 Xiaohong Zheng
Xiaohong Zheng Foyang Fan1
Foyang Fan1 Youfeng Zhou
Youfeng Zhou- 1College of Clinical Medicine for Obstetrics & Gynecology and Pediatrics, Fujian Medical University, Fujian Children’s Hospital (Fujian Branch of Shanghai Children’s Medical Center), Fuzhou City, China
- 2Research Department, Chigene (Beijing) Translational Medical Research Center Co., Ltd., Beijing, China
Objective: De novo variants in MAST4 are increasingly implicated in neurodevelopmental disorders (NDDs), but the associated phenotypic spectrum remains incompletely characterized. We report a Chinese child with global developmental delay (GDD) and a novel MAST4 variant, further delineating the genotype-phenotype correlations for this gene.
Methods: Clinical and genetic data were retrospectively analyzed for a proband diagnosed with a MAST4-related NDD at Fujian Children's Hospital. Trio-based whole-exome sequencing (WES) and subsequent Sanger sequencing were performed to identify and validate the pathogenic variant.
Results: The 4-year-old male proband exhibited GDD with intellectual, motor, and speech impairments. Brain MRI showed delayed myelination. WES revealed a heterozygous MAST4 missense variant (NM_001164664.2: c.4142G>T, p.Arg1381Leu), absent in population databases (gnomAD) and confirmed as de novo. The variant affects a highly conserved residue, supporting its likely pathogenicity. Phenotypic comparison with five previously reported cases confirmed core features of GDD and white matter abnormalities, though our patient lacked infantile spasms, underscoring clinical heterogeneity.
Conclusion: This study reinforces MAST4's role in NDDs and expands the genetic and phenotypic spectrum associated with this gene. The absence of infantile spasms in our case suggests variable expressivity, necessitating further functional studies to assess the variant's pathogenicity and MAST4's neurobiological mechanisms.
1 Introduction
Neurodevelopmental disorders (NDDs) represent a heterogeneous group of conditions characterized by impairments in cognition, communication, behavior, and motor function, typically manifesting in early childhood. With a global prevalence of 8%–20%, NDDs impose substantial lifelong burdens on affected individuals and healthcare systems (1–4). Current diagnostic challenges stem from phenotypic heterogeneity and unreliable biomarkers, often delaying intervention. Moreover, treatment strategies remain largely symptomatic rather than mechanistic, underscoring the urgent need to elucidate the molecular underpinnings of these disorders. Advances in genetic research have revealed the remarkable complexity of NDD pathogenesis, involving diverse risk genes (e.g., FMR1, SHANK3) and variant types (e.g., copy number variations, single-nucleotide variants) (5–7). Notably, variable expressivity and incomplete penetrance complicate genotype-phenotype correlations, suggesting that additional modifiers may contribute to disease mechanisms.
Among emerging NDD-associated genes, the MAST family (MAST1–4) encodes microtubule-associated serine/threonine kinases with critical roles in neural development. These proteins share 49%–64% sequence homology and contain four conserved domains: DUF1908, a serine/threonine kinase domain, an AGC kinase C-terminal domain, and a PDZ domain (8). MAST1 variants are linked to megacorpus callosum syndrome and ID (9), while MAST3 variants are associated with NDDs with or without seizures (10). In contrast, MAST2 remains poorly characterized. MAST4 has garnered particular interest due to its multifaceted roles in neurodevelopment. Firstly, MAST4 modulates microtubule dynamics and axonal integrity by activating Cdc42 via interaction with Tctex-1, a process critical for primary cilium resorption and synaptic signaling (11). Furthermore, through phosphorylation of FOXO1, MAST4 suppresses RTKN2 promoter binding, potentially influencing neuronal survival and synaptic plasticity (12). MAST4 is highly expressed in oligodendrocytes, suggesting a role in myelination, a hypothesis supported by frequent white matter abnormalities in patients with MAST4 variants (12, 13). Spatiotemporal expression analyses reveal that MAST4 is enriched in the medial prefrontal cortex and thalamic nuclei during embryogenesis, with persistent expression in hippocampal neurons, cerebellar Purkinje cells, and oligodendrocytes in adulthood (8, 13). This pattern aligns with its putative functions in circuit formation and maintenance. Clinical evidence further implicates MAST4 in NDDs, epilepsy, and hippocampal malformations (8, 14), though the phenotypic spectrum remains incompletely defined.
Here, we report a pediatric case featuring global developmental delay (GDD) and a novel MAST4 variant. We combine clinical phenotyping with neuroimaging and genetic analyses to expand the genotype-phenotype landscape of MAST4-related disorders.
2 Materials and methods
2.1 Study participant and clinical evaluation
The proband was a 4-year-old male who presented to the Neurology Clinic of Fujian Provincial Children's Hospital at 2 years and 1 month of age with global developmental delay (GDD), including deficits in fine/gross motor skills and speech. Clinical assessment included neurological examination, developmental evaluation with China Developmental Scale for Children (CDSC) (0–6 years), and neuroimaging with cranial MRI. Electroencephalography (EEG) was deferred due to no clinical seizure history. The patient received intermittent neurodevelopmental therapy (focused on motor coordination and speech habilitation) through the hospital's rehabilitation program.
2.2 Genetic testing and variant interpretation
Trio whole-exome sequencing was performed by Chigene Translational Medicine Research Center Co., Ltd. (Beijing, China) on peripheral blood-derived DNA (proband and parents) using the IDT xGen Exome Research Panel v2.0 on an Illumina NovaSeq 6,000 platform (150 bp paired-end reads, average 100 × coverage). Raw reads were processed (fastq), aligned to GRCh37/hg19 (BWA-MEM v0.7.17), and variants called following GATK (v4.2.6.1) best practices, including duplicate marking, base quality recalibration, and local realignment. Variants were annotated (ANNOVAR) and filtered against population databases (gnomAD/ExAC/1,000 Genomes), then prioritized based on predicted impact using multiple algorithms (SIFT/PolyPhen-2/CADD/REVEL for missense; SpliceAI/dbscSNV for splicing; GERP++/phyloP for conservation). Variants were classified per ACMG/AMP guidelines with ClinGen specifications, integrating population frequency, computational predictions, inheritance patterns, and phenotype correlation (HGMD/ClinVar/OMIM). The MAST4 variant (c.4142G>T) was validated by Sanger sequencing (ABI 3730xl).
3 Results
3.1 Clinical description
The patient, a 4-year-old boy, presented to our institution at 2 years and 1 month of age with global developmental delay affecting fine motor, gross motor, and speech domains. His clinical features included independent ambulation with gait instability, impaired manual dexterity (manifested by slow reaching/grasping and weak grip strength), and significant language delay (limited to single-word utterances with poor articulation). On physical examination, he was alert and responsive with stable vital signs. His head circumference measured 50.5 cm (within normal range), and his anterior fontanelle was closed. Skin examination revealed no jaundice, pallor, edema, or petechiae, with the absence of transverse palmar creases. Facial features included normal ocular motility without hypertelorism or ptosis, along with a mildly flattened nasal bridge. Oropharyngeal structures were intact, without a high-arched palate or cyanosis. Cardiopulmonary and abdominal assessments were unremarkable. Neurological evaluation demonstrated preserved visual and auditory responses, absent startle reflex, mild hypotonia, and symmetrically normal deep tendon reflexes (knee and Achilles ++). Plantar responses (Babinski sign), ankle clonus, and meningeal signs (Kernig's and Brudzinski's signs) were all negative.
The child was born at term with a normal birth weight of 3.1 kg. During the neonatal period, he exhibited heightened startle reflexes and frequent, unexplained crying spells. His motor developmental milestones were as follows: head lifting in the prone position by 2–3 months, stable head control by 4 months, independent sitting by 10 months, crawling by 17 months, and unaided walking by 19 months. At 2 years and 1 month of age, he was referred to our outpatient clinic for evaluation of developmental delays in fine motor skills, gross motor function, and language abilities. A cranial MRI at that time revealed inadequate myelination (Figure 1).
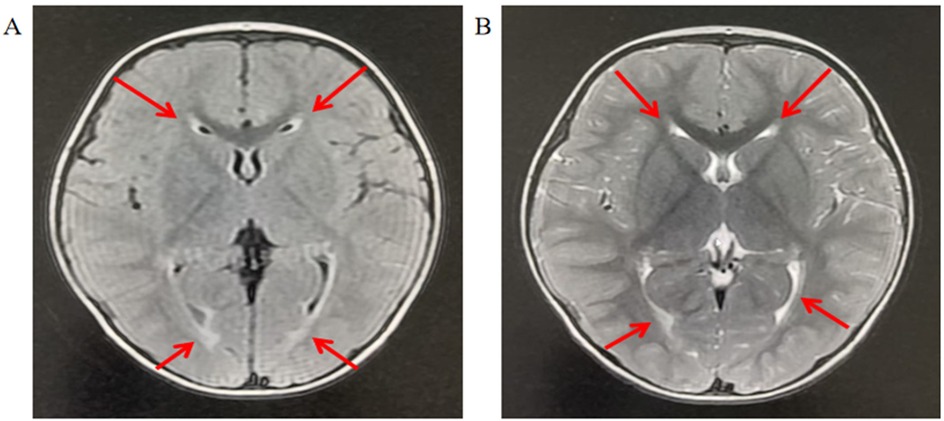
Figure 1. Brain MRI of the patient indicates myelination dysplasia. (A) T2-weighted imaging: The arrowhead denotes a region displaying abnormally persistent hyperintensity, suggesting delayed or disrupted myelination. This finding is consistent with hypomyelination, characterized by reduced myelin content and disorganized axonal maturation. (B) T1-weighted imaging: The arrow indicates a region with persistently hyperintense signal, further corroborating hypomyelination as immature white matter retains high T1 signal intensity until full myelination is achieved. The T1–T2 signal discrepancy (“T1 bright, T2 bright” paradox) is pathognomonic for developmental white matter abnormalities.
Developmental assessment using the CDSC (0–6 years) for Children at 2 years 1 month and 3 years 6 months revealed persistent delays across all domains (Table 1). This assessment evaluates gross motor skills, fine motor skills, adaptive behavior, language competence, and personal-social behavior. The developmental quotient (DQ) serves as an indicator of overall developmental status, with classifications as follows: DQ ≥ 85 (normal development), 76–84 (borderline delay), 55–75 (mild delay), 40–54 (moderate delay), 25–39 (severe delay), and ≤24 (profound delay). In this case, the patient demonstrated persistent developmental delays, with both DQ scores falling below 70, consistent with mild developmental delay. This sustained subthreshold performance underscores the need for targeted clinical interventions to address deficits across multiple functional domains.

Table 1. China developmental scale for children (CDSC) (0–6 years).
3.2 Molecular data
Whole-exome sequencing identified a heterozygous MAST4 missense variant (NM_001164664.2: c.4142G>T, p.Arg1381Leu) in the proband. Trio analysis confirmed its de novo origin (PS2), with Sanger sequencing demonstrating the variant in the proband (heterozygous G/T peaks) and wild-type genotypes (G/G) in both parents (Figure 2). Pedigree analysis revealed an autosomal dominant inheritance pattern with no family history of neurodevelopmental disorders (Figure 3). The variant was absent in major population databases [gnomAD v4.0 (807,162 alleles; 62,784 East Asian), ExAC, and ESP6500], with an allele frequency <0.0001% (PM2_Supporting). Computational tools unanimously predicted pathogenicity: SIFT (deleterious, 0.02), PolyPhen-2 (probably damaging, 0.998), REVEL (pathogenic, 0.88), and CADD (Phred 28.5) (PP3). The variant meets ACMG-AMP criteria for Likely Pathogenic classification (PS2 + PM2_Supporting + PP3). No other pathogenic variants or CNVs were detected that could explain the phenotype.
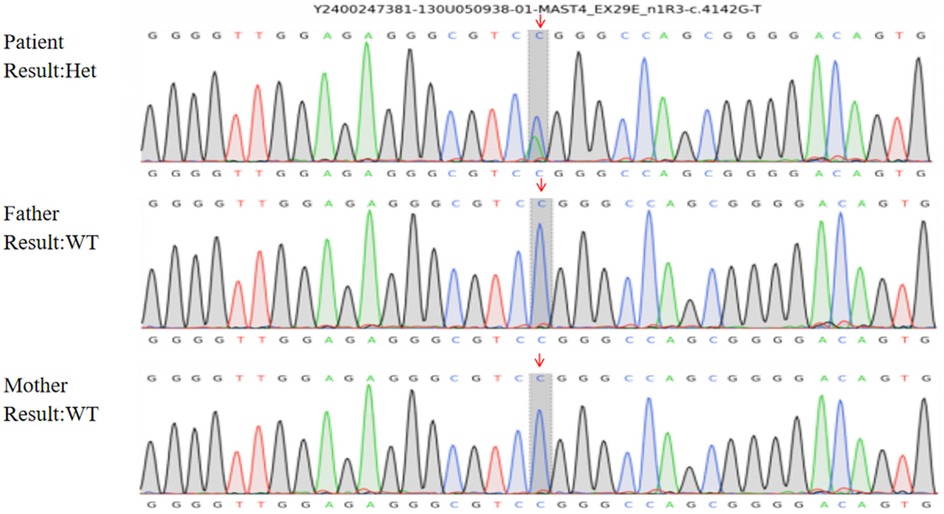
Figure 2. Sanger sequencing confirmation of the MAST4 variant. Chromatogram showing the heterozygous c.4142G>T (p.Arg1381Leu) variant in the proband (indicated by overlapping G/T peaks at position 4,142). Both parents show wild-type sequences (G/G), confirming the de novo origin of this variant.
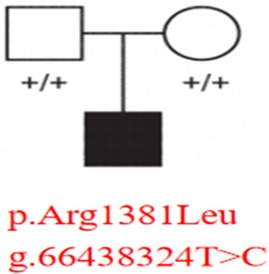
Figure 3. Pedigree analysis of the MAST4 variant inheritance. Squares (male) and circles (female) represent family members, with filled symbols indicating affected status. The proband carries the heterozygous MAST4 p.Arg1381Leu variant (g.66438324T>C), while both unaffected parents show wild-type genotypes (“+/+”).
Multiple sequence alignment revealed exceptional evolutionary conservation of Arg1381 across vertebrate species, suggesting critical functional importance (Figure 4). Structural modeling localized the variant to a disordered loop region (residue 1,381) (Figure 5).
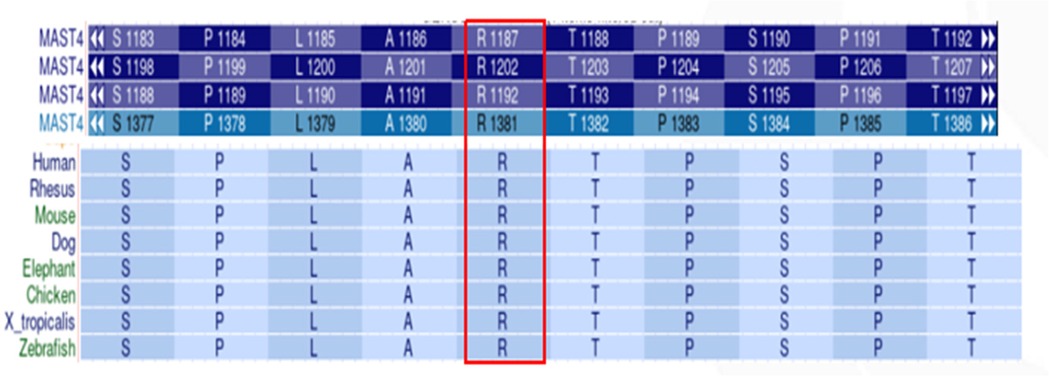
Figure 4. Evolutionary conservation analysis of MAST4 p.Arg1381. Multiple sequence alignment shows high arginine residue conservation at position 1,381 across vertebrate species.
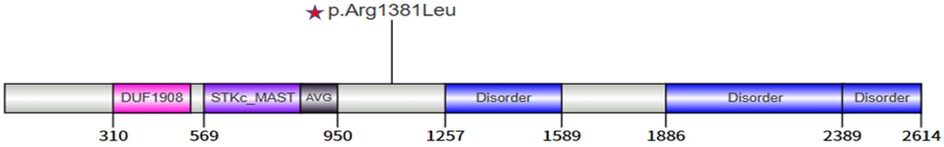
Figure 5. Domain architecture of MAST4 protein showing the location of the p.Arg1381Leu variant.
AlphaFold2-based modeling (Google DeepMind) revealed that wild-type MAST4 Arg1381 forms stabilizing hydrogen bonds with Ala1380 and Thr1382 in the kinase domain (Figure 6A). While the p.Arg1381Leu variant maintained similar hydrogen bond numbers, the substitution of a positively charged arginine with hydrophobic leucine likely impairs function through (Figure 6B): (1) disruption of local electrostatic balance, potentially affecting substrate binding; and (2) steric interference with kinase domain accessibility. This charge and structural alteration at a highly conserved residue may compromise protein-protein interactions and catalytic activity, suggesting a plausible mechanism by which this variant contributes to neurodevelopmental pathology through structure-function perturbation. Together, these findings provide strong evidence for the pathogenic potential of this de novo MAST4 variant in the patient's neurodevelopmental disorder.
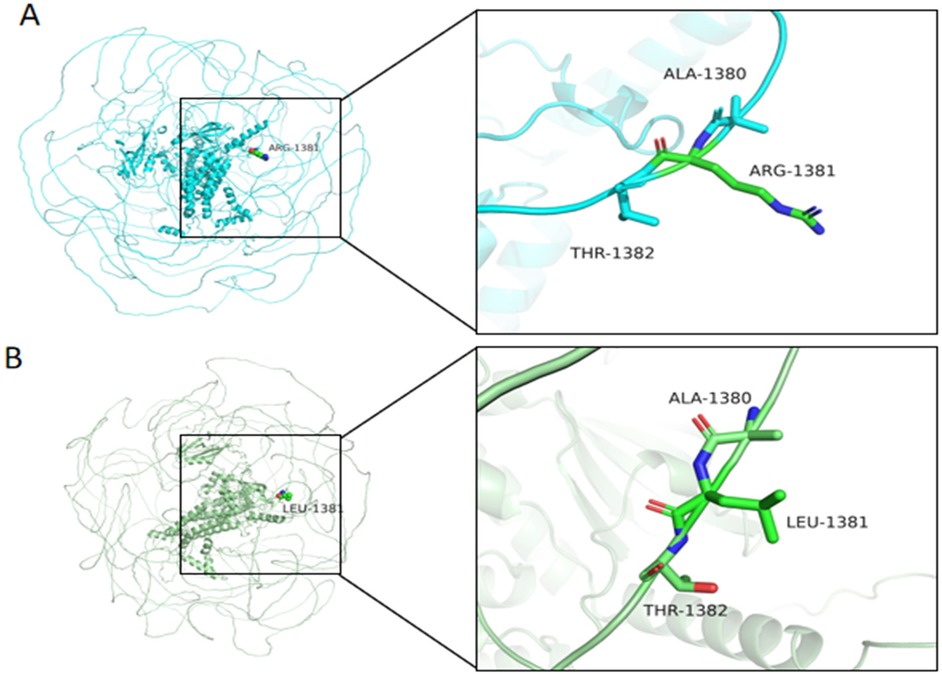
Figure 6. Structural comparison of wild-type and mutant MAST4 protein at residue 1,381. (A) Wild-type MAST4 structure showing Arg1381 forming hydrogen bonds (dashed lines) with adjacent residues Ala1380 and Thr1382. (B) Mutant MAST4 (p.Arg1381Leu) structure demonstrating altered local conformation, potentially affecting protein stability or partner binding. Residue labels indicate positions 1,380–1,382 in both structures. Cyan, WT, green, mutant.
3.3 Literature review
3.3.1 MAST4 in neurodevelopmental disorders and epilepsy
Through a systematic review of PubMed literature using the search term “MAST4 gene variant”, we identified seven relevant publications, including two studies definitively linking MAST4 variants to neurodevelopmental disorders. Table 2 summarizes the clinical characteristics of patients with MAST4 variants from previous reports and the current case. Cases 1–4 were derived from the prior study (8), while Case 5 represents the novel clinical presentation reported in this investigation. The seminal study (8) identified four children harboring de novo MAST4 missense variants (p.Ile898Thr, p.Thr1471Ile, and p.Ser2552Trp), all exhibiting global developmental delay and intellectual disability, with three patients demonstrating infantile spasms—two with drug-resistant epilepsy and one achieving seizure remission on levetiracetam. Structural brain anomalies, including ventriculomegaly and corpus callosum hypoplasia, were observed in a subset of cases. These variants, absent from all major population databases (gnomAD/ExAC/ESP6500), were unanimously predicted as pathogenic by multiple computational algorithms (SIFT/PolyPhen-2/REVEL/CADD). The clinical presentations consistently featured neurodevelopmental impairments coupled with diverse seizure phenotypes, including drug-responsive focal seizures and developmental epileptic encephalopathy. Landoulsi et al. (15) support MAST4's role in epileptogenesis through its critical functions in microtubule-associated kinase activity and neuronal signaling pathways. The collective evidence positions MAST4 as a clinically relevant gene in the overlapping spectra of neurodevelopmental disorders and epilepsy, with particular importance for cases presenting with infantile spasms and developmental delay.
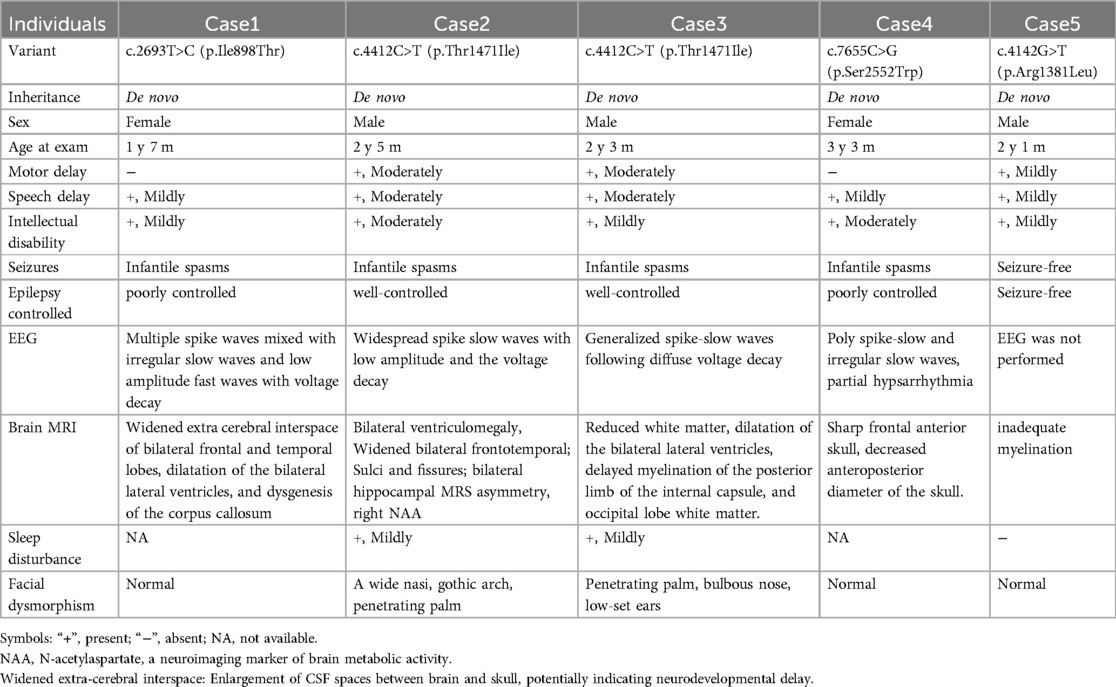
Table 2. The clinical features of the affected individual with the MAST4 variants.
3.3.2 MAST4 involvement in non-neurological conditions
The remaining five articles revealed MAST4's pleiotropic roles beyond the nervous system. In developmental disorders, MAST4 variants were found to inhibit the Wnt/β-catenin signaling pathway, leading to enamel hypoplasia through functional interactions with known tooth agenesis-associated genes (e.g., WNT10A, AXIN2) (16). In oncology, variants in MAST4 have been implicated in the progression of acral melanoma, potentially facilitating tumor cell metastasis through alterations in adhesion, motility, and invasiveness (17). Additionally, MAST4 expression levels were integrated into kinase profiling for myelodysplastic syndromes (MDS), demonstrating prognostic value for disease progression risk stratification and therapeutic target responsiveness (18). In prostate cancer research, MAST4 has been identified as a participant in chromosomal translocations within the 5q region of the VCaP cell line, a process driven by chromothripsis. This highlights MAST4's potential role in contributing to genomic instability, as chromosomal translocations are a form of chromosomal instability that can lead to cancer development (19).
To summarize, MAST4 variants exhibit remarkable pleiotropy, contributing to neurodevelopmental disorders with epilepsy (8, 15), enamel defects (16), cancer metastasis (17), MDS risk stratification (18), and chromosomal rearrangements in malignancies (19). However, our patient lacked infantile spasms and displayed delayed myelination as the primary neuroimaging feature, contrasting with the previously reported structural abnormalities. This phenotypic divergence suggests either variable expressivity of MAST4 variants or modulation by genetic modifiers. This multifactorial involvement underscores the need for mechanistic studies to delineate tissue-specific pathogenic pathways and explore therapeutic targeting strategies.
4 Discussion
4.1 MAST4 expression and function
MAST4, a member of the MAST kinase family, is crucial for neurodevelopment as it regulates axonal guidance and synaptic plasticity via its kinase domain. This regulation is part of the complex molecular mechanisms that ensure proper neural network formation and function. Its predominant expression in cerebellar Purkinje cells, hippocampal regions, and white matter-enriched areas (20), along with embryonic enrichment in the dorsal midline thalamic nuclei and medial prefrontal cortex (8, 21), underscores its importance in early neural circuit formation. Animal studies reveal seizure-induced upregulation of MAST4 in the murine hippocampus (22), suggesting involvement in epileptogenic neuroplasticity. Functionally, MAST4 modulates neuronal survival via FOXO1 phosphorylation and regulates ciliary dynamics through Tctex-1/Cdc42 interactions (8, 10), establishing it as a multifunctional orchestrator of neural development and homeostasis. All of these suggest that MAST4 variants disrupt cellular self-renewal, proliferation, and differentiation (12).
4.2 Variant novelty and conservation
The pathogenicity of MAST4 variants in NDDs remains poorly characterized, as no MAST4-related disorders are currently cataloged in OMIM. Although the Human Gene Mutation Database (HGMD) lists 23 MAST4 variants classified as disease-causing (DM), their clinical significance lacks validation in large cohorts or functional studies. The de novo MAST4 missense variant (c.4142G>T, p.Arg1381Leu) identified in our patient offers several lines of evidence supporting its potential pathogenicity: (1) The variant is absent in gnomAD v4.0, ExAC, and ESP6500, suggesting strong negative selection; (2) Arg1381 is highly conserved across vertebrates (Figure 4), implying functional indispensability in the MAST4 kinase domain; (3) Concordant deleterious predictions from SIFT (deleterious), PolyPhen-2 (probabaly damaging), REVEL (0.885), and CADD (28.5) further bolster its disease association. Structural modeling localizes p.Arg1381Leu to a critical loop region within the kinase domain (Figure 5). The substitution of a charged arginine with a hydrophobic leucine likely destabilizes local protein conformation, impairing MAST4's interaction with downstream substrates (Figure 6). Unlike MAST1 variants, which are associated with megalencephaly and corpus callosum anomalies, our patient's MAST4 variant correlates with delayed myelination and developmental delay, without structural malformations. This divergence may reflect differential substrate specificity among MAST kinases, with MAST4 preferentially influencing white matter integrity via distinct signaling cascades.
4.3 Case interpretation and phenotypic comparison
Our patient presents a distinct clinical manifestation compared to previously reported MAST4 cases, characterized by a unique combination of significant global developmental delay (DQ < 70) without epileptic manifestations and predominant delayed myelination on MRI (Figure 1), contrasting with the typical structural abnormalities seen in other cases. This phenotypic divergence likely results from the interplay between variant-specific effects and epigenetic modulation. The p.Arg1381Leu variant may disrupt distinct protein interaction networks and substrate recognition, creating a unique perturbation pattern. Importantly, epigenetic mechanisms appear to play a protective role against seizures while potentially exacerbating myelination defects. The adenosine-mediated DNA hypomethylation pathway (23) and histone modification cascades may maintain an anti-epileptic state, while disproportionate disruption of Wnt/β-catenin signaling (24) and mTOR-mediated OPC differentiation (25) likely contribute to the myelination delay. The involvement of SWI/SNF and CHD chromatin remodelers (26) in oligodendrocyte differentiation, along with the known neurodevelopmental roles of H3K4 trimethylation and H3K27 demethylation (27), suggests complex epigenetic modulation of myelination defects. Although direct evidence linking MAST4 to these pathways requires further investigation, the kinase domain's potential to phosphorylate chromatin modifiers and its known regulation of cytoskeletal dynamics suggest plausible mechanistic connections. These findings highlight three key aspects of MAST4 pathobiology: domain-specific effects, epigenetic buffering capacity, and developmental stage-dependent neural vulnerability, underscoring the need for integrated genetic-epigenetic analyses.
4.4 Implications for future research
Our findings highlight three critical avenues for further investigation: First, constructing MAST4 p.Arg1381Leu mutant mouse models to systematically delineate its spatiotemporal effects on myelination and synaptic plasticity, with a focus on oligodendrocyte differentiation and axon-glia interactions. Second, integrating whole-genome sequencing and chromatin conformation capture (Hi-C) to identify MAST4-interacting epigenetic regulators (e.g., HDAC4, MECP2) and uncover molecular drivers of phenotypic heterogeneity. Third, developing targeted therapies, including allosteric small-molecule modulators or AAV-based gene editing strategies, to restore MAST4 kinase functionality. These efforts will deepen mechanistic insights and accelerate translational applications for MAST4-related disorders.
5 Conclusion
De novo variants in MAST4 are emerging as important genetic contributors to neurodevelopmental disorders, typically presenting with global developmental delay and white matter abnormalities. This case report broadens the known phenotypic spectrum by characterizing a novel p.Arg1381Leu variant and its unique association with myelination deficits in the absence of epilepsy—a departure from previously reported cases.The observed phenotypic heterogeneity likely reflects a complex interplay between the primary variant and modifier gene effects, particularly through epigenetic regulation of MAST4 expression during neurodevelopment. For instance, variability in DNA methylation at SOX10-binding enhancers may influence oligodendrocyte progenitor cell (OPC) migration in a region-specific manner, while H3K27ac-mediated chromatin remodeling could mitigate the consequences of MAST4 haploinsufficiency on axonal guidance pathways. This multilayered regulatory framework may explain both the developmental stage-specific expression of white matter abnormalities and the apparent resilience against epileptogenesis in this patient. Future directions should include: (1) Establishing multicenter cohorts to better delineate genotype-phenotype correlations. (2) Assessing functional impact through in vitro kinase activity assays and conditional knockout models. (3) Applying multi-omics approaches—such as single-cell methylome and 3D chromatin architecture profiling—to investigate epigenetic compensatory mechanisms in glial cells. (4) Developing brain organoid models using CRISPR-dCas9-based epigenetic editing to simulate modifier gene effects on neuron-glia interactions. (5) Conducting longitudinal follow-up to track neurodevelopmental trajectories and therapeutic responses.These efforts will refine molecular diagnostic strategies and pave the way toward personalized treatment approaches for MAST4-related neurodevelopmental disorders.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Ethics Committee of Fujian Children's Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants' legal guardians/next of kin.
Author contributions
XZ: Data curation, Writing – original draft, Conceptualization. FF: Conceptualization, Methodology, Writing – original draft. BL: Data curation, Investigation, Writing – original draft. YX: Investigation, Supervision, Writing – original draft. MP: Data curation, Writing – original draft. YZ: Data curation, Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
We thank the patients and their families for their cooperation and participation in our study.
Conflict of interest
Author MP was employed by company Chigene (Beijing) Translational Medical Research Center Co., Ltd. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. GBD. 2019 Mental Disorders Collaborators. Global, regional, and national burden of 12 mental disorders in 204 countries and territories, 1990–2019: a systematic analysis for the global burden of disease study 2019. Lancet Psychiatry. (2022) 9:137–50. doi: 10.1016/S2215-0366(21)00395-3
2. GBD. 2019 Diseases and Injuries Collaborators. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: a systematic analysis for the global burden of disease study 2019. Lancet. (2020) 396:1204–22. doi: 10.1016/S0140-6736(20)30925-9
3. Olusanya BO, Nair MKC. Premature mortality in children with developmental disabilities. Lancet Glob Health. (2019) 7:e1601–2. doi: 10.1016/S2214-109X(19)30419-X
4. Baxter AJ, Brugha TS, Erskine HE, Scheurer RW, Vos T, Scott JG. The epidemiology and global burden of autism spectrum disorders. Psychol Med. (2015) 45:601–13. doi: 10.1017/S003329171400172X
5. Satterstrom FK, Kosmicki JA, Wang J, Breen MS, De Rubeis S, An J-Y, et al. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell. (2020) 180:568–84.e23. doi: 10.1016/j.cell.2019.12.036
6. Coe BP, Stessman HAF, Sulovari A, Geisheker MR, Bakken TE, Lake AM, et al. Neurodevelopmental disease genes implicated by de novo mutation and copy number variation morbidity. Nat Genet. (2019) 51:106–16. doi: 10.1038/s41588-018-0288-4
7. Pinto D, Delaby E, Merico D, Barbosa M, Merikangas A, Klei L, et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am J Hum Genet. (2014) 94:677–94. doi: 10.1016/j.ajhg.2014.03.018
8. Zhang X, Xiao N, Cao Y, Peng Y, Lian A, Chen Y, et al. De novo variants in MAST4 related to neurodevelopmental disorders with developmental delay and infantile spasms: genotype-phenotype association. Front Mol Neurosci. (2023) 16:1097553. doi: 10.3389/fnmol.2023.1097553
9. Hecher L, Johannsen J, Bierhals T, Buhk J-H, Hempel M, Denecke J. The clinical picture of a bilateral perisylvian syndrome as the initial symptom of mega-corpus-callosum syndrome due to a MAST1-gene mutation. Neuropediatrics. (2020) 51:435–9. doi: 10.1055/s-0040-1710588
10. Spinelli E, Christensen KR, Bryant E, Schneider A, Rakotomamonjy J, Muir AM, et al. Pathogenic MAST3 variants in the STK domain are associated with epilepsy. Ann Neurol. (2021) 90:274–84. doi: 10.1002/ana.26147
11. Sakaji K, Ebrahimiazar S, Harigae Y, Ishibashi K, Sato T, Yoshikawa T, et al. MAST4 Promotes primary ciliary resorption through phosphorylation of Tctex-1. Life Sci Alliance. (2023) 6(11):e202301947. doi: 10.26508/lsa.202301947
12. Gongol B, Marin TL, Jeppson JD, Mayagoitia K, Shin S, Sanchez N, et al. Cellular hormetic response to 27-hydroxycholesterol promotes neuroprotection through AICD induction of MAST4 abundance and kinase activity. Sci Rep. (2017) 7(1):13898. doi: 10.1038/s41598-017-13933-9
13. Sun L, Gu S, Li X, Sun Y, Zheng D, Yu K, et al. Identification of a novel human MAST4 gene, a new member of the microtubule associated serine-threonine kinase family. Mol Biol (Mosk). (2006) 40(5):808–15. doi: 10.1134/S0026893306050062
14. Hibar DP, Adams HHH, Jahanshad N, Chauhan G, Stein JL, Hofer E, et al. Novel genetic loci associated with hippocampal volume. Nat Commun. (2017) 8:13624. doi: 10.1038/ncomms13624
15. Landoulsi Z, Laatar F, Noé E, Mrabet S, Ben Djebara M, Achaz G, et al. Clinical and genetic study of Tunisian families with genetic generalized epilepsy: contribution of CACNA1H and MAST4 genes. Neurogenetics. (2018) 19:165–78. doi: 10.1007/s10048-018-0550-z
16. Semjid D, Ahn H, Bayarmagnai S, Gantumur M, Kim S, Lee JH. Identification of novel candidate genes associated with non-syndromic tooth agenesis in Mongolian families. Clin Oral Investig. (2023) 28:56. doi: 10.1007/s00784-023-05415-2
17. Lim Y, Lee DY. Identification of genetic mutations related to invasion and metastasis of acral melanoma via whole-exome sequencing. J Dermatol. (2021) 48:999–1006. doi: 10.1111/1346-8138.15841
18. Yao C-Y, Lin C-C, Wang Y-H, Kao C-J, Tsai C-H, Hou H-A, et al. Kinome expression profiling improves risk stratification and therapeutic targeting in myelodysplastic syndromes. Blood Adv. (2024) 8:2442–54. doi: 10.1182/bloodadvances.2023011512
19. Teles Alves I, Hiltemann S, Hartjes T, van der Spek P, Stubbs A, Trapman J, et al. Gene fusions by chromothripsis of chromosome 5q in the VCaP prostate cancer cell line. Hum Genet. (2013) 132:709–13. doi: 10.1007/s00439-013-1308-1
20. Garland P, Quraishe S, French P, O'Connor V. Expression of the MAST family of serine/threonine kinases. Brain Res. (2008) 1195:12–9. doi: 10.1016/j.brainres.2007.12.027
21. Molnár Z, Garel S, López-Bendito G, Maness P, Price DJ. Mechanisms controlling the guidance of thalamocortical axons through the embryonic forebrain. Eur J Neurosci. (2012) 35:1573–85. doi: 10.1111/j.1460-9568.2012.08119.x
22. French PJ, O'Connor V, Jones MW, Davis S, Errington ML, Voss K, et al. Subfield-specific immediate early gene expression associated with hippocampal long-term potentiation in vivo. Eur J Neurosci. (2001) 13:968–76. doi: 10.1046/j.0953-816x.2001.01467.x
23. Younus I, Reddy DS. Epigenetic interventions for epileptogenesis: a new frontier for curing epilepsy. Pharmacol Ther. (2017) 177:108–22. doi: 10.1016/j.pharmthera.2017.03.002
24. Gaesser JM, Fyffe-Maricich SL. Intracellular signaling pathway regulation of myelination and remyelination in the CNS. Exp Neurol. (2016) 283(Pt B):501–11. doi: 10.1016/j.expneurol.2016.03.008
25. François L, Romagnolo A, Luinenburg MJ, Anink JJ, Godard P, Rajman M, et al. Identification of gene regulatory networks affected across drug-resistant epilepsies. Nat Commun. (2024) 15(1):2180. doi: 10.1038/s41467-024-46592-2
26. Berry K, Wang J, Lu QR. Epigenetic regulation of oligodendrocyte myelination in developmental disorders and neurodegenerative diseases. F1000Res. (2020) 9:1000–105. doi: 10.12688/f1000research.20904.1
Keywords: MAST4, developmental delay, myelination dysplasia, de novo variant, whole-exome sequencing
Citation: Zheng X, Fan F, Lei B, Xu Y, Peng M and Zhou Y (2025) Genetic and clinical insights into MAST4-related neurodevelopmental disorders. Front. Pediatr. 13:1603050. doi: 10.3389/fped.2025.1603050
Received: 31 March 2025; Accepted: 13 June 2025;
Published: 27 June 2025.
Edited by:
Nan Bao, Shanghai Jiao Tong University, ChinaReviewed by:
Serap Bilge, MGH Institute of Health Professions, United StatesTomoki Nomakuchi, Children’s Hospital of Philadelphia, United States
Copyright: © 2025 Zheng, Fan, Lei, Xu, Peng and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Youfeng Zhou, enlmdXhAeWVhaC5uZXQ=