 Xiaoyan Wang
Xiaoyan Wang Xiuli Chen
Xiuli Chen Ting Chen
Ting Chen Rongrong Xie
Rongrong Xie Qi Lin Chen
Qi Lin Chen Fengyun Wang
Fengyun Wang- Department of Endocrinology, Genetics and Metabolism, Children's Hospital of Soochow University, Suzhou, China
Familial glucocorticoid deficiency (FGD) is a rare autosomal recessive disorder characterized by isolated glucocorticoid deficiency. Mutations of MC2R, MRAP, STAR, NNT, and TXNRD2 have been implicated in FGD pathogenesis. To date, only four families with TXNRD2-associated familial Glucocorticoid Deficiency Type 5 (FGD5) have been reported worldwide. We report a patient with clinical features consistent with FGD5, increasing the total number of reported cases. Including this case, 11 probands across five independent kindreds have now been identified globally. Functional studies demonstrated that the novel compound heterozygous variants (c.1391A > G; p.H464R and c.1141C > T; p.R381W) reduce TXNRD2 protein levels in a heterologous expression system. This case expands the genetic spectrum of FGD5 and suggests a potential association between TXNRD2 variants and electrocardiographic abnormalities. Our findings underscore the importance of TXNRD2 in adrenal redox homeostasis and provide new insights for FGD5 diagnosis.
Background
Familial glucocorticoid deficiency (FGD), a rare hereditary endocrine disorder, manifests as selective adrenal unresponsiveness to adrenocorticotropic hormone (ACTH), resulting in isolated cortisol deficiency with preserved aldosterone synthesis. This condition typically presents in early childhood with life-threatening complications including hypoglycemic seizures, growth retardation, and characteristic mucocutaneous hyperpigmentation secondary to excessive ACTH-driven melanocyte stimulation. The characterized comprises undetectable serum cortisol, markedly elevated ACTH levels, and normal renin-aldosterone axis activity (1). FGD is characterized by undetectable serum cortisol, markedly elevated ACTH levels, and normal renin-aldosterone axis activity. It is primarily recognized by glucocorticoid deficiency alone, with other less common clinical manifestations including salt loss and hypoaldosteronism, primary hypothyroidism, hypogonadism, and growth defects (2).
Molecular studies have delineated nine causative genes associated with FGD, which can be functionally classified into three categories: ACTH Receptor Complex Defects: Pathogenic variants in MC2R (encoding melanocortin 2 receptor) and its accessory protein MRAP account for approximately 45% of cases. Mitochondrial Redox Dysregulation: Mutations affecting antioxidant defense systems, including NNT, TXNRD2, and GPX1, contribute to adrenal oxidative stress and steroidogenic failure. Steroid Biosynthesis Impairment: Rare variants in STAR and CYP11A1 disrupt cholesterol transport and initial steroidogenesis steps. The TXNRD2 gene encodes a mitochondrial selenoprotein essential for maintaining thioredoxin-mediated redox homeostasis. TXNRD2 catalyzes the NADPH-dependent reduction of oxidized thioredoxin, a critical antioxidant system protecting adrenal cells from reactive oxygen species(ROS) during steroidogenesis. Since the inaugural description of TXNRD2-linked FGD in 2014, global reports remain scarce, with distinctive clinical features. The distinctive clinical features include: neonatal-onset hypermelanosis involving mucosal surfaces, profound glucocorticoid insufficiency, exaggerated ACTH elevation, and intact zona glomerulosa function.
We report a case of thioredoxin reductase 2 (TXNRD2)-linked familial glucocorticoid deficiency-5 (FGD5) that occurred in China, increasing the total number of reported probands with this mutation and providing additional phenotypic information of this rare syndrome. In this case, a Chinese family harboured compound heterozygous variants (c.1391A > G; p.H464R and c.1141C > T; p.R381W). Our findings may expand the genetic and clinical spectrum of FGD5. We subsequently provide a review of pathogenic mutations currently documented in TXNRD2, including their specific localization across the gene's structural organization and corresponding protein domains, as well as their associated clinical manifestations.
Materials and methods
Ethical compliance and clinical examination
All procedures were approved by the Institutional Ethics Board of Children's Hospital of Soochow University(2021CS003), and informed consent was obtained from the patient's parents. Routine blood tests, serum electrolytes, serum biochemistry, thyroid function, plasma cortisol, ACTH, aldosterone, angiotensin II, renin, 24-h urine free cortisol, cellular/humoral immunity, magnetic resonance imaging (MRI) of the brain, adrenal ultrasound, testosterone, and bone age were assessed. All clinical information was collected and reviewed, and all clinical information was collected and investigated. All the procedures were performed in accordance with the Declaration of Helsinki (2013 revision).
Whole exome sequencing
Blood samples were collected from the patient and his family. Genomic DNA (gDNA) was extracted from the whole blood of the affected children and their parents. DNA samples from the case was fragmented using a Scientz08-III automated sonicator (NingBo Scientz Biotechnology, Ningbo, China) DNA samples were extracted using a DNA extraction kit (CWE2100 Blood DNA kit V2; Beijing Kangwei Century Biotechnology Co., Ltd., Beijing, China) on a 96-channel automatic nucleic acid extraction machine (Beijing Kangwei Century Biotechnology Co., Ltd.). DNA samples (750 ng) were fragmented into 150–200 bp via ultrasound treatment for 35 min (running for 3 s with 1-second intervals) with 50% ultrasonic intensity at 4˚C. High-throughput sequencing was performed on a HiSeq 2,500 system (Illumina, San Diego, CA, USA), and the data were sequenced using a sequencer (Illumina).
Data reading and bioinformatics analysis were performed following Control Software assessment (The Genome Analysis Toolkit; http://www.roadinstitute.org/gsa/wiki/index.php/Home_Page). The obtained sequences were separately Basic Local Alignment Search Tool–matched with GenBank (http://www.ncbi.nlm. nih.gov/genbank) sequences to confirm the variant sites; suspected variant sites were searched using MITOMAP (www. mitomap.org/MITOMAP). For polymorphism detection, gene variants were detected and verified via Sanger sequencing.
For the validation of the panel, parents were analyzed. The detected pathogenicity of rare variants (minor allele frequency ≤0.01) was evaluated according to the recommendations of the American College of Medical Genetics and Genomics (ACMG) for variant classification and reporting (3). Population data were determined using public genomic databases, including the 1,000 Genomes Project (https://www.internationalgenome.org), the Genome Aggregation Database (gnomAD; http://gnomad.broadinstitute.org/about), and the Database of Single Nucleotide Polymorphisms (dbSNP; https://www.ncbi.nlm.nih.gov/SNP/).
Other criteria considered were the type of variant (e.g., frameshift, nonsense, or essential splice variants), and clinical, functional, and genotype–phenotype data from the literature and disease databases, including the Human Gene Mutation Database Professional (http://www.hgmd.cf.ac.uk/ac/index.php) and PubMed (https://www.ncbi.nlm.nih.gov/pubmed). If such variants had not been previously reported, they were evaluated to predict their possible functional significance using in silico prediction tools including SIFT (https://sift.jcvi.org), PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/), and Mutation Taster (http://www.mutationtaster.org).
RNA extraction and TXNRD2 qPCR
Total RNA was extracted from blood using the Cell Total RNA Isolation Kit (RC113-01, Vazyme, Nanjing, China) and subsequently reverse transcribed to cDNA through HiScript II 1st Strand cDNA Synthesis Kit (R212-01, Vazyme, Nanjing, China). Gene expression levels were normalized using glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Sample quality was controlled using a Nanodrop ND-1000 spectrophotometer. Quantitative qPCR data of mRNA products were analysed by the 2-ΔΔCT method. The primers used for qPCR were as follows: TXNRD2 (forward: 5'-TTGAGGTCTATCACGCCC-3'; reverse:5'-CTTGAGTAACTTCGCCTGC-3') and GAPDH (forward: 5'-CTGGGCTACACTGAGCACC-3’; reverse: 5'-AAGTGGTCGTTGAGGGCAATG-3').
Western blot
Peripheral blood mononuclear cells (PBMCs) were isolated using density gradient centrifugation through whole blood. The proteins were extracted from PBMC using the RIPA lysis and extraction buffer (P0013B, Beyotime, Shanghai, China). The protein levels were quantified using a bicinchoninic acid (BCA) assay kit (P0010; Beyotime, Shanghai, China). The membrane was blocked with 5% skim milk for two hours and then incubated overnight at 4°C with GAPDH Mouse Monoclonal Antibody (1:1,000, AF0006, Beyotime, China) and TXNRD2 Polyclonal Antibody (1:1,000, 16,360-1-AP, Proteintech, USA). After washing, the membrane was incubated for one hour at room temperature with horseradish peroxidase (HRP)-conjugated goat anti-mouse/rabbit IgG (H + L) (1:5,000, A0216/A0208, Beyotime, China) as appropriate. Detection was performed using an enhanced chemiluminescence (ECL) system.
Protein structural modeling
To predict the structural changes of the identified mutation, Both wild-type and mutant TXNRD2 pdb files were predicted by AlphaFold developed by the Deepmind team, the data were realized and presented using Chimera software. The thermodynamic stability was predicted using the DynaMut online prediction tool (https://biosig.lab.uq.edu.au/dynamut/).
Statistical analyses
Data from independent experiments were presented as mean ± SEM. qPCR results were analyzed using a nonparametric test, and p < 0.05 were considered statistically significant. The significance between groups was calculated using GraphPad Prism 8 (GraphPad, USA).
Results
Case presentation
A 7-year-old Chinese male presented to our institution in May 2024 with acute gastroenteritis. During hospitalization, generalized hyperpigmentation was noted on physical examination. The patient had exhibited progressive skin discolouration of the lips and fingertips over the preceding week and had a history of congenital cutaneous hypermelanosis since birth. The patient had no history of drug exposure, trauma, dysphagia, vomiting, or chronic medical conditions (neurological, cardiac, renal, or hepatic).
Perinatal history: Born full-term via cesarean section for nuchal cord, with no prenatal/natal asphyxia or neonatal distress.
Developmental milestones: Age-appropriate.
Family history: No consanguinity or tuberculosis exposure (Figure 1).
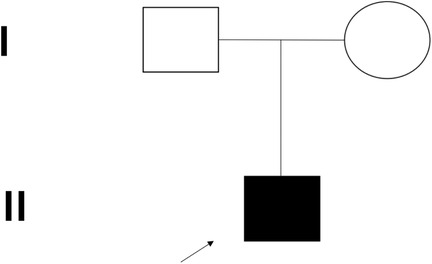
Figure 1. The family pedigrees of the FGD5 (TXNRD2 c.1391A>G and c.1141C>T).
Physical Examination: Height 130 cm, weight 30 kg. Heart rate 116 bpm, blood pressure 104/80 mmHg, respiratory rate 26/min. Diffuse hyperpigmentation with accentuation on extensor surfaces (elbows, knees), palmar creases, and buccal mucosa. Normal genitalia, lacrimation, and primitive reflexes. No dysmorphic features or neurologic deficits.
Testosterone: <0.24 nmol/L (reference: 0–1.02 nmol/L) and normal serum electrolytes. The 17ohp level was 262.10 pg/ml (reference: <1,100 pg/ml). Subsequent investigations revealed low serum cortisol (morning: <22 nmol/L, reference 138–690 nmol/L; afternoon: <22 nmol/L, reference 69–345 nmol/L) and very high adrenocorticotropic hormone (ACTH) levels (>278 ng/L, reference <46.37 ng/L) suggesting glucocorticoid deficiency. 24-hour urine free cortisol: 73.40 nmol/24 h (reference: 138.20–1,207.9 nmol/24 h), His serum aldosterone level was normal.
Other tests: Normal complete blood count, renal/liver function, renin-angiotensin system, cellular/humoral immunity, and serum electrolytes. Bone age was about 6 years old (Figure 2). Timeline of the disease course (Figure 3). Electrocardiographic Findings: Initial 12-lead electrocardiogram revealed a prolonged corrected QT interval with discernible U waves in precordial leads (Figure 4). 24-hour Holter Monitoring: Predominant sinus rhythm with marked diurnal heart rate variability (range: 68–142 bpm), intermittent cardiac arrhythmias including: Sporadic premature atrial contractions (70 events/24 hr), Paroxysmal atrial tachycardia (3 episodes, maximum duration 8 beats), Dynamic QT interval prolongation with nocturnal accentuation (maximum QTc 520 ms between 01:00 and 03:00). Magnetic resonance imaging of his brain and adrenal ultrasound were normal. Transthoracic echocardiography (TTE) revealed preserved biventricular systolic function (LVEF 66%) with no evidence of valvular abnormalities, chamber dilation, or regional wall motion abnormalities. Contrast-enhanced computed tomography angiography (CTA) of the thoracoabdominal vasculature demonstrated: normal aortic arch branching pattern, no aneurysmal dilation.
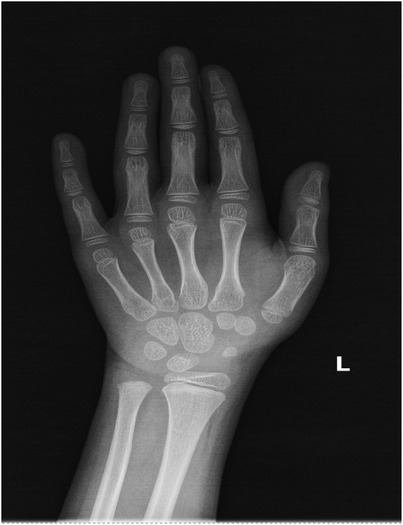
Figure 2. The bone age of the FGD5 (TXNRD2 c.1391A>G and c.1141C>T).
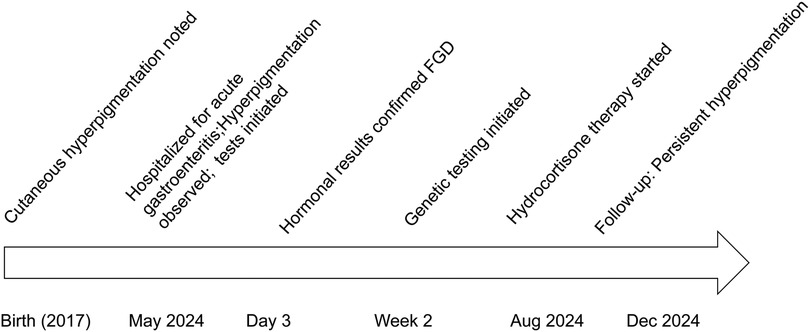
Figure 3. Timeline of the disease course.
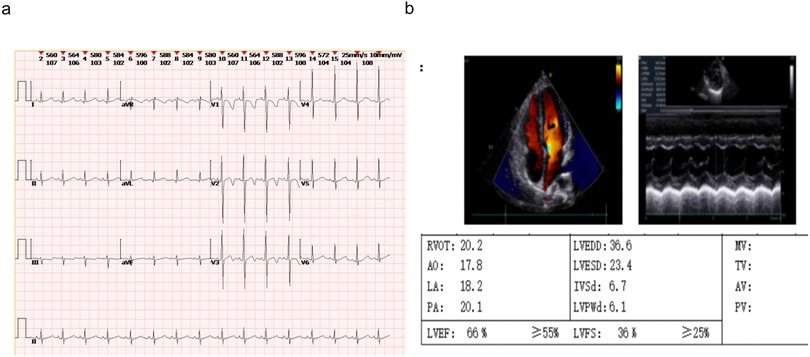
Figure 4. The ECG and echocardiography of the FGD5 (TXNRD2 c.1391A>G and c.1141C>T). (a) ECG, (b) echocardiography.
Genetic testing
Trio whole-exome sequencing (WES) was performed to screen for potential pathogenic genetic factors. As the TXNRD2 gene mutation (c.1391A > G; p.H464R and c.1141C > T; p.R381W) had not been previously reported or included in normal controls or disease databases in this context, Sanger sequencing was also performed on the parents, who were found to carry one variant each in the heterozygous state (Figure 5). According to the ACMG guidelines, one mutation (c.1391A > G; p.H464R) is likely pathogenic (LP), and another mutation (c.1141C > T; p.R381W) is Variant of unknown significance (VUS).
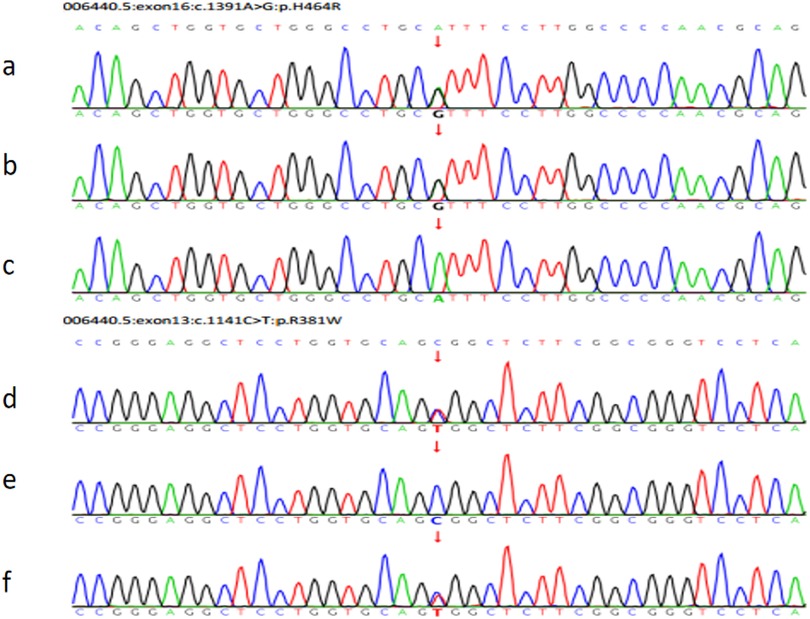
Figure 5. The pedigree of the FGD5 family with TXNRD2 (c.1391A>G and c.1141C>T). (a) The patient's TXNRD2 c.1391A>G; (b) The father's TXNRD2 c.1391A>G; (c) The mother's TXNRD2 c.1391A (wild type). (d) The patient's TXNRD2 c.1141C>T; (e) The father's TXNRD2 c.1141C (wild type); (f) The mother's TXNRD2 c.1141C>T.
Functional analysis results
Quantitative PCR and western blotting demonstrated significantly reduced TXNRD2 mRNA expression compared to heterozygote carrier parents (Figure 6b), with a corresponding decrease in TXNRD2 protein levels. Further analysis revealed a marked reduction in TXNRD2 gene expression in the patient's (P) peripheral blood relative to both the father (F) and mother (M) (Figure 6b). Consistently, Western blot assays demonstrated substantially lower TXNRD2 protein expression in P's peripheral blood mononuclear cells (PBMCs) compared to those of F and M (Figure 6c).
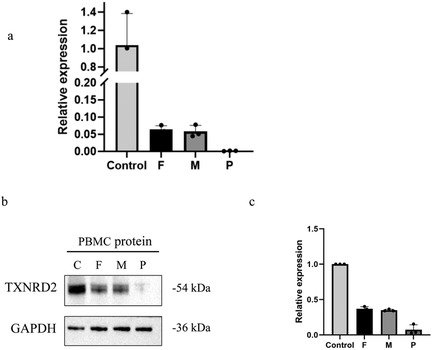
Figure 6. Evaluation the effect of the variant by experiments. (a) RT-qPCR demonstrates the effect of mutations on the mRNA expression level of TXNRD2 genes. (b,c) Effects of mutants on TXNRD2 protein levels were detected by Western blot. P: The patient (c.1391A>G p.H464R and c.1141C>T p.R381W), F: the father's TXNRD2 c.1391A>G, M: The mother's TXNRD2 c.1141C>T and control human. Three technical replicates were performed.
Treatments
Based on the results of gene sequencing and clinical features, the patient was considered to have FGD5 caused by the TXNRD2 gene mutation. Hydrocortisone(HC) acetate is the first choice for children, and the recommended HC dose is 8∼10 mg/(m2·d), administered according to the circadian rhythm. The purpose is to ensure normal growth and development and maintain electrolyte balance. The current hydrocortisone dosage for our children is 7.5 mg (8:00), 2.5 mg (16:00), 2.5 mg (22:00), the corticosteroid in the morning rose to 571 nmol/L, and the ACTH decreased to 177.6 ng/L in the morning, and the specific hormone situation is referred to Table 1, and there was no significant improvement in skin pigmentation after 3 months of treatment.
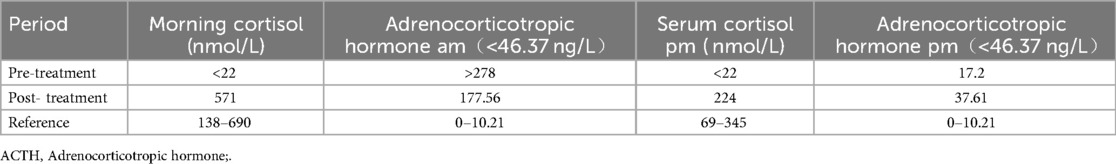
Table 1. The hormone of of the FGD5 (TXNRD2 c.1391A > G and c.1141C > T).
Protein structural modeling
Using protein structural modeling analysis, we compared the structural differences between the wild-type TXNRD2 protein and the two types of mutant TXNRD2 proteins. The tertiary and quaternary structure of TXNRD2 p.H464R did not change compared to the wild type, but the DynaMut website predicted that this mutation may lead to reduced protein stability. TXNRD2 p.Arg381Trp mutation tertiary structure at this location loses 2 hydrogen bonds formed with glutamate at position 347. Predictions on the DynaMut website show that the mutation has an uncertain effect on protein stability (Figure 7).
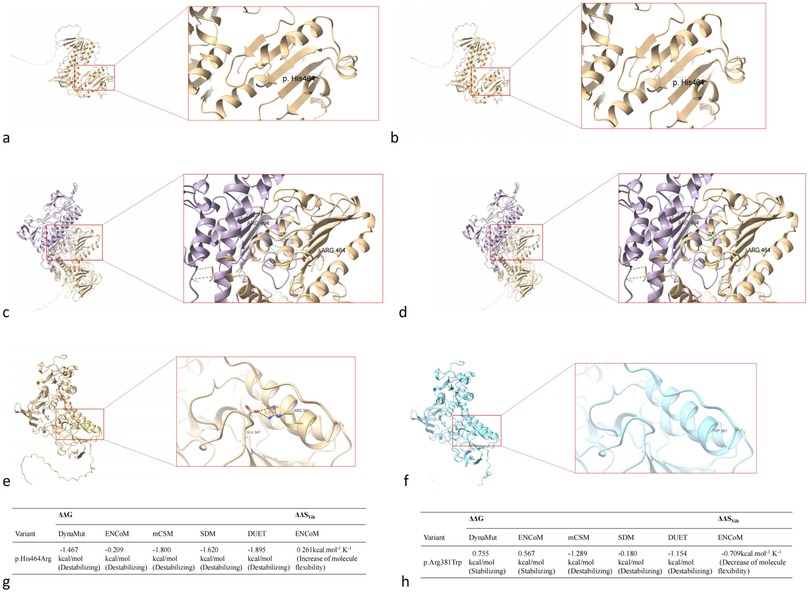
Figure 7. Impact of TXNRD2 gene mutation (c.1391A > G p.H464R and c.1141C > T p.R381W) on protein structure. (a) the tertiary structure of TXNRD2 p.H464R, (b) the tertiary structure of TXNRD2 p.H464 wild type, (c) the quaternary structure of TXNRD2 p.H464R, (d) the quaternary structure of TXNRD2 p.H464 wild type, (e) the tertiary structure of TXNRD2 p.A381 T, (f) the tertiary structure of TXNRD2 p.A381 wild type, (g) the DynaMut website predicted of TXNRD2 p.H464R, (h) the DynaMut website predicted of TXNRD2 p.A381 T.
Discussion
FGD exhibits genetic heterogeneity, with five main subtypes classified according to their associated genes: FGD1 [melanocortin-2 receptor associated protein (MC2R)], FGD2 [melanocortin receptor accessory protein 2 (MRAP)], FGD3 [steroidogenic acute regulatory protein (STAR)], FGD4 [nicotinamide nucleotide transhydrogenase (NNT)], and FGD5 (TXNRD2) (4). While MC2R and MRAP mutations account for approximately 45%–50% of cases, TXNRD2-associated FGD5 remains exceptionally rare. Table 2 summarizes characteristics of 4 previously reported FGD5 families with TXNRD2 variants thus far (5–8) To date, only 10 probands across four independent kindreds with TXNRD2 mutations have been reported globally. Our case represents the fifth documented family and the eleventh proband. Because FGD5 cases are rare, the gene was not reported in the initial report of this patient. Therefore, it is also a reminder to clinicians that the raw data need to be re-analyzed when the genetic report of a patient with glucocorticoid deficiency presents a negative report. TXNRD2 is one of three thioredoxin reductases, which in mammals are cytosolic (TXNRD1), mitochondrial (TXNRD2), and testis-specific thioredoxin reductase (TXNRD3). Mitochondrial thioredoxin reductase (TXNRD2), and mitochondrial thioredoxin and peroxide reducing proteins III and V are essential for the mitochondrial scavenging of reactive oxygen species (ROS), the excess of which can lead to oxidative stress and cell death (9).
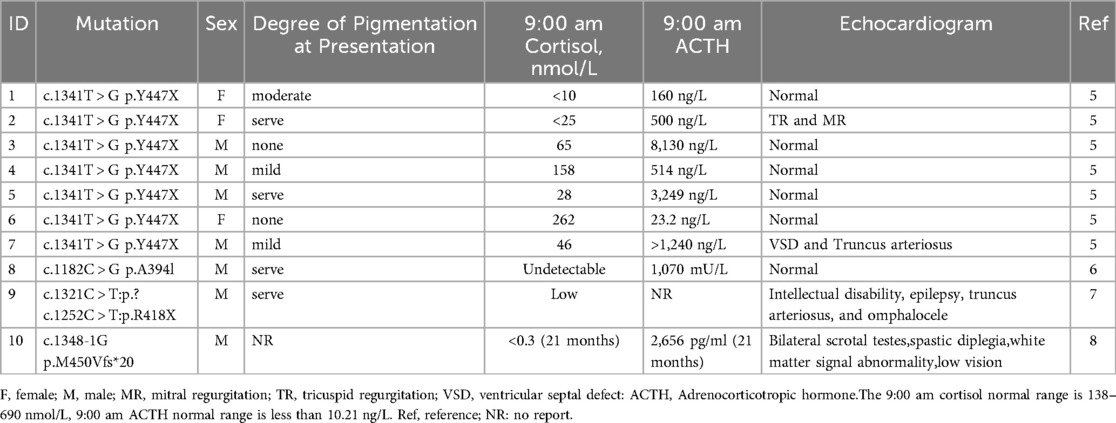
Table 2. Clinical details of FGD5 cases reported in the previous literature.
The thioredoxin (Trx) system consists of Trx, thioredoxin reductase (TR), and reduced nicotinamide adenine dinucleotide phosphate (10). TRX is an important antioxidant molecule that plays a prominent role in redox reactions by resisting the cell death caused by multiple stresses. Trx, thioredoxin reductase (TrxR) and NADPH together form the thioredoxin reductase system, which is involved in many physiological processes (11).
The first step in steroid hormone synthesis is the catalytic conversion of cholesterol-20, 22-desmolase (CYP11A or P450ssc) from cholesterol to pregnenolone (12). This enzyme binds to the inner membrane of mitochondria in all steroid-producing tissues. The adrenal cortisol production pathway is dependent on the energy supply of NADH and NADPH. Thus, the mitochondrial respiratory chain and ROS system are critical to steroid production, and mitochondrial oxidative stress disorder is associated with adrenal gland disease. TXNRD2 messenger RNA levels are high in the adrenal cortex. Loss of NNT gene function leads to decreased NADPH production, resulting in defects in antioxidant defenses, loss of mitochondrial DNA integrity, and reduced mitochondrial mass. in vitro models suggests that TXNRD2 deficiency leads to increased mitochondrial superoxide production, which is not fully compensated for by the glutathione system, resulting in oxidative stress impediment and steroid production. Deficiency in TXNRD2 impairs the functional integrity of the thioredoxin antioxidant system, leading to the accumulation of reactive oxygen species (ROS). Consequently, this redox imbalance perturbs the glutathione (GSH) metabolic network, as evidenced by a significant shift in the GSH/GSSG equilibrium ratio, reflecting systemic oxidative stress burden. Therefore, TXNRD2 and NNT mutations result in glucocorticoid deficiency, but the exact mechanism underlying this relationship is unknown (13).
TXNRD2 mutations demonstrate tissue-selective pathogenicity, primarily targeting adrenal fasciculata while sparing other high-energy organs—a stark contrast to systemic mitochondrial disorders caused by TXNRD2 defects (e.g., cardiomyopathy in encephalopathic forms). TXNRD2 deficiency disrupts glutathione regeneration, potentiating lipid peroxidation and cytochrome P450scc inactivation (14). Due to cardiomyocytes being highly dependent on mitochondrial energy metabolism, TXNRD2 may play a key role in the progression of heart failure in patients with ischemic heart disease, hereditary cardiomyopathy, or other diseases. In mice, TXNRD2 is essential for normal cardiac development and function, with mutations leading to mitochondrial degeneration, which in turn reduces myocardial contractility (15).
Gene mutations in the TXNRD2 have been linked to rare cases of dilated cardiomyopathy (1.3%) (16), and heterozygous carriers of TXNRD2 gene mutations have also been reported to have dilated cardiomyopathy, but no individuals with pure or variant TXNRD2 exhibited any signs of cardiomyopathy or conduction disease (17).
Notably, our patient exhibited electrocardiogram abnormalities including corrected QT interval prolongation and premature atrial contractions. Although previous reports of TXNRD2-associated FGD5 did not describe cardiac manifestations, this observation aligns with emerging evidence of TXNRD2's critical role in myocardial redox homeostasis. Cardiomyocytes, being highly dependent on mitochondrial energy metabolism, require efficient reactive oxygen species scavenging systems. Murine models with TXNRD2 deficiency develop fatal dilated cardiomyopathy through mechanisms involving mitochondrial degeneration and impaired contractility. While human heterozygous carriers typically show normal cardiac function, our findings suggest that biallelic TXNRD2 mutations might predispose to subclinical conduction abnormalities, warranting longitudinal cardiac monitoring in FGD5 patients. Heterozygous carriers of TXNRD2 mutations have normal adrenal function, and haplotype insufficiency of TXNRD2 does not result in adrenal phenotypic abnormalities. The details of our case are consistent with those in previous cases of the TXNRD2 mutation with only glucocorticoid deficiency and no mineralocorticoid deficiency.
The compound heterozygous variants identified in this case (c.1391A > G; p.H464R and c.1141C > T; p.R381W) occur in critical functional domains. The p.H464R substitution affects a conserved residue in the selenocysteine insertion sequence domain, essential for catalytic activity. The p.R381W variant disrupts a structurally important region adjacent to the flavin adenine dinucleotide binding pocket. Functional studies confirmed these mutations reduce TXNRD2 protein stability and expression, consistent with previous reports of pathogenic TXNRD2 variants causing impaired enzyme dimerization and NADPH binding. Our case harboured compound heterozygous variants in TXNRD2 and abnormal ECG findings, which have not been reported previously, but the related mechanism is unknown.
Diagnostically, this case underscores the importance of re-evaluating negative genetic reports in patients with glucocorticoid deficiency. Initial testing frequently focuses on MC2R and MRAP mutations, potentially overlooking rare causes like TXNRD2 defects. mineralocorticoid status, sexual development, and thyroid function remain normal in this case at the time of writing, and long-term follow-up is still required. Long-term management includes: Hydrocortisone replacement (10.8 mg/m2/day),biannual cardiac monitoring for conduction abnormalities, annual assessment of adrenal function and electrolytes,the parents reported improved energy levels but expressed concern regarding persistent hyperpigmentation. FGD is a treatable condition that is often missed due to nonspecific findings. If the diagnosis of this disease is delayed, it can lead to adrenal crisis and even death caused by severe infection. Once diagnosed, lifelong follow-up and treatment are required. Therapeutically, our patient required higher-than-standard hydrocortisone dosing (10.8 mg/m2/day vs. recommended 8–10 mg/m2/day) to normalize adrenocorticotropic hormone levels, suggesting potential interindividual variability in glucocorticoid sensitivity among FGD5 patients. This observation warrants further investigation into genotype-phenotype correlations and optimal dosing strategies for TXNRD2-associated adrenal insufficiency.
Summary
This study expands the TXNRD2 variant spectrum and highlights the importance of considering FGD5 in patients with isolated glucocorticoid deficiency, particularly those with ECG abnormalities. Reanalysis of negative genetic reports using updated databases is crucial for diagnosis. Long-term monitoring for cardiac complications is recommended.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
The studies involving humans were approved by This study was approved by the Institutional Ethics Board of Children's Hospital of Soochow University (2021CS003). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants' legal guardians/next of kin. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
XW: Conceptualization, Writing – original draft, Writing – review & editing. XC: Writing – review & editing. TC: Conceptualization, Writing – review & editing. RX: Investigation, Writing – review & editing. QC: Conceptualization, Writing – review & editing. HW: Formal analysis, Writing – original draft, Writing – review & editing. FW: Conceptualization, Formal analysis, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was funded by a Summit Project of Clinical Medicine (ML13100523).
Acknowledgements
We thank the study participants for generously donating their time and information. Moreover, we thank the families for their consent to publish this report.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2025.1585582/full#supplementary-material
References
1. Mazur A, Ostański M, Kalina M. Rodzinny niedobór glikokortykosteroidów [familial glucocorticoid deficiency]. Pediatr Endocrinol Diabetes Metab. (2007) 13(2):91–4. Polish.17880814
2. Malikova J, Flück CE. Novel insight into etiology, diagnosis and management of primary adrenal insufficiency. Horm Res Paediatr. (2014) 82(3):145–57. doi: 10.1159/000363107
3. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
4. Maharaj A, Maudhoo A, Chan LF, Novoselova T, Prasad R, Metherell LA, et al. Isolated glucocorticoid deficiency: genetic causes and animal models. J Steroid Biochem Mol Biol. (2019) 189:73–80. doi: 10.1016/j.jsbmb.2019.02.012
5. Prasad R, Chan LF, Hughes CR, Kaski JP, Kowalczyk JC, Savage MO, et al. Thioredoxin reductase 2 (TXNRD2) mutation associated with familial glucocorticoid deficiency (FGD). J Clin Endocrinol Metab. (2014) 99(8):E1556–63. doi: 10.1210/jc.2013-3844
6. Patjamontri S, Lucas-Herald AK, McMillan M, Prasad R, Metherell LA, McGowan R, et al. Thioredoxin reductase 2 (TXNRD2) variant as A cause of micropenis, undescended testis and selective glucocorticoid deficiency. Horm Res Paediatr. (2024) 97(5):509–51. doi: 10.1159/000535528
7. Maddirevula S, Alzahrani F, Al-Owain M, Al Muhaizea MA, Kayyali HR, AlHashem A, et al. Autozygome and high throughput confirmation of disease genes candidacy. Genet Med. (2019) 21(3):736–42. doi: 10.1038/s41436-018-0138-x
8. Brachet C, Laemmle A, Cools M, Sauter K-S, De Baere E, Vanlander A, et al. Insight into the role of TXNRD2 in steroidogenesis through a novel homozygous TXNRD2 splice variant. Eur J Endocrinol. (August 2024) 191(2):144–55. doi: 10.1093/ejendo/lvae090
9. Lu J, Holmgren A. The thioredoxin antioxidant system. Free Radic Biol Med. (2014) 66:75–87. doi: 10.1016/j.freeradbiomed.2013.07.036
10. Liu Z. Antioxidant activity of the thioredoxin system. Biophys Rep. (2023) 9(1):26–32. doi: 10.52601/bpr.2023.230002
11. Tuladhar A, Hondal RJ, Colon R, Hernandez EL, Rein KS. Effectors of thioredoxin reductase: brevetoxins and manumycin-A. Comp Biochem Physiol C Toxicol Pharmacol. (2019) 217:76–86. doi: 10.1016/j.cbpc.2018.11.015
12. Katsumata N, Ohtake M, Hojo T, Ogawa E, Hara T, Sato N, et al. Compound heterozygous mutations in the cholesterol Side-chain cleavage enzyme gene (CYP11A) cause congenital adrenal insufficiency in humans. J Clin Endocrinol Metab. (2002) 87(8):3808–13. doi: 10.1210/jcem.87.8.8763
13. Pons Fernández N, Moriano Gutiérrez A, Taberner Pazos B, Tarragon Cros A, Díez Gandía E, Zuñiga Cabrera Á. A novel mutation in the NNT gene causing familial glucocorticoid deficiency, with a literature review. Ann Endocrinol (Paris). (2024) 85(1):70–81. doi: 10.1016/j.ando.2023.05.011
14. Yoshioka J. Thioredoxin reductase 2 (TXNRD2) regulates mitochondrial integrity in the progression of age-related heart failure. J Am Heart Assoc. (2015) 4(7):e002278. doi: 10.1161/JAHA.115.002278
15. Kiermayer C, Northrup E, Schrewe A, Walch A, de Angelis MH, Schoensiegel F, et al. Heart-specific knockout of the mitochondrial thioredoxin reductase (TXNRD2) induces metabolic and contractile dysfunction in the aging myocardium. J Am Heart Assoc. (2015) 4(7):e002153. doi: 10.1161/JAHA.115.002153
16. Rajapreyar I, Sinkey R, Pamboukian SV, Tita A. Did a shared thioredoxin-reductase gene mutation lead to maternal peripartum cardiomyopathy and fatal dilated cardiomyopathy in her son? A case report. Case Rep Womens Health. (2020) 26:e00196. doi: 10.1016/j.crwh.2020.e00196
Keywords: FGD5, TXNRD2, exome sequencing, qPCR, glucocorticoid deficiency
Citation: Wang X, Chen X, Chen T, Xie R, Chen QL, Wu H and Wang F (2025) Case report and literature review: novel TXNRD2 compound heterozygous variants in familial glucocorticoid deficiency type 5. Front. Pediatr. 13:1585582. doi: 10.3389/fped.2025.1585582
Received: 28 February 2025; Accepted: 30 June 2025;
Published: 14 July 2025.
Edited by:
Enrique Medina-Acosta, State University of Northern Rio de Janeiro, BrazilReviewed by:
Tatiana Novoselova, Queen Mary University of London, United KingdomAvinaash Vickram Maharaj, Queen Mary University of London, United Kingdom
Copyright: © 2025 Wang, Chen, Chen, Xie, Chen, Wu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haiying Wu, aGFpbmV3aHlAMTYzLmNvbQ==