 Luca Bosisio1
Luca Bosisio1 Matteo Cataldi2
Matteo Cataldi2 Marina Grandis1,3Barbara Tappino4
Marina Grandis1,3Barbara Tappino4 Monica Traverso5Francesco Germano1,6
Monica Traverso5Francesco Germano1,6 Lino Nobili1,2
Lino Nobili1,2 Chiara Fiorillo1,2*
Chiara Fiorillo1,2*
- 1Department of Neuroscience, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health, Department of Neuroscience (DINOGMI), University of Genoa, Genoa, Italy
- 2Child Neuropsychiatry Unit, IRCCS Istituto Giannina Gaslini, Genoa, Italy
- 3Neurology Clinic, IRCCS, Ospedale Policlinico San Martino, Genova, Italy
- 4Central Laboratory of Analysis, IRCCS Istituto Giannina Gaslini, Genoa, Italy
- 5UOC Medical Genetic, IRCCS Istituto Giannina Gaslini, Genoa, Italy
- 6Unit of Neurology and Neurophisiolgy, EO Ospedali Galliera, Genoa, Italy
Aim: Parsonage-Turner syndrome, also known as neuralgic amyotrophy affects the brachial plexus and includes idiopathic (INA) and rare hereditary forms (HNA). Mutations in the SEPTIN9 gene, which encodes a cytoskeletal GTPase, have been implicated in HNA. While Parsonage-Turner syndrome is typically adult-onset, with stress often acting as a trigger, the presentation in children is less acknowledged.
Methods: We report a case of 9-year-old girl with brachial plexus neuritis who carries a SEPTIN9 missense mutation inherited from her father. We conducted a literature review to explore early-onset cases and gain insight into the disease's progression over time.
Results: Patient presented with episodic intense pain and severe weakness in her right upper limb since age 5 years. Central nervous system involvement and inflammatory polyneuropathy were excluded. Neurological assessment showed weakness and muscle atrophy in the right shoulder girdle. Dysmorphic features, such as long nasal bridge, hypertelorism, and epicanthal folds, were also noted. Her father reported a similar episode in the past without investigations. SEPTIN9 gene sequencing revealed the missense mutation (c.262C>T; p.Arg88Trp) in both individuals. The review of 109 patients with hereditary neuropathy linked to SEPTIN9 mutations revealed a mean age of onset at 13 years, though the average time from symptom onset to diagnosis was 22 years. The syndrome typically follows a relapsing-remitting course, but monophasic and progressive forms are also described.
Conclusion: Clinicians should consider HNA in children with asymmetric upper limb weakness and dysmorphic features, especially with a family history of upper limb neuralgia. Early diagnosis can improve long-term outcomes and avoid unnecessary tests.
Introduction
Neuralgic amyotrophy (NA) is a rare neurological disease affecting the brachial plexus, first identified in 1948 by Parsonage and Turner (1).
The disorder may present monophasic, relapsing-remitting or, less frequently, slowly progressive course. Subjects typically experience an abrupt onset of intense pain in the upper limbs, predominantly in scapulohumeral and arm regions, which is then followed, after days to weeks, by the development of muscle weakness and, in some cases, muscle atrophy. Attacks are usually unilateral but may also occur bilaterally (2, 3). Two variants of NA have been described: the idiopathic neuralgic amyotrophy (INA) and the hereditary neuralgic amyotrophy (HNA), an autosomal dominant disorder (4, 5). From the genetic point of view, several missense mutations and pathogenic duplications of SEPTIN9 gene have been reported (2, 6). Here we describe the clinical, electrophysiological, and genetic data of a pediatric HNA patient and provide a literature review of neuropathy phenotypes associated with SEPTIN9 mutation.
Methods
Case report
Clinical data collection, neurological examination, and nerve conduction studies were conducted at Gaslini Children Hospital by pediatric neurologists (LB, MC and CF). Genetic studies were conducted by geneticists (BT, MT) by Sanger sequencing of the coding regions of the SEPTIN9 gene.
Search strategy
We performed a comprehensive search of PubMed/MEDLINE. The search terms were a) “SEPTIN9”, and B) “SEPT9”.
Inclusion and exclusion criteria
We selected studies published in peer-reviewed journals. We included papers with at least one patient carrying a SEPTIN9variant and presenting with a peripheral nervous system (PNS) disorder. Only original case reports and case series were considered, as they provide individual-level clinical details. In contrast, review articles and meta-analyses were excluded to avoid duplication and ensure consistency in the data extraction process. Studies that were not available online or from which critical clinical or genetic data could not be extracted were also excluded.
Study selection
Two reviewers (LB and MC) analyzed 652 manuscripts by title and abstracts from January 1993 to June 2023. Although SEPTIN9 mutations were first recognized as causative for hereditary neuralgic amyotrophy in 2005, we extended the search period back to January 1993 to identify earlier reports of clinically relevant cases that may have been genetically characterized in later studies or retrospectively associated with SEPTIN9. A total of 41 studies potentially eligible for inclusion were selected. After full-text evaluation, 15 manuscripts were included (2–16). Eligibility was assessed according to inclusion and exclusion criteria. Any discrepancies between the reviewers’ selection were resolved by a third reviewer (CF) consensus. If multiple publications shared the same pedigree, we evaluated only the latest paper or the one we considered most accurate for the purposes of our review. HNA symptomatic cases not genetically tested but with a family history of SEPTIN9 gene mutation were equally considered variant carriers.
Data extraction and synthesis
We extracted the following clinical features for each patient: sex, diagnosis, disease family history, disease onset and relapse age, diagnosis age, clinical features, lower limbs involvement, dysmorphic features, neurophysiological examination, disease course, residual neurological signs or symptoms and genetic variant.
Results
Case presentation
A 9-year-old Italian girl with normal development and good health came to our institute the age of 5 years for 2 previous episodes of pain and weakness on the right shoulder girdle and arm occurred in the past months. These episodes were described, without any physical or infectious triggers, lasting several weeks and with spontaneous remission of the pain but with residual limitation of the right arm elevation. Neurological examination showed mild bilateral atrophy, and hypotonia of the scapular girdle, predominant in the right side. Muscle strength in right deltoids and levator scapula muscles was reduced (Medical Research Council scale for muscle strength, MRC, 4) with difficulties in elevation of the right arm (Figures 1B,C). Minor dysmorphic features were noted including long nasal bridge, small oral openings, hypertelorism, epicanthal folds, and neck skin folds (Figure 1A). Cranial nerves were spared. Muscle strength was normal in lower limbs as well as sensation and coordination. Cognitive functions, vision and hearing were normal. Pain or paresthesia were not present during the evaluation. Nerve conduction studies (NCS), performed several months after the last pain episode, showed normal motor conduction velocities and Compound Muscle Action Potential (CMAP) amplitudes in the median, ulnar, tibial, and peroneal nerves bilaterally, as well as normal sensory conduction velocities in the median, ulnar, and sural nerves on both sides. Giving the normal NCS study and neglecting the unspecific pain symptom reported in such a small age patient, we suspected an inherited myopathy, but genetic tests (CGH-array and NGS genetic panel for congenital myopathy) were negative.
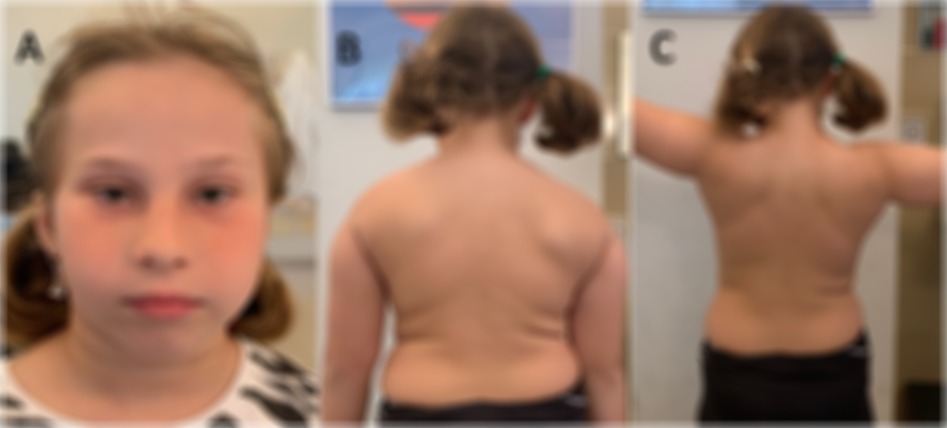
Figure 1. Clinical features of the patient at age 9 years. (A) The minor dysmorphic features including hypotelorism, shortened palpebral fissures, epicanthal fold, long nasal bridge, and neck skin folds; (B,C) the neuromuscular findings including bilateral winged scapula, right sloped shoulder and lesser strength in elevation of the right arm.
During the follow up visits, hypotonia and atrophy of the scapular girdle persisted, and we also detected atrophy of both pectoral muscles (more evident on the right side), hyporeflexia of the biceps and triceps reflexes and mild hyperlordosis. No sensory disturbances or pain were observed. A repeat nerve conduction study was performed at the age of 8, confirming normal conduction velocities and amplitudes of both sensory and motor potentials in the upper and lower limbs. However, an asymmetry of the F response from the ulnar nerves was detected, with a lower persistence of the F waves in the affected side. Electromyography (EMG) revealed signs of chronic denervation of right deltoid muscle including presence of polyphasic MUP with increased duration.
A closer look at the family history revealed that also the child's father suffered from two episodes of upper limb pain and weakness in the past: the first occurred at age 10 years and it was clinically characterized by muscle weakness in his right upper limb. At the age of 36 years, he had a relapse apparently triggered by a mild compression applied to his left arm during sleep. Paresthesias, pain and weakness in the left hand were detected. The patient clinically improved over months but a mild atrophy of the left thenar eminence remained. In suspicion of an early onset of a HNA, we performed a Sanger sequencing for SEPTIN9 gene, which showed a heterozygous pathogenic variant (c.262C>T; p.Arg88Trp) (RefSeq: NM_006640.5 in GenBank) in both child and father.
Review of HNA patients with SEPTIN9 mutations
We found 32 pedigrees (109 patients) with mutations in the SEPTIN9 gene and a HNA clinical neurological phenotype, with the only exception of one family reported as Charcot-Marie-Tooth (CMT) (8). The mean age at disease onset, extrapolated by available retrospective information, was 13 years while the mean age at syndrome diagnosis was 35 years. In only 22 cases the disease course was available: we observed a clear prevalence of relapsing and remitting disorder over the monophasic or primarily progressive form. Clinical manifestations exclusively involved the upper limbs, except for three cases: two patients who received a diagnosis of CMT and one HNA patient with lower limbs involvement. The most common symptoms in the cohort included shoulder weakness followed by upper limbs pain. Fourteen patients had residual signs, mostly shoulder or scapular girdle atrophy, during remission stages. The nerve conduction studies or EMG investigation was performed on 14 patients, data were available in Table 1. Dysmorphic features were clearly reported in 45 patients. The most common signs were hypertelorism, skin folds in the neck and arms, and short stature (Table 1; Supplementary Table S1). Therapy history was available in only 7 patients: 3 patients received treatment with intravenous immunoglobulins with benefit in reducing the attack duration (7, 13, 15). The remaining 4 patients were treated with other drugs, and one of the two patients treated with steroids showed a positive effect from the treatment (3, 9). As per genotype, the most common mutations are the recurrent missense c.262C>T (Arg88Trp) and several duplications of the SEPTIN9 gene or parts of it (Table 1). Rarer mutations are the 6missense 278 C>T p.Ser93Phe and the private c.-131G>C in the untranslated region of the gene identified in the first HNA family (11). Lately another variant of unknown significance c.1406T>C p.Val469Ala has been described in two patients with clinical diagnosis of CMT (8). Additional details and related references are available in the Supplementary Table S1.
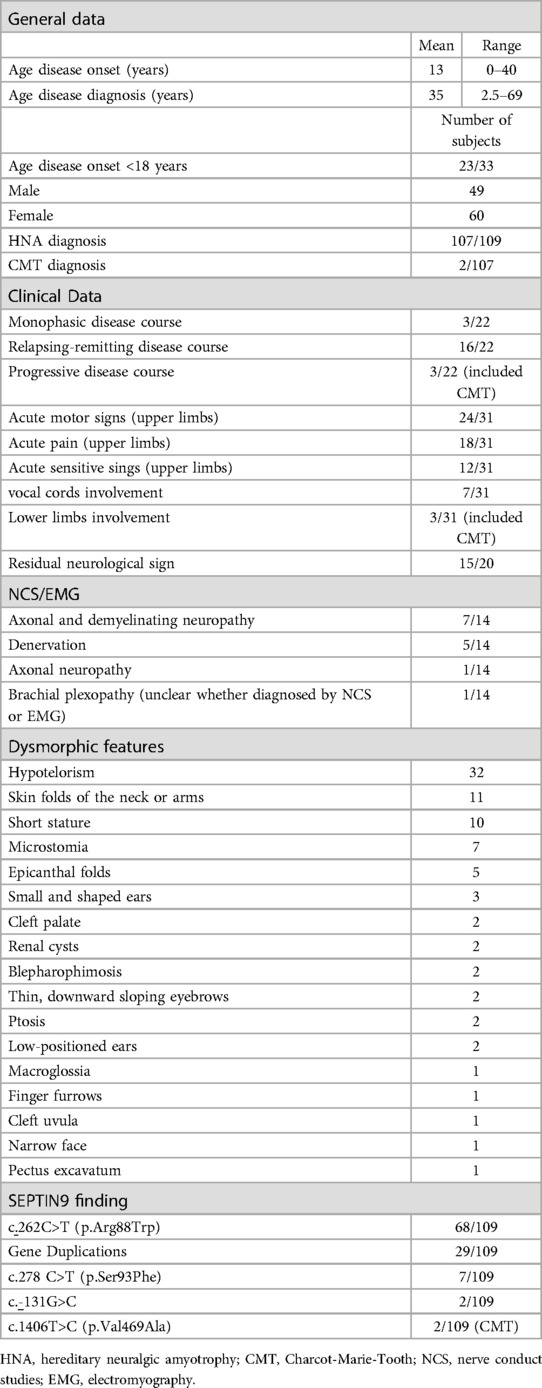
Table 1. General, clinical, genetic, neurophysiological, and dysmorphic features of the 109 SEPTIN9 patients included in our literature review.
Discussion
Parsonage-Turner syndrome is typically considered a disorder of the adult age. Accordingly, in the review of Van Alfen et al. describing 44 patients with HNA, a median age onset of 28 years is reported (17). Conversely, in our revision the age at disease onset was frequently reported in childhood (23/33) including 12 patients with symptoms before 6 years of age. (Table 1; Supplementary Table S1) The average time from clinical onset to syndrome diagnosis is 22 years. This could be a risk factor toward a worse clinical outcome, especially related to an increased risk for exposure to external triggers that may facilitate a relapses such as physical exertion, emotional stress or infectious illness. According to the proposed guidelines and HNA core features (18), our patient showed the typical HNA phenotype due to the SEPTIN9 mutation: a pediatric onset, a family history suggestive of brachial neuralgia, and the presence of typical dysmorphic features (hypertelorism and skin folds in the neck). Moreover, despite her young age and the absence of disease relapses, the child suffered from residual atrophy even after recovering from the first neuritis episode.
Dysmorphic features could be a helpful marker in suspecting HNA; in fact, they were found in the entire population, except for two patients (43/45). These include long nasal bridge, small oral openings (microstomia), hypertelorism, epicanthal folds, and neck skin folds and are easilyrecognizable even in small children. Although nerve conduction studies (NCS) and electromyography (EMG) are essential for characterizing peripheral nerve involvement in neuralgic amyotrophy, they are not always performed in pediatric patients during the initial evaluation, particularly when symptoms are mild or have already resolved. Invasive procedures such as needle EMG are often deferred in very young children unless clearly indicated. In our case, a full electrodiagnostic study including EMG was completed at follow-up, confirming chronic denervation.
This was also reflected in our review, where NCS and EMG investigations proved useful in demonstrating denervation in the upper limbs, with sparing of the lower limbs; indeed, lower limb involvement was reported in only one patient with HNA and in two cases diagnosed with Charcot-Marie-Tooth disease. Most of mutations identified in our review are missense mutations or duplication of SEPTIN9 gene, with different size and location often with unique breakpoints (2). Notably, SEPTIN9 is rich in Alu sequences that might have mediated the occurrence of duplications. Apparently, there is no genotype-phenotype correlation, and both missense mutation or duplications have a similar clinical outcome.
A further SEPTIN9 missense variant c.1406T>C (p.Val469Ala), has been recently described in a autosomal dominant family with a CMT1 phenotype (8) without dysmorphic features. If this shall be confirmed, it is possible that SEPTIN9 gene is involved in a broader spectrum of neuropathies (8). How SEPTIN9 mutations cause HNA is still debated. SEPTIN9 belongs to the family of genes coding for Septins, a group of conserved GTP-binding proteins involved in cytoarchitecture regulation. Septins combine to form oligomeric complexes that assemble into filaments; and the Septin 9 protein promotes microtubules binding (19, 20). It has been hypothesized that mutations block SEPTIN9 filaments assembly in the cytoskeleton and this pathomechanism is a common base in the molecular etiology of other neuropathies (21).
This study has some limitations. Although we conducted a comprehensive literature review, it is possible that some relevant articles were missed, particularly those published in non-indexed journals or not available in English. Furthermore, our search was limited to studies published up to June 2023, and additional cases or relevant findings may have been reported since then. Future updates of the literature may help refine the phenotypic spectrum and natural history of HNA associated with SEPTIN9 mutations.
Conclusion
Despite the rarity of Parsonage Turner brachial plexopathy in children, the typical clinical features combined with family history and the presence of dysmorphic features should lead the clinician to search for a SEPTIN9 gene mutation.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Comitato Etico Regione Liguria. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
LB: Data curation, Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. MC: Data curation, Formal analysis, Investigation, Methodology, Writing – original draft. MG: Data curation, Investigation, Writing – review & editing. BT: Data curation, Investigation, Writing – review & editing. MT: Data curation, Formal analysis, Investigation, Writing – original draft. FG: Data curation, Investigation, Writing – review & editing. LN: Funding acquisition, Supervision, Writing – review & editing. CF: Conceptualization, Investigation, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. CF was supported by Italian Ministry of Health, Ricerca Finalizzata NET-2019-12370049 Grant.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2025.1589397/full#supplementary-material
References
1. Parsonage MJ, Aldren Turner JW. Neuralgic amyotrophy the shoulder-girdle syndrome. Lancet. (1948) 251(6513):973–8. doi: 10.1016/S0140-6736(48)90611-4
2. Collie AMB, Landsverk ML, Ruzzo E, Mefford HC, Buysse K, Adkins JR, et al. Non-recurrent SEPT9 duplications cause hereditary neuralgic amyotrophy. J Med Genet. (2010) 47(9):601–7. doi: 10.1136/jmg.2009.072348
3. Serin HM, Yılmaz S, Kanmaz S, Şimşek E, Aktan G, Tekgül H, et al. A rare cause of brachial plexopathy: hereditary neuralgic amyotrophy. Turk Pediatri Ars. (2019) 54(3):189–91. doi: 10.5152/TurkPediatriArs.2018.5837
4. Hoque R, Schwendimann RN, Kelley RE, Bien-Willner R, Sivakumar K. Painful brachial plexopathies in SEPT9 mutations: adverse outcome related to comorbid states. J Clin Neuromuscul Dis. (2008) 9(4):379–84. doi: 10.1097/CND.0b013e318166ee89
5. Neubauer K, Boeckelmann D, Koehler U, Kracht J, Kirschner J, Pendziwiat M, et al. Hereditary neuralgic amyotrophy in childhood caused by duplication within the SEPT9 gene: a family study. Cytoskelet Hoboken NJ. (2019) 76(1):131–6. doi: 10.1002/cm.21479
6. Hannibal MC, Ruzzo EK, Miller LR, Betz B, Buchan JG, Knutzen DM, et al. SEPT9 Gene sequencing analysis reveals recurrent mutations in hereditary neuralgic amyotrophy. Neurology. (2009) 72(20):1755–9. doi: 10.1212/WNL.0b013e3181a609e3
7. Chuk R, Sheppard M, Wallace G, Coman D. Pediatric hereditary neuralgic amyotrophy: successful treatment with intravenous immunoglobulin and insights into SEPT9 pathogenesis. Child Neurol Open. (2016) 3:2329048X16668970. doi: 10.1177/2329048X16668970
8. Grosse GM, Bauer C, Kopp B, Schrader C, Osmanovic A. Identification of a rare SEPT9 variant in a family with autosomal dominant charcot-marie-tooth disease. BMC Med Genet. (2020) 21(1):45. doi: 10.1186/s12881-020-0984-7
9. Jürgensen L, Fagerberg C, Kibæk M, Brasch-Andersen C. [Neuralgic amyotrophy is an overlooked diagnosis by sudden onset of shoulder pain]. Ugeskr Laeger. (2016) 178(34):V04160292.
10. Klein CJ, Wu Y, Cunningham JM, Windebank AJ, Dyck PJB, Friedenberg SM, et al. SEPT9 mutations and a conserved 17q25 sequence in sporadic and hereditary brachial plexus neuropathy. Arch Neurol. (2009) 66(2):238–43. doi: 10.1001/archneurol.2008.585
11. Kuhlenbäumer G, Hannibal MC, Nelis E, Schirmacher A, Verpoorten N, Meuleman J, et al. Mutations in SEPT9 cause hereditary neuralgic amyotrophy. Nat Genet. (2005) 37(10):1044–6. doi: 10.1038/ng1649
12. Laccone F, Hannibal MC, Neesen J, Grisold W, Chance PF, Rehder H. Dysmorphic syndrome of hereditary neuralgic amyotrophy associated with a SEPT9 gene mutation–a family study. Clin Genet. (2008) 74(3):279–83. doi: 10.1111/j.1399-0004.2008.01022.x
13. Leshinsky-Silver E, Ginzberg M, Dabby R, Sadeh M, Lev D, Lerman-Sagie T. Neonatal vocal cord paralysis—an early presentation of hereditary neuralgic amyotrophy due to a mutation in the SEPT9 gene. Eur J Paediatr Neurol. (2013) 17(1):64–7. doi: 10.1016/j.ejpn.2012.08.006
14. Souza Pd, Gonçalves EA, Badia BdM, Farias IB, Libardi Silva LH, Yanagiura MT, et al. Teaching NeuroImages: slowly progressive hypertrophic brachial plexopathy due to SEPT9 mutation. Neurology. (2020) 95(1):e109–10. doi: 10.1212/WNL.0000000000009739
15. Ueda M, Kawamura N, Tateishi T, Sakae N, Motomura K, Ohyagi Y, et al. Phenotypic spectrum of hereditary neuralgic amyotrophy caused by the SEPT9 R88W mutation. J Neurol Neurosurg Psychiatry. (2010) 81(1):94–6. doi: 10.1136/jnnp.2008.168260
16. Zeng T-F, Ding X, Yang D, Wu CC, Xie DD, Zhang WM. Clinical reasoning: a 26-year-old woman with recurrent pain, weakness, and atrophy in bilateral upper limbs during pregnancy and puerperium. Neurology. (2023) 100(13):631–7. doi: 10.1212/WNL.0000000000201705
17. Van Alfen N, Van Engelen BGM. The clinical spectrum of neuralgic amyotrophy in 246 cases. Brain. (2006) 129(2):438–50. doi: 10.1093/brain/awh722
18. Kuhlenbäumer G, Stögbauer F, Timmerman V, De Jonghe P. Diagnostic guidelines for hereditary neuralgic amyotrophy or heredofamilial neuritis with brachial plexus predilection. Neuromuscul Disord. (2000) 10(7):515–7. doi: 10.1016/S0960-8966(00)00109-7
19. Kim MS, Froese CD, Estey MP, Trimble WS. SEPT9 occupies the terminal positions in septin octamers and mediates polymerization-dependent functions in abscission. J Cell Biol. (2011) 195(5):815–26. doi: 10.1083/jcb.201106131
20. Kuzmić M, Castro Linares G, Leischner Fialová J, Iv F, Salaün D, Llewellyn A, et al. Septin-microtubule association via a motif unique to isoform 1 of septin 9 tunes stress fibers. J Cell Sci. (2022) 135(1):jcs258850. doi: 10.1242/jcs.258850
Keywords: Parsonage-Turner syndrome, SEPTIN9, children neuropathy, HNA, mutation
Citation: Bosisio L, Cataldi M, Grandis M, Tappino B, Traverso M, Germano F, Nobili L and Fiorillo C (2025) Case Report: Parsonage-Turner syndrome due to SEPTIN9 mutation: report of an Italian family with childhood onset and review of the literature. Front. Pediatr. 13:1589397. doi: 10.3389/fped.2025.1589397
Received: 8 May 2025; Accepted: 10 July 2025;
Published: 7 August 2025.
Edited by:
German Moris, SESPA, SpainReviewed by:
Natalie Winter, Hertie Institute for Clinical Brain Research (HIH), GermanyJames Meiling, Mayo Clinic, United States
Copyright: © 2025 Bosisio, Cataldi, Grandis, Tappino, Traverso, Germano, Nobili and Fiorillo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chiara Fiorillo, Y2hpYXJhLmZpb3JpbGxvQGVkdS51bmlnZS5pdA==