 Ting Tang
Ting Tang Shuqi Wu
Shuqi Wu Chang Peng
Chang Peng- Department of Pediatrics, Guizhou Children’s Hospital, Affiliated Hospital of Zunyi Medical University, Zunyi, Guizhou, China
Pulmonary arterial hypertension (PAH) is a rare and severe condition that has been linked to hereditary factors. Mutations in the gene encoding bone morphogenetic protein receptor 2 (BMPR2) have been identified as a cause of heritable PAH. We report the discovery of a novel point mutation combined with a deletion insertion mutation (c.621+2T>C/c.621+5_621+11delinsA) in the BMPR2 gene of an 11-year-old PAH patient lacking a family history of genetic disease (Clinical trial number: not applicable). This report expands the genetic landscape and offers a scientific foundation for early disease detection, personalized treatment strategies, and genetic counseling.
1 Introduction
Pulmonary arterial hypertension (PAH) is a cluster of diseases marked by a relentless rise in pulmonary vascular resistance and progressive right heart failure, which can be triggered by mutations in specific genes (1). The bone morphogenetic protein receptor 2 (BMPR2) gene, which codes for a protein known as bone morphogenetic protein receptor II, crucial for sustaining pulmonary vascular equilibrium, is mutated in approximately 75% of cases of heritable pulmonary arterial hypertension (HPAH) (2). When the BMPR2 gene is altered, it initiates a cascade of pathological changes, including vasoconstriction, heightened inflammatory responses, endothelial cell dysfunction, and eventually PAH (3). PAH in pediatric patients constitutes a unique clinical syndrome characterized by a more rapid progression of the disease compared to adult-onset cases. Genetic susceptibility is particularly significant in this context. More than 70% of heritable PAH cases and approximately 20% of idiopathic pediatric PAH cases are associated with pathogenic variants in the BMPR2 gene, which predominantly consist of loss-of-function mutations that disrupt the signaling pathways of bone morphogenetic proteins (BMPs). Recent research highlights the profound clinical implications of BMPR2 mutations within pediatric populations: individuals with these mutations present symptoms 5.8 years earlier than those without (median age 6.4 vs. 12.2 years), and they face a 3.2-fold increased risk of right ventricular failure within the first two years post-diagnosis (4, 5). The relationship between genotype and phenotype also influences therapeutic outcomes, as patients with BMPR2 deficiency exhibit diminished vasoreactivity to nitric oxide and necessitate an earlier transition to prostacyclin analog therapy (6). PAH is a rare yet severe condition that truncates life expectancy. Consequently, the early identification, diagnosis, and treatment of PAH can significantly enhance the prognosis of this potentially fatal disease (7). In this report, we present the case of a boy who was admitted to the Department of Pediatric Respiratory and Cardiovascular Medicine at our hospital and diagnosed with PAH due to a mutation in the BMPR2 gene. Whole-exome testing revealed a novel mutation site (c.621+2T>C/c.621+5_621+11delinsA) within the BMPR2 gene, and the clinical data are detailed herein.
2 Clinical data
The patient, a 11-year-old male, was admitted to the hospital due to recurrent episodes of wheezing and shortness of breath lasting over three months, accompanied by a ten-day history of coughing, panic, palpitations, and chest tightness. The child began experiencing wheezing and shortness of breath following an early morning jog more than three months ago, accompanied by chest discomfort, palpitations, occasional dizziness, pallor, and profuse sweating. These symptoms have been alleviated by rest over the past three months. Over the last three months, the child has exhibited symptoms of wheezing and shortness of breath, particularly subsequent to ascending a flight of stairs for approximately three minutes. This is accompanied by palpitations, chest tightness, and sweating, all of which alleviate with rest. The child was admitted to another hospital for further treatment 10 days ago after developing a cough, which was characterized by monosyllabic sounds and white sputum.
The patient's history, encompassing drug and solvent exposure, as well as comprehensive clinical, serologic, laboratory, and imaging assessments, did not indicate any connective tissue diseases, portal hypertension, human immunodeficiency virus infection, or other conditions linked to pulmonary hypertension. Notably, the patient had undergone “repair of ventricular septal defect” at seven months of age, yet no pulmonary hypertension or residual shunt was detected during the five-year postoperative follow-up. There was no history of infectious diseases such as hepatitis or tuberculosis. No history of food or drug allergies was reported. The patient denied any history of blood transfusions. The patient born full-term via Cesarean section, with no history of asphyxia or resuscitation, and no history of mechanical ventilation or exposure to high oxygen concentrations. The birth history, feeding history, and developmental history were all unremarkable. There were no instances of consanguineous marriages in the family tree, both parents were previously healthy, and there was no history of inherited metabolic diseases. However, the patient's grandfather's brother had a history of cardiac disease diagnosed during his adolescence within the family, though the specific diagnosis remains unclear.
Laboratory tests conducted at our hospital indicated the following for the myocardial infarction biomarkers: myoglobin at 21 ng/ml, high-sensitivity troponin T at 20.4 ng/L, and N-terminal pro-B-type natriuretic peptide (NT-proBNP) at 2,477 pg/ml. Echocardiography revealed an aneurysmal dilatation of the pulmonary artery, severe pulmonary hypertension, and postoperative repair of a ventricular septal defect with no atrial, ventricular, or arterial shunting. There was also evidence of right heart enlargement and bilobed pulmonary valve malformations, with an estimated pulmonary systolic pressure of approximately 133 mmHg (normal reference range: 15–30 mmHg) based on tricuspid regurgitation. The left ventricular ejection fraction was 69% (normal reference range: 55%–70%), and the fractional shortening was 39% (normal reference range: 25%–45%). The electrocardiogram showed sinus tachycardia and right ventricular hypertrophy with strain. Considering that cardiac catheterization is an invasive procedure and there are ethical concerns associated with it, it is not the primary examination for pediatric patients. Therefore, this examination was not carried out on this pediatric patient.
Upon obtaining informed consent, blood samples were collected from the patient and both parents for whole-exome sequencing (100 × coverage) and mitochondrial genome sequencing (3,000 × coverage) (8). The genetic analysis revealed minor variants (Single Nucleotide Variants/Insertions and Deletions) (SNVs/InDels) in the BMPR2 gene: c.621+2T>C/c.621+5_621+11delinsA, which were classified as pathogenic by ACMG classification (American College of Medical Genetics and Genomics) (Clinical trial number: not applicable).These variants are linked to familial primary pulmonary hypertension (type I), with or without hereditary hemorrhagic telangiectasia, an autosomal dominant disorder. As shown in Figure 1 and Table 1. Particularly, the mutations c.621+2T>C and c.621+5_621+11delinsA, situated within the intronic region, may influence the gene's structure and function. Such changes could lead to alterations in the transcribed RNA, which in turn affects protein synthesis, potentially disrupting essential biological processes including cell growth, differentiation, and metabolism (9). Furthermore, we employed four distinct software tools to forecast the impact of these alterations on donor/acceptor splice sites. Within the SPIDEX software's predictive model, a positive score signifies the enhancement of splicing, whereas a negative score signifies its inhibition. The magnitude of the absolute value correlates with the variant's influence on splicing; the greater the absolute value, the more pronounced the effect. A score of −15.26 suggests that the genetic variant is likely to exert a significant negative impact on splicing, potentially leading to diminished splicing efficiency or the emergence of aberrant splicing events. In the context of SpliceAI's predictive analysis, scores of 0.04 and 0.09 suggest that the variant might marginally improve the recognition of splice sites, albeit with a negligible effect. The MutationTaster software's prediction, denoted by the term “Disease causing”, implies that the variant could impair gene function, thereby possibly precipitating disease onset. The number 1 signifies the prediction's confidence level, with lower numbers reflecting greater confidence in the prediction. Consequently, the variant is strongly suspected to be disease-inducing. Lastly, the Genomic Evolutionary Rate Profiling (GERP) software's prediction, marked by “Conserved”, indicates that the gene's position is conserved across various species, signifying minimal evolutionary change. A score of 5.92 suggests that the higher the score, the more conserved the position is, and it may be crucial for biological functionality (10). As illustrated in Table 2.

Figure 1. Gene sequencing map (transcript version number: NM_LC24052700033001AA1).
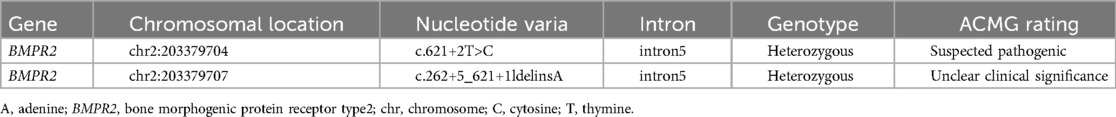
Table 1. Sequencing verification results in BMPR2 gene mutation.
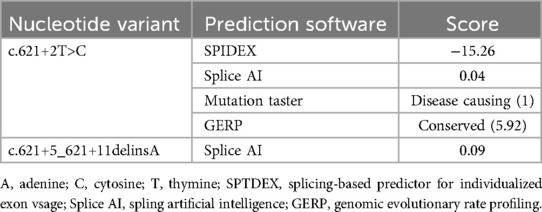
Table 2. Prediction of protein functional damage in BMPR2 gene mutation.
3 Discussion
PAH, a cluster of disorders, is marked by symptoms such as breathlessness, fatigue, and fainting (11). It is characterized by a significant dilation of the pulmonary vasculature's internal diameter, elevated pulmonary artery pressure, and hypertrophy of the right ventricle (12). The pathophysiological alterations impede the normal antegrade movement of blood within the pulmonary circulation. As a result, persistent disruptions in pulmonary hemodynamics can culminate in the emergence of right-heart failure, via mechanisms including heightened pulmonary vascular resistance leading to augmented right-ventricular afterload, right-ventricular hypertrophy, and subsequent decompensation (13). In recent times, a growing body of research has concentrated on the established fact that mutations in BMPR2 are associated with pathogenicity in PAH (14). The identified genes responsible include Neurogenic locus notch homolog protein 3 (NOTCH3), BMPR2, Actin Alpha 2, Smooth Muscle (ACTA2), Vascular Endothelial Growth Factor Receptor 2 (VEGFR2), T-box transcription factor 4 (TBX4), and SRY-box transcription factor 17 (SOX17), which facilitate the emergence and advancement of pulmonary hypertension via distinct mechanisms (15). The protein produced by the BMPR2 gene, as detailed in this report, is a pivotal element of the bone morphogenetic proteins (BMPs) signaling pathway, playing a crucial role in the formation of the cardiovascular system during embryonic stages (16). In individuals with PAH, mutations in the BMPR2 gene disrupt the normal BMPs signaling, thereby impacting the proper development and upkeep of the pulmonary vasculature (17). The BMPR2 protein also operates within the transforming growth factor β (TGF-β) pathway. It regulates the phosphorylation of downstream Smad proteins, which are pivotal mediators in the TGF-β signaling cascade (18). The protein influences various cellular responses, including cell proliferation, differentiation, and apoptosis, within the context of pulmonary arterial hypertension pathogenesis. By interacting with non-BMP-specific elements of the TGF-β pathway, the BMPR2 protein contributes to the overall complexity of the molecular mechanisms underlying the disease (19). These mutations encompass a spectrum of types, such as missense mutations, nonsense mutations, and splice site variants, all of which can result in defective BMPR2 protein function. This dysfunction can subsequently lead to heightened pulmonary arterial pressure and structural anomalies within the tissues (20).
In this study, we identified novel intronic shear mutation sites and deletion and insertion mutations (c.621+2T>C, c.621+5_621+11delinsA) in the BMPR2 gene. Numerous mutations previously associated with pulmonary hypertension include nonsense mutations (c.631C>T, c.76C>T; p.Gln26Ter), missense mutations (c.604A>T, c.794A>G, c.797G>C, and c.806G>T), shear mutations (c.852+1G>C, c.853-1G>C, c.621+1G>A), promoter deletions and insertion mutations (c.621+5_621+11delinsA), promoter mutations (c.-575A>3ET, c.-586dupT), and shift mutations (c.612del A, c.690-691del AGins T, and c.673-679del CGTCCAG). In this report, we have found that the mutation of 621+2T>C, an intron, adheres to the GT-AG rule. The GT-AG rule is essential for the splicing of eukaryotic pre-mRNA, wherein introns are delineated by the presence of GT (or GU in RNA) at the 5′ terminus and AG at the 3′ terminus. The spliceosome employs these specific sequences to facilitate the processing of pre-mRNA into mature mRNA, which is subsequently translated. In the course of our investigation into BMPR2 mutations, we hypothesize that the mutation impacts the GT-AG splicing sites within the BMPR2 gene. Given the pivotal role of BMPR2 in the TGF-β signaling pathway, which is instrumental in the regulation of pulmonary arterial cells, the resultant mutant protein diminishes the phosphorylation of downstream small mothers against decapentaplegic (SMADs) (21). This impairment disrupts the signaling cascade, leading to aberrant cellular behaviors such as unchecked proliferation and resistance to apoptosis, which are characteristic of heritable pulmonary arterial hypertension (PAH) (18). Although this mutation does not directly alter the protein's amino acid sequence, it indirectly affects gene expression and protein function by disrupting the gene splicing process. The post-shear insertion change of c.621+5_621+11delinsA may impact the structure and function of the gene (22). We believe that the incomplete sequence of the BMPR2 gene could lead to defective BMPR2 function. We correlated the features of our pediatric patient with established BMPR2-related PAH phenotypes from the literature. The patient's early onset, severe echocardiographic findings, treatment response, and nuances of family history, along with the presence of pulmonary artery aneurysmal dilation, align with known patterns of BMPR2-associated PAH (23). For long-term care, we adhered to the guidelines of the American College of Chest Physicians (ACCP) and incorporated genetic counseling for BMPR2 mutation carriers. This includes emphasizing autosomal-dominant inheritance, suggesting cascade testing for first-degree relatives, regular cardiopulmonary screening, and specific monitoring for asymptomatic carriers. The discovery of the mutation informs personalized prevention, drug selection, and early transplantation evaluation. However, since pathogenicity relies on bioinformatics, we will communicate its limitations during counseling and integrate relatives’ test results with clinical phenotypes for ongoing risk assessment of asymptomatic individuals (24).
PAH, a condition that not only endangers patients' health but also imposes significant psychological and financial strains on their families and society, has long been a formidable challenge for the medical community due to its intricate and hereditary characteristics (9). The genetic complexity of PAH means that the identification of causative genes is an ongoing process necessitating advanced genomic research (25). Research has indicated that individuals with PAH and BMPR2 mutations tend to experience more severe symptoms at a younger age and face a heightened mortality risk compared to those without such mutations (26). Recent studies reveal that 10%–15% of childhood-onset PAH cases are caused by de novo (newly occurring) BMPR2 mutations, even without family history (27). Consequently, genetic testing is especially crucial for PAH patients. This report uncovers a novel mutation locus, which will facilitate future clinical research and the identification of new therapeutic targets (28). The genetic tests detailed in this report can serve as a guide for clinicians in early detection and intervention, potentially enhancing the quality of life and prognosis for patients. Considering that this investigation constitutes the inaugural identification of the novel BMPR2 mutation, the proposed applications within these domains are speculative in nature. They delineate potential avenues for subsequent research endeavors. It is explicitly stated that these concepts necessitate further scrutiny and validation. For example, cohort-based investigations are indispensable for establishing dependable biomarkers for early detection, and meticulously designed clinical trials are imperative to explore personalized treatment modalities. The recently identified BMPR2 mutation, despite its comprehensive implications being unverified, may influence early disease detection, personalized treatment approaches, and genetic counseling (29). In the context of early detection, it could potentially steer the discovery of biomarkers, where alterations in pertinent protein or metabolite levels can be utilized to accurately evaluate treatment efficacy and modify regimens as required. For personalized treatment, it might inform the selection of drugs targeting the affected pathways. In response to the condition of the pediatric patient in this instance, medications such as sildenafil and propranolol were administered during the hospitalization period. During the subsequent telephone follow - up, not only did the pediatric patient not exhibit any significant adverse reactions to the medications, but also clear signs of improvement became evident. The shortness of breath, which was previously limiting the patient's activity, was markedly reduced. The patient could now engage in light physical activities without experiencing the same degree of breathlessness. Symptoms such as chest discomfort, palpitations, and dizziness have all been alleviated to varying degrees, indicating that the treatment was efficacious. With respect to genetic counseling, it augments the knowledge for predicting inheritance patterns and guiding family-based testing, thus holding significance in these critical areas. Nevertheless, the report acknowledges certain limitations, including insufficient data, recall bias, restricted study scope, and limited practical application. It is imperative to acknowledge the constraints inherent in this singular case study. The primary disadvantage is the diminished statistical power resulting from the limited sample size. This limitation impedes our capacity to generalize the observed outcomes to the larger population of individuals with heritable PAH. It is essential to note that due to unforeseen and uncontrollable factors, we lack a few perspectives from the patient and their family. Notwithstanding these constraints, the thorough examination of this specific case has yielded distinctive insights that may function as a foundation for additional research endeavors. With the continuous advancement of gene editing technology and bioinformatics, we anticipate offering more effective and safer treatment options for PAH patients, ultimately aiming to achieve long-term management or even a cure for the disease.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
The studies involving humans were approved by Ethics Review Committee of the Affiliated Hospital of Zunyi Medical University. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the minor(s)' legal guardian for the publication of any potentially identifiable images or data included in this article.
Author contributions
TT: Conceptualization, Investigation, Supervision, Validation, Writing – original draft, Writing – review & editing. SW: Investigation, Resources, Visualization, Writing – review & editing. CP: Conceptualization, Data curation, Supervision, Writing – original draft. LW: Conceptualization, Investigation, Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Generative AI was used in the creation of this manuscript.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Dodson MW, Brown LM, Elliott CG. Pulmonary arterial hypertension. Heart Fail Clin. (2022) 14(3):255–69. doi: 10.1016/j.hfc.2018.02.003
2. Tatius B, Wasityastuti W, Astarini FD, Nugrahaningsih DAA. Significance of BMPR2 mutations in pulmonary arterial hypertension. Respir Investig. (2021) 59(4):397–407. doi: 10.1016/j.resinv.2021.03.011
3. Pfarr N, Szamalek-Hoegel J, Fischer C, Hinderhofer K, Nagel C, Ehlken N, et al. Hemodynamic and clinical onset in patients with hereditary pulmonary arterial hypertension and BMPR2 mutations. Respir. (2011) 12(1):99. doi: 10.1186/1465-9921-12-99
4. Rosenzweig EB, Abman SH, Adatia I, Beghetti M, Bonnet D, Haworth S, et al. Paediatric pulmonary arterial hypertension: updates on definition, classification, diagnostics and management. Eur Respir J. (2019) 53(1):1801916. doi: 10.1183/13993003.01916-2018
5. Morrell NW, Aldred MA, Chung WK, Elliott CG, Nichols WC, Soubrier F, et al. Genetics and genomics of pulmonary arterial hypertension. Eur Respir J. (2019) 53(1):1801899. doi: 10.1183/13993003.01899-2018
6. Rosenzweig EB, Morse JH, Knowles JA, Chada KK, Khan AM, Roberts KE, et al. Clinical implications of determining BMPR2 mutation status in a large cohort of children and adults with pulmonary arterial hypertension. J Heart Lung Transplant. (2008) 27(6):668–74. doi: 10.1016/j.healun.2008.02.009
7. Zhang ZQ, Zhu SK, Wang M, Wang XA, Tong XH, Wan JQ, et al. New progress in diagnosis and treatment of pulmonary arterial hypertension. J Cardiothorac Surg. (2022) 17(1):216. doi: 10.1186/s13019-022-01947-y
8. Gelinas SM, Benson CE, Khan MA, Berger RMF, Trembath RC, Machado RD, et al. Whole exome sequence analysis provides novel insights into the genetic framework of childhood-onset pulmonary arterial hypertension. Genes (Basel). (2020) 11(11):1328. doi: 10.3390/genes11111328
9. Song J, Hinderhofer K, Kaufmann LT, Benjamin N, Fischer C, Grünig E, et al. BMPR2 promoter variants effect gene expression in pulmonary arterial hypertension patients. Genes (Basel). (2020) 11(10):1168. doi: 10.3390/genes11101168
10. Gräf S, Haimel M, Bleda M, Hadinnapola C, Southgate L, Li W, et al. Identification of rare sequence variation underlying heritable pulmonary arterial hypertension. Nat Commun. (2018) 9(1):1416. doi: 10.1038/s41467-018-03672-4
11. Luna-López R, Ruiz Martín A, Escribano Subías P. Pulmonary arterial hypertension. Med Clin (Barc). (2022) 158(12):622–9. doi: 10.1016/j.medcli.2022.01.003
12. de Jesus Perez VA. Molecular pathogenesis and current pathology of pulmonary hypertension. Heart Fail Rev. (2016) 21(3):239–57. doi: 10.1007/s10741-015-9519-2
13. Lan NSH, Massam BD, Kulkarni SS, Lang CC. Pulmonary arterial hypertension: pathophysiology and treatment. Diseases. (2018) 6(2):38. doi: 10.3390/diseases6020038
14. Ma L, Chung WK. The role of genetics in pulmonary arterial hypertension. J Pathol. (2017) 241(2):273–80. doi: 10.1002/path.4833
15. Southgate L, Machado RD, Gräf S, Morrell NW. Molecular genetic framework underlying pulmonary arterial hypertension. Nat Rev Cardiol. (2020) 17(2):85–95. doi: 10.1038/s41569-019-0242-x
16. Morrell NW. Role of bone morphogenetic protein receptors in the development of pulmonary arterial hypertension. Adv Exp Med Biol. (2010) 661:251–64. doi: 10.1007/978-1-60761-500-2_16
17. Ye D, Liu Y, Pan H, Feng Y, Lu X, Gan L, et al. Insights into bone morphogenetic proteins in cardiovascular diseases. Front Pharmacol. (2023) 14:1125642. doi: 10.1007/978-1-60761-500-2_16
18. Hiepen C, Jatzlau J, Hildebrandt S, Kampfrath B, Goktas M, Murgai A, et al. BMPR2 acts as a gatekeeper to protect endothelial cells from increased TGFβ responses and altered cell mechanics. PLoS Biol. (2019) 17(12):e3000557. doi: 10.1371/journal.pbio.3000557
19. Xu B, Xu G, Yu Y, Lin J. The role of TGF-β or BMPR2 signaling pathway-related miRNA in pulmonary arterial hypertension and systemic sclerosis. Arthritis Res Ther. (2021) 23(1):288. doi: 10.1186/s13075-021-02678-6
20. Welch CL, Chung WK. Genetics and other omics in pediatric pulmonary arterial hypertension. Chest. (2020) 157(5):1287–95. doi: 10.1016/j.chest.2020.01.013
21. Scott A, Petrykowska HM, Hefferon T, Gotea V, Elnitski L. Functional analysis of synonymous substitutions predicted to affect splicing of the CFTR gene. J Cyst Fibros. (2012) 11(6):511–7. doi: 10.1016/j.jcf.2012.04.009
22. Boucly A, Gerges C, Savale L, Jaïs X, Jevnikar M, Montani D, et al. Pulmonary arterial hypertension. Presse Med. (2023) 52(3):104168. doi: 10.1016/j.lpm.2023.104168
23. Liu D, Wu WH, Mao YM, Yuan P, Zhang R, Ju FL, et al. BMPR2 mutations influence phenotype more obviously in male patients with pulmonary arterial hypertension. Circ Cardiovasc Genet. (2012) 5(5):511–8. doi: 10.1161/CIRCGENETICS.111.962209
24. Badesch DB, Abman SH, Simonneau G, Rubin LJ, McLaughlin VV. Medical therapy for pulmonary arterial hypertension: updated ACCP evidence-based clinical practice guidelines. Chest. (2007) 131(6):1917–28. doi: 10.1378/chest.06-2674
25. Ntiloudi D, Kasinos N, Kalesi A, Vagenakis G, Theodosis-Georgilas A, Rammos S. Diagnosis and management of pulmonary hypertension: new insights. Diagnostics (Basel). (2024) 14(18):2052. doi: 10.3390/diagnostics14182052
26. Taha F, Southgate L. Molecular genetics of pulmonary hypertension in children. Curr Opin Genet Dev. (2022) 75:101936. doi: 10.1016/j.gde.2022.101936
27. Levy M, Eyries M, Szezepanski I, Ladouceur M, Nadaud S, Bonnet D, et al. Genetic analyses in a cohort of children with pulmonary hypertension. Eur Respir J. (2016) 48(4):1118–26. doi: 10.1183/13993003.00211-2016
28. Dai L, Du L. Genes in pediatric pulmonary arterial hypertension and the most promising BMPR2 gene therapy. Front Genet. (2022) 13:961848. doi: 10.3389/fgene.2022.961848
Keywords: pulmonary arterial hypertension, BMPR2 mutation, deletion insertion mutation, children, diagnosis
Citation: Tang T, Wu S, Peng C and Wang L (2025) Case Report: Pulmonary arterial hypertension in children caused by a new mutation in the BMPR2 gene. Front. Pediatr. 13:1572733. doi: 10.3389/fped.2025.1572733
Received: 7 February 2025; Accepted: 30 April 2025;
Published: 27 May 2025.
Edited by:
Sudhiranjan Gupta, VISN 17 Center of Excellence for Research on Returning War Veterans, United StatesReviewed by:
Joanna Kwiatkowska, Medical University of Gdansk, PolandSelena Ferrian, Independent Researcher, Palo Alto, United States
Megan Griffiths, University of Texas Southwestern Medical Center, United States
Copyright: © 2025 Tang, Wu, Peng and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chang Peng, cGVuZ2NoYW5nXzIwMDZAMTI2LmNvbQ==