 Chuan Gan1
Chuan Gan1 Yuanyuan Wu
Yuanyuan Wu Tao Qin
Tao Qin- 1Department of Infectious Diseases Children’s Hospital of Chongqing Medical University, National Clinical Research Center for Child Health and Disorders, Ministry of Education Key Laboratory of Child Development and Disorders, Chongqing Key Laboratory of Child Rare Diseases in Infection and Immunity, The First Batch of Key Disciplines on Public Health in Chongqing, Chongqing, China
- 2Health Medicine Center, The Second Affiliated Hospital of Chongqing Medical University, Chongqing Medical University, Chongqing, China
Background: Gaucher disease (GD) is a rare autosomal recessive disorder caused by mutations in the glucocerebrosidase1 (GBA1) gene. Reports on the clinical presentations of various types of GD in Chinese children are scarce, and there is limited research addressing co-occurrence of GD with bacterial (including tuberculosis), viral, or fungal, infections. Pediatric GD typically manifests with greater severity due to developmental vulnerability of organ systems and immature immunity, leading to heightened infection risks. Unlike non-GD children, those with GD exhibit multiorgan involvement (e.g., hepatosplenomegaly, cytopenias) that predisposes them to opportunistic infections. In this study, we describe the clinical features and infection risks associated with different types of GD in Chinese children.
Methods: This study was done in Children's hospital of Chongqing Medical University. Seventeen patients aged <18 years, diagnosed with GD from January 2008 to December 2019, were enrolled. Clinical symptoms, laboratory results, mutation genotypes, and imaging data were collected for analysis.
Results: Of the 17 patients, 9 were diagnosed with Type 2 GD, while 4 each had Type 1 and 3 GD. Median (interquartile range) age of onset was 7 (3.0–18.5) months. Approximately two-thirds of patients experienced malnutrition, and most exhibited hepatosplenomegaly and hematological abnormalities. Anemia was the most frequent hematological disorder, followed by thrombocytopenia, with almost half developing leukopenia. Liver function abnormalities were common, particularly in Type 2 GD, and characterized by elevated aspartate aminotransferase and glutamyl transpeptidase levels, prolonged prothrombin time, and decreased albumin. Patients with Type 2 GD had increased susceptibility to infections, with respiratory failure from severe infections a leading cause of death. Genome sequencing revealed a novel deletion mutation (c.787_c.788 delAA) in the GBA1 gene associated with Type 2 GD.
Conclusion: In pediatric patients with Gaucher disease, Type 1 GD is associated with worse hematological impairment, while Type 2 GD involves significant hepatic insufficiency and heightened susceptibility to infections.
Impact of this article
• Clinical manifestations of Gaucher disease (GD) vary significantly.
• Almost half of pediatric patients with all types of GD develop leukopenia.
• Hepatic insufficiency is related to both the type and severity of GD in pediatric patients.
• Patients with Type 2 GD have an increased susceptibility to infections, particularly with gram-negative bacteria.
1 Background
Gaucher disease (GD, OMIM #230800) is a rare autosomal recessive disorder caused by mutations in the glucocerebrosidase1 (GBA1) gene, leading to decreased β-glucocerebrosidase (GCase) activity and consequent issues with lysosomal storage (1). The consequences of this deficiency are generally attributed to the accumulation of the GCase substrate, GlcCer, in macrophages, inducing their transformation into Gaucher cells (2). Gaucher cells subsequently infiltrate the bone marrow, spleen, liver, and multiple systemic organs, eliciting corresponding pathological changes. GD can be categorized into three subtypes based on the presence of neuronopathy and the rate of disease progression. Type 1 GD is non-neuronopathic and the most prevalent form worldwide. Type 2 GD, the acute neuronopathic form, manifests in infancy and patients with this form of GD have a life expectancy of less than 2 years. Type 3 GD is an adolescent onset form with subacute neuronopathy. However, this traditional classification is increasingly being challenged by a growing body of evidence indicating a correlation between Type 1 GD and neurological impairments, such as Parkinson's disease and peripheral neuropathies (3, 4). GD diagnosis relies on detecting reduced GCase activity or direct sequencing of the GBA1 gene. Despite the importance of timely diagnosis and treatment, GD is often missed or misdiagnosed, due to its rarity and heterogeneous symptoms. The incidence of GD is low, with an estimated rate of 1/40,000–1/60,000 births in the general population (2, 5). Clinicians, especially generalists, are often unfamiliar with the early clinical features of GD. Additionally, the signs and symptoms of GD are highly variable, making it challenging to differentiate the disease from other conditions. In a survey by Mistry et al., when patients presented with typical features, only 20% of hematologists or oncologists considered GD in their differential diagnosis (6). Another study by Mehta et al. found that almost one in six patients remained undiagnosed for 7 years or more after initial consultation (7). Hematologists, hepatologists, and pediatricians are the main specialists to whom patients first present (8, 9). Gaucher disease patients in China experience elevated misdiagnosis incidence, substantial diagnostic delays, and financially burdensome treatment expenses (10). Although timely diagnosis can minimize the impact of misdiagnoses and avoid unnecessary invasive diagnostic procedures, as well as being crucial for optimal therapy and patient management, particularly since enzyme replacement and substrate reduction therapies have proven effective in treating certain types of GD. Due to prolonged diagnostic delays coupled with restricted access and prohibitive costs of therapies, Chinese patients often lose confidence in future treatment prospects (10).
Gaucher disease patients may exhibit functional immunodeficiency, clinically manifesting as impaired antimicrobial host defense, delayed resolution of infections, increased infection susceptibility, and increased risk of autoimmune disorders (11–13). Additionally, they demonstrate predisposition to recurrent infections (14). Zahran et al. reported that children with GD1 exhibit a significant expansion of activated T cells in peripheral blood, correlating with heightened infection risks (15). However, systematic data on infection susceptibility in Gaucher disease remain limited, and no study has yet compared infection risk across the different GD subtypes. Therefore, we analyzed Chinese pediatric patients to enhance awareness among hematologists, pediatricians, and other clinicians regarding the distinctive clinical and immunological profile of this rare disorder, with the aim of informing timely diagnosis, treatment selection, and long-term management.
2 Methods
2.1 Patients
This study included 17 patients aged <18 years, diagnosed with GD between January 2008 and December 2019 at the Children's Hospital of Chongqing Medical University. Diagnoses were based on GBA1 activity in peripheral leukocytes, Bone marrow cytology and/or gene sequencing, and were confirmed by Sanger sequencing. Exome capture was conducted using a GenCap Liquid Capture Kit (MyGenostics, MD, USA), and sequencing performed using the Illumina HiSeq 2500 platform. Average sequencing read-depth was 300×, with >90% of the exome covered at ≥20×. The study was approved by the Ethics Committee of the Children's Hospital of Chongqing Medical University [Approval No. 2025 Lunar Review (Research) No. (034)] and conducted in accordance with the Declaration of Helsinki.
2.2 Clinical data
Clinical symptoms, laboratory results, mutation genotypes, and imaging data were extracted from electronic medical records. GD types were classified by three pediatric specialists and follow-up was conducted through clinical visits or telephone counseling, with two patients lost to follow-up. Pneumonia was diagnosed according to the radiologists’ report, based on pulmonary infiltrates or opacities. Three or more episodes of respiratory infections within a year were defined as recurrent respiratory tract infections. Upper respiratory tract infection was defined as presenting with symptoms of upper respiratory infection and a record of visiting the outpatient department. Urinary infection was defined as symptoms of urinary irritability and/or abnormal urinalysis findings, along with a positive urine culture result.
2.3 Statistical analysis
Continuous variables are described using mean and standard deviation or median with interquartile range (IQR) values. Categorical variables are expressed as counts and/or percentages. One-way ANOVA in GraphPad Prism 5 was used to analyze differences between continuous variables, with P values ≤ 0.05 considered statistically significant.
3 Results
3.1 Demographic characteristics of patients with GD
From 2008 to 2019, 17 pediatric GD patients were enrolled: Type 1 (n = 4), Type 2 (n = 9), Type 3 (n = 4); Female predominance: 64.7% (11/17). Median (IQR) age of onset was 7 (3.0–18.5) months. Malnutrition affected 70.6% (12/17) of patients: 75% of those with Type 1 (3/4), 66.7% with Type 2 (6/9), and 75% with Type 3 (3/4) GD. Fifteen patients received at least one follow-up via telephone or outpatient clinic, and a high mortality rate was observed among patients with Type 2 GD. One patient with Type 1 GD died 3 years after diagnosis, while no deaths occurred in the Type 3 GD group during the follow-up period. Hepatosplenomegaly and hematological abnormalities were common; anemia (94.1%) and thrombocytopenia (82.4%) were the most frequent hematological findings, followed by leukopenia (47.1%). Patients with Type 2 GD also more frequently exhibited growth retardation relative to those with Types 1 and 3, with a tendency towards short stature (Table 1).
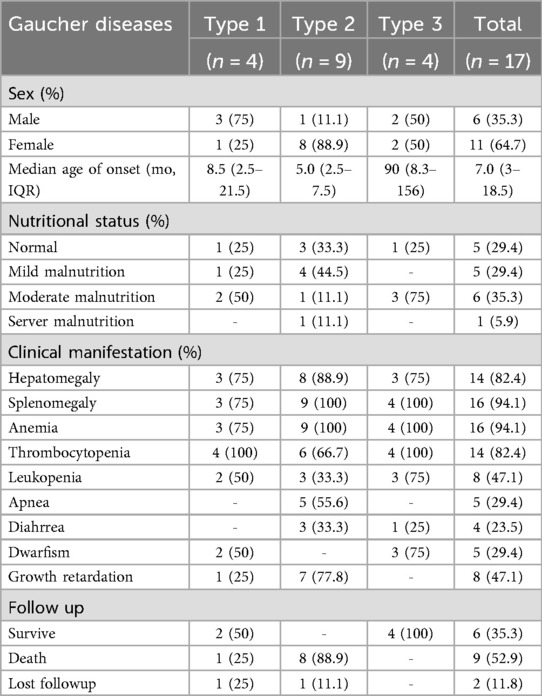
Table 1. Demographic features of Gaucher diseases in children.
3.2 Differences in laboratory findings among patients with the three types of GD
Significant differences in hematological and hepatic function were observed across GD subtypes (Table 2). Patients with Type 1 GD had severe hematological impairment: low leukocyte (3.6 ± 1.7 × 109/L) and hemoglobin levels (58.3 ± 20 g/L), while those with Type 2 GD showed pronounced hepatic dysfunction: increased aspartate aminotransferase (AST; 96.6 ± 47.7 mmol/L) and gamma-glutamyl transferase (GGT; 58.4 ± 79.1 U/L), and decreased albumin (ALB; 34.9 ± 7.1 g/L). Activated partial thromboplastin time (APTT) was prolonged in all groups, but the difference did not reach significance. Prothrombin time (PT) was significantly prolonged in patients with Type 2 GD (13.7 ± 6.09 s). These observations indicate that hematological deficits dominated in Type 1, while hepatic insufficiency correlated with Type 2 severity. Moreover, patients with type 1 exhibited significantly higher levels of alkaline phosphatase (ALP) and lactate dehydrogenase (LDH) than those with types 2 and 3. This finding may reflect the fact that both enzymes are not exclusively hepatic; they are also associated with the hematopoietic and skeletal systems.
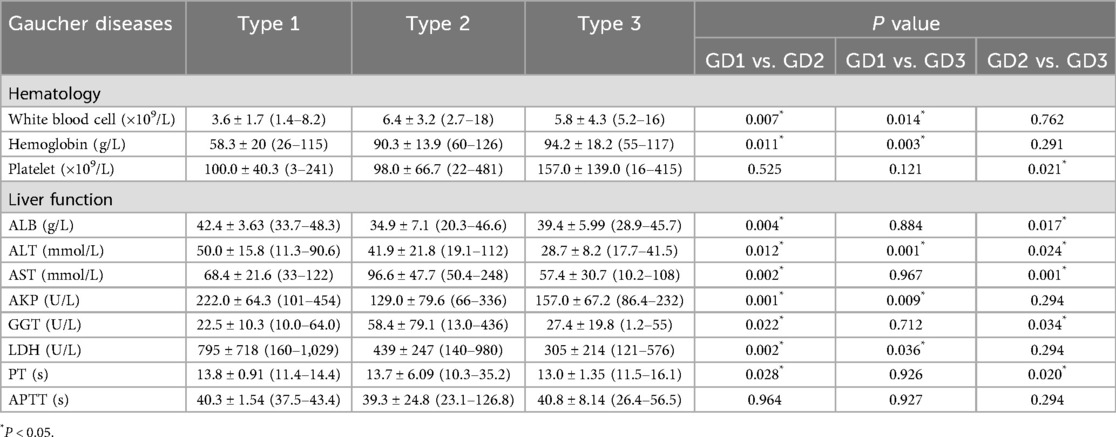
Table 2. The hematology and biochemistry findings in each group.
3.3 Inflammation and infection in patients with GD
A high infection rate (9/17, 52.9%) was observed, particularly in patients with Type 2 GD (8/9, 88.9%). Respiratory infections were most common, accounting for 77.8% (7/9) of cases. Additionally, one patient experienced a urinary tract infection. A rare case of lymphadenopathy due to Mycobacterium bovis following Bacillus Calmette-Guerin (BCG) vaccination was detected in one patient (Table 3). Bacterial infections were predominant, particularly gram-negative bacteria, including E. coli, K. pneumoniae, A. baumannii, and Moraxella catarrhalis. One patient had combined infections with adenovirus, parainfluenza virus, and C. albicans. Overall, patients with Type 2GD had an increased susceptibility to infections.
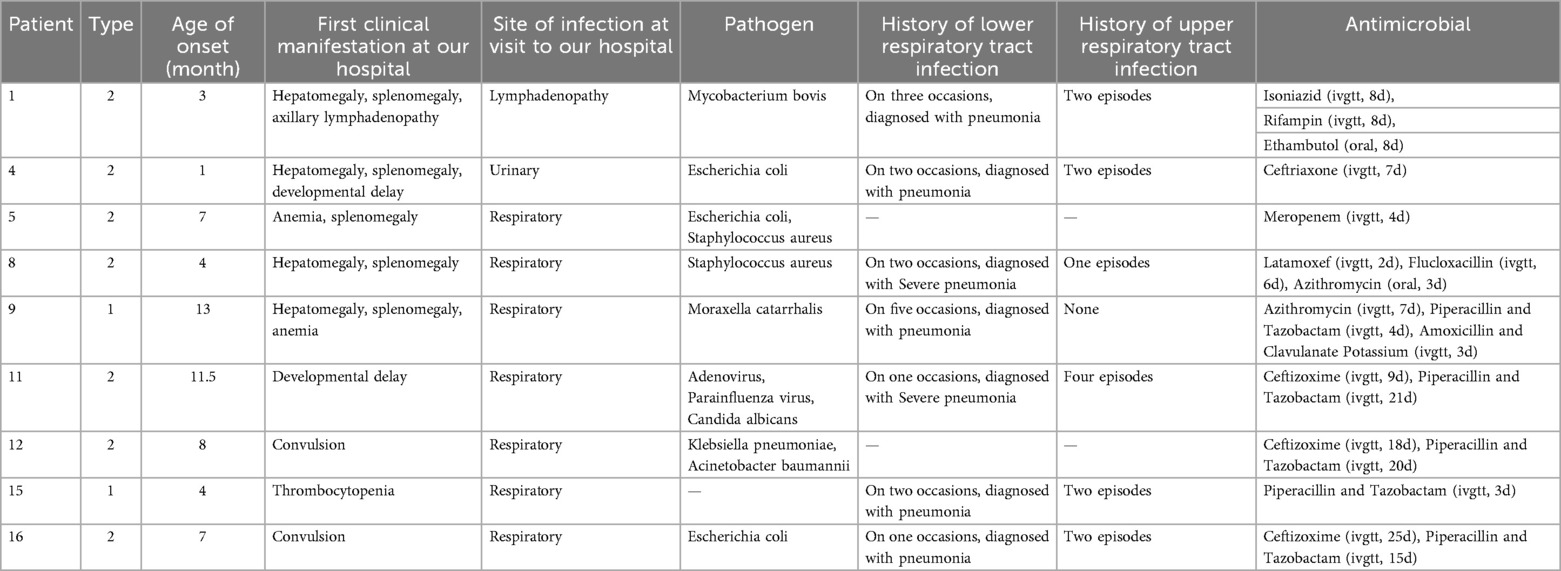
Table 3. Clinical manifestations and infection profiles in Gaucher's disease patients.
3.4 Immunoglobulins and lymphocyte subsets
Five patients underwent immunoglobulin testing, with no significant abnormalities observed in IgG, IgM, and IgE in any of them. One patient had an increase in IgA, while another patient had a decrease in this immunoglobulin. Additionally, four patients underwent lymphocyte subtype analysis, of which two had elevated double-negative T cells (Table 4).
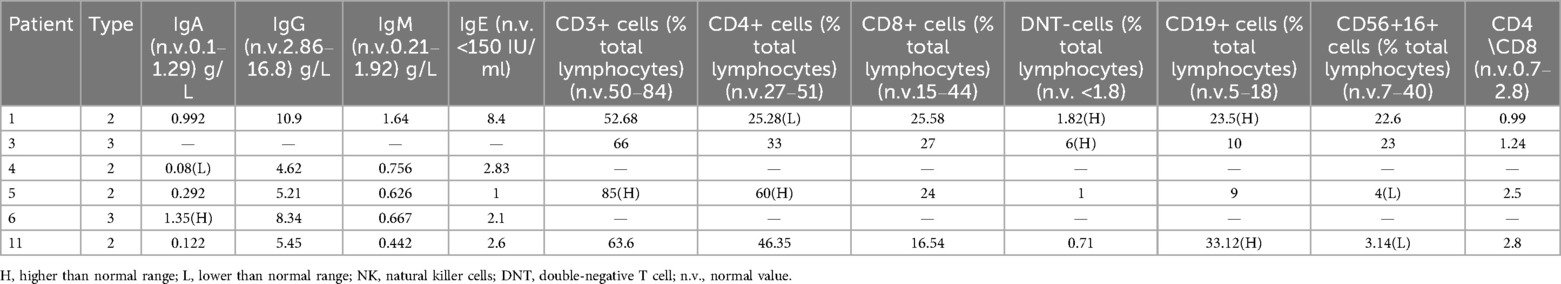
Table 4. Immunoglobulin, lymphocyte subsets for the patient in the study.
3.5 Bone marrow cytology examination results
Bone marrow cytology examinations were completed for 12 patients: two with Type 1, seven with Type 2, and three with Type 3 GD. The granulocyte series/erythroid series ratio was lower in all patients with Type 1 and higher in all those with Type 2 GD, except for one patient with Type 2 GD, who exhibited an inversion. One patient each with Type 1 and Type 2 GD had abnormal granulocyte morphology, while no abnormalities were observed in patients with Type 3 disease; however, all three patients with Type 3 GD exhibited abnormal erythroid morphology, suggesting that this may be a common finding (Table 5). A significant reduction in platelets was observed in two patients, one with Type 1 and the other with Type 2 GD; no platelet abnormalities were detected in patients with Type 3 disease.
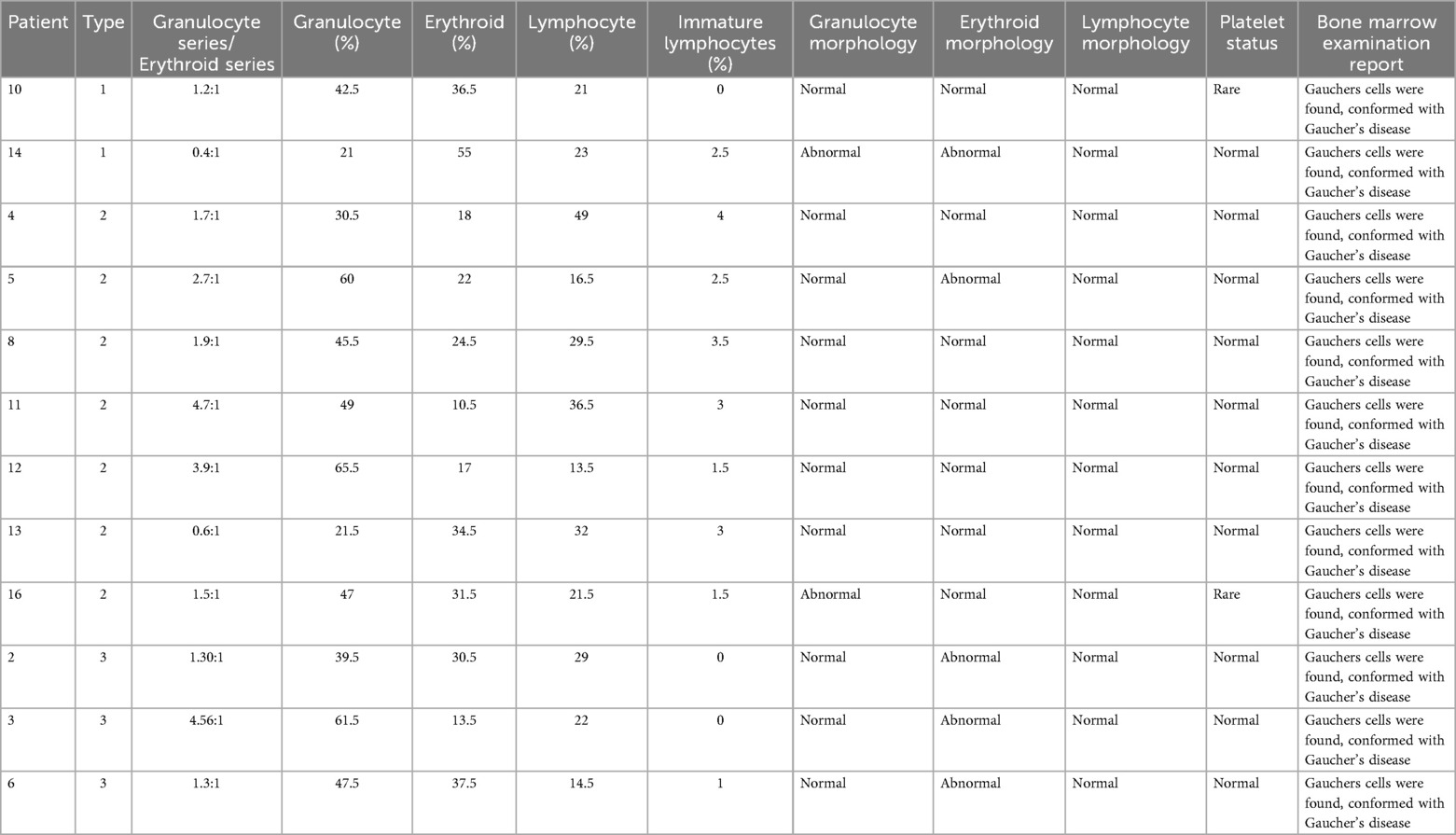
Table 5. Bone marrow cytology in GD patients.
3.6 Imaging characteristics of patients with GD
Abdominal ultrasonography indicated hepatosplenomegaly without liver fibrosis. Chest computed tomography (CT) scans in eight patients revealed abnormalities in six of them, with the most frequent being consolidated patches in bilateral lung lobes. One patient exhibited multiple patch shadows in the left lobe, with bilateral pleural effusions (Figures 1a,b). Cranial magnetic resonance imaging (MRI) revealed encephalatrophy in five of six patients, with two having arachnoid cysts (Figures 1c,d); whether or not arachnoid cysts are a complication of GD requires further investigation.
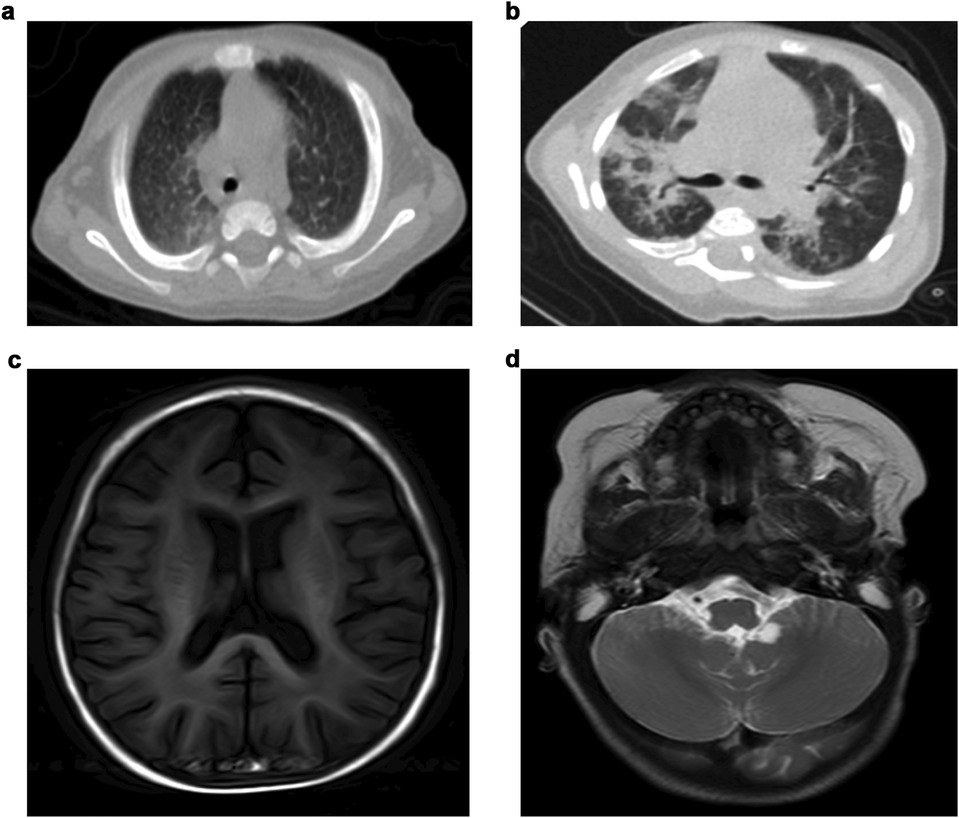
Figure 1. Imaging characteristics of GD patients.
3.7 Genotype and prognosis of patients with GD
Gene sequencing of samples from eight patients revealed the c.1448T > C (p.L483P) mutation was common (6/8). Other mutations detected included c.475C > T (p.R159W), c.501-1G > C, c.1255G > A (p.D419N), c.787_c.788delAA, c.689T > G (p.V230G), c.460G > A (p.G154R), and c.1081G > C (p.D361H) (Table 6). A novel GBA1 mutation genotype (c.787–788 delAA, p.L263V fs*9; c.1448T > C, p.L483P) was identified in a patient who initially presented with neurological disorders at an early stage; motor development retardation onset occurred at 2 months, followed by a rapid disease progression that culminated in the patient's death within only 6 months.

Table 6. Genotype of GBA gene in GD patients.
None of the patients had received enzyme replacement therapy or substrate reduction therapy. Nine of fifteen patients died during the follow-up period. All patients in the Type 2 GD group succumbed within 6 months, due to rapid progression of neuropathy and/or respiratory failure from infection. One patient in the Type 1 GD group died 3 years after disease onset, due to respiratory and heart failure. High mortality was observed, particularly in patients with Type 2 GD. Patients with Type 3 GD exhibited chronic progression during regular follow-up.
4 Discussion
Our study presents a comprehensive characterization of Chinese children with GD, detailing their clinical manifestations, biochemical markers, immunophenotypes, genotypes, and systemic organ involvement, together with systematic longitudinal follow-up of disease progression and prognosis. Type 1 GD patients exhibited pronounced hematologic impairment, whereas type 2 patients showed marked hepatic injury. For the first time, we demonstrate subtype-specific differences in infection risk among Chinese pediatric cohorts, revealing a significantly higher susceptibility and risk in type 2 patients. These findings underscore the importance of tailored infection surveillance and management strategies, particularly for individuals with type 2 GD.
The hematological manifestations of GD include splenomegaly, thrombocytopenia, anemia, and cytopenia; these disorders are attributable to hypersplenism, bone marrow infiltration by Gaucher cells, or intrinsic hematopoietic cell deficiencies (16–18). Occasionally, the phenomenon of erythrophagocytosis has been reported in GD (19, 20), suggesting immune dysfunction in the context of hematological abnormalities. Kaplan et al. found that anemia and severe hepatosplenomegaly were more common in young children, while thrombocytopenia was prevalent in patients up to 18 years old. Type 1 GD is associated with more severe hematological impairment than that detected in other GD types (21). Patients with Type 2 GD exhibit similar findings, with anemia and thrombocytopenia appearing later and/or with moderate severity (22). In the current study, patients in the Type 1 GD group exhibited worse hematological impairment than those in the other groups. In one case, Type 1 GD was comorbid with beta-thalassemia, and the patient presented with severe anemia, which could have interfered with appropriate interpretation of anemia in Type 1 GD. Almost half of patients in this study exhibited leukopenia, which is generally an uncommon hematological impairment. Long-term monitoring of a larger patient cohort is necessary to better evaluate the hematological characteristics of patients with GD.
Neurological impairment is common in Types 2 and 3 GD and can include brainstem degeneration, fine motor dysfunction, hypertonia, rigidity, and seizures (23, 24). Ophthalmological abnormalities, such as horizontal saccade and strabismus, are among the earliest central nervous system features in patients with GD, followed by hypertonia, rigidity, opisthotonus, swallowing impairment, and seizures (25, 26). Apnea caused by laryngeal spasms is a symptom of late stage of Type 2 GD and contributes to shortened life expectancy (27). In our study, neuronopathy was more severe in patients with Type 2 GD than in the Type 3 GD group. Symptoms including psychomotor retardation, seizures, and strabismus were observed in both groups, while hypertonia/hypotonia, opisthotonos, and laryngeal spasms were characteristic signs in the Type 2 GD group (data not shown).
Recent research has confirmed that neuronopathy also occurs in the non-neuronopathic form of GD (i.e., Type 1 GD); In a study, microstructural alterations of brain white matter were discovered in children with Type 1 GD using diffusion tensor magnetic resonance imaging (28), and the researchers compared the topological properties of the brain network in patients with Type 1 GD with those of healthy controls. Children with GD showed abnormal small-world topology and altered distribution of motor- and sensory-related regions throughout the course of the disease (29). These findings support the notion that GD should be considered a continuum, rather than as a set of discrete categories (8).
Bone impairment in GD includes osteopenia, osteonecrosis, bone crises, chronic bone pain, pathological fractures, lytic lesions, and skeletal deformities, which are prevalent symptoms of Types 1 and 3 GD (30–32). Kaplan et al. reported that approximately 80% of pediatric patients had at least one radiological skeletal abnormality at diagnosis (21). Erlenmeyer flask deformity (49%) and bone marrow infiltration (38%) were the two most common radiological manifestations, with older children more prone to severe skeletal problems. In another report, 63% of patients with GD aged <10 years presented with Erlenmeyer flask deformity (33). Importantly, children with GD who manifest bone pain are often incorrectly characterized as having growing pains (34).
In our cohort, the cytological findings from bone marrow examinations exhibited significant differences across the various subtypes of GD. Patients with type 1 consistently demonstrated a uniformly low myeloid-to-erythroid (M/E) ratio, while all type 2 patients presented with an elevated M/E ratio. Additionally, type 3 patients consistently displayed abnormal erythrocyte morphology. The potential correlation between these cellular aberrations and distinct skeletal manifestations warrants further investigation.
Hepatic involvement in GD includes hepatomegaly, liver fibrosis, and hepatocellular carcinoma. In this study, we found that liver dysfunction severity correlated with GD type, with significant abnormalities detected in patients with Type 2 GD (35–37). Patlas et al. analyzed the clinical features of 103 children with GD and found that all exhibited hepatomegaly (38). Serai et al. evaluated liver stiffness in patients with Type 1 GD using magnetic resonance elastography and discovered a significant correlation between liver stiffness and disease severity (39). Starosta et al. investigated hepatic impairment in 42 patients with GD, finding that the rates of liver enzyme abnormalities, hepatomegaly, and steatosis were 68%, 67%, and 8%, respectively; six patients underwent liver biopsy, among which three had liver fibrosis and two had steatohepatitis; one patient developed hepatocellular carcinoma (40). Several studies have observed prolongation of PT and APTT in both adults and children (41–43). In the current study, the severity of liver dysfunction was related to disease type. Patients with Type 2 GD exhibited elevated AST and GGT, but a decrease in ALB. Further, both PT and APTT were prolonged in all three groups, with significantly elevated APTT in the Type 2 GD group, indicating inferior liver function. Our data indicate that liver dysfunction severity correlates with GD type, with Type 2 GD associated with significant abnormalities.
In GD, lung lesions are caused either by Gaucher cell infiltration, leading to interstitial lung disease, including pulmonary fibrosis, pulmonary arterial hypertension, restrictive ventilatory impairment, and intrapulmonary arterial-venous shunts, or by repetitive aspiration pneumopathies, which lead to chronic lung disease (22, 44). Pulmonary involvement is severe in patients with Type 2 GD, often presenting as recurrent respiratory infections. Similarly, most patients with Type 2 GD in our study reported bacterial respiratory infections. These findings align with current evidence indicating that GD can confer increased susceptibility to infection and a propensity for recurrent infectious episodes (14). The heightened infection risk in GD may be attributable to dysfunction of immune cells, including monocytes, macrophages, dendritic cells, T cells, and B cells, as well involvement of cytokines, such as MCP1/CCL2, CXCL8/IL8, CXCL1, IL-1β, IFNγ, and TNFα, are implicated in GD progression (45–50). Gene-knockout mice exhibit impaired thymic maturation (51, 52) and a loss of macrophage function (53). An Egyptian pediatric study reported an increased in CD8⁺ T lymphocytes alongside a reduction in NK cells in GD patients (54). Although these alterations in immune cells and mediators may contribute to heightened susceptibility, the relationship between subtype-specific immunological profiles and differential infection risk in GD remains to be elucidated.
Patients with GD may present with hypergammaglobulinemia. Studies have reported elevated IgG, IgM, and IgA levels in GD patients, with IgG elevation being the most persistent and pronounced. Following enzyme replacement therapy, IgG levels typically rise further, whereas IgA and IgM usually normalize or decrease (55–58). Regrettably, no patients in our cohort received ERT and no significant immunoglobulin elevation was observed. These findings suggest that immunoglobulin dysregulation may be uncommon among Chinese pediatric GD patients. However, given the limited sample size, larger prospective studies with systematic immunoglobulin profiling are warranted to clarify the true prevalence of immunoglobulin abnormalities in this population.
GD may exhibit autoimmune lymphoproliferative syndrome (ALPS)-like characteristics and defects in FAS-mediated apoptosis. Although double-negative T cells are generally considered specific to ALPS, elevated levels of double-negative T cells have also been observed in patients with GD. In our study, lymphocyte subset analysis was performed on samples from four patients, of which 2 (50%) had elevated levels of double-negative T cells, consistent with a report by Maurizio Miano and colleagues (59). This finding also suggests an immunophenotypic overlap between GD and ALPS, indicating that genetic diagnosis is the gold standard for differentiation between these conditions. Over 400 GBA1 mutations have been identified, with some linked to specific phenotypes and ethnic groups (60, 61). The N370S mutation is highly prevalent in the Ashkenazi Jewish and Caucasian populations and is considered “neuroprotective,” as patients with this mutation, in both homozygous and heterozygous states, are less likely to develop neurological impairment (34, 62). The L444P mutation is common in Asian countries and associated with neurological impairment (63–66). Further, patients with Type 3 GD homozygous for the D409H mutation present with characteristic heart valve damage (67–69). Overall, although GD diagnosis depends on GBA1 mutation, the relationships between genotypes and phenotypes appear to be weak.
ERT has significantly improved most manifestations of GD1 and has enhanced the quality of life for patients (70). Additionally, it may provide benefits for patients with GD3 who experience chronic visceral involvement (8). However, pediatric Gaucher disease remains under-recognized in China, with only a small percentage of affected children receiving ERT. Therefore, systematic collection and analysis of long-term clinical data from a larger cohort is essential to facilitate early recognition and accurate diagnosis, enabling timely initiation of ERT and ultimately improving patient outcomes.
5 Conclusion
In Chinese children with Gaucher disease, Type 1 GD may be associated with more pronounced hematological impairment, whereas Type 2 GD is potentially characterized by significant liver damage and heightened susceptibility to infections. Timely diagnosis and appropriate management are crucial for improving the quality of life in pediatric patients with GD in China. Given the clinical and immunophenotypic overlap between GD and IEI, genetic diagnosis is the gold standard.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
The studies involving humans were approved by Ethics Committee of the Children's Hospital of Chongqing Medical University. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants' legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
CG: Data curation, Investigation, Methodology, Writing – original draft. YW: Investigation, Methodology, Supervision, Writing – review & editing. TQ: Methodology, Project administration, Resources, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was funded by the talent cultivation program of Children's Hospital of Chongqing Medical University and Natural Science Foundation of Chongqing, CSTB2024NSCQ-MSX029.
Acknowledgments
We thank all patients involved in the study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ALB, albumin; ALPS, autoimmune lymphoproliferative syndrome; APTT, activated partial thromboplastin time; AST, aspartate aminotransferase; GGT, gamma-glutamyl transferase; GD, Gaucher disease; GCase, β-glucocerebrosidase; IEI, inborn errors of immunity; IQR, interquartile range; PT, prothrombin time; GlcCer, glucosylceramide; GH/IGF-1, growth hormone and insulin-like growth factor 1; ERT, enzyme-replacement therapy.
References
1. Pastores GM, Hughes DA. Gaucher disease. In: Adam MP, Ardinger HH, Pagon RA, editors. GeneReviews®. Seattle (WA): University of Washington, Seattle (2000). p. 1993–2020.
2. Stirnemann J, Belmatoug N, Camou F, Serratrice C, Froissart R, Caillaud C, et al. A review of Gaucher disease pathophysiology, clinical presentation and treatments. Int J Mol Sci. (2017) 18(2):441. doi: 10.3390/ijms18020441
3. Thomas AS, Mehta A, Hughes DA. Gaucher disease: haematological presentations and complications. Br J Haematol. (2014) 165(4):427–40. doi: 10.1111/bjh.12804
4. Mistry PK, Lopez G, Schiffmann R, Barton NW, Weinreb NJ, Sidransky E. Gaucher disease: progress and ongoing challenges. Mol Genet Metab. (2017) 120(1–2):8–21. doi: 10.1016/j.ymgme.2016.11.006
5. Aerts JMFG, Kuo CL, Lelieveld LT, Boer DEC, van der Lienden MJC, Overkleeft HS, et al. Glycosphingolipids and lysosomal storage disorders as illustrated by Gaucher disease. Curr Opin Chem Biol. (2019) 53:204–15. doi: 10.1016/j.cbpa.2019.10.006
6. Mistry PK, Sadan S, Yang R, Yee J, Yang M. Consequences of diagnostic delays in type 1 Gaucher disease: the need for greater awareness among hematologists-oncologists and an opportunity for early diagnosis and intervention. Am J Hematol. (2007) 82(8):697–701. doi: 10.1002/ajh.20908
7. Mehta A, Belmatoug N, Bembi B, Deegan P, Elstein D, Göker-Alpan Ö, et al. Exploring the patient journey to diagnosis of Gaucher disease from the perspective of 212 patients with Gaucher disease and 16 Gaucher expert physicians. Mol Genet Metab. (2017) 122(3):122–9. doi: 10.1016/j.ymgme.2017.08.002
8. Özdemir GN, Gündüz E. Gaucher disease for hematologists. Turk J Haematol. (2022) 39(2):136–9. doi: 10.4274/tjh.galenos.2022.2021.0683
9. Weinreb NJ, Goker-Alpan O, Kishnani PS, Longo N, Burrow TA, Bernat JA, et al. The diagnosis and management of Gaucher disease in pediatric patients: where do we go from here? Mol Genet Metab. (2022) 136(1):4–21. doi: 10.1016/j.ymgme.2022.03.001
10. Qi X, Xu J, Shan L, Li Y, Cui Y, Liu H, et al. Economic burden and health related quality of life of ultra-rare Gaucher disease in China. Orphanet J Rare Dis. (2021) 16(1):358. doi: 10.1186/s13023-021-01963-6
11. Matta MC, Soares DC, Kerstenetzky MS, Freitas ACP, Kim CA, Torres LC. CD4? CD25 high Foxp3? Treg deficiency in a Brazilian patient with Gaucher disease and lupus nephritis. Hum Immunol. (2016) 77:196–200. doi: 10.1016/j.humimm.2015.11.018
12. Giraldo P, Pérez-López J, Nuńẽz R, de la Puebla RF, Luño E, Saura-Grau S. Patients with type 1 Gaucher disease in Spain: a cross-sectional evaluation of health status. Blood Cells Mol Dis. (2016) 56:23–30. doi: 10.1016/j.bcmd.2015.10.001
13. Bettman N, Avivi I, Rosenbaum H, Bisharat L, Katz T. Impaired migration capacity in monocytes derived from patients with Gaucher disease. Blood Cells Mol Dis. (2015) 55(2):180–6. doi: 10.1016/j.bcmd.2014.12.003
14. Şoroğlu CV, Berkay EG. Old disease-new reflections: Gaucher, immunity, and inflammation. J Cell Mol Med. (2024 Oct) 28(20):e70087. doi: 10.1111/jcmm.70087
15. Zahran AM, Eltayeb AA, Elsayh KI, Saad K, Ahmad FA, Ibrahim AIM. Activated and memory T lymphocytes in children with Gaucher disease. Arch Immunol Ther Exp. (2017) 65(3):263–9. doi: 10.1007/s00005-016-0421-y
16. Di Rocco M, Andria G, Deodato F, Giona F, Micalizzi C, Pession A. Early diagnosis of Gaucher disease in pediatric patients: proposal for a diagnostic algorithm. Pediatr Blood Cancer. (2014) 61(11):1905–9. doi: 10.1002/pbc.25165
17. Charrow J, Andersson HC, Kaplan P, Kolodny EH, Mistry P, Pastores G, et al. The Gaucher registry: demographics and disease characteristics of 1698 patients with Gaucher disease. Arch Intern Med. (2000) 160(18):2835–43. doi: 10.1001/archinte.160.18.2835
18. Hughes DA, Pastores GM. Haematological manifestations and complications of Gaucher disease. Curr Opin Hematol. (2013) 20(1):41–7. doi: 10.1097/MOH.0b013e32835a9148
19. Bove KE, Daugherty C, Grabowski GA. Pathological findings in Gaucher disease type 2 patients following enzyme therapy. Hum Pathol. (1995) 26(9):1040–5. doi: 10.1016/0046-8177(95)90097-7
20. Sengers RC, Lamers KJ, Bakkeren JA, Schretlen ED, Trijbels JM. Infantile Gaucher's disease: glucocerebrosidase deficiency in peripheral blood leukocytes and cultured fibroblasts. Neuropadiatrie. (1975) 6(4):377–82. doi: 10.1055/s-0028-1091678
21. Kaplan P, Andersson HC, Kacena KA, Yee JD. The clinical and demographic characteristics of nonneuronopathic Gaucher disease in 887 children at diagnosis. Arch Pediatr Adolesc Med. (2006) 160(6):603–8. doi: 10.1001/archpedi.160.6.603
22. Mignot C, Doummar D, Maire I, De Villemeur TB, French Type 2 Gaucher Disease Study Group. Type 2 Gaucher disease: 15 new cases and review of the literature. Brain Dev. (2006) 28(1):39–48. doi: 10.1016/j.braindev.2005.04.005
23. Tylki-Szymańska A, Vellodi A, El-Beshlawy A, Cole JA, Kolodny E. Neuronopathic Gaucher disease: demographic and clinical features of 131 patients enrolled in the International Collaborative Gaucher Group Neurological Outcomes Subregistry. J Inherit Metab Dis. (2010) 33(4):339–46. doi: 10.1007/s10545-009-9009-6
24. Kraoua I, Sedel F, Caillaud C, Froissart R, Stirnemann J, Chaurand G, et al. A French experience of type 3 Gaucher disease: phenotypic diversity and neurological outcome of 10 patients. Brain Dev. (2011) 33(2):131–9. doi: 10.1016/j.braindev.2010.02.005
25. Campbell PE, Harris CM, Harris CM, Sirimanna T, Vellodi A. A model of neuronopathic Gaucher disease. J Inherit Metab Dis. (2003) 26(7):629–39. doi: 10.1023/B:BOLI.0000005619.14180.5c
26. Tsai LP, Sue WC, Hwu WL, Lin KH, Wang TR. Oculomotor apraxia in a case of Gaucher’s disease with homozygous T1448C mutation. Zhonghua Min Guo Xiao Er Ke Yi Xue Hui Za Zhi. (1996) 37(1):52–5.8936012
27. Forster J, Chambers JP, Peters SP, Lee RE, Glew RH. Acute neuropathic Gaucher disease in a black infant. J Pediatr. (1978) 93(5):823–4. doi: 10.1016/S0022-3476(78)81093-2
28. Kang H, Zhang M, Ouyang M, Guo R, Yu Q, Peng Q, et al. Brain white matter microstructural alterations in children of type I Gaucher disease characterized with diffusion tensor MR imaging. Eur J Radiol. (2018) 102:22–9. doi: 10.1016/j.ejrad.2018.02.014
29. Zhang M, Wang S, Hu D, Kang H, Ouyang M, Zhang Y, et al. Altered brain functional network in children with type 1 Gaucher disease: a longitudinal graph theory-based study. Neuroradiology. (2019) 61(1):63–70. doi: 10.1007/s00234-018-2104-3
30. Clarke LA, Hollak CE. The clinical spectrum and pathophysiology of skeletal complications in lysosomal storage disorders. Best Pract Res Clin Endocrinol Metab. (2015) 29(2):219–35. doi: 10.1016/j.beem.2014.08.010
31. Marcucci G, Zimran A, Bembi B, Kanis J, Reginster J-Y, Rizzoli R, et al. Gaucher disease and bone manifestations. Calcif Tissue Int. (2014) 95(6):477–94. doi: 10.1007/s00223-014-9923-y
32. Oliveri B, González D, Quiroga F, Silva C, Rozenfeld P. A comprehensive study of bone manifestations in adult Gaucher disease type 1 patients in Argentina. Calcif Tissue Int. (2019) 104(6):650–7. doi: 10.1007/s00223-019-00536-x
33. Grabowski GA, Zimran A, Ida H. Gaucher disease types 1 and 3: phenotypic characterization of large populations from the ICGG Gaucher registry. Am J Hematol. (2015) 90(Suppl 1):S12–8. doi: 10.1002/ajh.24063
34. Wenstrup RJ, Roca-Espiau M, Weinreb NJ, Bembi B. Skeletal aspects of Gaucher disease: a review. Br J Radiol. (2002) 75(Suppl 1):A2–12. doi: 10.1259/bjr.75.suppl_1.750002
35. Lachmann RH, Wight DG, Lomas DJ, Fisher NC, Schofield JP, Elias E, et al. Massive hepatic fibrosis in Gaucher’s disease: clinicopathological and radiological features. QJM. (2000) 93(4):237–44. doi: 10.1093/qjmed/93.4.237
36. Bohte AE, van Dussen L, Akkerman EM, Nederveen AJ, Sinkus R, Jansen PLM, et al. Liver fibrosis in type I Gaucher disease: magnetic resonance imaging, transient elastography and parameters of iron storage. PLoS One. (2013) 8(3):e57507. doi: 10.1371/journal.pone.0057507
37. Xu R, Mistry P, McKenna G, Emre S, Schiano T, Bu-Ghanim M, et al. Hepatocellular carcinoma in type 1 Gaucher disease: a case report with review of the literature. Semin Liver Dis. (2005) 25(2):226–9. doi: 10.1055/s-2005-871201
38. Patlas M, Hadas-Halpern I, Abrahamov A, Elstein D, Zimran A. Spectrum of abdominal sonographic findings in 103 pediatric patients with Gaucher disease. Eur Radiol. (2002) 12(2):397–400. doi: 10.1007/s003300101031
39. Serai SD, Naidu AP, Andrew Burrow T, Prada CE, Xanthakos S, Towbin AJ. Correlating liver stiffness with disease severity scoring system (DS3) values in Gaucher disease type 1 (GD1) patients. Mol Genet Metab. (2018) 123(3):357–63. doi: 10.1016/j.ymgme.2017.10.013
40. Starosta RT, Vairo FPE, Dornelles AD, Basgalupp SP, Siebert M, Pedroso MLA, et al. Liver involvement in patients with Gaucher disease types I and III. Mol Genet Metab Rep. (2020) 22:100564. doi: 10.1016/j.ymgmr.2019.100564
41. Hollak CE, Levi M, Berends F, van Overs MH. Coagulation abnormalities in type 1 Gaucher disease are due to low-grade activation and can be partly restored by enzyme supplementation therapy. Br J Haematol. (1997) 96(3):470–6. doi: 10.1046/j.1365-2141.1997.d01-2076.x
42. Katz K, Tamary H, Lahav J, Soudry M, Cohen IJ. Increased operative bleeding during orthopaedic surgery in patients with type I Gaucher disease and bone involvement. Bull Hosp Jt Dis. (1999) 58(4):188–90.10711366
43. Mitrovic M, Sumarac Z, Antic D, Bogdanovic A, Elezovic I, Vukosavljevic D, et al. Markers of coagulation activation and enhanced fibrinolysis in Gaucher type 1 patient: effects of enzyme replacement therapy. Blood Cells Mol Dis. (2012) 49(1):58–9. doi: 10.1016/j.bcmd.2012.03.003
44. Mistry PK, Sirrs S, Chan A, Pritzker MR, Duffy TP, Grace ME, et al. Pulmonary hypertension in type 1 Gaucher’s disease: genetic and epigenetic determinants of phenotype and response to therapy. Mol Genet Metab. (2002) 77(1-2):91–8. doi: 10.1016/S1096-7192(02)00122-1
45. Camou F, Viallard JF. Extended remission of B-cell lymphoma with monoclonal gammopathy in a patient with type 1 Gaucher disease treated with enzyme replacement therapy. Blood Cells Mol Dis. (2012) 48(1):51–2. doi: 10.1016/j.bcmd.2011.09.005
46. Liu J, Halene S, Yang M, Iqbal J, Yang R, Mehal WZ, et al. Gaucher disease gene GBA functions in immune regulation. Proc Natl Acad Sci U S A. (2012) 109(25):10018–23. doi: 10.1073/pnas.1200941109
47. Pandey MK, Grabowski GA. Immunological cells and functions in Gaucher disease. Crit Rev Oncog. (2013) 18(3):197–220. doi: 10.1615/CritRevOncog.2013004503
48. Pandey MK, Grabowski GA, Köhl J. An unexpected player in Gaucher disease: the multiple roles of complement in disease development. Semin Immunol. (2018) 37:30–42. doi: 10.1016/j.smim.2018.02.006
49. Vitner EB, Farfel-Becker T, Ferreira NS, Leshkowitz D, Sharma P, Lang KS, et al. Induction of the type I interferon response in neurological forms of Gaucher disease. J Neuroinflammation. (2016) 13(1):104. doi: 10.1186/s12974-016-0570-2
50. Ługowska A, Hetmańczyk-Sawicka K, Iwanicka-Nowicka R, Fogtman A, Cieśla J, Purzycka-Olewiecka JK, et al. Gene expression profile in patients with Gaucher disease indicates activation of inflammatory processes. Sci Rep. (2019) 9(1):6060. doi: 10.1038/s41598-019-42584-1
51. Boven LA, van Meurs M, Boot RG, Mehta A, Boon L, Aerts JM, et al. Gaucher cells demonstrate a distinct macrophage phenotype and resemble alternatively activated macrophages. Am J Clin Pathol. (2004) 122(3):359–69. doi: 10.1309/BG5VA8JRDQH1M7HN
52. Mucci JM, Rozenfeld P. Pathogenesis of bone alterations in Gaucher disease: the role of immune system. J Immunol Res. (2015) 2015:192761. doi: 10.1155/2015/192761
53. Mistry PK, Liu J, Yang M, Nottoli T, McGrath J, Jain D, et al. Glucocerebrosidase gene-deficient mouse recapitulates Gaucher disease displaying cellular and molecular dysregulation beyond the macrophage. Proc Natl Acad Sci U S A. (2010) 107(45):19473–8. doi: 10.1073/pnas.1003308107
54. Zahran AM, Saad K, Elsayh KI, Abdou MAA, Abo-Elgheet AM, Eloseily EM, et al. Upregulation of cytotoxic T-cells in pediatric patients with Gaucher disease. Sci Rep. (2022) 12:4977. doi: 10.1038/s41598-022-08843-4
55. Vairo F, Alegra T, Dornelles A, Mittelstadt S, Netto CBO, Schwartz IVD. Hyperimmunoglobulinemia in pediatric Gaucher patients in Southern Brazil. Pediatr Blood Cancer. (2012) 59:339. doi: 10.1002/pbc.24091
56. Arikan-Ayyildiz Z, Yuce A, Uslu-Kizilkan N, Demir H, Gurakan F. Immuno globulin abnormalities and effects of enzyme replacement therapy in children with Gaucher disease. Pediatr Blood Cancer. (2011) 56:664–6. doi: 10.1002/pbc.22863
57. Wine E, Yaniv I, Cohen IJ. Hyperimmunoglobulinemia in pediatric-onset type 1 Gaucher disease and effects of enzyme replacement therapy. J Pediatr Hematol Oncol. (2007) 29(7):451–7. doi: 10.1097/MPH.0b013e31806451d3
58. Marzouk I, Deghady A, Omar OM, Ashour RS. Hyperimmunoglobulinemia and IgG subclass abnormalities in children with Gaucher disease. J Pediatr Hematol Oncol. (2019) 41(7):e416–20. doi: 10.1097/MPH.0000000000001574
59. Miano M, Madeo A, Cappelli E, Lanza F, Lanza T, Stroppiano M, et al. Defective FAS-mediated apoptosis and immune dysregulation in Gaucher disease. J Allergy Clin Immunol Pract. (2020) 8(10):3535–42. doi: 10.1016/j.jaip.2020.06.065
60. Gupta N, Oppenheim IM, Kauvar EF, Tayebi N, Sidransky E. Type 2 Gaucher disease: phenotypic variation and genotypic heterogeneity. Blood Cells Mol Dis. (2011) 46(1):75–84. doi: 10.1016/j.bcmd.2010.08.012
61. Ryan E, Seehra GK, Sidransky E. Mutations, modifiers and epigenetics in Gaucher disease: blurred boundaries between simple and complex disorders. Mol Genet Metab. (2019) 128(1-2):10–3. doi: 10.1016/j.ymgme.2019.08.006
62. Koprivica V, Stone DL, Park JK, Callahan M, Frisch A, Cohen IJ, et al. Analysis and classification of 304 mutant alleles in patients with type 1 and type 3 Gaucher disease. Am J Hum Genet. (2000) 66(6):1777–86. doi: 10.1086/302925
63. Ida H, Rennert OM, Kawame H, Maekawa K, Eto Y. Mutation prevalence among 47 unrelated Japanese patients with Gaucher disease: identification of four novel mutations. J Inherit Metab Dis. (1997) 20(1):67–73. doi: 10.1023/A:1005313724361
64. Bendikov-Bar I, Ron I, Filocamo M, Horowitz M. Characterization of the ERAD process of the L444P mutant glucocerebrosidase variant. Blood Cells Mol Dis. (2011) 46(1):4–10. doi: 10.1016/j.bcmd.2010.10.012
65. Wan L, Hsu CM, Tsai CH, Lee CC, Hwu WL, Tsai FJ. Mutation analysis of Gaucher disease patients in Taiwan: high prevalence of the RecNciI and L444P mutations. Blood Cells Mol Dis. (2006) 36(3):422–5. doi: 10.1016/j.bcmd.2006.02.001
66. Kang L, Wang Y, Gao X, Qiu W, Ye J, Han L, et al. Genotypes and phenotypes in 20 Chinese patients with type 2 Gaucher disease. Brain Dev. (2018) 40(10):876–83. doi: 10.1016/j.braindev.2018.06.006
67. Pasmanik-Chor M, Laadan S, Elroy-Stein O, Zimran A, Abrahamov A, Gatt S, et al. The glucocerebrosidase D409H mutation in Gaucher disease. Biochem Mol Med. (1996) 59(2):125–33. doi: 10.1006/bmme.1996.0077
68. George R, McMahon J, Lytle B, Clark B, Lichtin A. Severe valvular and aortic arch calcification in a patient with Gaucher’s disease homozygous for the D409H mutation. Clin Genet. (2001) 59(5):360–3. doi: 10.1034/j.1399-0004.2001.590511.x
69. Kurolap A, Del Toro M, Spiegel R, Gutstein A, Shafir G, Cohen IJ, et al. Gaucher disease type 3c: new patients with unique presentations and review of the literature. Mol Genet Metab. (2019) 127(2):138–46. doi: 10.1016/j.ymgme.2019.05.011
Keywords: Gaucher disease, clinical feature, infection risk, Chinese children, gene mutation
Citation: Gan C, Wu Y and Qin T (2025) Clinical features and infection risks of Chinese children with different types of Gaucher disease. Front. Pediatr. 13:1595394. doi: 10.3389/fped.2025.1595394
Received: 4 July 2025; Accepted: 25 August 2025;
Published: 5 September 2025.
Edited by:
Pilar Giraldo, University of Zaragoza, SpainReviewed by:
Ladan Mafakher, Ahvaz Jundishapur University of Medical Sciences, IranMargarita Ivanova, Lysosomal and Rare Disorders Research and Treatment Center, United States
Copyright: © 2025 Gan, Wu and Qin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuanyuan Wu, eXVhbnl1YW53dUBob3NwaXRhbC5jcW11LmVkdS5jbg==; Tao Qin, YmluZ2VyMjAxMTA5MDFAMTYzLmNvbQ==