 Vincenza Gragnaniello1,2
Vincenza Gragnaniello1,2 Silvia Carraro3
Silvia Carraro3 Tiziana Zangardi4Chiara Cazzorla2Daniela Gueraldi2
Tiziana Zangardi4Chiara Cazzorla2Daniela Gueraldi2 Alberto B. Burlina1,2*
Alberto B. Burlina1,2*
- 1Division of Inherited Metabolic Diseases, Department of Women's and Children's Health, University of Padova, Padova, Italy
- 2Division of Inherited Metabolic Diseases, Department of Women's and Children's Health, University Hospital of Padova, Padova, Italy
- 3Unit of Pediatric Allergy and Respiratory Medicine, Women’s and Children’s Health Department, University of Padova, Padova, Italy
- 4Pediatric Emergency Department, Department for Women’s and Children’s Health, University of Padua, Padua, Italy
Gaucher Disease Type 3 (GD3) is a rare lysosomal storage disorder characterized by both visceral and neurological involvement. Pulmonary manifestations can significantly impact prognosis and quality of life. This case report highlights the challenges in managing severe pulmonary involvement in GD and explores novel treatment approaches. We present a case of a patient with GD3, diagnosed through neonatal screening, who developed severe lung disease despite early initiation of enzyme replacement therapy (ERT). The patient, carrying compound heterozygous variants in the GBA1 gene (p.Leu483Pro, [p.His294Gln + p.Asp448His]), experienced respiratory distress requiring oxygen therapy from the age of 4 months. High-resolution computed tomography revealed a typical interstitial lung disease pattern. Despite ERT and a marked reduction in storage biomarkers, pulmonary symptoms persisted, accompanied by elevated inflammatory markers. We implemented a treatment regimen of systemic corticosteroids followed by hydroxychloroquine, resulting in clinical improvement. Furthermore, we observed a decrease in inflammatory biomarkers, such as TNF-alpha and Pp38 MAPK levels, providing insights into possible pathogenic mechanisms. This case underscores the limitations of ERT in addressing pulmonary manifestations of GD and highlights the need for personalized treatment strategies. It also emphasizes the importance of further research into the pathogenesis of pulmonary damage in Gaucher disease to develop more effective therapies for these challenging cases. The positive response to anti-inflammatory and immunomodulatory therapies suggests a potential role for these approaches in managing GD-related lung disease.
1 Introduction
Gaucher disease (GD) is an autosomal recessive disorder caused by mutations in the GBA1 gene, which encodes the lysosomal enzyme glucocerebrosidase (GCase). GCase catalyzes the hydrolysis of glucosylceramide (GluCer) to ceramide and glucose. Its deficiency results in the accumulation of GluCer within lysosomes, especially in macrophages (Gaucher cells) (1–3).
GD presents with a wide phenotypic spectrum, ranging from non-neuronopathic forms (GD1) to acute (GD2) or chronic (GD3) neuronopathic forms. GD3 can manifest with visceral involvement characterized by hepatosplenomegaly, cytopenia, bone marrow infiltration, and skeletal involvement, as well as slowly progressive neurological deterioration of variable onset and severity. Neurological manifestations may include supranuclear horizontal ophthalmoplegia, progressive myoclonic epilepsy, cerebellar ataxia, spasticity, and developmental delay (4–6).
Enzyme replacement therapy (ERT) is effective in treating non-neurological manifestations of GD3. While some evidence suggests initiating therapy for symptomatic patients (7), other studies indicate that specific treatment with ERT should be considered for all GD3 patients (8, 9). This recommendation extends to presymptomatic siblings of GD3 patients, particularly those with specific genotypes such as homozygous or compound heterozygous p.Leu483Pro and p.Asp448His variants (10).
Pulmonary involvement in GD can range from clinically asymptomatic with normal or mild radiographic changes to severe respiratory symptoms with significant radiographic findings (11). Infiltrative lung involvement, including interstitial lung diseases and rapidly progressive lung consolidation, is caused by Gaucher cells infiltrating the perivascular, peribronchial, septal regions, and alveolar air spaces.
Chest X-rays may show diffuse interstitial and nodular infiltrates, with honeycombing. However, the diagnostic gold standard is high-resolution computed tomography (HRCT), which typically reveals ground-glass opacities, sometimes superimposed with interlobular and intralobular septal thickening.
Despite the availability of enzyme replacement therapy (ERT), the course of the lung disease is associated with high morbidity and mortality. Patients frequently require long-term oxygen therapy and sometimes artificial ventilation (12).
In these cases, a deeper understanding of the pathogenesis of the damage and the use of personalized therapies is necessary. We describe the case of a patient with GD3 identified through neonatal screening, who suffered from severe interstitial lung disease poorly responsive to early ERT. The patient was successfully treated with anti-inflammatory and immunomodulatory therapy.
2 Case description
The patient is the third child born to non-consanguineous parents; the father is of Balkan origin, and the mother is Italian. At birth, extended neonatal screening, active in Northeast Italy for lysosomal diseases since 2015 (13, 14), showed reduced GCase activity (0.84 µmol/L/h, normal value >2.5) and elevated Lyso-Gb1 values (1,436 nmol/L, normal value 5.64–33.31) on dried blood spot (DBS), suggesting GD.
The diagnosis was confirmed by reduced GCase activity in lymphocytes (0.08 µmol/h/mg protein, normal value 0–1.93 µmol/h/mg protein) and elevated Lyso-Gb1 levels in plasma (182 µmol/L, normal value <1.93 µmol/L). Molecular testing of the GBA1 gene, become available when the child was 2 months old, revealed compound heterozygosity for the allele c.882T > G + c.1342G > C (p.His294Gln + p.Asp448His) and the allele c.1448T > C (p.Leu483Pro). Both alleles have been associated with acute and chronic neurovisceral phenotypes of Gaucher disease (15, 16) (for methods, see Supplementary Materials).
2.1 Clinical course
At the first assessment, the newborn showed no signs or symptoms of the disease, specifically no cytopenia (Hb 13.3 g/dl, PLT 207,000/mm3) and no visceromegaly on abdominal ultrasound. He presented no neurological signs or ocular abnormalities, and EEG, ABR, and brain MRI were unremarkable.
At 3 months, the patient had persistently elevated biomarkers (plasma Lyso-Gb1 180.7 µmol/L), mild splenomegaly on abdominal ultrasound (longitudinal diameter 6 cm, >90 pc), and mild axial hypotonia with partial head control. Hematological parameters remained normal (Hb 10.2 g/dl, PLT 358,000/mm3). Considering the disease signs and the genetic results suggestive of a severe form (8–10), ERT was initiated at 60 U/kg every other week, along with ambroxol 25 mg/kg in five doses/day as chaperon therapy (17). Biochemical response was good (reduction of Lyso-Gb1 of 64% after 1 month, Figure 1) and spleen volume remained unchanged. Psychomotor development progressed normally (Bayley III: cognitive 100, language 97, motor 88) and no neurological symptoms appeared.
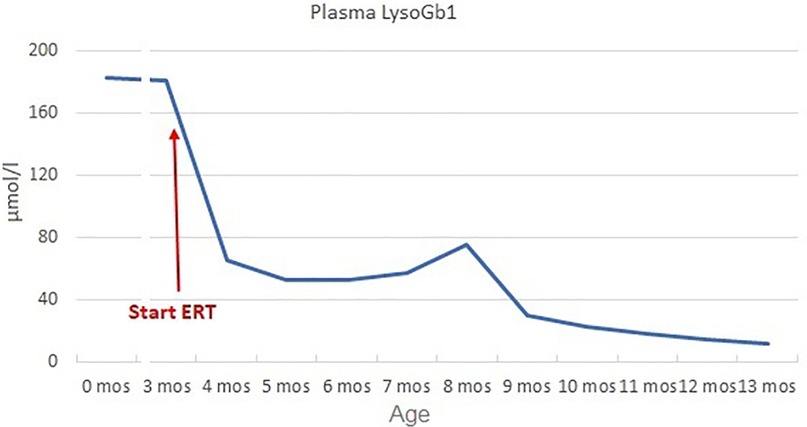
Figure 1. Plasma LysoGb1 trend. LysoGb1 levels rapidly decreased after the initiation of ERT (from 180.7 to 65.4 umol/L, −64% after 1 month), then slowly reduced until reaching 13.9 umol/L at 13 months of age (+10 months of ERT).
2.2 Pulmonary manifestations
During the second month of ERT, the patient presented with dyspnea, tachypnoea (RR 50–60 breaths per minute), and oxygen desaturation. Pulse oximetry monitoring revealed a lowest SatO2 of 85% in the supine position. Chest auscultation appeared normal. Chest x-ray was normal, and nasopharyngeal aspirate was negative for main respiratory viruses (adenovirus, bocavirus, enterovirus, metapneumovirus, parechovirus, parainfluenza, rhinovirus, SARS-CoV-2, RSV). Cardiological evaluation, including ECG and echocardiography, ruled out a cardiogenic cause of desaturation.
In the following days, the patient suffered from tachypnoea and dyspnea. The patient was continuously monitored with a pulse oximeter and intermittent oxygen therapy was initiated when saturation consistently fell below 92%. These desaturation episodes were more frequent when the patient was in a supine position and were not associated with feeding. Although the patient did not have increased respiratory secretion, ambroxol therapy was discontinued for a week with no changes, so it was reintroduced. EEG showed no electrical correlates. Polysomnography showed tonic desaturation with a mean pulse oximetry SatO2 of 90%. Nocturnal transcutaneous pCO2 monitoring did not show hypercapnia (median pCO2 46 mmHg, range 42–49). Chest HRCT revealed heterogeneous pulmonary density with a “mosaic” pattern, characterized by confluent “ground-glass” areas, predominantly distributed in the perihilar and subpleural regions. Furthermore, the peribronchal interstitium was mildly thickened along the perihilar segmental branches, particularly in the right upper lobe. These findings were suggestive of alveolar interstitial disease typical of GD (Figure 2). The ERT dose was increased to 90 U/kg/week based on isolated reports suggesting improved efficacy with high-dose ERT (18).

Figure 2. Chest HRCT: heterogeneous pulmonary density with a “mosaic” pattern, characterized by the presence of confluent “ground-glass” areas, suggestive of alveolar interstitial disease.
However, the severe pulmonary involvement [ILD staging 4 according to European protocols (19)] was poorly responsive to high-dose ERT. We hypothesized that the damage could be amplified by the inflammatory state induced by accumulation in Gaucher cells. Indeed, despite ERT, the patient exhibited elevated markers of systemic inflammation (TNF-alpha 28.5 ng/L, normal value < 8.1) and cellular stress. Specifically, the level of Pp38 MAPK in peripheral blood mononuclear cells (PBMCs) (20) was considerable (4.8 times higher than control).
For these reasons, at the age of 8 months, we initiated steroid therapy with oral methylprednisolone 2 mg/kg/day, tapered by 0.25 mg/kg/week and stopped after 8 weeks, at the age of 10 months (21). The patient experienced clinical improvement, with improvements in home pulse oximetry readings and reduced oxygen requirements at home (ILD staging 3). Concurrently, TNF-alpha levels decreased (9.6 ng/L at 1 month, 13.2 ng/L at 2 months, Figure 3) and Pp38 MAPK levels in PBMCs decreased by 72% at the end of the cycle (2 months) (Figure 4).
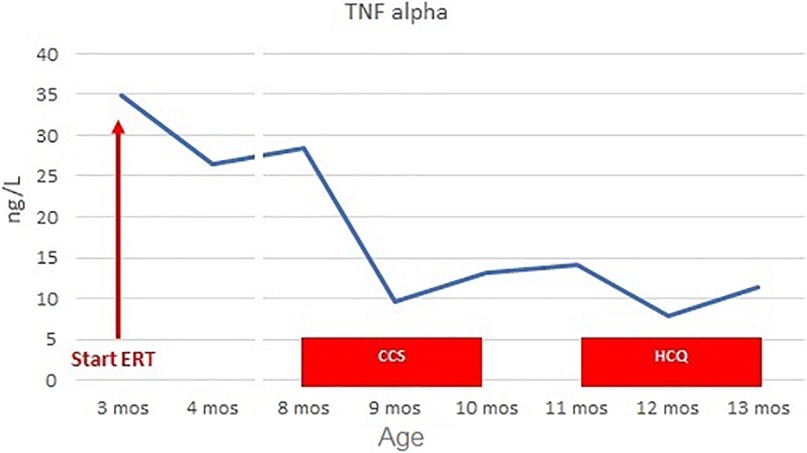
Figure 3. TNF-alpha levels before and during CCS/HCQ therapy. TNF-alpha levels decreased from 28.5 ng/L (normal value <8.1) to 9.6 ng/L during CCS therapy. They slightly increased to 14.1 ng/L at the end of therapy, then decreased again during HCQ treatment to 7.8 ng/L after the first month and 11.3 ng/L after the second month.
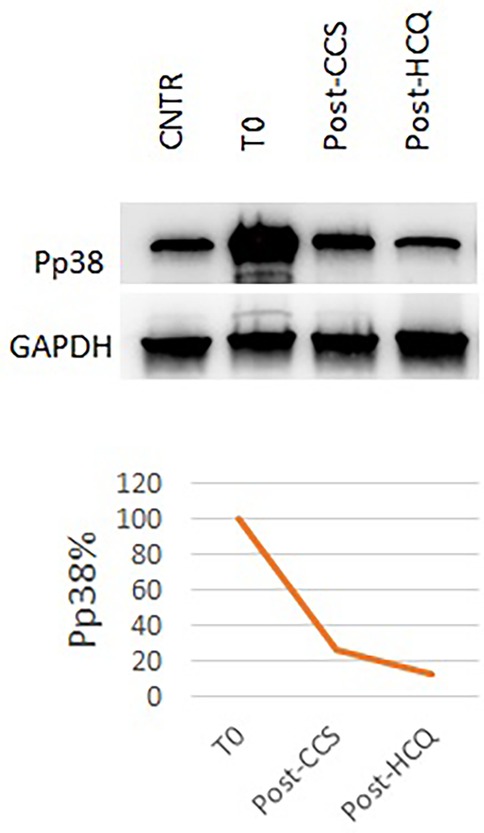
Figure 4. Pp38 levels before and during CCS/HCQ therapy. P-p38 levels decreased by 72% after CCS treatment and underwent a further slight reduction (−88% compared to baseline) after HCQ treatment, reaching values equal to the healthy control.
However, clinical benefits began to diminish as we tapered the steroid dose. Polysomnography at the end of steroid tapering showed no significant improvements from baseline (mean pulse oximetry SatO2 of 88.6%). Due to the transient benefit from steroid therapy, at 11 months of life, the patient started hydroxychloroquine 6 mg/kg/day, in accordance with European protocols for managing interstitial lung disease in children. Before starting therapy, ECG and ophthalmological examination were normal; chest x-ray showed a slight accentuation of the interstitial pattern in the lower lung.
After 2 months (age of 13 months), the patient showed clinical improvement (ILD staging 1). Chest x-ray showed only a slight accentuation of the perihilar texture and polysomnography was normal (mean pulse oximetry SatO2 of 98%). TNF-alpha levels slightly decreased (T0 14.1 ng/L, T1 month 7.8 ng/L, T2 months 11.3 ng/L, Figure 3). P-p38 levels in PBMCs decreased by 88% compared to the baseline values at the start of steroid therapy, reaching levels comparable to those of the control (Figure 4). Given the observed functional improvements, we chose not to repeat CT scans in the short term due to concerns about cumulative radiation exposure in a pediatric patient.
The only adverse event observed was an increase in urea (from 3.20 mmol/L to 7.70–7.90 mmol/L, normal value 1.8–6.4), with consistently normal creatinine levels (20–21 µmol/L, normal value 15–31).
3 Discussion
We describe the case of a patient with GD3, diagnosed through neonatal screening and early treated with ERT and chaperone therapy. Despite this, the patient developed severe lung disease with respiratory distress, requiring oxygen therapy.
Pulmonary involvement is more frequent in GD2 and GD3 patients (1, 22, 23), especially in those carrying the p.Leu483Pro variant, as our patient. Consistent with our case, primary lung disease is likely to occur at early ages, even in subjects with no overt neurological disease. It is related to a higher disease severity (11, 22, 24).
While improvements in pulmonary function tests and some clinical parameters have been reported in individuals receiving high-dose ERT, the general experience is that pulmonary manifestations are slow to respond to enzyme treatment, and some damage may not be reversed (12, 25, 26). Several cases of neuronopathic GD with progressive respiratory symptoms despite ERT have been described, necessitating tracheotomy and artificial ventilation (27). Few cases of lung transplantation have also been reported (26).
The inadequate response to ERT is likely attributable to poor accessibility of the infused enzyme to the lungs, as confirmed by post-mortem examinations. Burrow et al. reported on a 12.5-year-old GD type 3 patient who had received 11 years of ERT. The autopsy revealed clusters of macrophages and very elevated glucosylceramide (GluCer) and glucosylsphingosine (GluS) levels in the lungs and lymph nodes, while the liver and spleen were clear of storage cells and had nearly normal GluCer and GluS levels. The pathological features of the lungs were essentially identical to those of untreated GD3 patients, suggesting that ERT may not effectively penetrate (28). This highlights the need for additional therapeutic approaches and strategies based on a better understanding of lung damage pathogenesis.
It is known that in GD, chronic constitutive macrophage activation may lead to persistent immune system stimulation and systemic inflammation (29, 30). In a previous study, we demonstrated that TNF-alpha levels and P-p38 are sensitive markers of inflammation and cellular stress in Gaucher disease, showing alterations even in the presymptomatic phase (20). Other Authors have reported elevated plasma levels of TNF-alpha and other cytokines in GD patients, with highest concentrations in those with neuronopathic forms (31). Our patient also showed increased TNF-alpha levels without concomitant infections.
TNF-alpha is a pleiotropic cytokine that regulates several pathways, including the MAPK pathway (32). We investigated the activation of the proinflammatory kinase p38 in our patient, which was strongly elevated compared to the control before steroid therapy. Previous in vivo studies have demonstrated p38 activation in GD mice, showing increased P-p38 levels in brain tissues of neuronopathic mice and other affected organs such as liver and lung across all types of GD (33). Overactive p38 contributes to the generation of various inflammatory mediators, amplifying the inflammatory milieu (34).
The effect of steroids in other inflammatory lung diseases has been already reported, including their effects on cytokine production and MAPK pathways. It has been demonstrated that the extent of TNF-induced p38 MAPK phosphorylation is reduced in cells treated with dexamethasone. Indeed, the MAPK deactivator—mitogen-activated protein kinase phosphatase 1 (MKP-1)—is a corticosteroid-inducible gene (35–37). Consistently, our patient showed a reduction in TNF-alpha levels and P38 activation after steroid therapy. In particular, TNF-alpha levels were notably reduced after the first month of therapy, coinciding with clinical improvement. However, they subsequently increased during the tapering phase, concurrent with a return to baseline clinical conditions.
Due to the transient clinical benefit with corticosteroids, we used hydroxychloroquine for long-term treatment. Indeed, it is generally well-tolerated with relatively few side effects.
Interestingly, hydroxychloroquine appears to have an unfavorable effect on lysosomal function, inhibiting lysosomal acidification and preventing autophagosome degradation (38). Nevertheless, it proved effective in our patient. This observation, coupled with the further improvement in inflammation and cellular stress markers, leads us to hypothesize that the mechanism of action is primarily related to the immunomodulatory effect of the drug. The exact mechanism of action for its immunomodulatory effect is not fully understood, but it seems to include decreased macrophage-mediated cytokine production, especially IL-1 and IL-6 (19, 39).
Our patient showed an optimal clinical response after 2 months of therapy, with normal ventilatory pattern on polysomnography and further improvement in inflammatory biomarkers.
In conclusion, pulmonary involvement is crucial in determining the prognosis in neuronopathic Gaucher disease and can greatly influence the patient's quality of life. Although controlling respiratory symptoms is important, it is extremely challenging to achieve with currently available therapies. Our case underscores the potential role of inflammation in the pathogenesis of lung damage and the possible benefits of targeting the inflammatory pathways involved. The positive response to steroid therapy and hydroxychloroquine opens avenues for further research into these treatment modalities.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
The requirement of ethical approval was waived by Comitato Etico per la Sperimentazione Clinica della Provincia di Padova for the studies involving humans because Ethical committee approval not required for case reports. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants' legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
VG: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Writing – original draft. SC: Conceptualization, Formal analysis, Investigation, Writing – review & editing. TZ: Formal analysis, Investigation, Writing – review & editing. CC: Investigation, Writing – review & editing. DG: Investigation, Writing – review & editing. AB: Project administration, Resources, Supervision, Validation, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. The authors declare that this study received funding from the University of Padova. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article, or the decision to submit it for publication.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2025.1604433/full#supplementary-material
References
1. Beutler E, Grabowsky GA. Gaucher disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Basis of Inherited Diseases. New York, NY: McGraw-Hill (2001). p. 3635–68.
2. Grabowski GA. Gaucher disease and other storage disorders. Hematology. (2012) 2012(1):13–8. doi: 10.1182/asheducation.V2012.1.13.3797921
3. Cox TM, Cachón-González MB. The cellular pathology of lysosomal diseases. J Pathol. (2012) 226(2):241–54. doi: 10.1002/path.3021
4. Kaplan P, Andersson HC, Kacena KA, Yee JD. The clinical and demographic characteristics of nonneuronopathic gaucher disease in 887 children at diagnosis. Arch Pediatr Adolesc Med. (2006) 160(6):603. doi: 10.1001/archpedi.160.6.603
5. Grabowski GA. Phenotype, diagnosis, and treatment of Gaucher’s disease. Lancet. (2008) 372(9645):1263–71. doi: 10.1016/S0140-6736(08)61522-6
6. Cox T. Gaucher disease: clinical profile and therapeutic developments. Biologics. (2010) 4:299–313. doi: 10.2147/btt.s7582
7. Zimran A, Elstein D. Management of Gaucher disease: enzyme replacement therapy. Pediatr Endocrinol Rev. (2014) 12(1):82–7.25345089
8. Stirnemann J, Belmatoug N, Camou F, Serratrice C, Froissart R, Caillaud C, et al. A review of Gaucher disease pathophysiology, clinical presentation and treatments. Int J Mol Sci. (2017) 18(2):441. doi: 10.3390/ijms18020441
9. El-Beshlawy A, Tantawy AAG, Shawky RM, Elsayed SM, Marzouk IM, Elgawharyet S, et al. Diagnosis and management of patients with Gaucher disease: an Egyptian expert opinion. Egypt J Med Hum Genet. (2024) 25:88. doi: 10.1186/s43042-024-00552-z
10. Martins AM, Valadares ER, Porta G, Coelho J, Semionato Filho J, Pianovski MA, et al. Recommendations on diagnosis, treatment, and monitoring for Gaucher disease. J Pediatr. (2009) 155(4):S10–8. doi: 10.1016/j.jpeds.2009.07.004
11. de Farias L, Padilha I, dos Santos C, Maranhão C, de Miranda C. Pulmonary involvement in Gaucher disease. Radiologia Bras. (2017) 50(6):408–9. doi: 10.1590/0100-3984.2016.0081
12. Montanari C, Tagi VM, D’Auria E, Guaia V, Di Gallo A, Ghezzi M, et al. Lung diseases and rare disorders: is it a lysosomal storage disease? Differential diagnosis, pathogenetic mechanisms and management. Children. (2024) 11(6):668. doi: 10.3390/children11060668
13. Burlina AB, Polo G, Salviati L, Duro G, Zizzo C, Dardis A, et al. Newborn screening for lysosomal storage disorders by tandem mass spectrometry in North East Italy. J Inher Metab Disea. (2018) 41(2):209–19. doi: 10.1007/s10545-017-0098-3
14. Gragnaniello V, Cazzorla C, Gueraldi D, Puma A, Loro C, Porcù E, et al. Light and shadows in newborn screening for lysosomal storage disorders: eight years of experience in northeast Italy. Int J Neonatal Screen. (2023) 10(1):3. doi: 10.3390/ijns10010003
15. Santamaria R, Michelakakis H, Moraitou M, Dimitriou E, Dominissini S, Grossi S, et al. Haplotype analysis suggests a single Balkan origin for the Gaucher disease [D409H;H255Q] double mutant allele. Hum Mutat. (2008) 29(6):E58–67. doi: 10.1002/humu.20776
16. Schwartz IVD, Göker-Alpan Ö, Kishnani PS, Zimran A, Renault L, Panahloo Z, et al. Characteristics of 26 patients with type 3 Gaucher disease: a descriptive analysis from the Gaucher outcome survey. Mol Genet Metab Rep. (2018) 14:73–9. doi: 10.1016/j.ymgmr.2017.10.011
17. Ramadža DP, Zekušić M, Žigman T, Škaričić A, Bogdanić A, Mustać G, et al. Early initiation of ambroxol treatment diminishes neurological manifestations of type 3 Gaucher disease: a long-term outcome of two siblings. Eur J Paediatr Neurol. (2021) 32:66–72. doi: 10.1016/j.ejpn.2021.03.013
18. Arai N, Uematsu M, Abe Y, Fukuyo N, Wakusawa K, Kikuchi A, et al. High dose of enzyme replacement therapy was successful for the pulmonary involvement in a case of type 2 Gaucher disease. No To Hattatsu. (2010) 42(1):45–9.23858612
19. Bush A, Cunningham S, De Blic J, Barbato A, Clement A, Epaud R, et al. European protocols for the diagnosis and initial treatment of interstitial lung disease in children. Thorax. (2015) 70(11):1078–84. doi: 10.1136/thoraxjnl-2015-207349
20. Gragnaniello V, Gueraldi D, Saracini A, Velasquez Rivas D, Cazzorla C, Salviati L, et al. Natural history of inflammation and impaired autophagy in children with Gaucher disease identified by newborn screening. Mol Genet Metab Rep. (2025) 42:101187. doi: 10.1016/j.ymgmr.2025.101187
21. Gragnaniello V, Cazzorla C, Gueraldi D, Loro C, Massa P, Puma A, et al. Long-term follow-up of a patient with neonatal form of Gaucher disease. Am J Med Genet A. (2023) 191(7):1917–22. doi: 10.1002/ajmg.a.63196
22. Gawad Tantawy AA, Moneam Adly AA, Madkour SS, Salah El-Din NY. Pulmonary manifestations in young Gaucher disease patients: phenotype-genotype correlation and radiological findings. Pediatr Pulmonol. (2020) 55(2):441–8. doi: 10.1002/ppul.24544
23. Poletti V, Costabel U, Casoni GL, Bigliazzi C, Drent M, Olivieri D. Rare infiltrative lung diseases: a challenge for clinicians. Respiration. (2004) 71(5):431–43. doi: 10.1159/000080625
24. Santamaria F, Parenti G, Guidi G, Filocamo M, Strisciuglio P, Grillo G, et al. Pulmonary manifestations of Gaucher disease. Am J Respir Crit Care Med. (1998) 157(3):985–9. doi: 10.1164/ajrccm.157.3.9706057
25. Goitein O. Lung involvement and enzyme replacement therapy in Gaucher’s disease. QJM. (2001) 94(8):407–15. doi: 10.1093/qjmed/94.8.407
26. Rao AR, Parakininkas D, Hintermeyer M, Segura AD, Rice TB. Bilateral lung transplant in Gauchers type-1 disease. Pediatr Transplant. (2005) 9(2):239–43. doi: 10.1111/j.1399-3046.2005.00251.x
27. Tezuka Y, Fukuda M, Watanabe S, Nakano T, Okamoto K, Kuzume K, et al. Histological characterisation of visceral changes in a patient with type 2 Gaucher disease treated with enzyme replacement therapy. Blood Cells Mol Dis. (2018) 68:194–9. doi: 10.1016/j.bcmd.2016.11.006
28. Burrow TA, Sun Y, Prada CE, Bailey L, Zhang W, Brewer A, et al. CNS, lung, and lymph node involvement in Gaucher disease type 3 after 11years of therapy: clinical, histopathologic, and biochemical findings. Mol Genet Metab. (2015) 114(2):233–41. doi: 10.1016/j.ymgme.2014.08.011
29. Boot RG, Verhoek M, De Fost M, Hollak CEM, Maas M, Bleijlevens B, et al. Marked elevation of the chemokine CCL18/PARC in Gaucher disease: a novel surrogate marker for assessing therapeutic intervention. Blood. (2004) 103(1):33–9. doi: 10.1182/blood-2003-05-1612
30. Deegan PB, Moran MT, McFarlane I, Schofield JP, Boot RG, Aerts JMFG, et al. Clinical evaluation of chemokine and enzymatic biomarkers of Gaucher disease. Blood Cells Mol Dis. (2005) 35(2):259–67. doi: 10.1016/j.bcmd.2005.05.005
31. Michelakakis H, Spanou C, Kondyli A, Dimitriou E, Van Weely S, Hollak CEM, et al. Plasma tumor necrosis factor-a (TNF-a) levels in Gaucher disease. Biochim Biophys Acta. (1996) 1317(3):219–22. doi: 10.1016/S0925-4439(96)00056-7
32. Serfecz JC, Saadin A, Santiago CP, Zhang Y, Bentzen SM, Vogel SN, et al. C5a activates a pro-inflammatory gene expression profile in human Gaucher iPSC-derived macrophages. Int J Mol Sci. (2021) 22(18):9912. doi: 10.3390/ijms22189912
33. Kitatani K, Wada M, Perry D, Usui T, Sun Y, Obeid LM, et al. Activation of p38 mitogen-activated protein kinase in Gaucher’s disease. PLoS One. (2015) 10(8):e0136633. doi: 10.1371/journal.pone.0136633
34. Dean JLE, Sully G, Clark AR, Saklatvala J. The involvement of AU-rich element-binding proteins in p38 mitogen-activated protein kinase pathway-mediated mRNA stabilisation. Cell Signal. (2004) 16(10):1113–21. doi: 10.1016/j.cellsig.2004.04.006
35. Smoak K, Cidlowski JA. Glucocorticoids regulate tristetraprolin synthesis and posttranscriptionally regulate tumor necrosis factor alpha inflammatory signaling. Mol Cell Biol. (2006) 26(23):9126–35. doi: 10.1128/MCB.00679-06
36. King EM, Kaur M, Gong W, Rider CF, Holden NS, Newton R. Regulation of tristetraprolin expression by interleukin-1 beta and dexamethasone in human pulmonary epithelial cells: roles for nuclear factor-κ B and p38 mitogen-activated protein kinase. J Pharmacol Exp Ther. (2009) 330:575–85. doi: 10.1124/jpet.109.151423
37. Prabhala P, Bunge K, Ge Q, Ammit AJ. Corticosteroid-induced MKP-1 represses pro-inflammatory cytokine secretion by enhancing activity of tristetraprolin (TTP) in ASM cells. J Cell Physiol. (2016) 231(10):2153–8. doi: 10.1002/jcp.25327
38. Hassan AMIA, Zhao Y, Chen X, He C. Blockage of autophagy for cancer therapy: a comprehensive review. Int J Mol Sci. (2024) 25(13):7459. doi: 10.3390/ijms25137459
Keywords: Gaucher disease, glucocerebrosidase, newborn screening, interstitial lung disease, corticosteroid therapy, hydroxychloroquine, inflammation, enzyme replacement therapy
Citation: Gragnaniello V, Carraro S, Zangardi T, Cazzorla C, Gueraldi D and Burlina AB (2025) Case Report: Novel treatment approach for severe interstitial lung disease in type 3 Gaucher disease. Front. Pediatr. 13:1604433. doi: 10.3389/fped.2025.1604433
Received: 1 April 2025; Accepted: 26 May 2025;
Published: 11 June 2025.
Edited by:
Simona Sestito, University "Magna Graecia", ItalyReviewed by:
Julian Ramesh Vyas, Starship Children's Health, New ZealandMargarita Ivanova, Lysosomal and Rare Disorders Research and Treatment Center, United States
Copyright: © 2025 Gragnaniello, Carraro, Zangardi, Cazzorla, Gueraldi and Burlina. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alberto B. Burlina, YWxiZXJ0by5idXJsaW5hQHVuaXBkLml0