domain, a central catalytic core domain, and a C-terminal region (28, 29). The central catalytic core domain of eukaryotic TOP2B proteins, also known as the breakage-reunion domain, has a TOPRIM domain and a DNA-binding region and has catalytic properties (30). A total of 24 variants (including those in our study) are located in the N-terminal ATPase domain, the central catalytic core domain, and the C-terminal region (Figure 2d). Variants in the N-terminal ATPase domain and the C-terminal region are associated with developmental disorders, whereas variants in the central catalytic core domain are related to B-cell deficiency. Variants reported in patients with hearing loss are in both the central catalytic core domain and the C-terminal region (25). The variant in this study was in the central catalytic core domain, and our patient had both severe developmental disorders and B-cell deficiency. Our patient manifested as a typical triad: infantile spasms, hypsarrhythmia on EEG, and developmental arrest at the age of 7 months, which meets the diagnostic criteria for IESS. Developmental arrest included motor and speech developmental disorders in the patient. In addition to infantile spasms and developmental arrest, neurological involvement includes growth delay (short stature), intellectual disability, hypotonia, and autistic features. Among patients in reported studies, those with DAT have the youngest onset age. While neurodevelopmental disorders are frequently reported in patients with DAT, epilepsy is less commonly observed in this population (10, 11, 24). Tonic-clonic seizures were reported in a 6-year-3-month-old child with DAT (11); the clinical presentation did not fulfill the diagnostic criteria for IESS. IESS in this patient further expands the phenotype of TOP2B deficiency. Various combinations of anticonvulsants, including vigabatrin, lamotrigine, and vitamin B6, were administered to the patient, but her seizures remained refractory. Adrenocorticotropic hormone (ACTH) and oral corticosteroids were not used as first-line treatments in this case for two reasons: first, her comorbid immunodeficiency increased the risk of severe infections from immunosuppressive therapy; second, her epilepsy was genetically determined and often showed poor response to anticonvulsant medications.
DAT is a group of heterogeneous diseases, including Hoffman syndrome, BILU, AMS with immunodeficiency, and autosomal-dominant hereditary hearing loss. This heterogeneity may be due to variants located in different structural domains. An increasing number of studies indicate that TOP2B deficiency is associated with NDD.
TOP2B may be a causative gene for IESS.
Limitations
Although the TOP2B variant was predicted to be pathogenic through bioinformatics analysis, functional studies were not performed to validate this finding; due to the presence of central nervous system phenotypes (e.g., epilepsy and developmental delay), mitochondrial DNA testing was not conducted, leaving the potential contribution of mitochondrial gene variants unexamined. Further investigations are planned to address these limitations, including functional validation of the TOP2B variant and mitochondrial genome sequencing, to enhance the reliability of the current findings.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving humans were approved by Ethics board approval and consent was obtained for this work from the Ethics Committee at the Children's Hospital of Jiangnan University, Wuxi, Jiangsu, China. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants' legal guardians/next of kin. Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
G-qZ: Conceptualization, Data curation, Formal Analysis, Investigation, Validation, Writing – review & editing. YY: Data curation, Resources, Writing – review & editing. L-yY: Investigation, Methodology, Writing – review & editing. YH: Conceptualization, Data curation, Investigation, Methodology, Resources, Writing – review & editing. G-mL: Conceptualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
AMS, ablepharon-macrostomia syndrome; BILU, B-cell immunodeficiency, distal limb anomalies and urogenital malformations; CNVs, copy number variants; DEE, developmental epileptic encephalopathy; EEG, electroencephalogram; HGMD, human gene mutation database; IESS, infantile epileptic spasms syndrome; IVIG, intravenous immunoglobulin; NDDs, neurodevelopmental disorders; WES, whole-exome sequencing; VUS, variant of uncertain significance; WS, West syndrome.
References
1. Hanke A, Ziraldo R, Levene SD. DNA-topology simplification by topoisomerases. Molecules. (2021) 26:3375. doi: 10.3390/molecules26113375
2. Champoux JJ. DNA topoisomerases: structure, function, and mechanism. Annu Rev Biochem. (2001) 70:369–413. doi: 10.1146/annurev.biochem.70.1.369
3. Pommier Y, Nussenzweig A, Takeda S, Austin C. Human topoisomerases and their roles in genome stability and organization. Nat Rev Mol Cell Biol. (2022) 23:407–27. doi: 10.1038/s41580-022-00452-3
4. Segev A, Heady L, Crewe M, Madabhushi R. Mapping catalytically engaged TOP2B in neurons reveals the principles of topoisomerase action within the genome. Cell Rep. (2024) 43:113809. doi: 10.1016/j.celrep.2024.113809
5. Austin CA, Cowell IG, Khazeem MM, Lok D, Ng HT. TOP2B’s contributions to transcription. Biochem Soc Trans. (2021) 49:2483–93. doi: 10.1042/BST20200454
6. Austin CA, Lee KC, Swan RL, Khazeem MM, Manville CM, Cridland P, et al. TOP2B: the first thirty years. Int J Mol Sci. (2018) 19:2765. doi: 10.3390/ijms19092765
7. Papapietro O, Nejentsev S. Topoisomerase 2β and DNA topology during B cell development. Front Immunol. (2022) 13:982870. doi: 10.3389/fimmu.2022.982870
8. Papapietro O, Chandra A, Eletto D, Inglott S, Plagnol V, Curtis J, et al. Topoisomerase 2β mutation impairs early B-cell development. Blood. (2020) 135:1497–501. doi: 10.1182/blood.2019003299
9. Delint-Ramirez I, Konada L, Heady L, Rueda R, Jacome ASV, Marlin E, et al. Calcineurin dephosphorylates topoisomerase IIβ and regulates the formation of neuronal-activity-induced DNA breaks. Mol Cell. (2022) 82:3794–809.e8. doi: 10.1016/j.molcel.2022.09.012
10. Lam CW, Yeung WL, Law CY. Global developmental delay and intellectual disability associated with a de novo TOP2B mutation. Clin Chim Acta. (2017) 469:63–8. doi: 10.1016/j.cca.2017.03.022
11. Hiraide T, Watanabe S, Matsubayashi T, Yanagi K, Nakashima M, Ogata T, et al. A de novo TOP2B variant associated with global developmental delay and autism spectrum disorder. Mol Genet Genomic Med. (2020) 8:e1145. doi: 10.1002/mgg3.1145
12. Pavone P, Polizzi A, Marino SD, Corsello G, Falsaperla R, Marino S, et al. West syndrome: a comprehensive review. Neurol Sci. (2020) 41:3547–62. doi: 10.1007/s10072-020-04600-5
13. Mytinger JR. Definitions and diagnostic criteria for infantile spasms and West syndrome—historical perspectives and practical considerations. Semin Pediatr Neurol. (2021) 38:100893. doi: 10.1016/j.spen.2021.100893
14. Ng AC, Choudhary A, Barrett KT, Gavrilovici C, Scantlebury MH. Mechanisms of infantile epileptic spasms syndrome: what have we learned from animal models? Epilepsia. (2024) 65:266–80. doi: 10.1111/epi.17841
15. Gettings JV, Shafi S, Boyd J, Snead OC, Rutka J, Drake J, et al. The epilepsy surgery experience in children with infantile epileptic spasms syndrome at a tertiary care center in Canada. J Child Neurol. (2023) 38:113–20. doi: 10.1177/08830738231151993
16. Hadjinicolaou A, Briscoe Abath C, Singh A, Donatelli S, Salussolia CL, Cohen AL, et al. Timing the clinical onset of epileptic spasms in infantile epileptic spasms syndrome: a tertiary health center’s experience. Epilepsia. (2024) 65:984–94. doi: 10.1111/epi.17900
17. Snyder HE, Jain P, RamachandranNair R, Jones KC, Whitney R. Genetic advancements in infantile epileptic spasms syndrome and opportunities for precision medicine. Genes (Basel). (2024) 15:266. doi: 10.3390/genes15030266
18. Edery P, Le Deist F, Briard ML, Debré M, Munnich A, Griscelli C, et al. B cell immunodeficiency, distal limb abnormalities, and urogenital malformations in a three generation family: a novel autosomal dominant syndrome? J Med Genet. (2001) 38:488–93. doi: 10.1136/jmg.38.7.488
19. Hoffman HM, Bastian JF, Bird LM. Humoral immunodeficiency with facial dysmorphology and limb anomalies: a new syndrome. Clin Dysmorphol. (2001) 10:1–8. doi: 10.1097/00019605-200101000-00001
20. Tischkowitz M, Goodman F, Koliou M, Webster D, Edery P, Jones A, et al. Autosomal dominant B-cell immunodeficiency, distal limb anomalies and urogenital malformations (BILU syndrome)—report of a second family. Clin Genet. (2004) 66:550–5. doi: 10.1111/j.1399-0004.2004.00349.x
21. Hügle B, Hoffman H, Bird LM, Gebauer C, Suchowerskyj P, Sack U, et al. Hoffman syndrome: new patients, new insights. Am J Med Genet A. (2011) 155A:149–53. doi: 10.1002/ajmg.a.33678
22. Broderick L, Yost S, Li D, McGeough MD, Booshehri LM, Guaderrama M, et al. Mutations in topoisomerase IIβ result in a B cell immunodeficiency. Nat Commun. (2019) 10:3644. doi: 10.1038/s41467-019-11570-6
23. Erdős M, Lányi Á, Balázs G, Casanova JL, Boisson B, Maródi L. Inherited TOP2B mutation: possible confirmation of mutational hotspots in the TOPRIM domain. J Clin Immunol. (2021) 41:817–9. doi: 10.1007/s10875-020-00963-8
24. Çepni E, Börklü E, Avcı Ş, Kalaycı T, Eraslan S, Kayserili H. Revisiting TOP2B-related phenotypes: three new cases and literature review. Clin Genet. (2023) 104:251–8. doi: 10.1111/cge.14341
25. Xia W, Hu J, Ma J, Huang J, Jing T, Deng L, et al. Mutations in TOP2B cause autosomal-dominant hereditary hearing loss via inhibition of the PI3K-Akt signalling pathway. FEBS Lett. (2019) 593:2008–18. doi: 10.1002/1873-3468.13482
26. Knowledge on rare diseases and orphan drugs. Available at: https://www.orpha.net/en/disease/detail/567502 (Accessed December 09, 2024).
27. The Human Gene Mutation Database at the Institute of Medical Genetics in Cardiff. Available at: https://www.hgmd.cf.ac.uk/ac/index.php (Accessed December 09, 2024).
28. Ling EM, Baslé A, Cowell IG, van den Berg B, Blower TR, Austin CA. A comprehensive structural analysis of the ATPase domain of human DNA topoisomerase II beta bound to AMPPNP, ADP, and the bisdioxopiperazine, ICRF193. Structure. (2022) 30:1129–45.e3. doi: 10.1016/j.str.2022.05.009
29. West KL, Turnbull RM, Willmore E, Lakey JH, Austin CA. Characterisation of the DNA-dependent ATPase activity of human DNA topoisomerase IIbeta: mutation of Ser165 in the ATPase domain reduces the ATPase activity and abolishes the in vivo complementation ability. Nucleic Acids Res. (2002) 30:5416–24. doi: 10.1093/nar/gkf677
Keywords: case report, type II topoisomerase β, West syndrome, B-cell immunodeficiency, neurodevelopmental disorders
Citation: Zhu G-q, Yao Y, Yang L-y, Hua Y and Li G-m (2025) Infantile epileptic spasms syndrome as a new phenotype in TOP2B deficiency caused by a de novo variant: a case report and literature review. Front. Pediatr. 13:1542268. doi: 10.3389/fped.2025.1542268
Received: 9 December 2024; Accepted: 4 June 2025;
Published: 27 June 2025.
Edited by:
Adriano La Vecchia, University of Milano-Bicocca, ItalyReviewed by:
Amitha Ananth, University of Alabama at Birmingham, United StatesSandeep Negi, Post Graduate Institute of Medical Education and Research (PGIMER), India
Copyright: © 2025 Zhu, Yao, Yang, Hua and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ying Hua, dG9tYXRvMzMxMzE2QDE2My5jb20=; Guo-min Li, bGlndW9taW40ODZAc2luYS5jb20=