 Francesco La Torre1*
Francesco La Torre1* Giovanni Meliota2
Giovanni Meliota2 Adele Civino3
Adele Civino3 Angelo Campanozzi4
Angelo Campanozzi4 Valerio Cecinati5
Valerio Cecinati5 Enrico Rosati6Emanuela Sacco7Nicola Santoro8Ugo Vairo2Fabio Cardinale1
Enrico Rosati6Emanuela Sacco7Nicola Santoro8Ugo Vairo2Fabio Cardinale1
- 1Department of Pediatrics, Giovanni XXIII Pediatric Hospital, University of Bari, Bari, Italy
- 2Pediatric Cardiology, Giovanni XXIII Pediatric Hospital, Bari, Italy
- 3Pediatric Rheumatology and Immunology Unit, “Vito Fazzi” Hospital, Lecce, Italy
- 4Pediatric Unit, Department of Medical and Surgical Sciences, University of Foggia, Foggia, Italy
- 5Pediatric and Pediatric Oncohematology Unit, Santissima Annunziata Hospital, Taranto, Italy
- 6Neonatology and Intensive Care Unit, “Vito Fazzi” Hospital, Lecce, Italy
- 7Pediatric Unit, Fondazione IRCCS Casa Sollievo della Sofferenza, Foggia, Italy
- 8Pediatric and Oncohematology Unit, Azienda Ospedaliero Universitaria “Policlinico Consorziale”, Bari, Italy
Pediatric hyperinflammatory diseases, including Still's disease, Kawasaki disease (KD), multisystem inflammatory syndrome in children (MIS-C), and recurrent pericarditis (RP), represent a spectrum of conditions characterized by immune dysregulation and systemic inflammation. Each disorder exhibits distinct pathophysiological mechanisms and clinical features, yet their overlapping presentations often pose diagnostic challenges. Early and accurate differentiation is critical to mitigate complications such as macrophage activation syndrome (MAS), coronary artery aneurysms, and myocardial dysfunction. This narrative review explores the pathophysiology, diagnostic criteria, and management of these conditions, emphasizing the utility of advanced biomarkers, imaging modalities, and genetic testing. For Still's disease, the review highlights the transformative role of biologic therapies targeting IL-1 and IL-6 in reducing systemic inflammation and improving outcomes. In KD, timely administration of intravenous immunoglobulin (IVIG) and combination with high-dose steroids in high-risk patients is pivotal for preventing coronary complications. MIS-C, associated with SARS-CoV-2 infection, requires tailored immunomodulatory approaches, including corticosteroids and biologics, to address severe hyperinflammation and multiorgan involvement. RP management prioritizes NSAIDs, colchicine, and IL-1 inhibitors to reduce recurrence and corticosteroid dependence. The review advocates for a multidisciplinary approach, integrating standardized diagnostic algorithms and disease-specific expertise to optimize patient care. Future research directions include the identification of predictive biomarkers, exploration of novel therapeutic targets, and development of evidence-based treatment protocols to enhance long-term outcomes in pediatric inflammatory diseases.
1 Introduction
Pediatric autoinflammatory and hyperinflammatory conditions are characterized by dysregulated immune responses resulting in excessive systemic inflammation (1). These diseases encompass a spectrum of disorders, including Still's disease, Kawasaki disease (KD), multisystem inflammatory syndrome in children (MIS-C), and recurrent pericarditis (RP). While autoinflammation primarily involves innate immune dysregulation, hyperinflammation often arises from exaggerated activation of both innate and adaptive immunity, frequently triggered by infections, malignancies, or genetic predispositions (1). Despite their shared features, such as fever, elevated acute-phase reactants, and multiorgan involvement, each condition presents unique pathophysiological mechanisms, clinical manifestations, and therapeutic challenges.
Still's disease is a polygenic autoinflammatory disease characterized by systemic inflammation, presenting as intermittent fever, rash, and arthritis (2). Cytokine dysregulation, particularly involving IL-1 and IL-6, plays a central role in its pathogenesis, and complications such as macrophage activation syndrome (MAS) and interstitial lung disease (ILD) can be observed (2–6). Diagnosis relies on clinical evaluation and the exclusion of conditions such as leukemia, lymphoma, and other autoimmune or inflammatory disorders with overlapping features (7–10). Recent advances, including the introduction of IL-1 and IL-6 inhibitors, have improved outcomes, with IL-1 antagonists now considered first-line therapy (2, 8, 11–13).
KD is a systemic vasculitis predominantly affecting children under 5 years of age (14, 15). It presents with persistent fever, conjunctival hyperemia, mucocutaneous inflammation, and cervical lymphadenopathy (16, 17). Coronary artery involvement is the hallmark complication, potentially leading to aneurysm formation if untreated (14, 17–19). The disease is driven by vascular endothelial injury mediated by immune complex deposition and cytokine dysregulation (20–22).
MIS-C is a severe hyperinflammatory syndrome temporally related to SARS-CoV-2 infection (23). It is driven by a cytokine storm associated with excessive IL-1β, IL-6 and TNF-α activity, leading to systemic vasculitis and myocardial dysfunction (23). It is characterized by fever and multiorgan dysfunction, commonly involving cardiac, gastrointestinal, renal, and neurological systems, and laboratory evidence of inflammation (24, 25). Although MIS-C shares features with KD, such as fever and mucocutaneous involvement, it differs in the frequency of myocarditis and gastrointestinal symptoms (26, 27).
RP involves IL-1 dysregulation, leading to persistent repeated episodes of pericardial inflammation (28, 29). It is often idiopathic or secondary to autoinflammatory syndromes such as familial Mediterranean fever or Still's disease (30–32). Key clinical features include chest pain, pericardial effusion, and electrocardiographic changes (28–30).
The nonspecific and overlapping early presentations of these conditions often delay diagnosis, increasing the risk of severe complications. Distinguishing these conditions from infectious, oncological, or autoimmune diseases requires advanced biomarkers, imaging techniques, and genetic testing (33–36). In KD, incomplete presentations can mimic viral infections, requiring echocardiographic evaluation for coronary abnormalities (37). MIS-C shares clinical features with KD and sepsis, necessitating the use of biomarkers like N-Terminal Pro–B-Type Natriuretic Peptide (NT-proBNP) and cardiac troponins to confirm cardiac involvement (38). Differentiating Still's disease from malignancies or infections may involve bone marrow aspiration and infectious disease panels, while distinguishing RP from other inflammatory diseases often requires cardiac magnetic resonance (MRI)and computed tomography (CT) (32, 39).
Given the complexity of these diseases, a multidisciplinary approach is essential to optimize patient outcomes and address disease-specific challenges. This includes standardized diagnostic algorithms combining clinical, laboratory, and imaging data, as well as targeted use of biologics to reduce systemic inflammation and prevent complications. Training pediatric subspecialists, such as rheumatologists and cardiologists, is critical to ensuring disease-specific expertise. Efforts such as those in the Apulia region in southern Italy aim to develop integrated diagnostic and therapeutic care pathways tailored to regional healthcare systems.
This review aims to provide a comprehensive exploration of pediatric Still's disease, KD, MIS-C, and RP, with a focus on their distinct pathophysiological mechanisms, clinical presentations, and evidence-based management strategies. The authors seek to propose diagnostic and therapeutic algorithms for treatment escalation in these conditions, highlighting indications for the early initiation of biological therapies.
2 Methods
The authors formed a multidisciplinary working group within the Apulia region, comprising pediatric rheumatologists, immunologists, cardiologists, oncologists, and other specialists actively engaged in clinical care. The primary aim was to develop standardized diagnostic and therapeutic recommendations for managing hyperinflammatory conditions based on the best available evidence.
The authors conducted an online search for articles on pediatric Still's disease, KD, MIS-C, and RP. Relevant articles were retrieved from PubMed using various combinations of the search terms: “pediatric Still's disease” or “systemic juvenile idiopathic arthritis” OR “Kawasaki disease” OR “multisystem inflammatory syndrome in children” OR “pediatric recurrent pericarditis” AND “diagnosis” OR “therapy.” Articles were initially screened by title and abstract for relevance, followed by a full-text review. To broaden the scope of this narrative review, they included studies of all designs. Additionally, the authors screened the reference lists of selected articles to identify further relevant literature. Previously published review articles underwent full-text screening to identify gaps in the existing research and provide a more comprehensive discussion of this topic.
3 Still's disease
Systemic juvenile idiopathic arthritis (sJIA) is a subtype of juvenile idiopathic arthritis (JIA), accounting for 10%–15% of all JIA cases (2, 40–44). Recently, the EULAR/PRES recommendation defined systhemic juvenile idiopathic arthritis and adult-onset Still's disease (AOSD) as the same disease that should be designated by the same unique name, Still's disease (formerly called sJIA/AOSD) (8). Pediatric Still's disease affects males and females equally, with a peak incidence between 1 and 5 years of age (44, 45).
The hallmark features of Still's disease include persistent fevers lasting at least two weeks, an evanescent salmon-pink rash, arthralgia or arthritis, lymphadenopathy, hepatosplenomegaly, and serositis (Table 1) (2, 8, 46, 47). The course of the disease is variable, with good outcomes in patients with a monophasic course, other patients developing chronic, unremitting arthritis, and others experiencing a polyphasic course (2, 50–52). Despite extensive research, no biomarkers reliably predict which patients will experience a chronic, refractory disease course (45, 53–57).
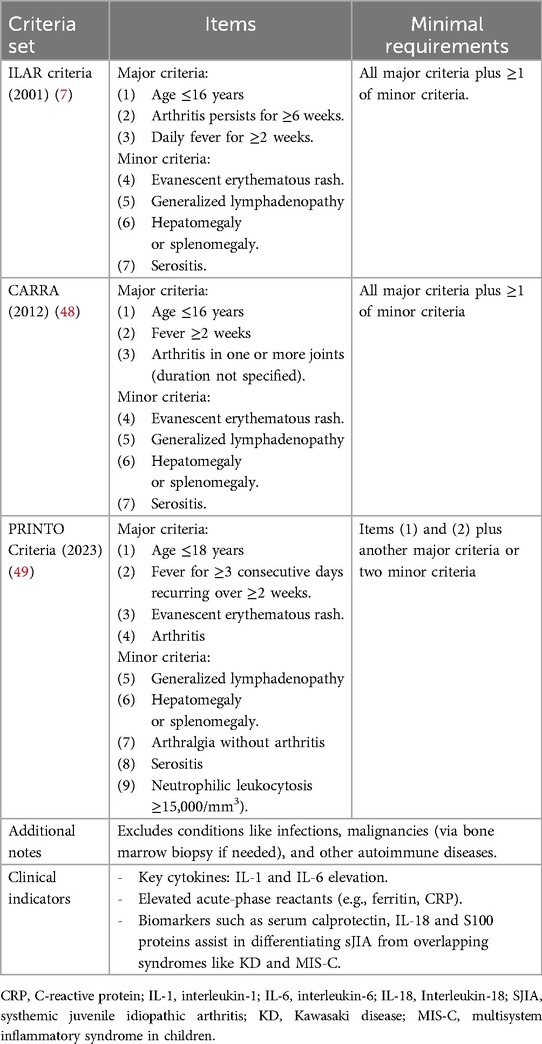
Table 1. Diagnostic criteria for SJIA based on ILAR, CARRA and PRINTO classifications.
Diagnosis relies on clinical criteria and the exclusion of other diseases (8). The International League of Associations for Rheumatology (ILAR) defines sJIA as arthritis lasting at least 6 weeks, with quotidian fever for 2 weeks and at least one of the following: evanescent erythematous rash, lymphadenopathy, hepatosplenomegaly, or serositis (Table 1) (7). The CARRA criteria differ from the ILAR criteria only in that the duration of arthritis is not specified (Table 1) (48). Newer PRINTO criteria exclude arthritis as an essential diagnostic feature, emphasizing the systemic nature of sJIA (49): fever for at least 3 consecutive days and reoccurring over a duration of at least 2 weeks and accompanied by one major criteria or two minor criteria. Major criteria are evanescent (non-fixed) erythematous rash and/or arthritis (49). Minor criteria are generalized lymph node enlargement and/or hepatomegaly and/or splenomegaly; serositis; arthralgia lasting 2 weeks or longer (in the absence of arthritis); and leukocytosis (≥15,000/mm3) with neutrophilia (Table 1) (49).
Differential diagnosis is challenging due to overlapping features with infections, oncological conditions, and autoimmune disorders (9, 10, 58–60). Biomarkers such as calprotectin (MRP8/14), IL-18, and S100 proteins help differentiate Still's disease from other conditions, including KD and MIS-C (8, 33, 35, 36).
The pathogenesis of Still's disease involves a biphasic immune response (2). The acute phase is driven by innate immune dysregulation, with IL-1 and IL-6 playing central roles in fever, rash, and systemic inflammation (2). Elevated ferritin, indicative of macrophage activation, is a common marker during this phase (2). Over time, IL-1 activation can stimulate adaptive immunity, transitioning to chronic arthritis characterized by synovial hyperplasia and cartilage destruction (2).
Historically, treatment relied on corticosteroids, particularly for systemic-phase disease. Advances in biologic DMARDs (bDMARDs) have transformed Still's disease management (12, 61, 62), with IL-1 inhibitors (e.g., anakinra, canakinumab) and IL-6 inhibitors (e.g., tocilizumab) achieving rapid systemic control, and reducing corticosteroid dependence (11, 63–69). In particular, anakinra has demonstrated significant efficacy in Still's disease, particularly in controlling inflammation and preventing disease progression (70, 71). Studies support its use as first-line monotherapy in new-onset, steroid-naïve Still's disease patients, showing rapid disease control and favorable long-term outcomes (72). Long-term safety data from the Pharmachild Registry indicate that anakinra is generally well tolerated, with a manageable safety profile, including a low incidence of severe adverse events (73). Similarly, canakinumab has proven effective in treating Still's disease, with studies demonstrating sustained remission and reduced flare rates (67, 74, 75). Canakinumab has also been investigated as a first-line steroid-free monotherapy, showing promising results in achieving inactive disease (76). Regarding safety, long-term studies indicate that canakinumab is well tolerated, with a favorable risk-benefit profile and a low incidence of severe infections or treatment-related complications (77). Tocilizumab is effective in Still's disease, with better outcomes when initiated early (78). Long-term studies confirm its sustained efficacy in controlling systemic and articular symptoms (79). While generally well tolerated, infections remain a concern (79).
Early initiation of biologics, ideally within 3 months of disease onset, maximizes efficacy and minimizes glucocorticoid exposure (11, 13, 70, 80). Notably, polymorphisms in the IL1RN gene, encoding the IL-1 receptor antagonist, may influence susceptibility and therapeutic responses to IL-1 inhibitors (81).
Corticosteroids remain essential for managing life-threatening systemic features, with pulse methylprednisolone or oral prednisone commonly employed (82).
Emerging treatments, including JAK inhibitors (e.g., ruxolitinib) (83), emapalumab (interferon-γ inhibitor) (84), and IL-18-targeted therapies (85), are under investigation for refractory cases and MAS-related hyperinflammation.
The sJIA Disease Activity Score (sJADAS) integrates systemic features, including joint count, global assessments, and acute-phase reactants [e.g., C-reactive protein (CRP), erythrocyte sedimentation rate (ESR)], to guide treatment and monitor disease progression (86, 87).
Complications include macrophage activation syndrome (MAS) and interstitial lung disease (ILD) (88, 89).
MAS usually refers to a secondary form of hemophagocytic lymphohistiocytosis (HLH) associated with autoimmunity, although there are other causes of secondary HLH, such as infections and malignancy (90). MAS is a life-threatening cytokine storm syndrome that affects 10% of pediatric Still's disease patients, with subclinical features in up to 40% (3, 90, 91). It presents with persistent, high-grade fever, hepatosplenomegaly, lymphadenopathy, and hemorrhagic manifestations (90) and carries an 8% mortality rate (92, 93).
Diagnostic criteria include ferritin >684 ng/ml and at least two additional abnormalities such as low platelet count (≤181 × 109/L), hypertriglyceridemia (>156 mg/dl), elevated aspartate aminotransferase (>48 U/L), or hypofibrinogenemia (≤360 mg/dl) (90) (Table 2). Early identification of MAS through careful monitoring of laboratory biomarkers in high-risk patients who show clinical exacerbation despite appropriate treatment is fundamental to preventing irreversible organ damage due to hyper-cytokinemia (90, 92, 95).

Table 2. Diagnostic criteria for HLH/MAS.
Distinguishing MAS from Still's disease flares or sepsis-like syndromes is challenging (92). In active Still's disease, laboratory findings typically reveal elevated platelet counts, fibrinogen levels, and ESR, whereas MAS is characterized by a decrease in these parameters, though CRP levels increase in both conditions (96, 97). Diagnostic and classification tools for HLH/MAS include the HScore for adults and the MH Score for pediatric patients, the latter of which aids in distinguishing between primary and secondary HLH (Table 2) (98, 99).
Genetic susceptibility to HLH/MAS has been identified, with macrophage activation triggered by inflammatory processes, malignancies, or viral infections, such as Epstyein-Barr (EBV), cytomegalovirus (CMV), SARS-CoV-2, adenovirus, HSV, and RSV (100–102). Due to overlapping clinical features, experts recommend genetic testing for primary HLH in cases of recurrent MAS in pediatric Still's disease patients (101).
High-dose pulse intravenous methylprednisolone (30 mg/kg/day for 3–5 days) remains first-line therapy for MAS (101). Intravenous immunoglobulin (IVIG) is also frequently added to methylprednisolone as part of the initial combination therapy, particularly in pediatric cases where MAS commonly coexists with infections (101).
Anti-IL-1 therapies, such as anakinra (4–12 mg/kg/day), have shown efficacy in second-line treatment of refractory MAS (101, 103–106). Tocilizumab, an anti-IL-6 agent, may also be considered for refractory MAS, particularly when anakinra is unavailable, though its efficacy appears somewhat lower (106). Emapalumab is emerging as a promising treatment for MAS leading to improvement of clinical and laboratory parameters with a favorable safety profile (84). To monitor disease activity and guide treatment, serial evaluation of leukocyte and platelet counts, aminotransferase and ferritin levels, and coagulation parameters are recommended (107, 108).
Emerging evidence links Still's diseaseto ILD, a rare but severe complication whose risk factors include early age of onset, high disease activity, extensive rash, MAS, trisomy 21, lymphopenia, eosinophilia, and biologic infusion reactions (4–6). Screening with pulmonary function tests and imaging (high-resolution computed tomography) is recommended for high-risk patients to enable early intervention (5).
In addition, cardiovascular ailments can occur in Still's disease patients, primarily manifesting as pericarditis, occurring in about 10% of Still's disease patients, with echocardiographic signs detected in over 30% of cases. However, it typically resolves with effective disease control (109, 110).
4 Kawasaki disease
KD is a systemic febrile vasculitis primarily affecting children under 5 years old (14, 15) and is the leading cause of acquired heart disease in developed countries (14, 15, 111). Global incidence varies widely, with Japan reporting the highest rates (260/100,000 children in 2012) compared to 25/100,000 in the USA and 10–15/100,000 in Europe (112–116). Boys are more frequently affected, and the disease is usually diagnosed before 5 years of age (14, 15, 115).
Diagnosis requires fever lasting at least 5 days and at least four of the following symptoms: rash, conjunctival injection, oral mucosal changes (e.g., strawberry tongue), extremity changes (e.g., erythema or swelling), and cervical lymphadenopathy (17, 117).
Incomplete or atypical KD, characterized by fewer than four clinical criteria, often occurs in infants under six months or children over five years, delaying diagnosis and increasing the risk of coronary complications (17, 117, 118). The recently updated statement of the American Heart Association for the management of Kawasaki disease confirms that for suspected incomplete KD is mandatory the presence of prolonged unexplained fever (at least 7 days for infants) and 2–5 clinical criteria associated with laboratory or echocardiographic findings as coronary involvement (Z-score of LAD CA or RCA ≥2.5) or elevated CRP (≥3 mg/dl), and/or ESR (≥40 mm/h), associated with at least three of the following items: anemia for age, leukocytosis (≥15,000 mmc), thrombocytosis (450,000 mmc), elevated ALT, leukocyturia (≥10/hpf), and hypoalbuminemia (≤3 g) (17, 117).
A recent proteomic and genetic study identified four distinct KD phenotypes: hepatic involvement, high neutrophil counts with cardiogenic shock risk, lymphoglandular expression, and early-onset KD associated with aneurysms (119). These findings highlight the need for individualized risk and treatment strategies.
KD Shock Syndrome (KDSS),a severe form associated with myocarditis, hypotension, and increased IVIG resistance, occurs in younger patients (mean age: 3.7 years) and has a mortality rate of 6.8% (120, 121).
Elevated cytokines, including IL-1β, TNF-α, and IL-17, drive systemic inflammation and endothelial activation, leading to vascular damage (20–22). Genetic studies highlight polymorphisms in immune regulatory genes, reinforcing the role of genetic predisposition (122).
Prompt treatment within 10 days of symptom onset significantly reduces coronary complications (117). IVIG (2 g/kg) is the cornerstone therapy, resolving inflammation in the great majority of cases, alongside high-dose aspirin (30–50 mg/kg/day, tapered to 3–5 mg/kg/day after fever resolution) (15, 117, 123). For IVIG-resistant cases, which represent approximately 10%–20% of cases, adjunctive therapies include corticosteroids and biologics (117, 124–126). Methylprednisolone pulses (30 mg/kg/day for three days) reduce coronary artery lesions (108), often in combination with IVIG (127). Biologic agents such as infliximab (anti-TNF) and anakinra are increasingly used for severe or refractory cases, with anakinra demonstrating efficacy in preventing coronary complications and controlling hyperinflammation (128–136).
Long-term outcomes depend on timely treatment. While most treated children recover without sequelae, 5%–8% of treated and 25% of untreated patients develop coronary aneurysms (14, 19, 34), with 50%–80% of giant aneurysms persisting up to 10 years after diagnosis (137). Coronary aneurysms are associated with inflammatory cell infiltration and endothelial remodeling, contributing to thrombosis and ischemia (15, 34). Advances in imaging and treatment have improved prognosis, leading to approximately 90% long-term survival (138). Echocardiography is crucial for detecting coronary artery involvement, and the novel Coronary Ectasia Diagnostic Index (CEDi) has shown high sensitivity and specificity for early identification (34). A CEDi value of ≥0.41 (left coronary) or ≥0.39 (right coronary) at admission is diagnostic, with 97% normalization within eight weeks (34).
MAS occurs in approximately 1% of KD cases (139, 140), though subclinical forms may be underdiagnosed. While rare, MAS in KD may be underdiagnosed, particularly in its subclinical forms. Diagnosis of MAS in KD is complicated by the overlap of symptoms with severe KD forms, such as IVIG-resistant KD and KDSS. Both conditions share features such as prolonged fever, thrombocytopenia, and hyperferritinemia, making it difficult to differentiate them clinically. Several studies suggest that the 2016 sJIA-MAS classification criteria (90), which emphasize hyperferritinemia (>684 ng/ml) and laboratory abnormalities such as thrombocytopenia, hypofibrinogenemia, elevated AST, and hypertriglyceridemia, may be more suitable for identifying MAS in KD compared to HLH-2009 criteria (139, 141). In a cohort of 719 KD patients, 1.1% were diagnosed with MAS using Ravelli's criteria (90), but only 0.42% met the HLH-2009 criteria, suggesting that the latter has low sensitivity for KD-associated MAS (139). A retrospective study of KD patients complicated by MAS found that affected individuals frequently exhibit fever > 5 days, oral mucosal changes, hepatomegaly, bilateral conjunctival injection, rash and extremity changes, thrombocytopenia, and elevated inflammatory markers such as ferritin and lactate dehydrogenase (139). Hemophagocytosis in bone marrow aspirates was observed in some but not all cases, reinforcing the notion that this feature is not required for diagnosis (139).
Early recognition and prompt treatment of MAS in KD are crucial to prevent severe complications, including multi-organ failure. The treatment of MAS in patients with KD is similar to that in Still's disease, and anakinra has shown success, reinforcing the potential role of IL-1 blockade in managing MAS in KD (142).
5 Multisystem inflammatory syndrome in children
MIS-C is a hyperinflammatory condition related to SARS-CoV-2 infection, typically manifesting 2–6 weeks after infection with a median presentation age of 8–9 years (23, 143).
MIS-C disproportionately affects Afro-Caribbean children and presents with severe systemic inflammation distinct from but overlapping with KD and other hyperinflammatory syndromes (26, 27, 59, 144–146).
The global incidence varies, with a rate of 0.11 cases per million person-months in the USA in 2023 (147) and a cumulative incidence of 3.27 per 100,000 children in southern Italy (148).
MIS-C is characterized by persistent intractable fever, gastrointestinal symptoms, coagulopathy, shock, mucocutaneous lesions, lymphadenopathy, and cardiovascular complications (24, 25, 149).
MIS-C diagnosis follows criteria from the World Health Organization (WHO). These criteria are characterized by the presence of fever ≥3 days in children or adolescents under 19 years old associated with elevation of inflammatory markers (PCR, ESR or procalcitonin), SARS-CoV-2 exposure, no obvious microbial cause of inflammation and two of the following clinical features: (1) Rash or bilateral non-purulent conjunctivitis or mucocutaneous inflammation signs; (2) hypotension or shock; features of myocardial dysfunction, pericarditis, valvulitis, or coronary abnormalities (including echocardiographic findings or elevated troponin/NT-proBNP); (3) evidence of coagulopathy [by prothrombin time [PT], partial thromboplastin time [PTT], elevated D-Dimers] and/or (4) acute gastrointestinal problems (diarrhea, vomiting, or abdominal pain (148, 150). Besides these, clinical findings include elevated D-dimer, NT-proBNP, cardiac troponins, and cardiac involvement, in particular left ventricular dysfunction (LVEF <50%), which is a prognostic feature present in 30%–60% of patients (26, 96, 151–156). LVEF, however, is reversible and typically normalizes at discharge (157–159). Left ventricular dysfunction can initially be assessed through basic electrocardiographic screening (160). This can be further supported by speckle-tracking echocardiographic evaluation, which measures left ventricular myocardial strain (161, 162), or by cardiac MRI, which can detect edema and fibrotic replacement with limited tissue involvement during the acute phase of the condition, providing additional insights into disease activity (160, 163).
The use of high-sensitivity troponins allows for the detection of nonspecific myocardial involvement, which may not necessarily be associated with myocarditis (164, 165).
Coronary artery aneurysms are less frequent in MIS-C compared to KD and the occurrence of complications, such as giant aneurysms, is rare (27, 166).
A study by Aydin et al. (96) reported the presence of serositis in 46.2% of MIS-C cases, with no statistically significant difference between the groups. Yashuara et al. (159) reported that mild mitral regurgitation, observed at onset in 50% of MIS-C cases, may serve as a soft marker indicative of early disease. Importantly, when appropriate treatment and supportive care are provided during the acute phase, cardiac involvement does not seem to adversely affect long-term prognosis (24).
Neurological symptoms such as headaches and encephalopathy further highlight the syndrome's systemic nature (167).
Unlike KD, MIS-C frequently presents with thrombocytopenia, reflecting coagulopathy (146).
Differential diagnosis of MIS-C is based on older age at onset (27, 168), pronounced myocardial dysfunction, and unique biomarker profiles such as elevated NT-proBNP, BNP, and cardiac troponins (27, 96, 151–153). Additionally, patients display higher fibrinogen levels and lymphopenia and relatively modest ferritin elevations compared to KD (96).
While MAS may develop during MIS-C, it generally follows a milder course with favorable outcomes compared to MAS in Still's disease (169).
Pathophysiologically, MIS-C involves a cytokine storm driven by elevated TNF-α, IFN-γ, IL-1β, and IL-6, causing widespread organ damage (23). Transcriptomic studies suggest MIS-C and KD lie on an inflammatory spectrum, with MIS-C representing a more severe manifestation driven by IL-15/IL-15RA dysregulation (26). Neutralizing autoantibodies targeting the interleukin-1 receptor antagonist (IL-1Ra) may account for increased IL-1β-signaling in MIS-C, suggesting a mechanistic role for IL-1 dysregulation in MIS-C pathogenesis (170). This discovery highlights potential therapeutic interventions, such as the use of anakinra (170). Importantly, these autoantibodies were observed less frequently in children with KD or other inflammatory disorders, emphasizing their specificity to MIS-C and enhancing our understanding of its unique pathophysiology (170).
Treatment of MIS-C targets systemic inflammation, myocardial dysfunction, and coagulopathy. Early intervention is fundamental to avoid life-threatening complications (171, 172). IVIG (2 g/kg) is usually the starting therapy, often combined with corticosteroids [intravenous (IV), 1–2 mg/kg/day] to enhance efficacy (25, 153, 173). At present, the use of a second dose of IVIG is not recommended due to the associated risks of hemolytic anemia and cardiovascular volume overload, particularly in patients with compromised left ventricular function (153). High-dose methylprednisolone (IV, 30 mg/kg/day for 3 days) is employed in life-threatening moderate to severe cases to control cytokine storm and prevent shock (25, 173). For refractory cases or in cases of myocarditis and in life-threatening severe patients, IL-1 inhibitor anakinra [>4 mg/kg/day, IV or subcutaneous (SC)] is effective in mitigating severe inflammation and preventing cardiovascular complications (25, 153, 173–175). Anticoagulation therapy with enoxaparin is recommended for thrombotic risk or significant coronary artery involvement or for patients with moderate or severe LV dysfunction (EF <35%) to prevent thromboembolic events (25, 173). Aspirin (ASA) therapy at an anti-aggregating dose (3–5 mg/kg/day) is usually recommended in patients who are not considered or after the acute phase as a replacement for anticoagulant therapy and continued for at least 8 weeks from the onset of fever. A percentage varying between 30% and 80% of MIS-C patients require intensive care due to hemodynamic instability or multiorgan dysfunction (171, 176). Despite severe acute presentations, most patients recover cardiac function within weeks, though long-term echocardiographic and MRI follow-ups are recommended for persistent abnormalities (152, 177). Early initiation of immunomodulatory therapies has reduced MIS-C mortality to 1.7%–2.6% (148, 171, 173, 176).
6 Recurrent pericarditis
Recurrent pericarditis occurs in approximately 22%–36% of pediatric patients after an initial episode of acute pericarditis (178, 179), with higher recurrence rates when colchicine is not included in initial treatment or when initial treatment included corticosteroids (180, 181). Although uncommon in children, RP is increasingly recognized due to improved diagnostic techniques and heightened awareness.
RP is characterized by episodes of sharp, pleuritic chest pain that worsens in a supine position and improves when sitting upright or leaning forward (30, 181). Additional findings include pericardial friction rub, present in 30%–40% of cases, and pericardial effusion, typically small to moderate in size but occasionally large (30, 181, 182). Systemic symptoms, such as fever, malaise, and elevated inflammatory markers, often accompany flares, mimicking systemic autoinflammatory syndromes (30).
Diagnosis requires at least one recurrence following a symptom-free interval of 4–6 weeks after an initial episode (30). Clinical criteria include two or more features of pericarditis: typical chest pain, pericardial effusion (imaged by echocardiography), pericardial friction rub, or electrocardiographic changes, such as PR segment depression or widespread concave ST elevation (30). Elevated CRP and ESR further support the diagnosis (30). Imaging techniques such as echocardiography are essential for evaluating effusion and ruling out alternative causes of chest pain (181).
Pediatric RP is idiopathic in most cases but can be linked to systemic diseases, such as Still's disease, or monogenic autoinflammatory syndromes, such as familial Mediterranean fever (FMF) and tumor necrosis factor receptor-associated periodic syndrome (TRAPS) in 6%–10% of patients (30).
Pathophysiologically, RP involves dysregulated inflammation driven by overexpression of IL-1 and other proinflammatory cytokines, leading to immune-mediated pericardial injury and fibrosis (28, 29). Chronic inflammasome activation may increase the risk of complications, such as constrictive pericarditis or cardiac tamponade (28, 29), though these remain rare in children (30).
Treatment of RP aims to alleviate symptoms, prevent recurrences, and minimize corticosteroid dependency. NSAIDs are first-line therapy (30, 181), with high doses of ibuprofen (30–50 mg/kg/day in three doses). Aspirin and indomethacin, though effective, are less favored in children due to hepatic (ASA) and neurological (indomethacin) side effects (181, 183–185). Indomethacin, though effective, is less favored in children due to neurological side effects (172, 174). As mentioned before, adding colchicine to NSAIDs significantly reduces recurrence rates (30, 180, 181, 185), and treatment should be continued for at least 6 months after recurrence (183). Recommended dosages are weight-based: 0.5 mg/day for children and adolescents under 70 kg and 1 mg/day for those over 70 kg (180, 181, 186).
Corticosteroids are reserved for cases refractory to NSAIDs and colchicine, but they should be added to aspirin/NSAIDs and colchicine and not used as standalone therapy (181). Low-dose regimens (0.2–0.5 mg/kg/day prednisone) with gradual tapering are preferred to minimize relapse risk and side effects (183). Notably, prolonged corticosteroid use increases recurrence rates and may interfere with colchicine efficacy (183).
Anakinra is highly effective for colchicine-resistant or corticosteroid-dependent RP (184, 187, 188). In particular, the study by Caorsi et al. (184) involving 57 pediatric patients showed that anakinra significantly reduced relapses in patients previously treated with NSAIDs (43%), NSAIDs + colchicine (17%), steroids (4%), NSAIDs + steroids (24%), and NSAIDs + colchicine + steroids (12%), even after steroid withdrawal. Early use of anakinra in colchicine-resistant cases before corticosteroid initiation facilitated faster disease control and reduced side effects (184). Importantly, the study highlighted that the first-line treatments (NSAIDs and colchicine) were underdosed, and even high doses of steroids failed to achieve complete control of pericarditis flares in 33% of patients (184). Furthermore, 94% of treated patients exhibited steroid dependence withdrawal (184). Notably, 10 patients from the study showed a prompt response to IL-1 blockers without prior steroid treatment withdrawal (184).
Typical doses of anakinra for RP treatment range from 1 to 2 mg/kg/day, administered subcutaneously (102, 180, 183, 184, 187). Treatment is generally continued for at least six months, with careful tapering to assess recurrence risk. In rare cases of anakinra intolerance, high-dose canakinumab may be considered, though results have been less consistent (184).
With appropriate treatment, the long-term prognosis of RP is favorable, though up to 36% of pediatric patients experience multiple relapses (178, 179, 185, 189). Early introduction of colchicine and IL-1 inhibitors is crucial to reducing recurrence risk (28, 30, 184). Chronic inflammation can lead to rare complications like constrictive pericarditis or tamponade, necessitating close monitoring with cardiac MRI in high-risk patients (182). Recurrent symptoms and corticosteroid side effects can adversely affect quality of life, underscoring the importance of steroid-sparing therapies to mitigate these impacts.
7 Expert opinion
7.1 Recommendations for a multidisciplinary care pathway
Managing pediatric hyperinflammatory diseases requires a multidisciplinary approach tailored to the specific condition and individual patient needs. Integrating evidence-based guidelines with clinical expertise is critical to achieving optimal outcomes.
For diagnostic workup, we recommend a standardized panel of first-level tests, including:
- Basic bloodwork: complete blood count, electrolytes, urea, creatinine, and blood glucose.
- Inflammatory markers: ESR, CRP, ferritin, and fibrinogen.
- Liver and coagulation Panels: transaminases, albumin, PT, PTT, international normalized ratio (INR).
- Infectious workup: viral multiplex swabs for common pathogens and antibody tests for EBV, CMV, and adenovirus and parvovirus B19.
- Protein and urine analysis.
Initial imaging includes chest x-ray, electrocardiography (ECG), and abdominal ultrasonography to assess systemic involvement. Echocardiography, while essential for cardiovascular assessment, is considered a second-line test reserved for patients with suspected cardiac complications.
For cases with inconclusive first-line results or specific clinical suspicion, second-line tests are recommended. These include:
- Infectious disease workup: tests for HIV, hepatitis B and C (HBV, HCV), Leishmania, and SARS-CoV-2.
- Advanced EBV Testing: If EBV antibodies are positive, quantitative polymerase chain reaction (PCR) testing is advised to assess viral load.
For patients with hyperinflammation or suspected MAS, advanced diagnostic panels are essential for accurate assessment and timely management. Key tests include lactate dehydrogenase (LDH) and triglycerides, which help identify hyperinflammatory states. In cases meeting MAS criteria, such as thrombocytopenia and elevated ferritin (>648 ng/ml), centralized sample analysis at specialized centers is strongly recommended to ensure standardized and reliable results. Second-level investigations, including perforin expression, interleukin-18 (IL-18), and CXCL-9 levels, should also be conducted at these specialized centers to maintain consistency and diagnostic accuracy (35, 190–193). Such advanced testing plays a critical role in guiding therapy for complex cases.
Effective management of MAS requires a multidisciplinary approach with frequent clinical reassessments, particularly in rapidly evolving cases. Centralized diagnostic workflows not only enhance accuracy but also facilitate cohesive decision-making among specialists.
7.2 Disease-specific recommendations
7.2.1 Still's disease
Differential diagnosis of Still's disease is critical to exclude oncological conditions. A bone marrow aspirate is recommended in cases of unexplained fever, bone pain, bicytopenia, or pancytopenia, and MRI bone marrow abnormalities, while minimally invasive lymph node biopsies may be necessary in cases of lymphadenopathy to guide therapy decisions. In cases of hyperinflammatory disease, the 2016 MAS Classification Criteria provide a first-line scoring system validated for sJIA, with good applicability to other inflammatory conditions (91, 194). The HLH-2004 criteria may also be used, particularly when there is suspicion of a genetic or familial predisposition (195) (Table 2). Referring to the 2022 EULAR recommendations, early MAS screening aids in identifying patients requiring further evaluation (101). Genetic testing, including clinical exome sequencing, should be considered in recurrent pediatric MAS cases.
A disease-specific, treat-to-target approach is essential for effective Still's disease management. Initiating IL-1 or IL-6 inhibitors as first-line therapy, even without corticosteroids, in agreement with the recent 2024 EULAR/PReS recommendations, is our recommended approach unless MAS is present (Figure 1) (8). In our experience and agreement with the literature, early use of biologics, such as short-acting anakinra during the differential diagnosis phase, improves long-term outcomes (2, 12, 13). Early intervention with first-line biologics, such as interleukin-1 inhibitors, during the initial inflammatory phase of Still's disease exploits a critical “window of opportunity” (2, 196). By targeting the innate immune system, this approach prevents its transition to adaptive immunity, thereby halting the progression from systemic to chronic articular Still's disease and potentially altering the disease trajectory (2, 196).
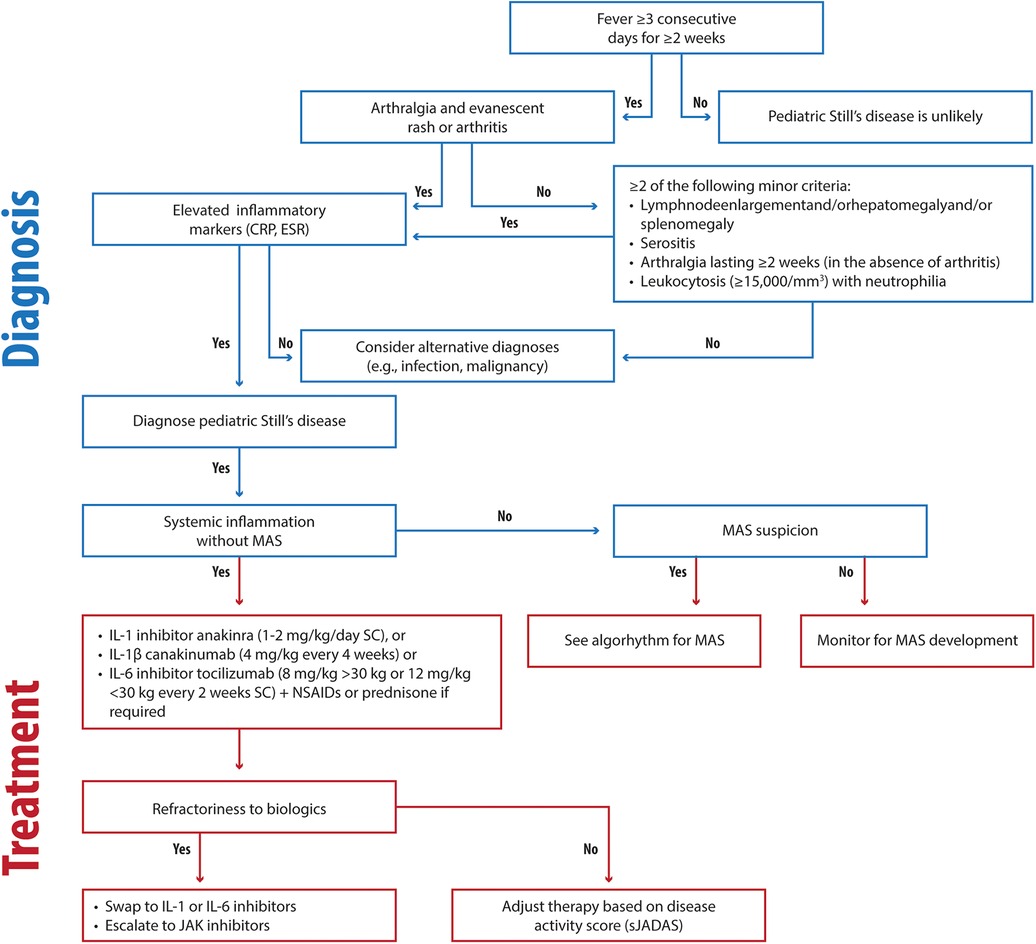
Figure 1. Clinical flowchart illustrating the diagnostic and therapeutic approach to Still's disease. CRP, C-reactive protein; ESR, Erythrocyte Sedimentation Rate; MAS, Macrophage Activation Syndrome; IV, intravenous; SC, subcutaneous.
We are not in favor of the administration of corticosteroids as a standalone initial therapy; instead, NSAIDs may be beneficial for brief initial treatment to modulate the autoinflammatory process and potentially prevent progression to autoimmunity. Our clinical experience shows that rapid initiation of biologics combined with tailored steroid tapering effectively resolves systemic symptoms and improves outcomes, even in MAS-complicated cases.
In our opinion, treatment goals should include achieving ≥50% improvement within three months and remission or low disease activity within six months. Fever resolution in systemic Still's diseaseshould occur within one week of starting therapy. We recommend monitoring disease activity using the sJADAS (86, 87).
In the 2023 consensus paper by Shakoory et al. (101), a broad agreement on the treatment of HLH/MAS was not reached. However, baseline therapy with high-dose glucocorticoids and anakinra is considered a reasonable starting point. Notably, methylprednisolone has a stronger anti-inflammatory effect compared to other glucocorticoids and may be better tolerated at the hepatic level. The selection of a specific glucocorticoid often depends on the clinician's expertise. For HLH, international guidelines recommend dexamethasone due to its rapid onset of action, typically within 2–3 days.
The utility of cyclosporin A (CsA) in MAS treatment remains a subject of debate. While corticosteroid therapy alone may suffice in some cases, certain studies have demonstrated the efficacy of CsA in managing hyperinflammation (197, 198). Furthermore, fibrinogen administration in MAS should be approached cautiously, as it may induce liver damage; thus, careful evaluation of the patient's hepatic status is essential.
We propose the following approach (Figure 2) for timely and effective MAS management in the absence of evidence of genetic disease:
- First-line therapy
- Methylprednisolone (IV, 30 mg/kg, max 1 g/dose with 3–5 consecutive doses) or dexamethasone (10 mg/m2) for 3–5 days (initially as boluses)
- prednisone (initially IV and then oral) at 2 mg/kg/day, divided into two doses, followed by tapering.
- Second-line therapy (based on ferritin levels)
- anakinra 4–12 mg/kg/day intravenously (IV) (2–3 mg/kg/dose every 6–24 h). A dose of 12 mg/kg should be administered via continuous infusion.
- ±cyclosporine 5–7 mg/kg/day divided into 2–3 doses (oral or IV).
- Advanced therapy
- Ruxolitinib (Jack 1/2 inhibitors), 5 mg twice a day (BIS) (<10 kg); 5 mg BIS (<25 kg); 10 mg twice a day (BIS) (≥25 kg) (199).
- Tocilizumab (anti-IL6), 8 mg/kg)
- MAS825 (IL-1 and IL-18 inhibitor) 10 mg/kg (max 700 mg) biweekly (200)
- Emapalumab (6 mg/kg/day on day 0, followed by 3 mg/kg every 3 days until day 15, and twice a week until day 28) (84).
- Low-dose etoposide should be reserved as a last option.
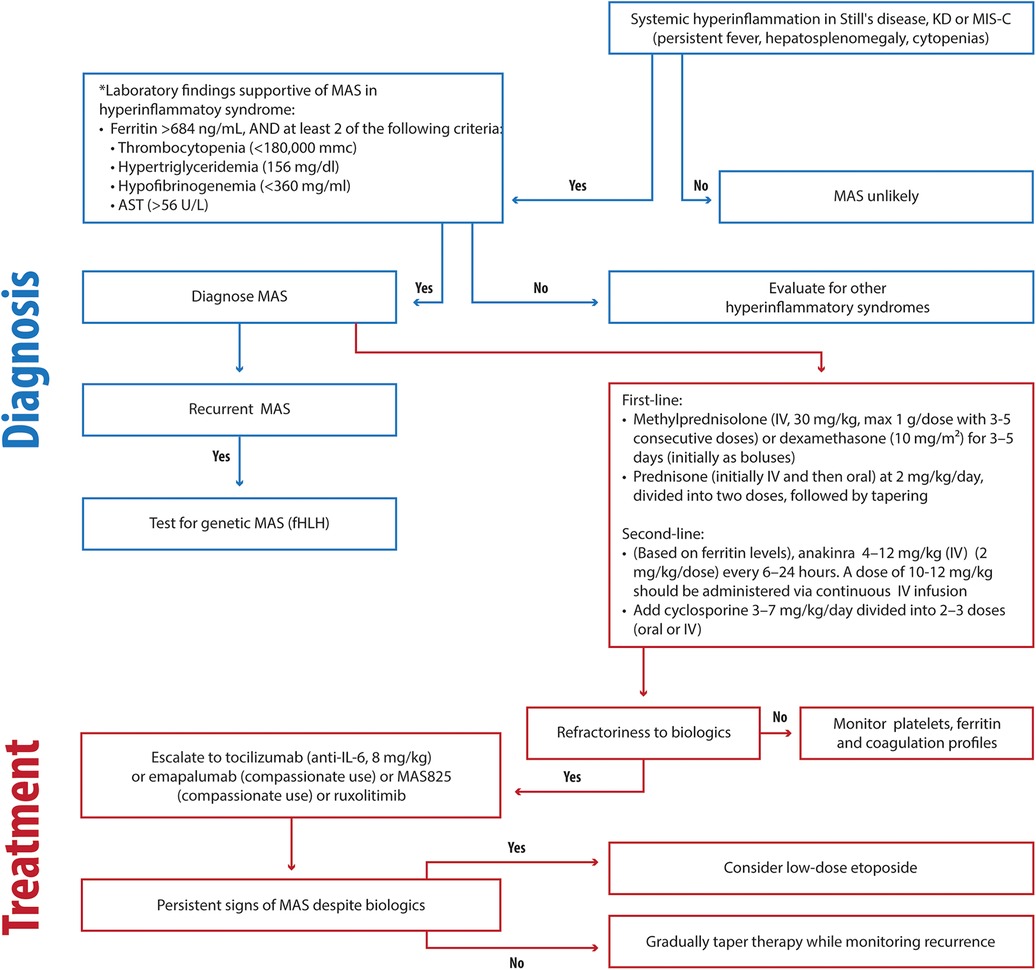
Figure 2. Clinical flowchart illustrating the diagnostic and therapeutic approach to MAS in hyperinflammatory disorders. KD, Kawasaki disease; MIS-C, Multisystem Inflammatory Syndrome in Children; fHLH, familial hemophagocytic lymphohistiocytosis; HLH, hemophagocytic lymphohistiocytosis; AST, Aspartate Aminotransferase.
Preference should be given to emapalumab or MAS825 when available. Ruxolitinib is typically used in the treatment of chronic hematological diseases due to its slow action.
7.2.2 Kawasaki disease
KD remains a clinical diagnosis characterized by fever, unilateral lymphadenopathy, rash, bilateral nonexudative conjunctival injection, swelling and erythema of the hands and feet, and oropharyngeal findings, including strawberry tongue and erythematous lips (17, 117). The algorithm for diagnosing complete or incomplete KD, with supportive laboratory features, is shown in Figure 3.
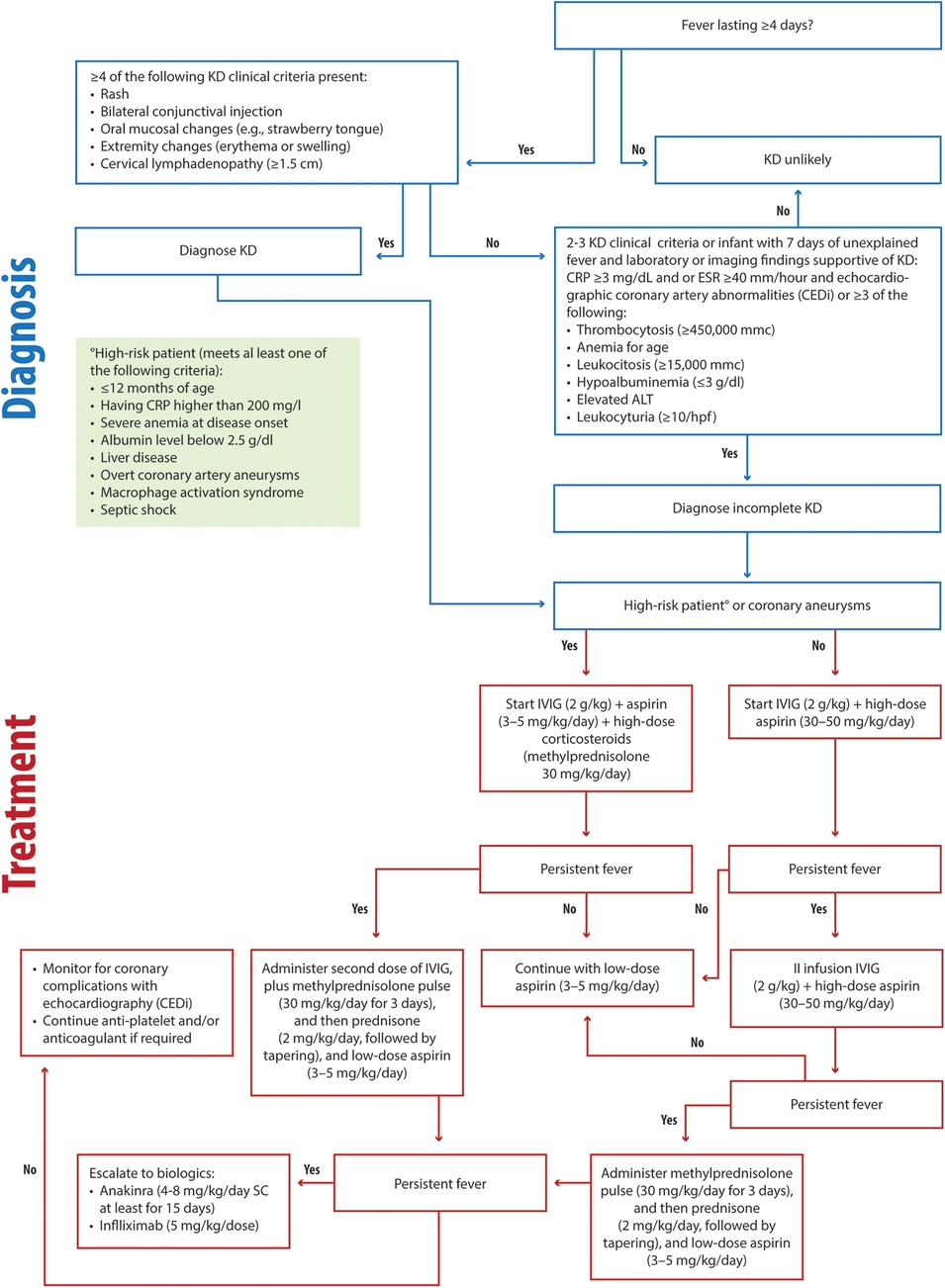
Figure 3. Clinical flowchart illustrating the diagnostic and therapeutic approach to KD. KD, Kawasaki disease RP, C-reactive protein; ESR, Erythrocyte Sedimentation Rate; ALT, alanine aminotransferase; IVIG, intravenous immunoglobulin.
Early administration of IVIG (2 g/kg) and aspirin (30–50 mg/kg/day) is critical for preventing coronary complications in KD patients, as recommended by current guidelines (17, 117) (Figure 3). For high-risk patients (children less than 12 months or children having CRP higher than 200 mg/L, severe anemia at disease onset, albumin level below 2.5 g/dl, liver disease, overt coronary artery aneurysms, macrophage activation syndrome or septic shock), we recommend IVIG (2 g/kg) combined with a bolus of methylprednisolone (30 mg/kg/day) and antiplatelet aspirin (3–5 mg/kg/day) (Figure 3). In low-risk patients, in cases of persistent fever, a second infusion of IVIG (2 g/kg) should be administered alongside aspirin (30–50 mg/kg/day) (Figure 3) (117). If the fever remains unresponsive, we recommend methylprednisolone (30 mg/kg/day) administration for three consecutive days, and then daily prednisone (2 mg/kg/day, with tapering) and continued anti-aggregating aspirin (3–5 mg/kg/day) (Figure 3).
In high-risk or IVIG-resistant cases, corticosteroids play a pivotal role. Our clinical experience demonstrates that timely corticosteroid use in high-risk patients reduces fever duration and halts coronary aneurysm progression. In these patients in second-line treatment, we recommend a second infusion of IVIG (2 g/kg) and methylprednisolone (30 mg/kg/day) administration for three consecutive days and then daily prednisone (2 mg/kg/day, with tapering), and continued anti-aggregating aspirin (3–5 mg/kg/day) (Figure 3). In persistent cases, infliximab (5 mg/kg) and anakinra (4–8 mg/kg/day for at least 15 days, SC) remain effective escalation options (Figure 3). Early risk stratification and monitoring using algorithms, such as CEDi and echocardiography, are invaluable for guiding management. The treatment of MAS in patients with KD should follow the same principles as in Still's disease.
7.2.3 Multisystem inflammatory syndrome in children
MIS-C presents a diagnostic challenge for the physician due to the nonspecific early symptoms, lack of pathognomonic findings, and potentially fatal complications (149). WHO case definitions are the best criteria for diagnosis of MIS-C patients (149, 150).
A study by La Torre et al. (148) evaluated 22 MIS-C patients with a mean age of 7.4 years in the Apulia region over a period of 6 months spanning from 1 November 2020 to 30 April 2021. The most prevalent symptoms were gastrointestinal involvement, cardiovascular involvement, features of KD, and MAS/HLH (present in 23% of cases). No patients were admitted to the Pediatric Intensive Care Unit (PICU) in this study (148). In severe presentations, treatment included IVIG infusions and methylprednisolone boluses (148). In cases of MAS, shock, myocarditis, or meningoencephalitis, endovenous anakinra (2 mg/kg, max 100 mg, every 6 h) was utilized as a first-line therapy (148). Combining high-dose steroids with anakinra demonstrated efficacy in restoring myocardial function, particularly in patients with MAS-like features or significant cardiac involvement, reducing the rates of admission to PICU (Figure 4). In our opinion, the rapid and flexible application of anakinra is noteworthy, as it is suited for “hybrid” conditions requiring immediate intervention. In fact, the prompt step-up treatment with IVIG, steroids, and anakinra, depending on the organ involvement or response to therapy, contributed to good outcomes in terms of PICU admission and long-term cardiac sequelae (148, 171).
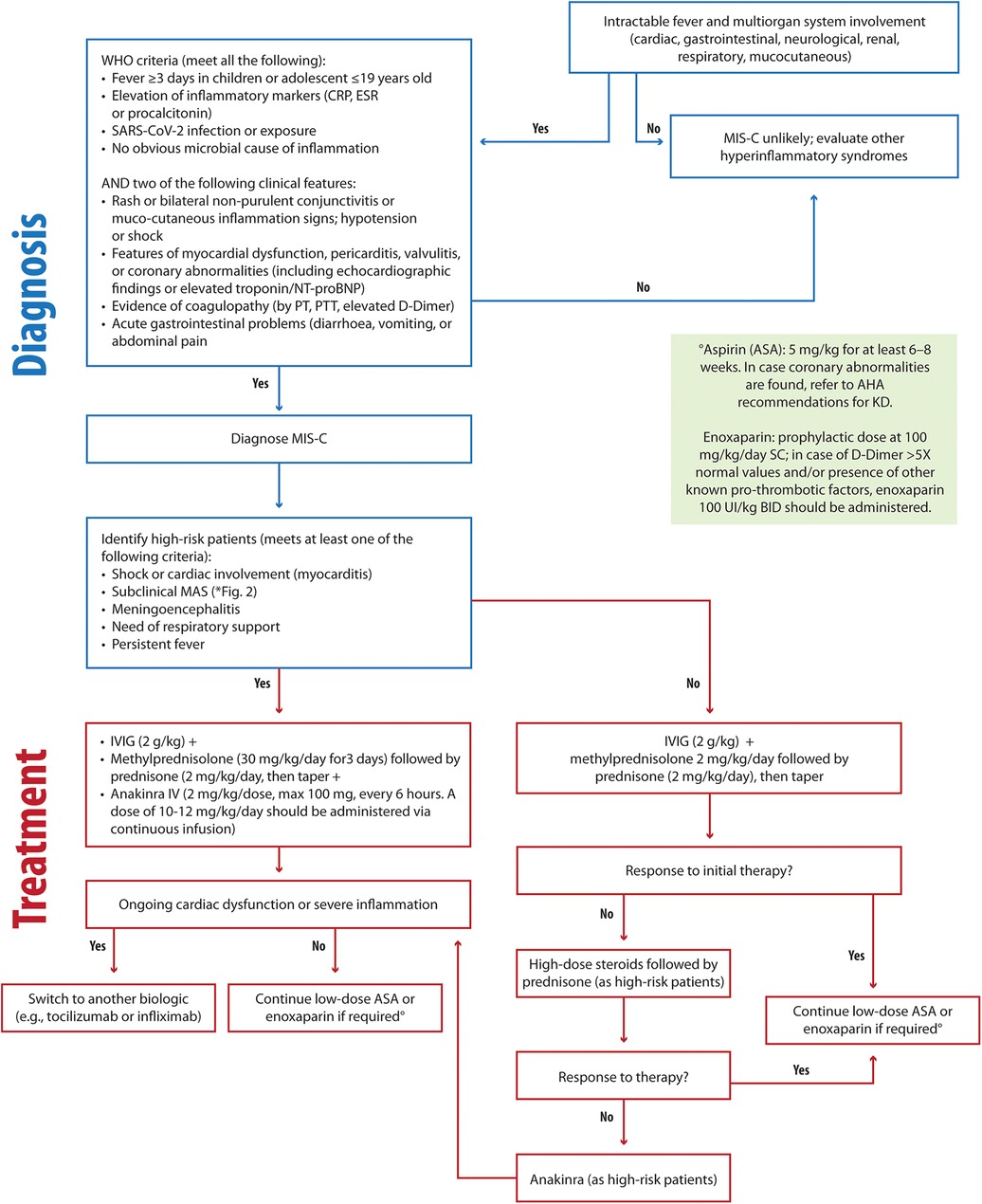
Figure 4. Clinical flowchart illustrating the diagnostic and therapeutic approach to MIS-C. MIS-C, Multisystem Inflammatory Syndrome in Children; CRP, C-reactive protein; ESR, Erythrocyte Sedimentation Rate; NT-proBNP, N-terminal pro-B-type natriuretic peptide; PT, prothrombin time; PTT, partial thromboplastin time; MAS, macrophage activation syndrome; AHA, Americal Heart Association; IVIG, intravenous immunoglobulin; ASA, acetylsalicylic acid.
To optimize care, we emphasize the importance of equipping cardiologists with specialized pediatric cardiology training and establishing dedicated registries to collect data from larger patient cohorts is essential for developing robust clinical guidelines.
7.2.4 Recurrent pericarditis
There are no clear differences regarding clinical presentation between acute and recurrent pericarditis. However, a symptom-free interval of 4–6 weeks and evidence of new pericardial inflammation are needed for the diagnosis (201). Effective treatment of acute pericarditis significantly reduces the risk of recurrence (201).
Colchicine is commonly employed in pericarditis management (28, 30, 184). Three studies are shown supporting the efficacy of colchicine in recurrences (202–204). A multicenter, double-blind study by Imazio et al. (194) demonstrated a reduced incidence of post-pericardiotomy syndrome with colchicine prophylaxis in surgical patients. However, due to a high incidence of adverse effects, routine colchicine use is not recommended for all surgical patients (205). IL-1 blockade with anakinra is beneficial for the treatment of recurrent pericarditis (184, 187, 188), as shown by several case series (206–208), the IRAP study (209) and the randomized controlled AIRTRIP (Anakinra-Treatment of Recurrent Idiopathic Pericarditis) trial (188).
For treating RP, we recommend the following approach (Figure 5):
- First-line therapy
- NSAIDs, including ibuprofen (30–50 mg/kg/day), aspirin (30–50 mg/kg/day), and colchicine (0.5 mg/day for children under 70 kg, 1 mg/day for children over 70 kg)
- Second-line therapy
- Steroids, particularly for cases unresponsive to NSAIDs and colchicine. Low-dose prednisone (0.2–0.5 mg/kg/day) is more effective in reducing recurrences, hospitalization duration, and side effects.
- Third-line therapy
- Anakinra 1–2 mg/kg/day, max 100 mg, SC) is recommended as a steroid-sparing option, with prolonged use proving effective in preventing relapses, even in refractory cases.
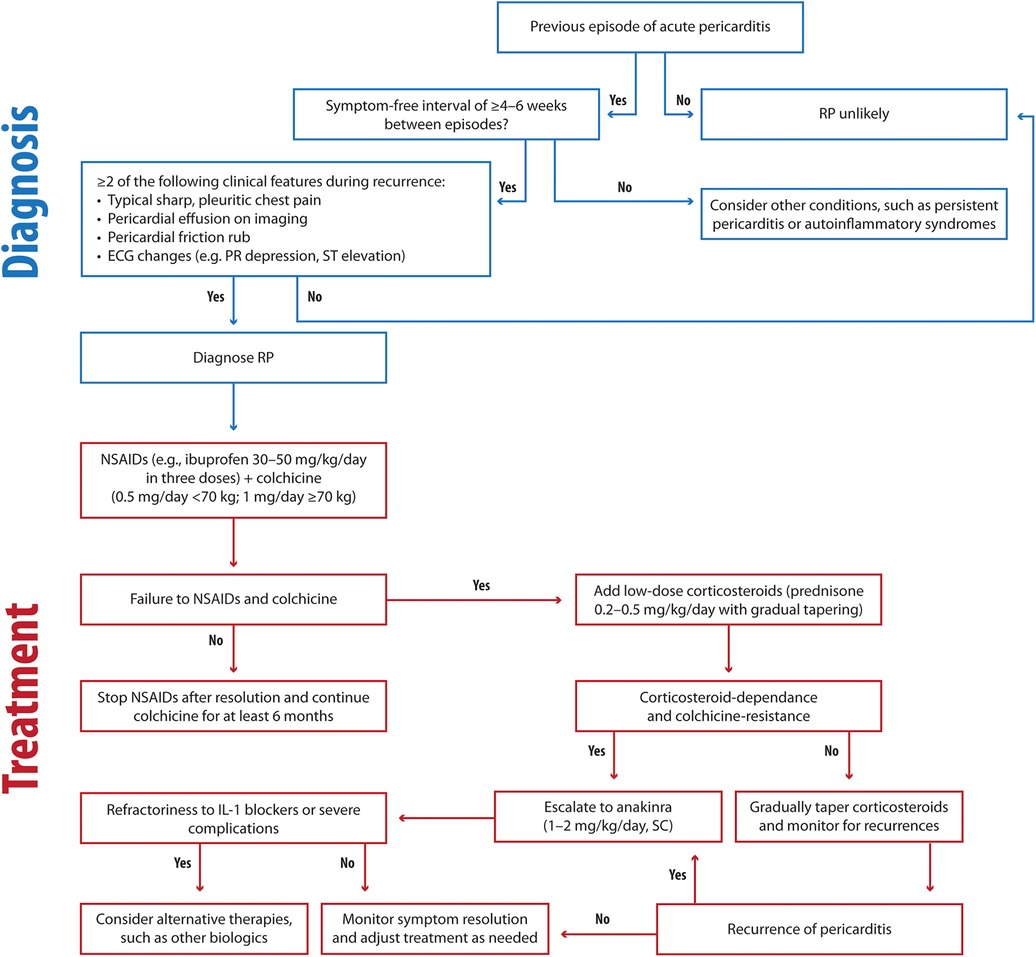
Figure 5. Clinical flowchart illustrating the diagnostic and therapeutic approach to RP. RP, recurrent pericarditis; ECG, electrocardiography; NSAIDs, Nonsteroidal anti-inflammatory drugs; SC: subcutaneous.
Comprehensive combination therapy further reduces recurrence risk while restricting intense physical activity until symptom resolution, guided by inflammatory markers, is essential.
The long-term prognosis for idiopathic RP is generally favorable, with cardiac tamponade and constrictive pericarditis remaining rare.
7.3 Open questions and unmet needs
Multidisciplinary collaboration among rheumatologists, cardiologists, and intensivists is essential, particularly in high-risk cases requiring complex, coordinated care. Despite advancements, several unresolved questions and challenges remain in the management of Still's disease. Key issues include the lack of predictive factors for therapeutic outcomes, uncertainty about the respective indications for IL-1 and IL-6 inhibitors, and the absence of consensus on tailoring treatment based on the presence or absence of arthritis. Diverging opinions on corticosteroid use, limited options for cases resistant to IL-1 and IL-6 inhibitors, and the lack of standardized protocols for tapering or discontinuing treatment further complicate management.
Many of these challenges also apply to conditions like KD, MIS-C, and RP. The lack of reliable predictive biomarkers hinders the precision of treatment approaches. Biomarkers that guide responses to IL-1 and IL-6 inhibitors are urgently needed. Advances in genetic testing and immune profiling hold potential for personalized treatment strategies but require further validation and integration into routine clinical care.
Anakinra has emerged as a promising option for reducing corticosteroid dependency across multiple inflammatory conditions, including Still's disease and RP. Challenges persist in managing colchicine-resistant and steroid-dependent RP cases, mitigating corticosteroid side effects, and addressing the impact of recurrences on quality of life. Anakinra provides a viable steroid-sparing alternative. However, head-to-head trials comparing anakinra and corticosteroids in pediatric populations are needed to establish optimal dosing and duration. Establishing effective strategies for such cases and enhancing long-term management approaches remain key areas for future research.
Similarly, the absence of consensus on tapering or withdrawing biologics complicates the long-term management of conditions, such as KD, MIS-C, and RP. Premature withdrawal may lead to disease flares, while prolonged use increases costs and the risk of side effects. Longitudinal studies are necessary to define safe and effective withdrawal protocols.
Additionally, patients unresponsive to IL-1 or IL-6 inhibitors represent a significant treatment gap.
Novel therapies, such as JAK inhibitors for Still's diseaseand biologics targeting IFN-γ or IL-18, show potential for refractory cases but require further research to confirm efficacy and safety.
Further research is needed to understand more in-depth the mechanisms of immune dysregulation underlying these conditions, along with the impact of genetic predispositions and environmental triggers.
Finally, as mentioned above, regional training programs in pediatric cardiology are urgently needed to address gaps in expertise. Conditions like MIS-C and KD underscore the importance of equipping centers with specialists skilled in managing inflammatory and autoimmune conditions. Such initiatives can enhance timely and accurate interventions, ultimately improving patient outcomes.
8 Conclusion
Despite advances in Still's disease management, challenges persist due to clinical variability, risk of complications, and the need for early intervention. Research into biomarkers, novel therapies, and personalized approaches remains vital to improving outcomes.
Hyperinflammatory diseases, such as Still's disease, KD, MIS-C, and RP, share overlapping features but differ in pathophysiology and clinical courses. Key findings include:
- Still's disease: IL-1 or IL-6 inhibitors as first-line therapy improve outcomes, guided by biomarkers like ferritin and sJADAS.
- KD: IVIG and aspirin prevent coronary complications; corticosteroids and anakinra aid in high-risk cases. Tools like CEDi enhance risk stratification.
- MIS-C: IVIG, corticosteroids, and anakinra reduce mortality, with biomarkers like NT-proBNP aiding diagnosis and management of cardiac involvement.
- RP: NSAIDs and colchicine remain foundational, while anakinra addresses colchicine-resistant cases, minimizing recurrence and corticosteroid use.
The integration of genetic testing and biomarker-guided treatments, such as serum calprotectin in Still's diseaseor NT-proBNP in MIS-C, offers precision in identifying complications like MAS and myocarditis. Imaging advances, including cardiac MRI, further enhance diagnostic and monitoring capabilities.
Future research should focus on immune dysregulation mechanisms and trials comparing existing and novel therapies, such as anakinra vs. corticosteroids or JAK inhibitors for refractory cases. Long-term management would benefit from safe withdrawal protocols for biologics to prevent flares while minimizing costs and side effects.
Developing regional expertise in pediatric inflammatory diseases through training programs will ensure timely, specialized care and improve outcomes. Addressing these research and clinical gaps will advance personalized medicine, reducing complications and enhancing quality of life.
Author contributions
FL: Conceptualization, Writing – original draft, Writing – review & editing. GM: Writing – review & editing. AC: Writing – review & editing. AC: Writing – review & editing. VC: Writing – review & editing. ER: Writing – review & editing. ES: Writing – review & editing. NS: Writing – review & editing. UV: Writing – review & editing. FC: Conceptualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. Editorial assistance was unconditionally supported by SOBI S.r l. The funder was not involved in the writing of this article or in the decision to submit it for publication.
Acknowledgments
Editorial assistance was provided by Valeria Benedusi, Valentina Attanasio and Aashni Shah (Polistudium s.r.l., Milan, Italy) and was unconditionally supported by SOBI S.r.l.
Conflict of interest
FL has received speakers' fees from Sobi and Novartis.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Marcuzzi A, Melloni E, Zauli G, Romani A, Secchiero P, Maximova N, et al. Autoinflammatory diseases and cytokine storms—imbalances of innate and adaptative immunity. Int J Mol Sci. (2021) 22:11241. doi: 10.3390/ijms222011241
2. Nigrovic PA. Review: is there a window of opportunity for treatment of systemic juvenile idiopathic arthritis? Arthritis Rheumatol. (2014) 66:1405–13. doi: 10.1002/art.38615
3. Ravelli A, Grom AA, Behrens EM, Cron RQ. Macrophage activation syndrome as part of systemic juvenile idiopathic arthritis: diagnosis, genetics, pathophysiology and treatment. Genes Immun. (2012) 13:289–98. doi: 10.1038/gene.2012.3
4. Belozerov KE, Solomatina NM, Isupova EA, Kuznetsova AA, Kostik MM. Systemic juvenile idiopathic arthritis–associated lung disease: a retrospective cohort study. World J Clin Pediatr. (2024) 13:88912. doi: 10.5409/wjcp.v13.i1.88912
5. Zhan W, Yang J, Qiu L, Yang K, Ye X, Shangguan Y, et al. Clinical characteristics and prognosis of interstitial lung disease in systemic juvenile idiopathic arthritis: a two-center retrospective observational cohort study. Pediatr Rheumatol Online J. (2024) 22:96. doi: 10.1186/s12969-024-01028-5
6. Gao F-Q, Zhang J-M, Li C-F. Clinical presentation and treatment of juvenile idiopathic arthritis combined with lung disease: a narrative review. Rheumatol Ther. (2023) 10:507–22. doi: 10.1007/s40744-023-00542-4
7. Petty RE, Southwood TR, Manners P, Baum J, Glass DN, Goldenberg J, et al. International league of associations for rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001. J Rheumatol. (2004) 31:390–2. Available at: https://www.jrheum.org/content/31/2/390.tab-e-letters14760812
8. Fautrel B, Mitrovic S, Matteis AD, Bindoli S, Antón J, Belot A, et al. EULAR/PReS recommendations for the diagnosis and management of still’s disease, comprising systemic juvenile idiopathic arthritis and adult-onset still’s disease. Ann Rheum Dis. (2024) 83:1614–27. doi: 10.1136/ard-2024-225851
9. Civino A, Bovis F, Ponzano M, Alighieri G, Prete E, Sorrentino S, et al. Development and initial validation of the ONCOREUM score to differentiate childhood cancer with arthropathy from juvenile idiopathic arthritis. J Pediatr. (2023) 253:219–24.e3. doi: 10.1016/j.jpeds.2022.09.046
10. Civino A, Alighieri G, Prete E, Caroleo AM, Magni-Manzoni S, Vinti L, et al. Musculoskeletal manifestations of childhood cancer and differential diagnosis with idiopathic arthritis (ONCOREUM): a multicentre, cross-sectional study. Lancet Rheumatol. (2021) 3:e507–16. doi: 10.1016/S2665-9913(21)00086-2
11. Pardeo M, Bracaglia C, De Benedetti F. Systemic juvenile idiopathic arthritis: new insights into pathogenesis and cytokine directed therapies. Best Pract Res Clin Rheumatol. (2017) 31:505–16. doi: 10.1016/j.berh.2018.02.002
12. Beukelman T, Tomlinson G, Nigrovic PA, Dennos A, Del Gaizo V, Jelinek M, et al. First-line options for systemic juvenile idiopathic arthritis treatment: an observational study of childhood arthritis and rheumatology research alliance consensus treatment plans. Pediatr Rheumatol. (2022) 20:113. doi: 10.1186/s12969-022-00768-6
13. Hedrich CM, Bruck N, Fiebig B, Gahr M. Anakinra: a safe and effective first-line treatment in systemic onset juvenile idiopathic arthritis (SoJIA). Rheumatol Int. (2012) 32:3525–30. doi: 10.1007/s00296-011-2249-4
14. Newburger JW, Takahashi M, Gerber MA, Gewitz MH, Tani LY, Burns JC, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the Committee on Rheumatic Fever, Endocarditis and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Circulation. (2004) 110:2747–71. doi: 10.1161/01.CIR.0000145143.19711.78
15. McCrindle BW, Rowley AH, Newburger JW, Burns JC, Bolger AF, Gewitz M, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a scientific statement for health professionals from the American Heart Association. Circulation. (2017) 135:e927–99. doi: 10.1161/CIR.0000000000000484
16. Yi X, Zhang W, Wang D, Wang X, Zhang F. Research perspective in the clinical management of Kawasaki disease. Front Pediatr. (2024) 12:1415941. doi: 10.3389/fped.2024.1415941
17. Marchesi A, Tarissi de Jacobis I, Rigante D, Rimini A, Malorni W, Corsello G, et al. Kawasaki disease: guidelines of the Italian society of pediatrics, part I—definition, epidemiology, etiopathogenesis, clinical expression and management of the acute phase. Ital J Pediatr. (2018) 44:102. doi: 10.1186/s13052-018-0536-3
18. Liu J, Yue Q, Qin S, Su D, Ye B, Pang Y. Risk factors and coronary artery outcomes of coronary artery aneurysms differing in size and emergence time in children with Kawasaki disease. Front Cardiovasc Med. (2022) 9:969495. doi: 10.3389/fcvm.2022.969495
19. Morishita KA, Goldman RD. Kawasaki disease recognition and treatment. Can Fam Physician. (2020) 66:577–9.32817030
20. Brodeur KE, Liu M, Ibanez D, de Groot MJ, Chen L, Du Y, et al. Elevation of IL-17 cytokines distinguishes Kawasaki disease from other pediatric inflammatory disorders. Arthritis Rheumatol. (2024) 76:285–92. doi: 10.1002/art.42680
21. Mahmoudinezhad Dezfouli SM, Salehi S, Khosravi S. Pathogenic and therapeutic roles of cytokines in Kawasaki diseases. Clin Chim Acta. (2022) 532:21–8. doi: 10.1016/j.cca.2022.05.015
22. Qiu Y, Zhang Y, Li Y, Hua Y, Zhang Y. Molecular mechanisms of endothelial dysfunction in Kawasaki-disease-associated vasculitis. Front Cardiovasc Med. (2022) 9:981010. doi: 10.3389/fcvm.2022.981010
23. Lin J, Harahsheh AS, Raghuveer G, Jain S, Choueiter NF, Garrido-Garcia LM, et al. Emerging insights into the pathophysiology of multisystem inflammatory syndrome associated with COVID-19 in children. Can J Cardiol. (2023) 39:793–802. doi: 10.1016/j.cjca.2023.01.002
24. Mannarino S, Raso I, Garbin M, Ghidoni E, Corti C, Goletto S, et al. Cardiac dysfunction in multisystem inflammatory syndrome in children: an Italian single-center study. Ital J Pediatr. (2022) 48:25. doi: 10.1186/s13052-021-01189-z
25. Henderson LA, Canna SW, Friedman KG, Gorelik M, Lapidus SK, Bassiri H, et al. American College of rheumatology clinical guidance for multisystem inflammatory syndrome in children associated with SARS-CoV-2 and hyperinflammation in pediatric COVID-19: version 3. Arthritis Rheumatol. (2022) 74:e1–e20. doi: 10.1002/art.42062
26. Ghosh P, Katkar GD, Shimizu C, Kim J, Khandelwal S, Tremoulet AH, et al. An artificial intelligence-guided signature reveals the shared host immune response in MIS-C and Kawasaki disease. Nat Commun. (2022) 13:2687. doi: 10.1038/s41467-022-30357-w
27. Tan C, Liu F. MIS-C is likely to be distinct from Kawasaki disease based on current studies: a narrative review. Pediatr Med. (2022) 5:10. doi: 10.21037/pm-20-113
28. Principi N, Lazzara A, Paglialonga L, Viafora F, Aurelio C, Esposito S. Recurrent pericarditis and interleukin (IL)-1 inhibitors. Int Immunopharmacol. (2024) 141:113017. doi: 10.1016/j.intimp.2024.113017
29. Karmali R, Kafil TS, Bayat A, Honnekeri B, Badwan O, Berglund F, et al. Recurrent pericarditis and paradigm shift in cardiovascular imaging and targeted therapeutics. JACC Adv. (2024) 3:101194. doi: 10.1016/j.jacadv.2024.101194
30. Shahid R, Jin J, Hope K, Tunuguntla H, Amdani S. Pediatric pericarditis: update. Curr Cardiol Rep. (2023) 25:157–70. doi: 10.1007/s11886-023-01839-0
31. Ercan Tutar H, Imamoglu A, Atalay S. Recurrent pericarditis as a manifestation of familial mediterranean fever. Circulation. (2000) 101:e71–2. doi: 10.1161/01.CIR.101.5.e71
32. Andreis A, Imazio M, Casula M, Avondo S, Brucato A. Recurrent pericarditis: an update on diagnosis and management. Intern Emerg Med. (2021) 16:551–8. doi: 10.1007/s11739-021-02639-6
33. Foell D, Saers M, Park C, Brix N, Glerup M, Kessel C, et al. A novel serum calprotectin (MRP8/14) particle-enhanced immuno-turbidimetric assay (sCAL turbo) helps to differentiate systemic juvenile idiopathic arthritis from other diseases in routine clinical laboratory settings. Mol Cell Pediatr. (2023) 10:14. doi: 10.1186/s40348-023-00168-0
34. Azzarelli A, Cucco C, Cortigiani L, Scalese M, Annoni V, Bovenzi F, et al. Coronary external diameter index for assessing coronary artery involvement in Kawasaki disease. Explor Cardiol. (2023) 1:103–13. doi: 10.37349/ec.2023.00011
35. Yasin S, Fall N, Brown RA, Henderlight M, Canna SW, Girard-Guyonvarc'h C, et al. IL-18 as a biomarker linking systemic juvenile idiopathic arthritis and macrophage activation syndrome. Rheumatology (Oxford). (2020) 59:361–6. doi: 10.1093/rheumatology/kez282
36. Aljaberi N, Tronconi E, Schulert G, Grom AA, Lovell DJ, Huggins JL, et al. The use of S100 proteins testing in juvenile idiopathic arthritis and autoinflammatory diseases in a pediatric clinical setting: a retrospective analysis. Pediatr Rheumatol Online J. (2020) 18:7. doi: 10.1186/s12969-020-0398-2
37. Li T, Feng J, Li N, Liu T. Correct identification of incomplete Kawasaki disease. J Int Med Res. (2021) 49:03000605211001712. doi: 10.1177/03000605211001712
38. Rivas M N, Arditi M. Kawasaki disease and multisystem inflammatory syndrome in children. Rheum Dis Clin North Am. (2023) 49:647–59. doi: 10.1016/j.rdc.2023.03.002
39. Hinze CH, Holzinger D, Lainka E, Haas J-P, Speth F, Kallinich T, et al. Practice and consensus-based strategies in diagnosing and managing systemic juvenile idiopathic arthritis in Germany. Pediatr Rheumatol Online J. (2018) 16:7. doi: 10.1186/s12969-018-0224-2
40. Cimaz R. Systemic-onset juvenile idiopathic arthritis. Autoimmun Rev. (2016) 15:931–4. doi: 10.1016/j.autrev.2016.07.004
41. Martini A, Lovell DJ, Albani S, Brunner HI, Hyrich KL, Thompson SD, et al. Juvenile idiopathic arthritis. Nat Rev Dis Primers. (2022) 8:5. doi: 10.1038/s41572-021-00332-8
42. McCurdy D, Parsa MF. Updates in juvenile idiopathic arthritis. Adv Pediatr. (2021) 68:143–70. doi: 10.1016/j.yapd.2021.05.014
43. Beukelman T, Ringold S, Davis TE, DeWitt EM, Pelajo CF, Weiss PF, et al. Disease-modifying antirheumatic drug use in the treatment of juvenile idiopathic arthritis: a cross-sectional analysis of the CARRA registry. J Rheumatol. (2012) 39:1867–74. doi: 10.3899/jrheum.120110
44. Polito C, Strano CG, Olivieri AN, Alessio M, Iammarrone CS, Todisco N, et al. Growth retardation in non-steroid treated juvenile rheumatoid arthritis. Scand J Rheumatol. (1997) 26:99–103. doi: 10.3109/03009749709115826
45. Russo RAG, Katsicas MM. Global damage in systemic juvenile idiopathic arthritis: preliminary early predictors. J Rheumatol. (2008) 35:1151–6. Available at: https://www.jrheum.org/content/35/6/1151.long18398937
46. Batthish M, Schneider R. Chapter 3—systemic juvenile idiopathic arthritis. In: Cimaz R, Lehman T, editors. Handbook of Systemic Autoimmune Diseases. Pediatrics in Systemic Autoimmune Diseases. Amsterdam: Elsevier (2016). p. 53–84. doi: 10.1016/B978-0-444-63596-9.00003-7
47. Vitale A, Della Casa F, Lopalco G, Pereira RM, Ruscitti P, Giacomelli R, et al. Development and implementation of the AIDA international registry for patients with still’s disease. Front Med (Lausanne). (2022) 9:878797. doi: 10.3389/fmed.2022.878797
48. DeWitt EM, Kimura Y, Beukelman T, Nigrovic PA, Onel K, Prahalad S, et al. Consensus treatment plans for new-onset systemic juvenile idiopathic arthritis. Arthritis Care Res (Hoboken). (2012) 64:1001–10. doi: 10.1002/acr.21625
49. Chen K, Zeng H, Togizbayev G, Martini A, Zeng H. New classification criteria for juvenile idiopathic arthritis. Int J Rheum Dis. (2023) 26:1889–92. doi: 10.1111/1756-185X.14813
50. Shoop-Worrall SJW, Kearsley-Fleet L, Thomson W, Verstappen SMM, Hyrich KL. How common is remission in juvenile idiopathic arthritis: a systematic review. Semin Arthritis Rheum. (2017) 47:331–7. doi: 10.1016/j.semarthrit.2017.05.007
51. Singh-Grewal D, Schneider R, Bayer N, Feldman BM. Predictors of disease course and remission in systemic juvenile idiopathic arthritis: significance of early clinical and laboratory features. Arthritis Rheum. (2006) 54:1595–601. doi: 10.1002/art.21774
52. Vitale A, Caggiano V, Lopalco G, Mayrink Giardini HA, Ciccia F, Almaghlouth IA, et al. Still’s disease continuum from childhood to elderly: data from the international AIDA network still’s disease registry. RMD Open. (2023) 9:e003578. doi: 10.1136/rmdopen-2023-003578
53. Spiegel LR, Schneider R, Lang BA, Birdi N, Silverman ED, Laxer RM, et al. Early predictors of poor functional outcome in systemic-onset juvenile rheumatoid arthritis: a multicenter cohort study. Arthritis Rheum. (2000) 43:2402–9. doi: 10.1002/1529-0131(200011)43:11%3C2402::AID-ANR5%3E3.0.CO;2-C
54. Schneider R, Lang BA, Reilly BJ, Laxer RM, Silverman ED, Ibanez D, et al. Prognostic indicators of joint destruction in systemic-onset juvenile rheumatoid arthritis. J Pediatr. (1992) 120:200–5. doi: 10.1016/s0022-3476(05)80427-5
55. Sandborg C, Holmes TH, Lee T, Biederman K, Bloch DA, Emery H, et al. Candidate early predictors for progression to joint damage in systemic juvenile idiopathic arthritis. J Rheumatol. (2006) 33:2322–9. Available at: https://www.jrheum.org/content/33/11/2322.long16960920
56. Bloom BJ, Alario AJ, Miller LC. Persistent elevation of fibrin D-dimer predicts longterm outcome in systemic juvenile idiopathic arthritis. J Rheumatol. (2009) 36:422–6. doi: 10.3899/jrheum.070600
57. Kaplan MM, Ekici Tekin Z, Çelikel E, Güngörer V, Karagöl C, Öner N, et al. Predictors of disease course in systemic juvenile idiopathic arthritis. Modern Rheumatology. (2024) 34:1238–45. doi: 10.1093/mr/roae036
58. Kanemasa H, Nanishi E, Takada H, Ishimura M, Nishio H, Honjo S, et al. Overlapping features in Kawasaki disease-related arthritis and systemic-onset juvenile idiopathic arthritis: a nationwide study in Japan. Front Pediatr. (2021) 9:597458. doi: 10.3389/fped.2021.597458
59. Boyarchuk O, Kovalchuk T. Overlapping clinical features of systemic juvenile idiopathic arthritis and SARS-CoV-2-related multisystem inflammatory syndrome in children. Reumatologia. (2023) 61:63–70. doi: 10.5114/reum/161185
60. Tamashiro MS, Aikawa NE, Campos LMA, Cristofani LM, Odone-Filho V, Silva CA. Discrimination of acute lymphoblastic leukemia from systemic-onset juvenile idiopathic arthritis at disease onset. Clinics (Sao Paulo). (2011) 66:1665–9. doi: 10.1590/S1807-59322011001000001
61. Hinze CH, Foell D, Kessel C. Treatment of systemic juvenile idiopathic arthritis. Nat Rev Rheumatol. (2023) 19:778–89. doi: 10.1038/s41584-023-01042-z
62. Nijhuis L, Swart JF, Prakken BJ, van Loosdregt J, Vastert SJ. The clinical and experimental treatment of juvenile idiopathic arthritis. Clin Exp Immunol. (2023) 213:276–87. doi: 10.1093/cei/uxad045
63. Peterson RG, Xiao R, Katcoff H, Fisher BT, Weiss PF. Effect of first-line biologic initiation on glucocorticoid exposure in children hospitalized with new-onset systemic juvenile idiopathic arthritis: emulation of a pragmatic trial using observational data. Pediatr Rheumatol. (2021) 19:109. doi: 10.1186/s12969-021-00597-z
64. Vitale A, Caggiano V, Sfikakis PP, Dagna L, Lopalco G, Ragab G, et al. Efficacy of canakinumab in patients with still’s disease across different lines of biologic therapy: real-life data from the international AIDA network registry for still’s disease. Front Med (Lausanne). (2023) 10:1256243. doi: 10.3389/fmed.2023.1256243
65. Vitale A, Caggiano V, Maggio MC, Lopalco G, Emmi G, Sota J, et al. Canakinumab as first-line biological therapy in still’s disease and differences between the systemic and the chronic-articular courses: real-life experience from the international AIDA registry. Front Med (Lausanne). (2022) 9:1071732. doi: 10.3389/fmed.2022.1071732
66. Sota J, Vitale A, Lopalco G, Pereira RMR, Giordano HF, Antonelli IPB, et al. Efficacy and safety of tocilizumab in adult-onset still’s disease: real-life experience from the international AIDA registry. Semin Arthritis Rheum. (2022) 57:152089. doi: 10.1016/j.semarthrit.2022.152089
67. De Matteis A, Bracaglia C, Pires Marafon D, Piscitelli AL, Alessio M, Naddei R, et al. Canakinumab in systemic juvenile idiopathic arthritis: real-world data from a retrospective Italian cohort. Rheumatology (Oxford). (2022) 61:1621–9. doi: 10.1093/rheumatology/keab619
68. Sota J, Rigante D, Ruscitti P, Insalaco A, Sfriso P, de Vita S, et al. Anakinra drug retention rate and predictive factors of long-term response in systemic juvenile idiopathic arthritis and adult onset still disease. Front Pharmacol. (2019) 10:918. doi: 10.3389/fphar.2019.00918
69. Sota J, Vitale A, Insalaco A, Sfriso P, Lopalco G, Emmi G, et al. Safety profile of the interleukin-1 inhibitors anakinra and canakinumab in real-life clinical practice: a nationwide multicenter retrospective observational study. Clin Rheumatol. (2018) 37:2233–40. doi: 10.1007/s10067-018-4119-x
70. Nigrovic PA, Mannion M, Prince FHM, Zeft A, Rabinovich CE, van Rossum MAJ, et al. Anakinra as first-line disease-modifying therapy in systemic juvenile idiopathic arthritis: report of forty-six patients from an international multicenter series. Arthritis Rheum. (2011) 63:545–55. doi: 10.1002/art.30128
71. Pardeo M, Pires Marafon D, Insalaco A, Bracaglia C, Nicolai R, Messia V, et al. Anakinra in systemic juvenile idiopathic arthritis: a single-center experience. J Rheumatol. (2015) 42:1523–27. doi: 10.3899/jrheum.141567
72. Mastrolia MV, Abu-Rumeileh S, Maccora I, Maniscalco V, Marrani E, Pagnini I, et al. Anakinra as first-line monotherapy for new-onset steroid-naïve sJIA patients. Expert Rev Clin Immunol. (2024) 20:1293–5. doi: 10.1080/1744666X.2024.2384703
73. Giancane G, Papa R, Vastert S, Bagnasco F, Swart JF, Quartier P, et al. Anakinra in patients with systemic juvenile idiopathic arthritis: long-term safety from the pharmachild registry. J Rheumatol. (2022) 49:398–407. doi: 10.3899/jrheum.210563
74. Brunner HI, Quartier P, Alexeeva E, Constantin T, Koné-Paut I, Marzan K, et al. Efficacy and safety of canakinumab in patients with systemic juvenile idiopathic arthritis with and without fever at baseline: results from an open-label, active-treatment extension study. Arthritis Rheumatol. (2020) 72:2147–58. doi: 10.1002/art.41436
75. Ruperto N, Brunner HI, Quartier P, Constantin T, Wulffraat N, Horneff G, et al. Two randomized trials of canakinumab in systemic juvenile idiopathic arthritis. N Engl J Med. (2012) 367:2396–406. doi: 10.1056/NEJMoa1205099
76. Horneff G, Klein A, Minden K, Kallinich T, Weller-Heinemann F, Trauzeddel R, et al. LBA0008 effectiveness of canakinumab for first line steroid free treatment in systemic juvenile idiopathic arthritis. Ann Rheum Dis. (2024) 83:238–9. doi: 10.1136/annrheumdis-2024-eular.LBA46
77. Iwata N, Nishimura K, Hara R, Imagawa T, Shimizu M, Tomiita M, et al. Long-term efficacy and safety of canakinumab in the treatment of systemic juvenile idiopathic arthritis in Japanese patients: results from an open-label phase III study. Mod Rheumatol. (2023) 33:1162–70. doi: 10.1093/mr/roac128
78. Yan X, Tang W, Zhang Z, Zhang Y, Luo C, Tang X. Tocilizumab in systemic juvenile idiopathic arthritis: response differs by disease duration at medication initiation and by phenotype of disease. Front Pediatr. (2021) 9:735846. doi: 10.3389/fped.2021.735846
79. Brunner HI, Ruperto N, Ramanan AV, Horneff G, Minden K, Calvo Penades I, et al. Long-term efficacy and safety of subcutaneous tocilizumab in clinical trials of polyarticular or systemic juvenile idiopathic arthritis. Rheumatology (Oxford). (2024) 63:2535–46. doi: 10.1093/rheumatology/keae180
80. Vastert SJ, de Jager W, Noordman BJ, Holzinger D, Kuis W, Prakken BJ, et al. Effectiveness of first-line treatment with recombinant interleukin-1 receptor antagonist in steroid-naive patients with new-onset systemic juvenile idiopathic arthritis: results of a prospective cohort study. Arthritis Rheumatol. (2014) 66:1034–43. doi: 10.1002/art.38296
81. Pardeo M, Rossi MN, Pires Marafon D, Sacco E, Bracaglia C, Passarelli C, et al. Early treatment and IL1RN single-nucleotide polymorphisms affect response to anakinra in systemic juvenile idiopathic arthritis. Arthritis Rheumatol. (2021) 73:1053–61. doi: 10.1002/art.41612
82. Vannucci G, Cantarini L, Giani T, Marrani E, Moretti D, Pagnini I, et al. Glucocorticoids in the management of systemic juvenile idiopathic arthritis. Paediatr Drugs. (2013) 15:343–9. doi: 10.1007/s40272-013-0038-0
83. He T, Xia Y, Luo Y, Yang J. JAK inhibitors in systemic juvenile idiopathic arthritis. Front Pediatr. (2023) 11:1134312. doi: 10.3389/fped.2023.1134312
84. De Benedetti F, Grom AA, Brogan P, Bracaglia C, Pardeo M, Marucci G, et al. Macrophage activation syndrome (MAS) in systemic juvenile idiopathic arthritis (sJIA): treatment with emapalumab, an anti-interferon gamma (IFNγ) monoclonal antibody. Blood. (2021) 138:2058. doi: 10.1182/blood-2021-147596
85. Canna SW, De Benedetti F. The 4th NextGen therapies of SJIA and MAS, part 4: it is time for IL-18 based trials in systemic juvenile idiopathic arthritis? Pediatr Rheumatol Online J. (2024) 21:79. doi: 10.1186/s12969-023-00867-y
86. Rosina S, Rebollo-Giménez AI, Tarantola L, Pistorio A, Vyzhga Y, El Miedany Y, et al. Defining criteria for disease activity states in systemic juvenile idiopathic arthritis based on the systemic juvenile arthritis disease activity score. Arthritis Rheumatol. (2024) 76:1446–54. doi: 10.1002/art.42865
87. Tibaldi J, Pistorio A, Aldera E, Puzone L, El Miedany Y, Pal P, et al. Development and initial validation of a composite disease activity score for systemic juvenile idiopathic arthritis. Rheumatology (Oxford). (2020) 59:3505–14. doi: 10.1093/rheumatology/keaa240
88. Shimizu M. Macrophage activation syndrome in systemic juvenile idiopathic arthritis. Immunol Med. (2021) 44:237–45. doi: 10.1080/25785826.2021.1912893
89. Petrongari D, Di Filippo P, Misticoni F, Basile G, Di Pillo S, Chiarelli F, et al. Lung involvement in systemic juvenile idiopathic arthritis: a narrative review. Diagnostics (Basel). (2022) 12:3095. doi: 10.3390/diagnostics12123095
90. Ravelli A, Minoia F, Davì S, Horne A, Bovis F, Pistorio A, et al. 2016 classification criteria for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: a European League Against Rheumatism/American College of Rheumatology/Paediatric Rheumatology International Trials Organisation Collaborative Initiative. Arthritis Rheumatol. (2016) 68:566–76. doi: 10.1002/art.39332
91. Ravelli A, Minoia F, Davì S, Horne A, Bovis F, Pistorio A, et al. Expert consensus on dynamics of laboratory tests for diagnosis of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. RMD Open. (2016) 2:e000161. doi: 10.1136/rmdopen-2015-000161
92. Minoia F, Davì S, Horne A, Demirkaya E, Bovis F, Li C, et al. Clinical features, treatment, and outcome of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: a multinational, multicenter study of 362 patients. Arthritis Rheumatol. (2014) 66:3160–9. doi: 10.1002/art.38802
93. Minoia F, Davì S, Horne A, Bovis F, Demirkaya E, Akikusa J, et al. Dissecting the heterogeneity of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. J Rheumatol. (2015) 42:994–1001. doi: 10.3899/jrheum.141261
94. Schwier NC, Stephens K, Johnson PN. Management of idiopathic viral pericarditis in the pediatric population. J Pediatr Pharmacol Ther. (2022) 27:595–608. doi: 10.5863/1551-6776-27.7.595
95. Çakan M, Karadağ ŞG, Tanatar A, Ayaz NA. The frequency of macrophage activation syndrome and disease course in systemic juvenile idiopathic arthritis. Mod Rheumatol. (2020) 30:900–4. doi: 10.1080/14397595.2019.1660026
96. Aydın F, Çelikel E, Ekici Tekin Z, Coşkun S, Sezer M, Karagöl C, et al. Comparison of baseline laboratory findings of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis and multisystem inflammatory syndrome in children. Int J Rheum Dis. (2021) 24:542–7. doi: 10.1111/1756-185X.14078
97. Triggianese P, Vitale A, Lopalco G, Mayrink Giardini HA, Ciccia F, Al-Maghlouth I, et al. Clinical and laboratory features associated with macrophage activation syndrome in still’s disease: data from the international AIDA network still’s disease registry. Intern Emerg Med. (2023) 18:2231–43. doi: 10.1007/s11739-023-03408-3
98. Minoia F, Bovis F, Davì S, Insalaco A, Lehmberg K, Shenoi S, et al. Development and initial validation of the macrophage activation syndrome/primary hemophagocytic lymphohistiocytosis score, a diagnostic tool that differentiates primary hemophagocytic lymphohistiocytosis from macrophage activation syndrome. J Pediatr. (2017) 189:72–8.e3. doi: 10.1016/j.jpeds.2017.06.005
99. Knaak C, Nyvlt P, Schuster FS, Spies C, Heeren P, Schenk T, et al. Hemophagocytic lymphohistiocytosis in critically ill patients: diagnostic reliability of HLH-2004 criteria and HScore. Crit Care. (2020) 24:244. doi: 10.1186/s13054-020-02941-3
100. Cetica V, Sieni E, Pende D, Danesino C, De Fusco C, Locatelli F, et al. Genetic predisposition to hemophagocytic lymphohistiocytosis: report on 500 patients from the Italian registry. J Allergy Clin Immunol. (2016) 137:188–96.e4. doi: 10.1016/j.jaci.2015.06.048
101. Shakoory B, Geerlinks A, Wilejto M, Kernan K, Hines M, Romano M, et al. The 2022 EULAR/ACR points to consider at the early stages of diagnosis and management of suspected haemophagocytic lymphohistiocytosis/macrophage activation syndrome (HLH/MAS). Arthritis Rheumatol. (2023) 75:1714–32. doi: 10.1002/art.42636
102. Otsuka R, Seino K. Macrophage activation syndrome and COVID-19. Inflamm Regen. (2020) 40:19. doi: 10.1186/s41232-020-00131-w
103. Lee J, Bae KS, Rhim JW, Lee S-Y, Jeong DC, Kang JH. Macrophage activation syndrome in children: update on diagnosis and treatment. Children. (2024) 11:755. doi: 10.3390/children11070755
104. Eloseily EM, Weiser P, Crayne CB, Haines H, Mannion ML, Stoll ML, et al. Benefit of anakinra in treating pediatric secondary hemophagocytic lymphohistiocytosis. Arthritis Rheumatol. (2020) 72:326–34. doi: 10.1002/art.41103
105. Kahn PJ, Cron RQ. Higher-dose anakinra is effective in a case of medically refractory macrophage activation syndrome. J Rheumatol. (2013) 40:743–4. doi: 10.3899/jrheum.121098
106. Grevich S, Shenoi S. Update on the management of systemic juvenile idiopathic arthritis and role of IL-1 and IL-6 inhibition. Adolesc Health Med Ther. (2017) 8:125–35. doi: 10.2147/AHMT.S109495
107. Griffin G, Shenoi S, Hughes GC. Hemophagocytic lymphohistiocytosis: an update on pathogenesis, diagnosis, and therapy. Best Pract Res Clin Rheumatol. (2020) 34:101515. doi: 10.1016/j.berh.2020.101515
108. Jordan MB, Allen CE, Greenberg J, Henry M, Hermiston ML, Kumar A, et al. Challenges in the diagnosis of hemophagocytic lymphohistiocytosis: recommendations from the North American Consortium for Histiocytosis (NACHO). Pediatr Blood Cancer. (2019) 66:e27929. doi: 10.1002/pbc.27929
109. Sachs RN, Talvard O, Lanfranchi J. Myocarditis in adult Still’s disease. Int J Cardiol. (1990) 27:377–80. doi: 10.1016/0167-5273(90)90295-g
110. Ward SC, Wiselka MJ, Nicholson KG. Still’s disease and myocarditis associated with recent mumps infection. Postgrad Med J. (1988) 64:693–5. doi: 10.1136/pgmj.64.755.693
111. Duarte R, Cisneros S, Fernandez G, Castellon D, Cattani C, Melo C A, et al. Kawasaki disease: a review with emphasis on cardiovascular complications. Insights Imaging. (2010) 1:223–31. doi: 10.1007/s13244-010-0035-6
112. Makino N, Nakamura Y, Yashiro M, Ae R, Tsuboi S, Aoyama Y, et al. Descriptive epidemiology of Kawasaki disease in Japan, 2011–2012: from the results of the 22nd nationwide survey. J Epidemiol. (2015) 25:239–45. doi: 10.2188/jea.JE20140089
113. Chang R-KR. The incidence of Kawasaki disease in the United States did not increase between 1988 and 1997. Pediatrics. (2003) 111:1124–5. doi: 10.1542/peds.111.5.1124
114. Holman RC, Curns AT, Belay ED, Steiner CA, Schonberger LB. Kawasaki syndrome hospitalizations in the United States, 1997 and 2000. Pediatrics. (2003) 112:495–501. doi: 10.1542/peds.112.3.495
115. Holman RC, Belay ED, Christensen KY, Folkema AM, Steiner CA, Schonberger LB. Hospitalizations for Kawasaki syndrome among children in the United States, 1997–2007. Pediatr Infect Dis J. (2010) 29:483–8. doi: 10.1097/INF.0b013e3181cf8705
116. Piram M. Epidemiology of Kawasaki disease in Europe. Front Pediatr. (2021) 9:673554. doi: 10.3389/fped.2021.673554
117. Jone P-N, Tremoulet A, Choueiter N, Dominguez SR, Harahsheh AS, Mitani Y, et al. Update on diagnosis and management of Kawasaki disease: a scientific statement from the American Heart Association. Circulation. (2024) 150:e481–500. doi: 10.1161/CIR.0000000000001295
118. Falcini F, Ozen S, Magni-Manzoni S, Candelli M, Ricci L, Martini G, et al. Discrimination between incomplete and atypical Kawasaki syndrome versus other febrile diseases in childhood: results from an international registry-based study. Clin Exp Rheumatol. (2012) 30:799–804. Available at: https://www.clinexprheumatol.org/abstract.asp?a=594923020938
119. Wang H, Shimizu C, Bainto E, Hamilton S, Jackson HR, Estrada-Rivadeneyra D, et al. Subgroups of children with Kawasaki disease: a data-driven cluster analysis. The Lancet Child & Adolescent Health. (2023) 7:697–707. doi: 10.1016/S2352-4642(23)00166-9
120. Kanegaye JT, Wilder MS, Molkara D, Frazer JR, Pancheri J, Tremoulet AH, et al. Recognition of a Kawasaki disease shock syndrome. Pediatrics. (2009) 123:e783–789. doi: 10.1542/peds.2008-1871
121. Gamez-Gonzalez LB, Moribe-Quintero I, Cisneros-Castolo M, Varela-Ortiz J, Muñoz-Ramírez M, Garrido-García M, et al. Kawasaki disease shock syndrome: unique and severe subtype of Kawasaki disease. Pediatr Int. (2018) 60:781–90. doi: 10.1111/ped.13614
122. Onouchi Y. The genetics of Kawasaki disease. Int J Rheum Dis. (2018) 21:26–30. doi: 10.1111/1756-185X.13218
123. Dusser P, Koné-Paut I. Still’s disease in the constellation of hyperinflammatory syndromes: a link with Kawasaki disease? J Clin Med. (2021) 10:3244. doi: 10.3390/jcm10153244
124. Tremoulet AH, Best BM, Song S, Wang S, Corinaldesi E, Eichenfield JR, et al. Resistance to intravenous immunoglobulin in children with Kawasaki disease. J Pediatr. (2008) 153:117–21. doi: 10.1016/j.jpeds.2007.12.021
125. Bar-Meir M, Kalisky I, Schwartz A, Somekh E, Tasher D, Group IK. Prediction of resistance to intravenous immunoglobulin in children with Kawasaki disease. J Pediatric Infect Dis Soc. (2018) 7:25–9. doi: 10.1093/jpids/piw075
126. Burns JC, Capparelli EV, Brown JA, Newburger JW, Glode MP. Intravenous gamma-globulin treatment and retreatment in Kawasaki disease. US/Canadian Kawasaki syndrome study group. Pediatr Infect Dis J. (1998) 17:1144–8. doi: 10.1097/00006454-199812000-00009
127. Chen S, Dong Y, Yin Y, Krucoff MW. Intravenous immunoglobulin plus corticosteroid to prevent coronary artery abnormalities in Kawasaki disease: a meta-analysis. Heart. (2013) 99:76–82. doi: 10.1136/heartjnl-2012-302126
128. Tremoulet AH, Jain S, Kim S, Newburger J, Arditi M, Franco A, et al. Rationale and study design for a phase I/IIa trial of anakinra in children with Kawasaki disease and early coronary artery abnormalities (the ANAKID trial). Contemp Clin Trials. (2016) 48:70–5. doi: 10.1016/j.cct.2016.04.002
129. Burns JC, Koné-Paut I, Kuijpers T, Shimizu C, Tremoulet A, Arditi M. Review: found in translation: international initiatives pursuing interleukin-1 blockade for treatment of acute Kawasaki disease. Arthritis Rheumatol. (2017) 69:268–76. doi: 10.1002/art.39975
130. Campbell AJ, Burns JC. Adjunctive therapies for Kawasaki disease. J Infect. (2016) 72(Suppl):S1–5. doi: 10.1016/j.jinf.2016.04.015
131. Burns JC, Roberts SC, Tremoulet AH, He F, Printz BF, Ashouri N, et al. Infliximab versus second intravenous immunoglobulin for treatment of resistant Kawasaki disease in the USA (KIDCARE): a randomised, multicentre comparative effectiveness trial. Lancet Child Adolesc Health. (2021) 5:852–61. doi: 10.1016/S2352-4642(21)00270-4
132. Go E, van Veenendaal M, Manlhiot C, Schneider R, McCrindle BW, Yeung RSM. Kawasaki disease and systemic juvenile idiopathic arthritis—two ends of the same Spectrum. Front Pediatr. (2021) 9:665815. doi: 10.3389/fped.2021.665815
133. Mastrolia MV, Abbati G, Signorino C, Maccora I, Marrani E, Pagnini I, et al. Early anti IL-1 treatment replaces steroids in refractory Kawasaki disease: clinical experience from two case reports. Ther Adv Musculoskelet Dis. (2021) 13:1759720X211002593. doi: 10.1177/1759720X211002593
134. Guillaume M-P, Reumaux H, Dubos F. Usefulness and safety of anakinra in refractory Kawasaki disease complicated by coronary artery aneurysm. Cardiol Young. (2018) 28:739–42. doi: 10.1017/S1047951117002864
135. Kabbaha S, Milano A, Aldeyab MA, Thorlund K. Infliximab as a second-line therapy for children with refractory Kawasaki disease: a systematic review and meta-analysis of randomized controlled trials. Br J Clin Pharmacol. (2023) 89:49–60. doi: 10.1111/bcp.15547
136. Wang L, He M, Wang W, Li S, Zhao G. Efficacy and safety of infliximab in the treatment of Kawasaki disease: a systematic review and meta-analysis. Eur J Pediatr. (2024) 183:1765–76. doi: 10.1007/s00431-024-05437-2
137. Kato T, Miura M, Kobayashi T, Kaneko T, Fukushima N, Suda K, et al. Analysis of coronary arterial aneurysm regression in patients with Kawasaki disease by aneurysm severity: factors associated with regression. J Am Heart Assoc. (2023) 12:e022417. doi: 10.1161/JAHA.121.022417
138. Lee JJY, Lin E, Widdifield J, Mahood Q, McCrindle BW, Yeung RSM, et al. The long-term cardiac and noncardiac prognosis of Kawasaki disease: a systematic review. Pediatrics. (2022) 149:e2021052567. doi: 10.1542/peds.2021-052567
139. Wang W, Gong F, Zhu W, Fu S, Zhang Q. Macrophage activation syndrome in Kawasaki disease: more common than we thought? Semin Arthritis Rheum. (2015) 44:405–10. doi: 10.1016/j.semarthrit.2014.07.007
140. Rhee S, Kim D, Cho K, Rhim JW, Lee S-Y, Jeong DC. Under-recognized macrophage activation syndrome in refractory Kawasaki disease: a wolf in sheep’s clothing. Children (Basel). (2022) 9:1588. doi: 10.3390/children9101588
141. Han SB, Lee SY, Jeong DC, Kang JH. Should 2016 criteria for macrophage activation syndrome be applied in children with Kawasaki disease, as well as with systemic-onset juvenile idiopathic arthritis? Ann Rheum Dis. (2016) 75:e44. doi: 10.1136/annrheumdis-2016-209395
142. Corinaldesi E, Fabi M, Scalabrini I, Praticò ER, Andreozzi L, Torcetta F, et al. Kawasaki disease complicated with macrophage activation syndrome: the importance of prompt diagnosis and treatment–three case reports. Rheumato. (2023) 3:201–9. doi: 10.3390/rheumato3040015
143. Caorsi R, Consolaro A, Speziani C, Sozeri B, Ulu K, Faugier-Fuentes E, et al. The HyperPed-COVID international registry: impact of age of onset, disease presentation and geographical distribution on the final outcome of MIS-C. J Autoimmun. (2024) 147:103265. doi: 10.1016/j.jaut.2024.103265
144. Chen M-R, Kuo H-C, Lee Y-J, Chi H, Li SC, Lee H-C, et al. Phenotype, susceptibility, autoimmunity, and immunotherapy between Kawasaki disease and coronavirus disease-19 associated multisystem inflammatory syndrome in children. Front Immunol. (2021) 12:632890. doi: 10.3389/fimmu.2021.632890
145. Pouletty M, Borocco C, Ouldali N, Caseris M, Basmaci R, Lachaume N, et al. Paediatric multisystem inflammatory syndrome temporally associated with SARS-CoV-2 mimicking Kawasaki disease (kawa-COVID-19): a multicentre cohort. Ann Rheum Dis. (2020) 79:999–1006. doi: 10.1136/annrheumdis-2020-217960
146. Ravelli A. MIS-C and Kawasaki disease: different illnesses or part of the same spectrum? Glob Pediatr. (2024) 8:100150. doi: 10.1016/j.gpeds.2024.100150
147. Yousaf AR. Notes from the field: surveillance for multisystem inflammatory syndrome in children—United States, 2023. MMWR Morb Mortal Wkly Rep. (2024) 73:225–8. doi: 10.15585/mmwr.mm7310a2
148. La Torre F, Elicio MP, Monno VA, Chironna M, Moramarco F, Campanozzi A, et al. Incidence and prevalence of multisystem inflammatory syndrome in children (MIS-C) in southern Italy. Children (Basel). (2023) 10:766. doi: 10.3390/children10050766
149. La Torre F, Taddio A, Conti C, Cattalini M. Multi-inflammatory syndrome in children (MIS-C) in 2023: is it time to forget about it? Children (Basel). (2023) 10:980. doi: 10.3390/children10060980
150. World Health Organization. Multisystem inflammatory syndrome in children and adolescents with COVID-19 (2020). Available at: https://www.who.int/publications/i/item/multisystem-inflammatory-syndrome-in-children-and-adolescents-with-covid-19 (last accessed December 16, 2024).
151. Khan RS, Ordog T, Hong SD, Schmitz AH, Thattaliyath B, Sharathkumar AA. Evolution of cardiovascular findings in multisystem inflammatory syndrome in children (MIS-C) across COVID-19 variants: common trends and unusual presentations. Pediatr Cardiol. (2024) 45:552–9. doi: 10.1007/s00246-023-03397-2
152. Valverde I, Singh Y, Sanchez-de-Toledo J, Theocharis P, Chikermane A, Di Filippo S, et al. Acute cardiovascular manifestations in 286 children with multisystem inflammatory syndrome associated with COVID-19 infection in Europe. Circulation. (2021) 143:21–32. doi: 10.1161/CIRCULATIONAHA.120.050065
153. Stasiak A, Kędziora P, Smolewska E. Complications of multisystem inflammatory syndrome associated with SARS-CoV-2 infection—many facets of one disease—a literature review based on a case report. J Clin Med. (2024) 13:4146. doi: 10.3390/jcm13144146
154. Feldstein LR, Tenforde MW, Friedman KG, Newhams M, Rose EB, Dapul H, et al. Characteristics and outcomes of US children and adolescents with multisystem inflammatory syndrome in children (MIS-C) compared with severe acute COVID-19. JAMA. (2021) 325:1074–87. doi: 10.1001/jama.2021.2091
155. Friedman KG, Harrild DM, Newburger JW. Cardiac dysfunction in multisystem inflammatory syndrome in children. J Am Coll Cardiol. (2020) 76:1962–4. doi: 10.1016/j.jacc.2020.09.002
156. Alsaied T, Tremoulet AH, Burns JC, Saidi A, Dionne A, Lang SM, et al. Review of cardiac involvement in multisystem inflammatory syndrome in children. Circulation. (2021) 143:78–88. doi: 10.1161/CIRCULATIONAHA.120.049836
157. Belhadjer Z, Méot M, Bajolle F, Khraiche D, Legendre A, Abakka S, et al. Acute heart failure in multisystem inflammatory syndrome in children in the context of global SARS-CoV-2 pandemic. Circulation. (2020) 142:429–36. doi: 10.1161/CIRCULATIONAHA.120.048360
158. Theocharis P, Wong J, Pushparajah K, Mathur SK, Simpson JM, Pascall E, et al. Multimodality cardiac evaluation in children and young adults with multisystem inflammation associated with COVID-19. Eur Heart J Cardiovasc Imaging. (2021) 22:896–903. doi: 10.1093/ehjci/jeaa212
159. Yasuhara J, Masuda K, Watanabe K, Shirasu T, Takagi H, Sumitomo N, et al. Longitudinal cardiac outcomes of multisystem inflammatory syndrome in children: a systematic review and meta-analysis. Pediatr Cardiol. (2023) 44:892–907. doi: 10.1007/s00246-022-03052-2
160. Wu EY, Campbell MJ. Cardiac manifestations of multisystem inflammatory syndrome in children (MIS-C) following COVID-19. Curr Cardiol Rep. (2021) 23:168. doi: 10.1007/s11886-021-01602-3
161. Liu K, Yu J, Song G. Global myocardial strain in multisystem inflammatory syndrome in children, Kawasaki disease, and healthy children: a network meta-analysis. Front Pediatr. (2022) 10:848306. doi: 10.3389/fped.2022.848306
162. Anagnostopoulou A, Dourdouna M-M, Loukopoulou S, Mpourazani E, Poulakis M, Karanasios E, et al. Longitudinal cardiac evaluation of children with multisystem inflammatory syndrome (MIS-C) following COVID-19 by conventional and speckle-tracking echocardiography. Pediatr Cardiol. (2024) 45:1110–9. doi: 10.1007/s00246-023-03375-8
163. Sirico D, Basso A, Sabatino J, Reffo E, Cavaliere A, Biffanti R, et al. Evolution of echocardiographic and cardiac magnetic resonance imaging abnormalities during follow-up in patients with multisystem inflammatory syndrome in children. Eur Heart J Cardiovasc Imaging. (2022) 23:1066–74. doi: 10.1093/ehjci/jeac096
164. Aakre KM, Meex SJR, Mills NL. Cardiac troponin reporting in children and young adults: time to change clinical practice. Circulation. (2023) 148:17–9. doi: 10.1161/CIRCULATIONAHA.123.065297
165. Eggers KM, Jernberg T, Lindahl B. Cardiac troponin elevation in patients without a specific diagnosis. J Am Coll Cardiol. (2019) 73:1–9. doi: 10.1016/j.jacc.2018.09.082
166. Haq K, Anyalechi EG, Schlaudecker EP, McKay R, Kamidani S, Manos CK, et al. Multiple MIS-C readmissions and giant coronary aneurysm after COVID-19 illness and vaccination: a case report. Pediatr Infect Dis J. (2023) 42:e64–9. doi: 10.1097/INF.0000000000003801
167. Henderson LA, Canna SW, Friedman KG, Gorelik M, Lapidus SK, Bassiri H, et al. American College of rheumatology clinical guidance for multisystem inflammatory syndrome in children associated with SARS-CoV-2 and hyperinflammation in pediatric COVID-19: version 2. Arthritis Rheumatol. (2021) 73:e13–29. doi: 10.1002/art.41616
168. Wang W-Y, Wang Y-J, An C-X, Zhao Q-J, Wang S-Y, Li W-Y, et al. Multisystem inflammatory syndrome (MIS-C) with SARS-CoV-2 omicron variant BA.2.38 in a four-year-old Chinese girl: a case report. Front Public Health. (2022) 10:1021200. doi: 10.3389/fpubh.2022.1021200
169. Buda P, Strauss E, Januszkiewicz-Lewandowska D, Czerwinska E, Ludwikowska K, Szenborn L, et al. Clinical characteristics of children with MIS-C fulfilling classification criteria for macrophage activation syndrome. Front Pediatr. (2022) 10:981711. doi: 10.3389/fped.2022.981711
170. Pfeifer J, Thurner B, Kessel C, Fadle N, Kheiroddin P, Regitz E, et al. Autoantibodies against interleukin-1 receptor antagonist in multisystem inflammatory syndrome in children: a multicentre, retrospective, cohort study. Lancet Rheumatol. (2022) 4:e329–37. doi: 10.1016/S2665-9913(22)00064-9
171. Taddio A, Della Paolera S, Abbagnato L, Agrusti A, Badolato R, Biscaro F, et al. Early anakinra treatment improves cardiac outcome of multisystem inflammatory syndrome in children, regardless of disease severity. Rheumatology (Oxford). (2024) 63:366–75. doi: 10.1093/rheumatology/kead381
172. Son MBF, Murray N, Friedman K, Young CC, Newhams MM, Feldstein LR, et al. Multisystem inflammatory syndrome in children—initial therapy and outcomes. N Engl J Med. (2021) 385:23–34. doi: 10.1056/NEJMoa2102605
173. Cattalini M, Taddio A, Bracaglia C, Cimaz R, Paolera SD, Filocamo G, et al. Childhood multisystem inflammatory syndrome associated with COVID-19 (MIS-C): a diagnostic and treatment guidance from the rheumatology study group of the Italian society of pediatrics. Ital J Pediatr. (2021) 47:24. doi: 10.1186/s13052-021-00980-2
174. Licciardi F, Covizzi C, Dellepiane M, Olivini N, Mastrolia MV, Lo Vecchio A, et al. Outcomes of MIS-C patients treated with anakinra: a retrospective multicenter national study. Front Pediatr. (2023) 11:1137051. doi: 10.3389/fped.2023.1137051
175. La Torre F, Calabrese G, Signorile K, Bizzoco F, Mastrorilli C, Strippoli A, et al. Efficacy of sildenafil and high-dose anakinra in an MIS-C patient with pulmonary vasculitis: a case report. Front Pediatr. (2022) 10:1015617. doi: 10.3389/fped.2022.1015617
176. Sık G, Inamlık A, Akçay N, Kesici S, Aygun F, Kendırlı T, et al. Mortality risk factors among critically ill children with MIS-C in PICUs: a multicenter study. Pediatr Res. (2023) 94:730–7. doi: 10.1038/s41390-023-02518-0
177. Bartoszek M, Małek ŁA, Barczuk-Falęcka M, Brzewski M. Cardiac magnetic resonance follow-up of children after pediatric inflammatory multisystem syndrome temporally associated with SARS-CoV-2 with initial cardiac involvement. J Magn Reson Imaging. (2022) 55:883–91. doi: 10.1002/jmri.27870
178. Ratnapalan S, Brown K, Benson L. Children presenting with acute pericarditis to the emergency department. Pediatr Emerg Care. (2011) 27:581–5. doi: 10.1097/PEC.0b013e31822251ba
179. Krasic S, Prijic S, Ninic S, Borovic R, Petrovic G, Stajevic M, et al. Predictive factors of recurrence after pediatric acute pericarditis. J Pediatr (Rio J). (2020) 97:335–41. doi: 10.1016/j.jped.2020.06.007
180. Imazio M, Brucato A, Pluymaekers N, Breda L, Calabri G, Cantarini L, et al. Recurrent pericarditis in children and adolescents: a multicentre cohort study. J Cardiovasc Med (Hagerstown). (2016) 17:707–12. doi: 10.2459/JCM.0000000000000300
181. Adler Y, Charron P, Imazio M, Badano L, Barón-Esquivias G, Bogaert J, et al. 2015 ESC guidelines for the diagnosis and management of pericardial diseases: the Task Force for the Diagnosis and Management of Pericardial Diseases of the European Society of Cardiology (ESC) Endorsed by: the European Association for Cardio-Thoracic Surgery (EACTS). Eur Heart J. (2015) 36:2921–64. doi: 10.1093/eurheartj/ehv318
182. Tombetti E, Giani T, Brucato A, Cimaz R. Recurrent pericarditis in children and adolescents. Front Pediatr. (2019) 7:419. doi: 10.3389/fped.2019.00419
183. Lilly LS. Treatment of acute and recurrent idiopathic pericarditis. Circulation. (2013) 127:1723–6. doi: 10.1161/CIRCULATIONAHA.111.066365
184. Caorsi R, Insalaco A, Bovis F, Martini G, Cattalini M, Chinali M, et al. Pediatric recurrent pericarditis: appropriateness of the standard of care and response to IL-1 blockade. J Pediatr. (2023) 256:18–26.e8. doi: 10.1016/j.jpeds.2022.11.034
185. Imazio M, Bobbio M, Cecchi E, Demarie D, Demichelis B, Pomari F, et al. Colchicine in addition to conventional therapy for acute pericarditis. Circulation. (2005) 112:2012–6. doi: 10.1161/CIRCULATIONAHA.105.542738
186. Riassunto Delle Caratteristiche Del Prodotto. Available at: https://api.aifa.gov.it/aifa-bdf-eif-be/1.0.0/organizzazione/1565/farmaci/9964/stampati?ts=RCP (last accessed December 18, 2024).
187. Finetti M, Insalaco A, Cantarini L, Meini A, Breda L, Alessio M, et al. Long-term efficacy of interleukin-1 receptor antagonist (anakinra) in corticosteroid-dependent and colchicine-resistant recurrent pericarditis. J Pediatr. (2014) 164:1425–31.e1. doi: 10.1016/j.jpeds.2014.01.065
188. Brucato A, Imazio M, Gattorno M, Lazaros G, Maestroni S, Carraro M, et al. Effect of anakinra on recurrent pericarditis among patients with colchicine resistance and corticosteroid dependence: the AIRTRIP randomized clinical trial. JAMA. (2016) 316:1906–12. doi: 10.1001/jama.2016.15826
189. Fowler NO. Recurrent pericarditis. Cardiol Clin. (1990) 8:621–6. doi: 10.1016/S0733-8651(18)30334-5
190. Wulffraat NM, Rijkers GT, Elst E, Brooimans R, Kuis W. Reduced perforin expression in systemic juvenile idiopathic arthritis is restored by autologous stem-cell transplantation. Rheumatology. (2003) 42:375–9. doi: 10.1093/rheumatology/keg074
191. Takahara T, Shimizu M, Nakagishi Y, Kinjo N, Yachie A. Serum IL-18 as a potential specific marker for differentiating systemic juvenile idiopathic arthritis from incomplete Kawasaki disease. Rheumatol Int. (2015) 35:81–4. doi: 10.1007/s00296-014-3059-2
192. Rodriguez-Smith JJ, Verweyen EL, Clay GM, Esteban YM, de Loizaga SR, Baker EJ, et al. Inflammatory biomarkers in COVID-19-associated multisystem inflammatory syndrome in children, Kawasaki disease, and macrophage activation syndrome: a cohort study. Lancet Rheumatol. (2021) 3:e574–84. doi: 10.1016/S2665-9913(21)00139-9
193. Hinze T, Kessel C, Hinze CH, Seibert J, Gram H, Foell D. A dysregulated interleukin-18-interferon-γ-CXCL9 axis impacts treatment response to canakinumab in systemic juvenile idiopathic arthritis. Rheumatology (Oxford). (2021) 60:5165–74. doi: 10.1093/rheumatology/keab113
194. Fordham NJ, Bartram J, Ghorashian S, O'Connor D, Taylor A, Sibson K, et al. What is the diagnostic yield of bone marrow aspiration to exclude leukaemia prior to systemic treatment in juvenile idiopathic arthritis? Br J Haematol. (2022) 199:447–51. doi: 10.1111/bjh.18413
195. Henter J-I, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. (2007) 48:124–31. doi: 10.1002/pbc.21039
196. Malcova H, Milota T, Strizova Z, Cebecauerova D, Striz I, Sediva A, et al. Interleukin-1 blockade in polygenic autoinflammatory disorders: where are we now? Front Pharmacol. (2020) 11:619273. doi: 10.3389/fphar.2020.619273
197. Devaux CA, Melenotte C, Piercecchi-Marti M-D, Delteil C, Raoult D. Cyclosporin A: a repurposable drug in the treatment of COVID-19? Front Med. (2021) 8:663708. doi: 10.3389/fmed.2021.663708
198. Gámez-González LB, Hamada H, Yamazaki-Nakashimada MA. Cyclosporin for treatment of refractory multisystemic inflammatory syndrome in a child. Cardiol Young. (2023) 33:800–2. doi: 10.1017/S1047951122002748
199. Keenan C, Nichols KE, Albeituni S. Use of the JAK inhibitor ruxolitinib in the treatment of hemophagocytic lymphohistiocytosis. Front Immunol. (2021) 12:614704. doi: 10.3389/fimmu.2021.614704
200. Caorsi R, Bertoni A, Matucci-Cerinic C, Natoli V, Palmeri S, Rosina S, et al. Long-term efficacy of MAS825, a bispecific anti-IL1β and IL-18 monoclonal antibody, in two patients with sJIA and recurrent episodes of MAS. Rheumatology (Oxford). (2025) 64:1528–33. doi: 10.1093/rheumatology/keae440
201. Chiabrando JG, Bonaventura A, Vecchié A, Wohlford GF, Mauro AG, Jordan JH, et al. Management of acute and recurrent pericarditis: jACC state-of-the-art review. J Am Coll Cardiol. (2020) 75:76–92. doi: 10.1016/j.jacc.2019.11.021
202. Imazio M, Brucato A, Cemin R, Ferrua S, Belli R, Maestroni S, et al. Colchicine for recurrent pericarditis (CORP): a randomized trial. Ann Intern Med. (2011) 155:409–14. doi: 10.7326/0003-4819-155-7-201110040-00359
203. Imazio M, Bobbio M, Cecchi E, Demarie D, Pomari F, Moratti M, et al. Colchicine as first-choice therapy for recurrent pericarditis: results of the CORE (COlchicine for REcurrent pericarditis) trial. Arch Intern Med. (2005) 165:1987–91. doi: 10.1001/archinte.165.17.1987
204. Imazio M, Belli R, Brucato A, Cemin R, Ferrua S, Beqaraj F, et al. Efficacy and safety of colchicine for treatment of multiple recurrences of pericarditis (CORP-2): a multicentre, double-blind, placebo-controlled, randomised trial. Lancet. (2014) 383:2232–7. doi: 10.1016/S0140-6736(13)62709-9
205. Imazio M, Belli R, Brucato A, Ferrazzi P, Patrini D, Martinelli L, et al. Rationale and design of the COlchicine for prevention of the post-pericardiotomy syndrome and post-operative atrial fibrillation (COPPS-2 trial): a randomized, placebo-controlled, multicenter study on the use of colchicine for the primary prevention of the postpericardiotomy syndrome, postoperative effusions, and postoperative atrial fibrillation. Am Heart J. (2013) 166:13–9. doi: 10.1016/j.ahj.2013.03.025
206. Abadie BQ, Cremer PC. Interleukin-1 antagonists for the treatment of recurrent pericarditis. BioDrugs. (2022) 36:459–72. doi: 10.1007/s40259-022-00537-7
207. Dincer Z T, Karup S, Yilmaz E, Corbali O, Azman FN, Melikoglu M, et al. Anakinra in idiopathic recurrent pericarditis: a comprehensive case series and literature review. Z Rheumatol. (2024) 83:587–96. doi: 10.1007/s00393-023-01471-x
208. Scott IC, Hajela V, Hawkins PN, Lachmann HJ. A case series and systematic literature review of anakinra and immunosuppression in idiopathic recurrent pericarditis. J Cardiol Cases. (2011) 4:e93–7. doi: 10.1016/j.jccase.2011.07.003
Keywords: hyperinflammation, still disease, Kawasaki disease, MIS-C multisystem inflammatory syndrome, recurrent pericarditis, biologics, IL-1, IL-6
Citation: La Torre F, Meliota G, Civino A, Campanozzi A, Cecinati V, Rosati E, Sacco E, Santoro N, Vairo U and Cardinale F (2025) Advancing multidisciplinary management of pediatric hyperinflammatory disorders. Front. Pediatr. 13:1553861. doi: 10.3389/fped.2025.1553861
Received: 31 December 2024; Accepted: 3 April 2025;
Published: 30 April 2025.
Edited by:
Lovro Lamot, University of Zagreb, CroatiaReviewed by:
Emanuela Del Giudice, Sapienza University of Rome, ItalyDragana Lazarevic, University Clinical Center Nis, Serbia
Copyright: © 2025 La Torre, Meliota, Civino, Campanozzi, Cecinati, Rosati, Sacco, Santoro, Vairo and Cardinale. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesco La Torre, bGF0b3JyZV9mcmFuY2VzY29AdmlyZ2lsaW8uaXQ=