 Xianfei Gao
Xianfei Gao Ying Li
Ying Li Xuerong Lan
Xuerong Lan Ping Zeng
Ping Zeng- Department of Pediatric, Allergy Immune and Rheumatology, Guangzhou Women and Children’s Medical Center, Guangzhou Medical University, Guangdong Provincial Clinical Research Center for Child Health, Guangzhou, China
Background: Systemic lupus erythematosus (SLE) is a complex autoimmune disease in which the immune system mistakenly attacks healthy cells and tissues. This condition can impact various organs and systems, including the hematological system, where aplastic anemia (AA) emerges as a particularly severe and rare complication. Relevant clinical manifestations and treatment experiences must be shared and updated urgently.
Methods: This manuscript presents the clinical features, examination, and treatment of two pediatric SLE patients with AA. And a systemic search of MEDLINE, EMBASE, LILACS, SciELO, and Scopus, using MeSH headings and keywords for “Aplastic Anaemia” and “Systemic Lupus Erythematosus”.
Results: Both of our cases were severe, resulting in one death. Our systematic literature review identified 32 eligible studies. We found a total of 38 patients, and along with the two patients from our case series, there were 9 out of 40 SLE patients who experienced alopecia areata as children. In all 40 patients, 52% (21 out of 40) developed AA at the onset of SLE. The infection rate was concerning, with 16 out of 40 patients affected. While 83% (33 out of 40) of the patients showed improved outcomes, all seven patients who died had contracted an infection.
Conclusion: SLE with AA is rare in both children and adults. Although the prognosis of this dangerous disease is optimistic in terms of data, it is still necessary to be vigilant, and early bone marrow examination, infection prevention, and on-time treatment are crucial.
Introduction
Aplastic anemia (AA) is a rare but serious condition characterized by the bone marrow's inability to produce enough blood cells. This leads to a deficiency in the production of red blood cells, white blood cells, and platelets. AA can be a complication of systemic lupus erythematosus (SLE), an autoimmune disease characterized by widespread inflammation and tissue damage across various organs (1). The development of aplastic anemia in patients with systemic lupus erythematosus involves a complex interplay of immunological, genetic, and environmental factors. Although AA has a low incidence, its severe implications in SLE patients necessitate heightened clinical awareness and careful management (2). AA presents with a range of non-specific symptoms that may overlap with other hematological and systemic disorders. Common symptoms include fatigue and weakness due to anemia, bleeding tendencies from thrombocytopenia, increased susceptibility to infections from neutropenia, fever, pallor, and dyspnea. It is important to note that Macrophage Activation Syndrome can cause similar cytopenias but is typically associated with elevated ferritin levels, hemophagocytosis in the bone marrow, and increased transaminases. Infections may result in either leukocytosis or leukopenia; however, inflammation markers like CRP (C-reactive protein) or ESR (erythrocyte sedimentation rate) are usually elevated, and blood cultures may be positive in cases of bacteremia or septicemia. Drug-induced cytopenias may resemble AA but can show signs such as eosinophilia or increased blast cells without significant marrow hypoplasia. In severe cases, macrocytic anemia with pancytopenia may be observed, along with hypersegmented neutrophils and macro-ovalocytes on blood smears and deficiencies in Vitamin B12 and folate levels. Diagnosing AA in the context of SLE is complex due to the overlapping symptoms and varying complications. There is a need for more case reports that illustrate clinical manifestations and treatment experiences to enhance understanding of aplastic anemia in SLE and to develop appropriate treatment strategies (3).
In this article, we present a case series of two pediatric patients with AA as a complication of SLE, accompanied by a literature review discussing the clinical manifestations, diagnosis, and treatment of SLE complicated by AA.
Case series
Case 1
A 9-year-old Chinese girl was admitted to our hospital with a cough and malar rash for 1 month, and fatigue and hair loss for 10 days. Her paternal grandmother had skin cancer, and her maternal grandmother suffered from rheumatic heart disease. At time of admission, dark brown pigmentation was discovered around the fingernails on both sides. However, she had no history of trauma, other illnesses, or exposure to pigments. Investigation revealed an HB (hemoglobin concentration) of 86 g/L, WBC (white blood cell) 1.3 × 109/L, Neutropenia 0.28 × 109/L, PLT (platelet) 16 × 109/L, ARC (absolute reticulocyte count) 0.006 × 1012/L, C3 (complement 3) 0.56 g/L, antinuclear antibody 1/80, urine RBC (red blood cell) 198/μl, 24-hour Urine protein 1.06 g/L, fibrinogen 4.86 g/L, ferritin 516.40 ng/ml, and CRP 38.3 mg/L. The patient suffered an Upper respiratory tract infection with Haemophilus influenzae in sputum culture and a normal chest CT. The patient was initially treated with MTX (methotrexate) 10 mg/week, prednisone 30 mg/day, Mercaptopurine 50 mg/day, and Cefuroxime for the infection. Bone marrow smear showed hypocellular marrow, and bone marrow biopsy showed cellularity of 10%, supporting AA. Due to bone marrow suppression, the patient discontinued mercaptopurine and began receiving intravenous methylprednisolone (starting with 500 mg daily), intravenous immunoglobulin (IVIG), granulocyte-colony-stimulating factor (G-CSF), and switched from MTX to mycophenolate mofetil (MMF). However, the HB kept on dropping to 67 g/L, PLT dropping to 13 × 109/L, and the patients suffering dizziness, petechiae, and bloody stool. RBC and PLT transfusions were conducted. On the 23rd day of admission, the patient improved with HB 93 g/L, WBC 5.1 × 109/L, and PLT 34 × 109/L. The patient received regular intravenous methylprednisolone at a dosage of 5 mg/kg for 3 days each month, along with cyclophosphamide administered at 5 mg/kg per day for 2 days each month, over a period of 7 months. Following this, the same dosages were given every 2 months for an additional 4 months. After this treatment, the patient discontinued intravenous methylprednisolone and cyclophosphamide because the SLEDAI (Systemic Lupus Erythematosus Disease Activity Index) score had reduced to 5, and there was a high frequency of infections. The patient was maintained on oral steroids (2 mg/day), hydroxychloroquine (6.5 mg/kg/day), and MMF (400 mg/kg/day). After 5 years, the patient maintained SLEDAI scores of less than four.
Case 2
A 9-year-old Chinese girl was admitted to our hospital with a half-month history of arthralgia in bilateral wrist and knee joints, a malar rash, and repeated fever (38.8℃) for 10 days. On evaluation she was noted to have oral ulcers, and labs showed an HB 83 g/L, WBC 2.7 × 109/L, PLT 62 × 109/L, C3 0.19 g/L, C4 (complement 4) 0.03 g/L, direct Coombs test 3+, antinuclear antibody titer of 1/640, anti-dsDNA (Anti-double-stranded DNA) antibody 1+. The SLE diagnosis was made, and the patient was initially treated with intravenous methylprednisolone and IVIG for 1 week, then MMF (0.75 g/day), regularly reduced oral steroids, and intravenous Benlysta (10 mg/kg) administered twelve times for 2 years. During this period, SLEDAI scored 2–5 points with low complement concentration, high anti-ds-DNA, or low platelet. However, at the age of 12, the patient took oral medications irregularly and did not have follow-up visits on time for about 1 year. Consequently, she experienced malar rash, pale lips, swelling of both eyelids, pitting edema of the limbs, chest pain, dizziness, fatigue, cough, and decreased urine output. The patient was hospitalized, and her evaluation revealed severe thrombocytopenia (54 × 109/L) and anemia (57 g/L). She had low levels of C3 (0.15 g/L), C4 (0.02 g/L), a direct Coombs test reading of 3+, and low albumin (29.1 g/L). The infectious workup, which included tests for Epstein–Barr Virus (EBV), cytomegalovirus (CMV), Human Immunodeficiency Virus (HIV), Hepatitis C Virus (HCV), Hepatitis B Virus (HBV), and blood cultures, returned negative results. On the first day of her visit, a bone marrow smear showed active proliferation of granulocytes and erythroid cells, with visible megakaryocytes. Cardiac ultrasound revealed moderate pericardial effusion, while a chest CT scan indicated pulmonary congestion, bilateral pleural effusion, a small amount of condensed inflammation in both lungs, an enlarged heart shadow, and pericardial effusion. The patient was started on intravenous methylprednisolone, IVIG, and intravenous human albumin. Due to renal failure (BUN of 32.46 mmol/L) and high uric acid levels (753 µmol/L), she underwent plasma exchange and hemodialysis. For the elevated uric acid, she was treated with azathioprine. Subsequently, her ANC decreased to 0.06 × 109/L and ARC to 0.003 × 1012/L, requiring RBC transfusions for critically low hemoglobin levels of 44 g/L and PLT transfusions for critically low levels of 2 × 109/L. Due to a lack of response, a second peripheral blood and bone marrow smear performed on the 27th day revealed AA with hypocellular marrow. A bone marrow biopsy was not performed as the patient had low platelet counts and was at risk of bleeding. Her ferritin level was 1,590.30 ng/ml, while transaminases, fibrinogen, ESR, and CRP were normal. Macrophage activation syndrome (MAS) was excluded. The patient then developed Trichosporon cutaneum, confirmed by blood culture, and was treated with meropenem, linezolid, and caspofungin. On the 31st day, her myelosuppression improved with PLT levels rising to 75 × 109/L, hemoglobin to 75 g/L, and WBC count to 10.2 × 109/L. However, she began experiencing seizures due to intracranial hemorrhage. As her generalized edema worsened, along with refractory electrolyte imbalances and hypertension, she suffered another seizure, experienced loss of consciousness, and developed multiple intracranial hemorrhagic lesions. Ultimately, she passed away due to multiple organ failure.
Systemic review of the literature
We performed a systemic search in several scientific databases (MEDLINE, EMBASE, LILACS, SciELO, and Scopus) using the following keywords: systemic lupus erythematosus, SLE, lupus, LES, aplastic anemia and aplastic anemia. We used MeSH-controlled vocabulary to index articles for MEDLINE. The study period ran from January 1950 through October 2024. Two authors independently screened all citations and abstracts to identify eligible studies. In addition, secondary references were obtained from the selected article. The inclusion criteria were English-language written and human articles. Both pediatric and adult SLE were included. Exclusion criteria were conference proceedings, abstracts, or editorials. Patients less than or equal to 18 years of age are defined as children, and adults are defined as older than 19.
Results
Based on the data collected from 32 publications and our presented cases, there are 40 cases of SLE with AA. Females were the dominant gender, with 35 patients. Among the 19 patients who developed AA after SLE, the duration from the onset age of SLE to AA varies from 0.5 to 22 years, as shown in Table 1. Children (patients aged ≤18 years) occupied about 9 (23%) of this rare condition. There was no noticeable difference among different races numerically.
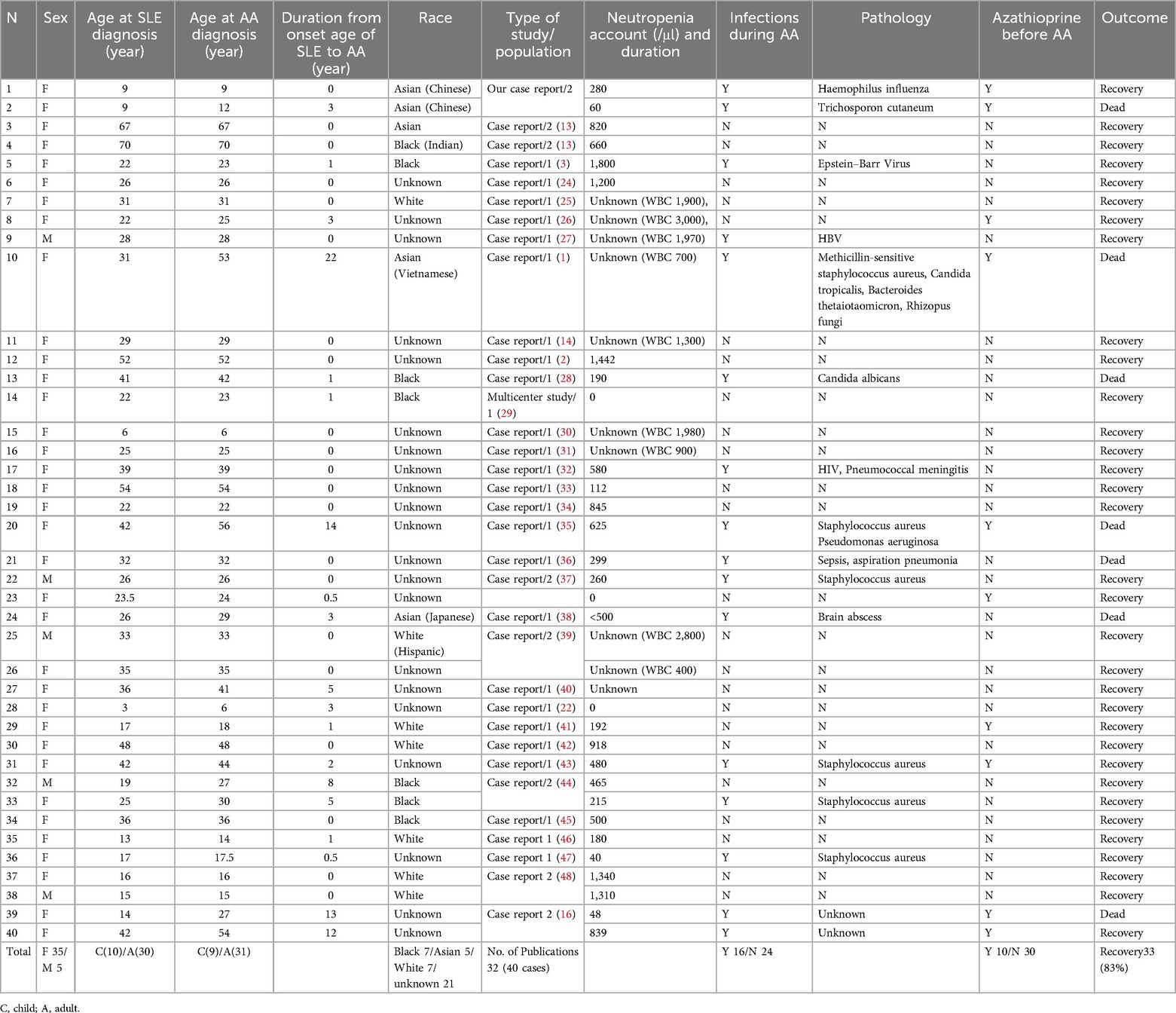
Table 1. Summary of published studies regarding SLE with aplastic anemia in pediatric patients, associated factors, and outcome.
Infections occurred in 16 of 40 patients (40%), with Staphylococcus aureus as the most frequent pathogen. The survival rate for infected patients (9 of 16 with infection = 56%) was lower than the overall survival rate (33 of 40 overall = 83%). Both pediatric patients from our case series had an azathioprine history. A summary and the literature review results show that 10 patients with an azathioprine history were found before AA onset (Table 1). The outcome for SLE with AA was promising, with a survival rate of 83%. Notably, all 7 cases with fatal outcomes (Patients No. 8, 11, 18, 19, 22, 37, 39) had infections during AA treatment.
The treatments are shown in Table 2. Corticosteroids were the primary medication in the treatment, administered to 95%, followed by blood transfusion at 55%, cyclosporine at 30%, HCQ at 28%, IVIg at 25%, etc. Only seven patients (18%) received standard SAA treatments. Standard SAA treatment was immunosuppressive therapy (IST) with ATG and cyclosporine, or hematopoietic stem cell transplant (4).
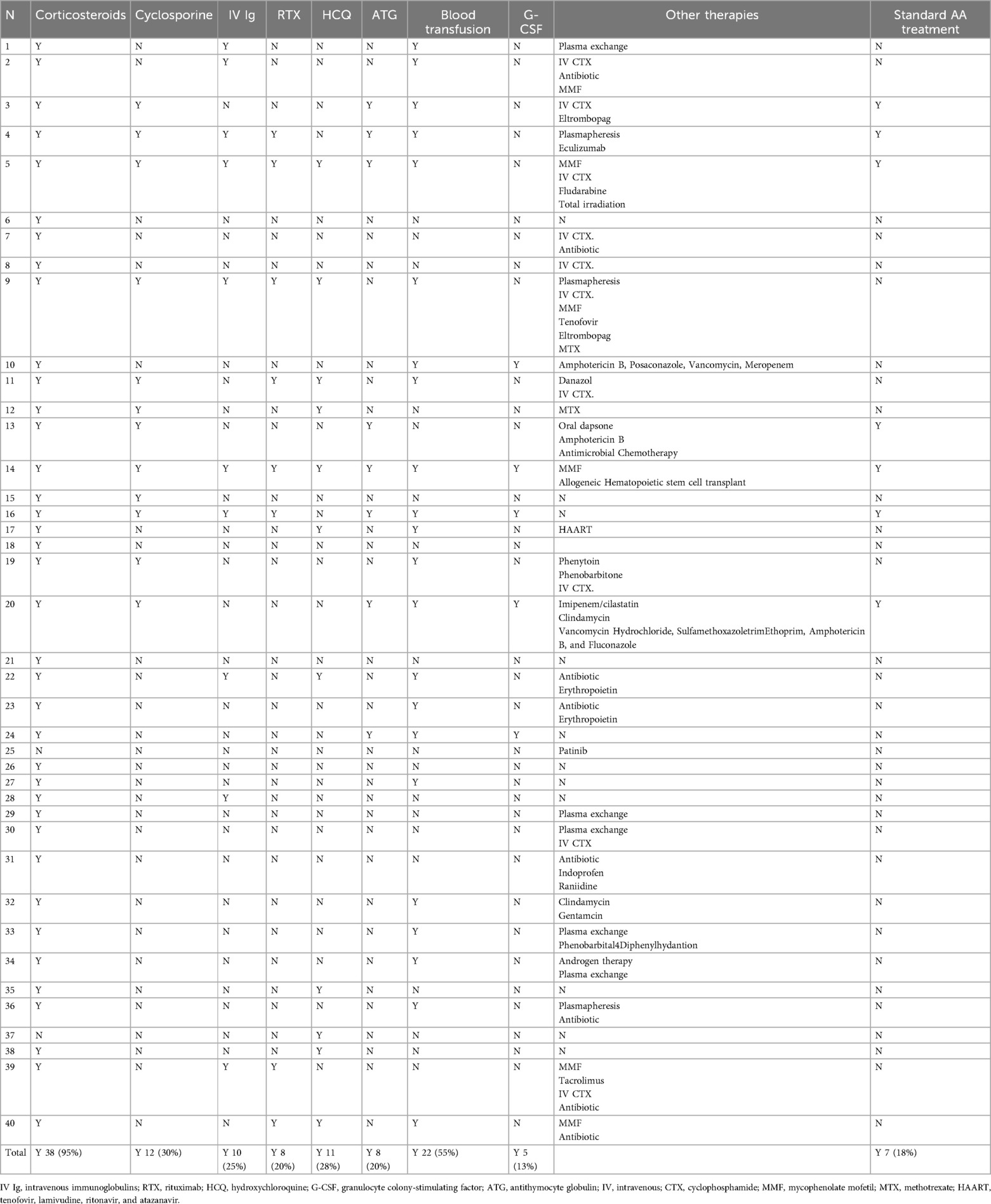
Table 2. Treatment of SLE with AA.
Discussion and conclusions
Acquired aplastic anemia is a rare, life-threatening hematological disorder characterized by bone marrow failure resulting in pancytopenia, or the reduction of red blood cells, white blood cells, and platelets. Even though the mechanism of acquired aplastic anemia is unclear, several factors have been identified as potential triggers. (1) Environmental factors. Exposure to certain toxins and chemicals, such as benzene and its derivatives, increases the risk of developing aplastic anemia (5). (2) Pharmacological Agents. Various medications have been associated with the onset of aplastic anemia. Notable culprits include nonsteroidal anti-inflammatory drugs (NSAIDs), antiepileptics, and certain antibiotics such as chloramphenicol. Chemotherapeutic agents, such as Azathioprine, and radiation have also been causative in some cases (6). (3) Viral Infections. Viral infections are another significant risk factor. Hepatitis viruses, Epstein–Barr Virus (EBV), Cytomegalovirus (CMV), and parvovirus B19 have been linked to aplastic anemia through mechanisms under investigation (7–10). (4) Immune System Dysregulation. The immune system's inappropriate attack on hematopoietic stem cells within the bone marrow is a critical mechanism in acquired aplastic anemia. SLE is one of the reasons. It involves autoreactive T-cells and the overproduction of cytokines like gamma-interferon and tumor necrosis factor-alpha (TNF-α), leading to hematopoietic suppression (11).
The revised Camitta criteria categorize AA into three classes based on BM cellularity, absolute neutrophil count (ANC), platelet count, and absolute reticulocyte count (ARC). Severe aplastic anemia (SAA) is identified by the presence of pancytopenia, with ANC below 0.5 × 109/L, platelet count below 20 × 109/L, and reticulocyte count below 20 × 109/L as measured by manual count, or below 60 × 109/L when using an automated analyzer. Additionally, diagnosis of AA also requires a hypocellular bone marrow with bone marrow cellularity of less than 25%, as measured by bone marrow core biopsy. Very severe aplastic anemia (vSAA) meets the same criteria, except for an ANC of less than 0.2 × 109/L. Non-Severe Aplastic Anemia (NSAA) patients have depressed blood counts but do not meet the specific criteria for SAA or VSAA (12).
The overlapping clinical features of SLE with AA necessitate a high index of suspicion for diagnosis. Diagnosing AA in patients with SLE is crucial yet often challenging, leading to delays in diagnosis and negative outcomes. Most studies in this field consist mainly of case reports; therefore, multicenter research is necessary to support statistical and clinical analysis (13).
In our study, the first case presented both SLE and AA occurring simultaneously at the onset. The underlying pathophysiological connection arises from autoimmunity and the immune-mediated destruction of hematopoietic progenitor cells. This comorbidity complicates diagnosis and treatment, emphasizing the necessity for a nuanced understanding of effective clinical management (14). Although bone marrow biopsy is invasive, it should be considered when an SLE patient presents with blood system-related symptoms (3).
Managing therapeutic interventions for SLE in patients with concurrent AA poses challenges for physicians. Even though 83% of the patients from our literature review achieved remission, infection is a significant threat to SLE patients with AA, as all the patients with fatal outcomes suffered from infection. Overall survival for patients with infections was only 38%. Both of the cases reported here suffered from infections, and only one of them survived.
Regardless of severity, supportive care is fundamental in managing aplastic anemia. This includes Transfusions, Infection Prevention, and Routine Monitoring. For SAA and VSAA, the combination of ATG and CsA is the frontline treatment for patients who are not candidates for hematopoietic stem cell transplantation (HSCT). ATG targets and depletes T-cells, reducing their attack on bone marrow, while CsA inhibits T-cell activation and proliferation. Eltrombopag, a thrombopoietin receptor agonist, is sometimes included in the treatment regimen to stimulate bone marrow and increase platelet production. HSCT offers a potential cure for eligible patients, especially younger ones with a matched sibling donor. The procedure involves replacing the damaged bone marrow with healthy cells from a donor. Advances in transplantation techniques have improved outcomes significantly. Patients with NSAA typically exhibit a less aggressive disease course. Treatment may involve watchful waiting and symptomatic management (15).
However, for specific SLE-induced AA, the treatment strategy often focuses more on SLE (16). Corticosteroids are commonly used as the first-line treatment for SLE (17). Calcineurin inhibitors (CNI) like cyclosporine effectively reduce immune-mediated destruction in AA and control SLE activity, making them crucial for managing both conditions concurrently. Azathioprine is a cornerstone drug in managing SLE and exerts immunosuppressive effects by inhibiting purine synthesis, thereby reducing the proliferation of lymphocytes. Thus, azathioprine use should be approached with caution, particularly in patients presenting with unexplained cytopenias (18). Biologic agents targeting specific pathways in SLE are emerging as potential therapeutic options in this dual-diagnosis scenario (19). Rituximab, a monoclonal antibody targeting CD20-positive B cells, reduces autoantibody production and may mitigate some autoimmune mechanisms contributing to AA (14). Antithymocyte globulin (ATG), often used in severe AA cases, can reduce bone marrow suppression. Combining ATG with CNI has shown efficacy; however, its impact on SLE must be continuously monitored (20). Plasma exchange reduces the autoimmunity burden by clearing the pathological factors, potentially improving marrow function over time, and sometimes offers an optimum treatment option. For patients with both SLE and AA, allogeneic HSCT can provide a potential cure, but this approach carries high risks, including graft vs. host disease and the potential for severe autoimmune flares (21).
Supportive care is also crucial for patients with SLE and AA. Regular blood transfusions for anemia, platelet transfusions for thrombocytopenia, and granulocyte colony-stimulating factor (G-CSF) for neutropenia can be critical in managing acute, life-threatening events. Given these patients' immunocompromised state, prophylactic antibiotics are essential to prevent infections, which can have catastrophic consequences (22, 23).
In conclusion, cytopenias are commonly seen at the onset of SLE, and bone marrow aspirate and biopsy should be considered for patients with multiple lineages affected in order to rule out AA. AA should be regarded when there are changes in other systems involved in SLE. The outcomes of these patients were numerically optimistic, but infection poses a lethal threat, and prophylactic antibiotics should be considered.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Scientific Ethics Committee Guangzhou Women and Children's Medical Center, Guangzhou Medical University. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants' legal guardians/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
XG: Writing – original draft, Writing – review & editing, Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization. YL: Investigation, Methodology, Writing – review & editing. XL: Conceptualization, Methodology, Writing – review & editing. PZ: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by the Clinical Doctoral Start-up Research Fund of Guangzhou Women and Children's Medical Center (award number 1600218, XG) and the Municipal and University (Hospital) Joint Funding Project of Guangzhou Municipal Science and Technology Bureau (award number 2024A03J0806, PZ).
Acknowledgments
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article or claimed by its manufacturer may not be guaranteed or endorsed by the publisher.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Yeter KC, Afkhami M, Brynes RK, Quismorio FP Jr. Aplastic anemia secondary to azathioprine in systemic lupus erythematosus: report of a case with normal thiopurine S-methyltransferase enzyme activity and review of the literature. Lupus. (2013) 22(14):1526–8. doi: 10.1177/0961203313504636
2. Seo SR, Lee SJ, Park DJ, Kim TJ, Park YW, Lee SS. Successful treatment using cyclosporine in a patient with rhupus complicated by aplastic anemia: a case report and review of the literature. Clin Exp Rheumatol. (2011) 29(4):708–11.21813067
3. Chalayer E, Ffrench M, Cathebras P. Aplastic anemia as a feature of systemic lupus erythematosus: a case report and literature review. Rheumatol Int. (2015) 35(6):1073–82. doi: 10.1007/s00296-014-3162-4
4. Red Blood Cell Disease (Anemia) Group, Chinese Society of Hematology, Chinese Medical Association. Guidelines for the diagnosis and management of aplastic anemia in China (2022). Zhonghua Xue Ye Xue Za Zhi. (2022) 43(11):881–8. doi: 10.3760/cma.j.issn.0253-2727.2022.11.001
5. Li H, Xu X, Wang D, Zeng L, Li B, Zhang Y, et al. miR-146b-5p regulates bone marrow mesenchymal stem cell differentiation by SIAH2/PPARγ in aplastic anemia children and benzene-induced aplastic anemia mouse model. Cell Cycle. (2020) 19(19):2460–71. doi: 10.1080/15384101.2020.1807081
6. Young NS. Acquired aplastic anemia. Ann Intern Med. (2002) 136(7):534–46. doi: 10.7326/0003-4819-136-7-200204020-00011
7. Alshaibani A, Dufour C, Risitano A, de Latour R, Aljurf M. Hepatitis-associated aplastic anemia. Hematol Oncol Stem Cell Ther. (2022) 15(2):8–12. doi: 10.1016/j.hemonc.2020.10.001
8. Chen T, Chen Y, Bao W, Lu W. T-lymphocyte subsets and Th1/Th2 cytokines in convalescent patients with Epstein–Barr virus-associated aplastic anemia. Hematology. (2020) 25(1):11–6. doi: 10.1080/16078454.2019.1702304
9. Hinojosa OA, Ammari O. Herpes simplex virus-associated aplastic anemia. Cureus. (2023) 15(2):e35320. doi: 10.7759/cureus.35320
10. Shehi E, Ghazanfar H, Fortuzi K, Gonzalez E, Zeana C. A rare case of parvovirus B19 infection manifesting as chronic aplastic anemia and neutropenia in a human immunodeficiency virus-infected patient. Cureus. (2020) 12(12):e12174. doi: 10.7759/cureus.12174
11. Votavova H, Belickova M. Hypoplastic myelodysplastic syndrome and acquired aplastic anemia: immune-mediated bone marrow failure syndromes (review). Int J Oncol. (2022) 60(1):7. doi: 10.3892/ijo.2021.5297
12. Camitta BM, Rappeport JM, Parkman R, Nathan DG. Selection of patients for bone marrow transplantation in severe aplastic anemia. Blood. (1975) 45(3):355–63. doi: 10.1182/blood.V45.3.355.355
13. Anderson E, Shah B, Davidson A, Furie R. Lessons learned from bone marrow failure in systemic lupus erythematosus: case reports and review of the literature. Semin Arthritis Rheum. (2018) 48(1):90–104. doi: 10.1016/j.semarthrit.2017.12.004
14. Alishiri GH, Saburi A, Bayat N, Saadat AR, Saburi E. The initial presentation of systemic lupus erythematosis with aplastic anemia successfully treated with rituximab. Clin Rheumatol. (2012) 31(2):381–4. doi: 10.1007/s10067-011-1878-z
15. Onishi Y. Aplastic anemia: history and recent developments in diagnosis and treatment. Int J Hematol. (2024) 119(3):217–9. doi: 10.1007/s12185-024-03715-1
16. Al-Ghazawi Z, Al-Farajat A, Toubasi AA, Tawileh HBA, Qteish A, Aladily TN, et al. Pancytopenia with aplastic anemia in systemic lupus erythematosus: case series and literature review. Rheumatol Int. (2024) 44(5):943–53. doi: 10.1007/s00296-024-05585-6
17. Ruiz-Irastorza G, Danza A, Khamashta M. Glucocorticoid use and abuse in SLE. Rheumatology. (2012) 51(7):1145–53. doi: 10.1093/rheumatology/ker410
18. Croyle L, Morand EF. Optimizing the use of existing therapies in lupus. Int J Rheum Dis. (2015) 18(2):129–37. doi: 10.1111/1756-185X.12551
19. Kronbichler A, Neumann I, Mayer G. Moderator’s view: the use of calcineurin inhibitors in the treatment of lupus nephritis. Nephrol Dial Transplant. (2016) 31(10):1572–6. doi: 10.1093/ndt/gfw288
20. Rosenfeld S, Follmann D, Nunez O, Young NS. Antithymocyte globulin and cyclosporine for severe aplastic anemia: association between hematologic response and long-term outcome. J Am Med Assoc. (2003) 289(9):1130–5. doi: 10.1001/jama.289.9.1130
21. Iftikhar R, Chaudhry QUN, Anwer F, Neupane K, Rafae A, Mahmood SK, et al. Allogeneic hematopoietic stem cell transplantation in aplastic anemia: current indications and transplant strategies. Blood Rev. (2021) 47:100772. doi: 10.1016/j.blre.2020.100772
22. Sumimoto S, Kawai M, Kasajima Y, Hamamoto T. Aplastic anemia associated with systemic lupus erythematosus. Am J Hematol. (1991) 38(4):329–31. doi: 10.1002/ajh.2830380415
23. Feng X, Scheinberg P, Samsel L, Rios O, Chen J, McCoy JP Jr, et al. Decreased plasma cytokines are associated with low platelet counts in aplastic anemia and immune thrombocytopenic purpura. J Thromb Haemost. (2012) 10(8):1616–23. doi: 10.1111/j.1538-7836.2012.04757.x
24. Ahmad SQ, Khan O, Zafar SI, Zafar SN. A case of systemic lupus erythematosus with aplastic anaemia. J Pak Med Assoc. (2011) 61(8):817–9.22356010
25. Roffe C, Cahill MR, Samanta A, Bricknell S, Durrant ST. Aplastic anaemia in systemic lupus erythematosus: a cellular immune mechanism? Br J Rheumatol. (1991) 30(4):301–4. doi: 10.1093/rheumatology/30.4.301
26. Walport MJ, Hubbard WN, Hughes GRV. Reversal of aplastic anaemia secondary to systemic lupus erythematosus by high-dose intravenous cyclophosphamide. Br Med J. (1982) 285(6344):769–70. doi: 10.1136/bmj.285.6344.769
27. Hadid B, Kodza A, Suresh SC, Feoktistov A. Refractory acquired amegakaryocytic thrombocytopenia with rapid progression to aplastic anaemia in SLE. Mediterr J Rheumatol. (2023) 34(4):537–43. doi: 10.31138/mjr.110823.raa
28. Meyerson MA, Cohen PR. Dapsone-induced aplastic anemia in a woman with bullous systemic lupus erythematosus. Mayo Clin Proc. (1994) 69(12):1159–62. doi: 10.1016/s0025-6196(12)65768-1
29. Chalayer E, Costedoat-Chalumeau N, Beyne-Rauzy O, Ninet J, Durupt S, Tebib J, et al. Bone marrow involvement in systemic lupus erythematosus. QJM. (2017) 110:701–11. doi: 10.1093/qjmed/hcx102
30. Thakur N, Chandra J, Dhingra B, Singh V. Pediatric lupus: varied haematological picture and presentation. Indian J Hematol Blood Transfus. (2015) 31(1):68–70. doi: 10.1007/s12288-014-0357-5
31. Liu W, Hu Z, Lin S, He J, Zhou Y. Systemic lupus erythematosis with severe aplastic anemia successfully treated with rituximab and antithymocyte globulin. Pak J Med Sci. (2014) 30(2):449–51.24772161
32. de Oliveira LR, Ferreira TC, Neves Fde F, Meneses AC. Aplastic anemia associated to systemic lupus erythematosus in an AIDS patient: a case report. Rev Bras Hematol Hemoter. (2013) 35(5):366–8. doi: 10.5581/1516-8484.20130100
33. Baumann P, Volkl A, Bauerle M, Schmidmaier R, Oduncu FS. Aplastic crisis as primary manifestation of systemic lupus erythematosus. Onkologie. (2011) 34(8-9):452–4. doi: 10.1159/000331069
34. Singh NP, Prakash A, Garg D, Makhija A, Pathania A, Prakash N, et al. Aplastic anemia complicating systemic lupus erythematosus: successful management with cyclosporine. Rheumatol Int. (2004) 24(1):40–2. doi: 10.1007/s00296-003-0318-z
35. Tabushi Y, Fukazawa T, Abe K, Kaneda K, Hirashima M, Young-Joon K, et al. A case of aplastic anemia in a patient with systemic lupus erythematosus. Mod Rheumatol. (2003) 13(2):177–80. doi: 10.3109/s10165-002-0219-0
36. Pavithran K, Raji NL, Thomas M. Aplastic anemia complicating systemic lupus erythematosus–report of a case and review of the literature. Rheumatol Int. (2002) 22(6):253–5. doi: 10.1007/s00296-002-0254-3
37. Tagoe C, Shah A, Yee H, Belmont HM. Aplastic anemia in systemic lupus erythematosus: a distinct presentation of acquired aplastic anemia? J Clin Rheumatol. (2001) 7(6):377–83. doi: 10.1097/00124743-200112000-00006
38. Morishita Y, Matsukawa Y, Kura Y, Takei M, Tomita Y, Nishinarita S, et al. Antithymocyte globulin for a patient with systemic lupus erythematosus complicated by severe pancytopenia. J Int Med Res. (1997) 25(4):219–23. doi: 10.1177/030006059702500409
39. Chute JP, Hoffmeister K, Cotelingam J, Davis TA, Frame JN, Jamieson T. Aplastic anemia as the sole presentation of systemic lupus erythematosus. Am J Hematol. (1996) 51(3):237–9. doi: 10.1002/(sici)1096-8652(199603)51:3%3C237::aid-ajh10%3E3.0.co;2-d
40. Marques JA, Rhim H, Distenfeld A. Inhibition of hematopoiesis by a plasma factor in a case of aplastic anemia associated with systemic lupus erythematosus. P R Health Sci J. (1995) 14(4):293–6.8637971
41. Bailey FA, Lilly M, Bertoli LF, Ball GV. An antibody that inhibits in vitro bone marrow proliferation in a patient with systemic lupus erythematosus and aplastic anemia. Arthritis Rheum. (1989) 32(7):901–5. doi: 10.1002/j.2326-5205.1989.tb00022.x
42. Winkler A, Jackson RW, Kay DS, Mitchell E, Carmignani S, Sharp GC. High-dose intravenous cyclophosphamide treatment of systemic lupus erythematosus-associated aplastic anemia. Arthritis Rheum. (1988) 31(5):693–4. doi: 10.1002/art.1780310518
43. Saal JG, Daniel PT, Berg PA. Indoprofen-induced aplastic anemia in active connective tissue disease detected by drug-specific lymphocyte transformation. Klin Wochenschr. (1986) 64(10):481–5. doi: 10.1007/BF01713174
44. Stricker RB, Shuman MA. Aplastic anemia complicating systemic lupus erythematosus: response to androgens in two patients. Am J Hematol. (1984) 17(2):193–201. doi: 10.1002/ajh.2830170211
45. Fitchen JJ, Cline MJ, Saxon A, Golde DW. Serum inhibitors of hematopoiesis in a patient with aplastic anemia and systemic lupus erythematosus. Recovery after exchange plasmapheresis. Am J Med. (1979) 66(3):537–42. doi: 10.1016/0002-9343(79)91097-0
46. McDuffie FC. Bone marrow depression after drug therapy in patients with systemic lupus erythematosus. Ann Rheum Dis. (1965) 24(3):289–92. doi: 10.1136/ard.24.3.289
47. Brooks BJ Jr, Broxmeyer HE, Bryan CF, Leech SH. Serum inhibitor in systemic lupus erythematosus associated with aplastic anemia. Arch Intern Med. (1984) 144(7):1474–7. doi: 10.1001/archinte.1984.00350190172030
Keywords: systemic lupus erythematosus, SLE, aplastic anaemia, AA, pediatric
Citation: Gao X, Li Y, Lan X and Zeng P (2025) Case Report: Rare aplastic anemia in pediatric systemic lupus erythematosus: a case series and systematic literature review. Front. Pediatr. 13:1602651. doi: 10.3389/fped.2025.1602651
Received: 30 March 2025; Accepted: 16 June 2025;
Published: 27 June 2025.
Edited by:
Giorgia Martini, University Hospital of Padua, ItalyReviewed by:
Jill de Jong, The University of Chicago, United StatesKathleen Overholt, Riley Children's Health, United States
Copyright: © 2025 Gao, Li, Lan and Zeng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ping Zeng, emVuZ3Bpbmd0b21AMTI2LmNvbQ==
†These authors share first authorship
‡ORCID:
Xianfei Gao
orcid.org/0009-0004-1540-3280
Ping Zeng
orcid.org/0009-0001-1588-1483