 Qingbao He1,2
Qingbao He1,2 Lei Zhang
Lei Zhang- 1Department of Minimally Invasive Urological Surgery, Children's Hospital Affiliated to Shandong University, Jinan, China
- 2Department of Minimally Invasive Urological Surgery, Jinan Children's Hospital, Jinan, China
Background: Junctional epidermolysis bullosa (JEB) is a rare inherited blistering disorder, and its urological spectrum remains poorly defined.
Case Presentation: A 19-month-old boy carrying compound heterozygous ITGB4 mutations (p.R252C, p.P305l) had a 17-month history of intermittent voiding. Ultrasound demonstrated focal papillomatous bladder-wall thickening, and cystoscopy showed scattered follicular mucosal changes without masses. Biopsies revealed mild oedema and chronic lymphocytic inflammation; no malignancy. Urine cultures were negative.
Conclusion: This case broadens the reported urological spectrum of ITGB4-related JEB by illustrating a papillomatous–follicular bladder phenotype. Early urological evaluation in patients with JEB presenting with unexplained urinary symptoms may facilitate timely, targeted management and help prevent chronic complications.
Introduction
Epidermolysis bullosa (EB) comprises a group of inherited mechanobullous disorders with an overall incidence of ≈1: 50,000 live births, of which junctional EB (JEB) accounts for 5%–10% (1, 2). According to the 2020 consensus reclassification (3), EB is subdivided into EB simplex, JEB, dystrophic EB and Kindler EB, each defined by the ultrastructural level of tissue cleavage and its causative genes. In addition to ITGB4-related JEB, bladder involvement has also been reported in JEB caused by laminin-332 gene variants (LAMA3, LAMB3, LAMC2), manifesting as urethral strictures, meatal stenosis or severe inflammatory cystitis (4, 5). JEB involves tissue separation at the lamina lucida within the basement membrane zone (6). It typically arises from mutations in genes encoding hemidesmosome components or associated integrins, compromising dermal-epidermal adhesion and causing widespread skin fragility, mucosal involvement, and multisystem complications (1).
The ITGB4 gene, encoding the β4 subunit of the α6β4 integrin complex (a vital hemidesmosome component anchoring basal keratinocytes), is particularly significant (7). ITGB4 mutations are classically linked to JEB with pyloric atresia (JEB-PA) (7), but phenotypic variability exists. ITGB4 mutations also occur in patients with JEB without pyloric atresia or with milder/late-onset skin issues alongside significant extracutaneous involvement, including severe uropathy (8).
Urological complications occur in various EB forms, and patients with JEB are particularly susceptible. Documented issues include meatal (urethral) stenosis (up to 11.6% in JEB-Herlitz), urinary retention (9.3%), hydronephrosis (7.0%), bladder-wall hypertrophy (4.6%), and cystitis (9). The urothelium, like epidermis, relies on robust cell-matrix adhesion; α6β4 integrin disruptions can predispose it to damage (10). While skin and gastrointestinal JEB manifestations are well-documented, and general bladder pathologies (mucosal blistering, wall thickening, fibrosis, polypoid masses) are anecdotally noted in EB (4), specific bladder phenotypes linked to ITGB4 mutations are largely uncharacterized.
We report a 19-month-old boy with junctional epidermolysis bullosa carrying compound-heterozygous ITGB4 variants who fulfils the diagnostic criteria for JEB with pyloric atresia (JEB-PA); the pyloric atresia was surgically corrected in the neonatal period. Here we present the clinical, imaging and genetic findings of this case and discuss their implications. We discuss the potential pathophysiological relevance of these changes, their relationship to the underlying ITGB4 deficiency, and how they further broaden the recognised genitourinary spectrum of ITGB4-related disease.
Case presentation
Patient history and clinical findings
Skin fragility had been mild and episodic since birth; consequently, a definitive diagnosis of epidermolysis bullosa was not established until comprehensive genetic testing at 19 months of age. A 19-month-old boy presented with a 17-month history of difficult, interrupted urine streams noted by caregivers, accompanied by occasional discomfort. Congenital pyloric atresia was corrected surgically neonatally. At 2 months, he developed intermittent urinary difficulty with white flocculent material in his urine. A basic urinalysis at that age showed mild pyuria and hematuria; urine culture and crystal analysis were not performed. Antibiotics provided temporary relief, but episodes recurred. An ultrasound at 6 months revealed bilateral ureteral dilation; voiding cystourethrography showed no vesicoureteral reflux. Ureteral dilation resolved by 12 months. At 19 months, ultrasound revealed a focal bladder wall thickening (∼0.7 cm) that protruded into the lumen, exhibiting a papillomatous appearance (Figure 1).
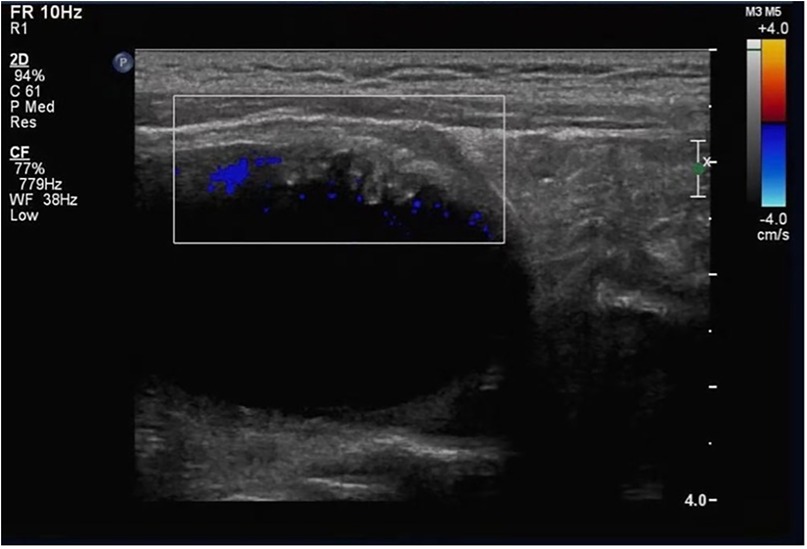
Figure 1. Ultrasound findings of the bladder wall. Localized papillomatous thickening of the bladder wall protruding into the bladder lumen, with visible vascular flow signal (color Doppler).
He is the son of nonconsanguineous, asymptomatic parents; his older sister is unaffected. Physical examination showed partial nail loss/thickening, occasional subungual blistering, and multiple oral mucosal ulcers, consistent with EB. Genital/external urinary exams were normal. Cardiorespiratory and abdominal examinations were unremarkable.
Laboratory tests
Urinalysis performed at 19 months revealed mild hematuria (red blood cells 306.5 cells µl−1) and pyuria (white blood cells 12.6 cells µl−1), with trace proteinuria; urine culture was negative.
Imaging
Bladder ultrasound showed localized wall thickening protruding into the lumen, with a papillomatous appearance (Figure 1). Color Doppler confirmed vascular flow.
Cystoscopy and histopathology
Cystoscopy revealed scattered, raised lesions exhibiting follicular mucosal changes on the bladder mucosa. Surrounding mucosa appeared pale and edematous. No solid masses were identified (Supplementary Figure 1). These projections were distinct from typical diffuse inflammatory changes. Bladder dome biopsies showed mild edema and chronic lymphocytic inflammatory infiltrate with vascular congestion. The urothelium was largely intact and no malignant cells were identified (Figure 2). Immunohistochemistry: Cytokeratin (+), Vimentin (+), PAX-8 (+), PAX-2 (+), few CyclinD1 (+) cells, SMA (−), BCL-2 (−), Ki67 <1%. Findings were consistent with chronic mucosal inflammation and no neoplasia; viral immunostains for human papillomavirus (p16) and polyomavirus SV40 were not performed owing to limited tissue.
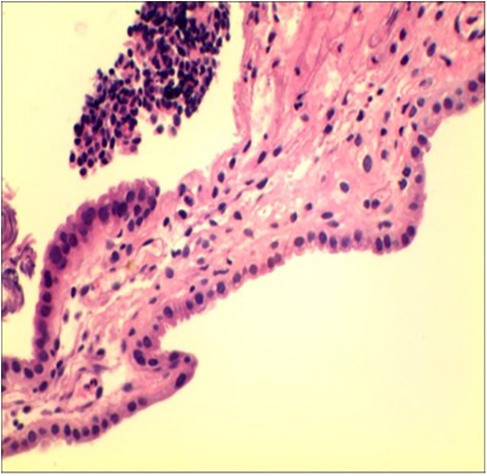
Figure 2. Histopathological examination of the bladder dome biopsy. Mild inflammation is observed, with edema and lymphocytic infiltration.
Genetic testing
Genetic analysis identified compound heterozygous ITGB4 mutations [c.754C>T (p.R252C) and c.914C>T (p.P305l)] (Supplementary Figure 2). Each mutation was inherited from an asymptomatic parent, consistent with autosomal recessive inheritance (Figures 3A,B; Supplementary Figures 3A,B). Bioinformatic tools (SIFT, PolyPhen-2, MutationTaster) suggested these variants were likely damaging. According to the American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP) criteria, c.754C>T (p.Arg252Cys) meets evidence codes PVS1, PM1, PM2 and PP3 and is therefore classified as pathogenic, whereas c.914C>T (p.Pro305Leu) meets PM2 and PP3 and is classified as likely pathogenic. Both variants were queried against multiple population and clinical databases. They are absent from gnomAD v3.1 and v4.0 (allele count = 0). c.754C>T (p.Arg252Cys) is recorded in ClinVar and HGMD as pathogenic, whereas c.914C>T (p.Pro305Leu) is not listed in either database. The complete absence of population frequency, together with existing clinical annotations, further supports their pathogenicity. In-silico pathogenicity was further assessed with SIFT v6.2 (J. Craig Venter Institute, Rockville, USA); PolyPhen-2 v2.2 (Harvard University, Cambridge, USA); and MutationTaster (Charité—Universitätsmedizin Berlin, Germany). Structural modelling suggested p.Arg252Cys might disrupt hydrogen bonds, destabilising the integrin α6β4 complex, and p.Pro305Leu might affect protein flexibility.
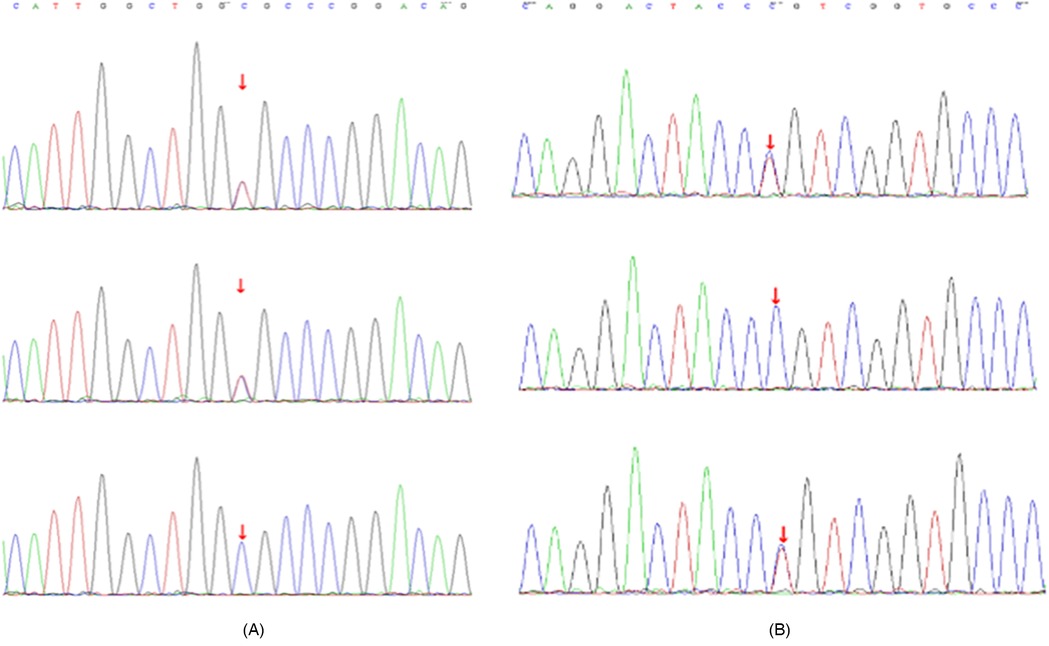
Figure 3. Sanger sequencing results of ITGB4 mutations. (A) Sanger sequencing showing the ITGB4 c.754C>T (p.R252C) mutation in the proband (top panel), father (middle panel), and mother (bottom panel). (B) Sanger sequencing showing the ITGB4 c.914C>T (p.P305l) mutation in the proband (top panel), father (middle panel), and mother (bottom panel).
Structural modeling suggested p.R252C might disrupt hydrogen bonds, destabilizing the integrin α6β4 complex (Figures 4A,B), and p.P305l might affect protein flexibility (Figures 5A,B).
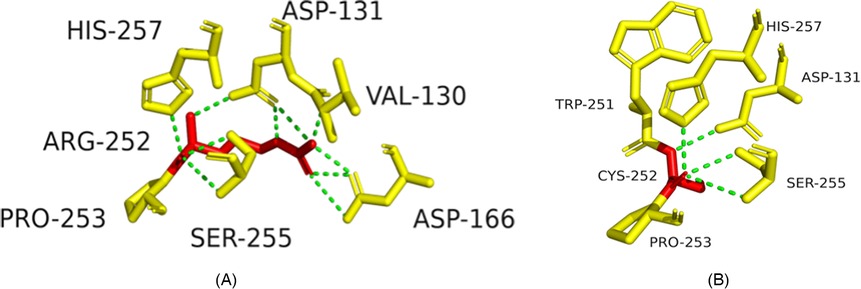
Figure 4. Structural impact of ITGB4 c.754C>T (p.R252C) mutation. (A) Wild-type ITGB4 structure showing key amino acid interactions. (B) Mutant ITGB4 structure with c.754C>T substitution, showing loss of hydrogen bonds.
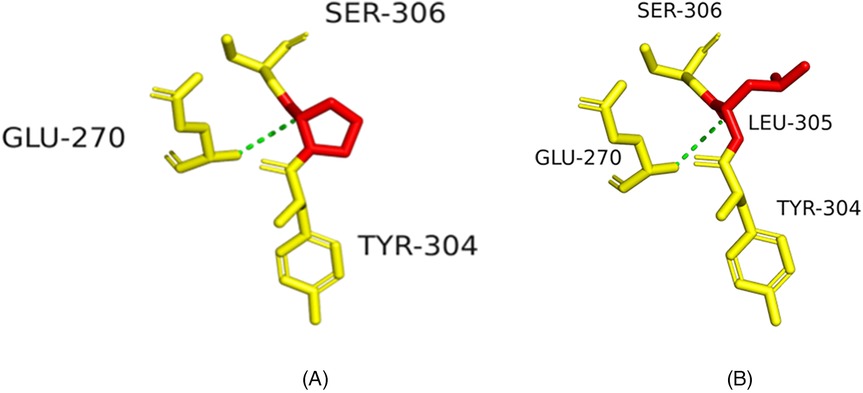
Figure 5. Structural impact of ITGB4 c.914C>T (p.P305l) mutation. (A) Wild-type ITGB4 structure showing key amino acid interactions. (B) Mutant ITGB4 structure with c.914C>T substitution, showing structural changes at LEU-305.
Discussion
This report describes a patient with JEB who developed a previously undescribed papillomatous–follicular bladder phenotype associated with compound heterozygous ITGB4 variants. These findings expand the current understanding of urological manifestations in JEB. Similar papillomatous changes, described as “papilla bullosa”, were reported in an 11-year-old girl (parental origin documented) with ITGB4-related JEB by Ilmiers et al. in 2016 (11). In addition, phenotypic heterogeneity associated with β4-integrin mutations has been highlighted and commented on by Bruckner-Tuderman in an editorial comparing two siblings with divergent JEB manifestations (12).
Contextualization with existing literature
Urological involvement in JEB is known. Previous reports focus on urethral strictures, hydronephrosis, urinary retention, general bladder wall hypertrophy, and cystitis (9). General bladder pathology in EB includes mucosal blistering, diffuse wall thickening, fibrosis, and occasional polypoid masses (4). A recent systematic review summarised 13 epidermolysis bullosa cases with vesicular or papillomatous cystitis, underscoring bladder mucosal vulnerability (13). The current case's papillomatous protrusion and accompanying follicular mucosal changes are morphologically distinct. Whereas “wall thickening” is a non-specific descriptor, a papillomatous projection denotes an organised outgrowth rather than simple hypertrophy. The follicular mucosal changes likely represent small, lymphoid-aggregate–related lesions that differ from larger polypoid formations (4). To our knowledge, this combination has not previously been described in the JEB literature.
Significance of ITGB4 mutations (p.R252c and p.P305l)
Compound-heterozygous ITGB4 variants p.R252C and p.P305l are central to this case. ITGB4 encodes the β4-integrin subunit, a key component of hemidesmosomes in epithelia such as the urothelium (6). Alterations at residue Arg252 are particularly noteworthy: Ellis et al. reported a patient with p.R252l who had mild cutaneous JEB-PA but severe obstructive uropathy (8), and Dang et al. documented a lethal JEB-PA neonate with p.R252C (14). These observations suggest that residue R252 is critical and that its alteration predisposes carriers to urological complications. Similar ITGB4-PA cases without bladder involvement have been reported (15). Reitelman et al. described an ITGB4-mutant patient with severe ureteric obstruction but no bladder lesions, further illustrating phenotypic variability (16). However, the specific “stalactite-like” bladder-wall thickening and “follicular” mucosal changes documented in our patient were not reported in those cases (8, 14). Ellis et al.'s patient had bladder-wall haemorrhage/blistering (8, 10) reported exuberant, de-epithelialised bladder mucosa in a child carrying different ITGB4 variants without pyloric atresia. This highlights that although R252 mutations are linked to uropathy, our patient's bladder manifestation is distinct. The p.P305l variant, far less characterised, was bio-informatically predicted as damaging and may further impair α6β4-integrin function. Although pyloric atresia defines the JEB-PA subtype, bladder involvement has been documented only sporadically. The coexistence of surgically corrected PA and a papillomatous–follicular bladder phenotype in our patient broadens the recognised genitourinary spectrum of ITGB4-related disease and underscores the need for systematic urological surveillance in JEB-PA.
Table 1 compares urological phenotypes.
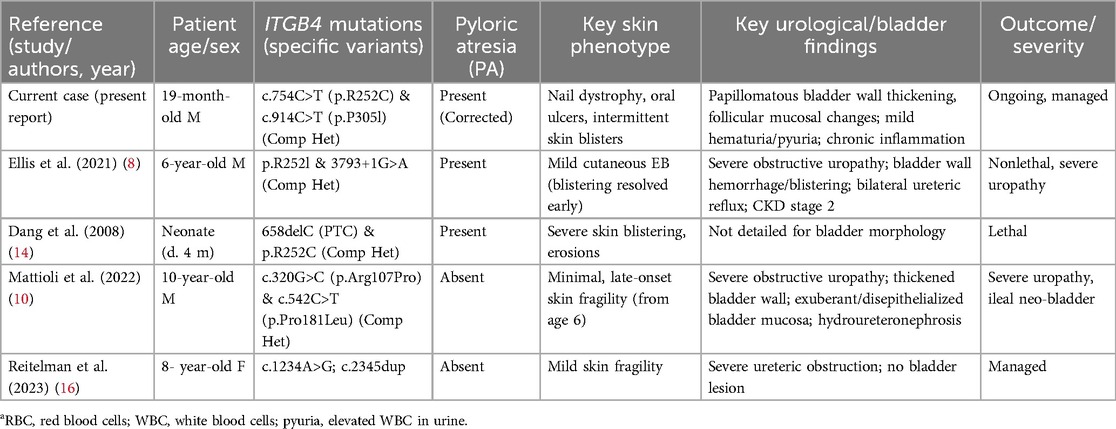
Table 1. Comparison of urological phenotypes in reported cases of JEB with ITGB4 mutationsa.
This comparison shows that while ITGB4 mutations (especially at R252) link to severe uropathy, our patient's specific bladder phenotypes appear novel.
Mechanistic insights
The novel phenotypes most plausibly arise from impaired urothelial integrity caused by dysfunctional α6β4 integrin. The ITGB4 variants (p.R252C, p.P305l) are predicted to destabilise the integrin complex, rendering the bladder mucosa vulnerable to mechanical stress, chronic inflammation and recurrent sub-clinical injury. Repeated insults may induce dysregulated repair, leading to chronic oedema, inflammatory infiltration and aberrant remodelling. The observed papillomatous projections could represent focal fibromuscular hyperplasia or organised granulation tissue, whereas the follicular mucosal changes suggest reactive lymphoid hyperplasia. Although urine cultures were negative, occult low-grade colonisation or sterile inflammation cannot be excluded. The widespread expression of α6β4 integrin in urogenital epithelium offers a biologically plausible link between integrin dysfunction, mucosal fragility and these secondary alterations (4).
Clinical implications
Recognizing these novel bladder phenotypes in patients with JEB, especially with ITGB4 mutations, may aid earlier diagnosis and management. This case reinforces recommendations for systematic urological evaluation (ultrasound, possibly cystoscopy) in patients with JEB with urinary symptoms or unexplained bladder anomalies (13, 17). Early identification could allow timely interventions to mitigate chronic irritation and prevent progressive dysfunction. A multidisciplinary approach is paramount.
Future directions
1. Verification of Phenotypic SpecificityCohort studies or case series are needed to determine if these bladder changes are specific to certain JEB genotypes (e.g., ITGB4 p.R252C) or broader manifestations.
2. Molecular Mechanism StudiesInvestigating how ITGB4 mutations lead to these specific bladder phenotypes using patient-derived cells or organoids could identify dysregulated pathways or biomarkers.
3. Exploration of Gene TherapyAdvances in gene therapy for EB, such as Beremagene geperpavec (B-VEC) for dystrophic EB and ex vivo LAMB3 correction for JEB (18), provide a rationale for developing ITGB4-targeting strategies, in line with recent insights into ITGB4 genetics and management (19). These approaches could potentially mitigate both cutaneous and systemic complications, including the bladder phenotype described here.
Limitations
This single case report's findings may not be generalizable. Long-term follow-up and additional cases are needed to determine the specificity, prevalence, and clinical relevance of these bladder changes in JEB.
Conclusion
This report describes a novel bladder phenotype in a patient with junctional epidermolysis bullosa (JEB) carrying compound heterozygous ITGB4 mutations, characterized by focal papillomatous bladder wall thickening and follicular mucosal changes. These findings broaden the recognized phenotypic spectrum of JEB and emphasize the importance of comprehensive urological assessment in patients presenting with urinary symptoms or unexplained bladder abnormalities. Early recognition and targeted intervention may help prevent chronic urological complications. Further investigation into the underlying molecular mechanisms may inform the development of future therapeutic strategies.
Data availability statement
The data underlying this article are available in the article itself; further information can be obtained from the corresponding author on reasonable request.
Ethics statement
The studies involving humans were approved by Ethics Committee of Children's Hospital Affiliated to Shandong University (Jinan Children's Hospital). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants' legal guardians/next of kin. Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
QH: Conceptualization, Investigation, Methodology, Writing – original draft, Writing – review & editing. MG: Conceptualization, Writing – review & editing. HW: Investigation, Writing – original draft. LZ: Conceptualization, Data curation, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
We thank the patient's family and the multidisciplinary team for their cooperation and support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2025.1555599/full#supplementary-material
References
1. Bardhan A, Bruckner-Tuderman L, Chapple ILC, Fine JD, Harper N, Has C, et al. Epidermolysis bullosa. Nat Rev Dis Primers. (2020) 6(1):78. doi: 10.1038/s41572-020-0210-0
2. Bruckner-Tuderman L, Has C. Epidermolysis bullosa: advances in research and treatment. J Dermatol Sci. (2019) 96(2):123–31. doi: 10.1016/j.jdermsci.2019.09.004
3. Has C, Bauer JW, Bodemer C, Bolling MC, Bruckner-Tuderman L, Diem A, et al. Consensus reclassification of inherited epidermolysis bullosa and other mechanobullous disorders. J Am Acad Dermatol. (2020) 82(3):614–22. doi: 10.1016/j.jaad.2019.10.148
4. Almaani N, Mellerio JE. Genitourinary tract involvement in epidermolysis bullosa. Dermatol Clin. (2010) 28(2):343–6. doi: 10.1016/j.det.2010.01.014
5. Mattioli G, Barabino A, Mascio A, Pintus C, Gambini C, Costanza F, et al. Severe obstructive uropathy in a child with junctional epidermolysis bullosa due to LAMA3 mutations. J Pediatr Urol. (2022) 18(3):348–52. doi: 10.1016/j.jpurol.2022.01.006
6. Uitto J, Pulkkinen L, Smith FJ. Junctional epidermolysis bullosa: unique clinical and molecular characteristics. Clin Dermatol. (2018) 36(3):366–74. doi: 10.1016/j.clindermatol.2018.03.007
7. Vill K, Koenig E, Hillmer A, Frommhold D, Winter L, Hennies HC, et al. ITGB4-related junctional epidermolysis bullosa with pyloric atresia: a genotypic and phenotypic update. Pediatr Dermatol. (2020) 37(2):285–93.
8. Ellis C, Eason C, Snyder A, Siegel M, Pai GS, Ryan E, et al. Novel missense p.R252L mutation of ITGB4 compounded with known 3793+1G>A mutation associated with nonlethal epidermolysis bullosa-pyloric atresia with obstructive uropathy. JAAD Case Rep. (2021) 11:63–8. doi: 10.1016/j.jdcr.2021.03.016
9. Fine JD, Johnson LB, Weiner M, Suchindran C. Genitourinary complications of inherited epidermolysis bullosa: experience of the national epidermylosis bullosa registry and review of the literature. J Urol. (2004) 172(5 Pt 1):2040–4. doi: 10.1097/01.ju.0000143200.86683.2c
10. Mattioli G, Diociaiuti A, Rossi S, Zambruno G, Carlucci M, Pisaneschi E, et al. ITGB4-mutated junctional epidermolysis bullosa without pyloric atresia presenting with severe urinary involvement and late-onset minimal skin fragility: diagnostic and therapeutic challenges. Acta Derm Venereol. (2022) 102:935. doi: 10.2340/actadv.v102.935
11. Gualandi F, Tadini G, Pisaneschi E, Kiritsi D, Hausser I, Zambruno G, et al. Papilla bullosa: a papillomatous bladder manifestation in junctional epidermolysis bullosa. J Pediatr Urol. (2016) 12(4):244–7.27480469
12. Bruckner-Tuderman L. Phenotype variation between siblings with junctional epidermolysis bullosa caused by β4-integrin mutations. Acta Derm Venereol. (2008) 88(5):429–30. doi: 10.2340/00015555-0483
13. Karacan M, Okur H, Yalcin B, Kocyigit A, Parlak N, Has C, et al. Genitourinary manifestations in epidermolysis bullosa: a systematic review of the literature and proposed follow-up protocol. Pediatr Dermatol. (2023) 40(1):112–20.
14. Dang N, Klingberg S, Rubin AI, Edwards M, Borelli S, Relic J, et al. Differential expression of pyloric atresia in junctional epidermolysis bullosa with ITGB4 mutations suggests that pyloric atresia is due to factors other than the mutations and not predictive of a poor outcome: three novel mutations and a review of the literature. Acta Derm Venereol. (2008) 88(5):438–48. doi: 10.2340/00015555-0484
15. Valencia M, Quintero L, Penchaszadeh VB, Martinez-Mir A, Zambruno G, Has C, et al. Junctional epidermolysis bullosa with pyloric atresia caused by ITGB4 mutations: two siblings with different outcomes. Orphanet J Rare Dis. (2021) 16(1):257.34088339
16. Tadini G, Castori M, Ermini M, Paradisi M, Zambruno G, Posteraro P, et al. Ureteric obstruction without bladder lesions in an ITGB4-related epidermolysis bullosa patient. J Pediatr Urol. (2023) 19(2):150–6.
17. Yilmaz R, Caksen H, Unal O, Odabas D. Kidney and urinary tract involvement in epidermolysis bullosa: is routine follow-up necessary? Dermatol Pract Concept. (2021) 11(3):e2021051. doi: 10.5826/dpc.1103a136
18. Laimer M, Prodinger C, Bauer JW. Current and future treatment options for epidermolysis bullosa. Clin Cosmet Investig Dermatol. (2024) 17:915–28. doi: 10.2147/CCID.S432321
Keywords: junctional epidermolysis bullosa, ITGB4 mutation, bladder involvement, papillomatous bladder thickening, follicular mucosal changes, rare disease
Citation: He Q, Gui M, Wang H and Zhang L (2025) A novel bladder phenotype in junctional epidermolysis bullosa: a case report. Front. Pediatr. 13:1555599. doi: 10.3389/fped.2025.1555599
Received: 5 January 2025; Accepted: 2 July 2025;
Published: 21 August 2025.
Edited by:
Hongjiang Qiao, Fourth Military Medical University, ChinaReviewed by:
Dedee Murrell, University of New South Wales, AustraliaMelinda Matyas, University of Medicine and Pharmacy Iuliu Hatieganu, Romania
Copyright: © 2025 He, Gui, Wang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lei Zhang, emhhbmd5aXl1T0tAMTI2LmNvbQ==