 Lijuan Zhang
Lijuan Zhang Fei Wang
Fei Wang Yanfang Zhu
Yanfang Zhu Hongxiao Zhang
Hongxiao Zhang Yahong Liu
Yahong Liu- Department of Pediatrics, The Second Hospital of Lanzhou University, Lanzhou, China
Treacher Collins syndrome (TCS) is a craniofacial malformation caused by the abnormal development of the first and second pharyngeal arches during embryogenesis. While pathogenic variants in POLR1B, POLR1C, and POLR1D are implicated, the TCOF1 gene represents the primary causative locus. This case report describes a novel heterozygous frameshift variant, TCOF1:NM_001135243.2:c.3559delG (p.Ala1187GlufsTer21), identified in a child with TCS. This truncation disrupts the C-terminal nuclear localization signal (NLS), specifically ablating a phosphorylation site at residues 1,199–1,200. Consequently, ribosome biosynthesis and craniofacial neural crest cell development are impaired, culminating in characteristic clinical manifestations, including downslanting palpebral fissures, depressed nasal bridge, marked malar hypoplasia, mandibular hypoplasia, and microtia. Although the mother carried the same variant, she exhibited no clinical symptoms, suggesting incomplete penetrance. This variant is the first internationally reported instance. Its identification reinforces the central pathogenic role of TCOF1 in TCS, underscores the functional significance of the treacle protein's NLS, and expands the variant database. Penetrance variability complicates genetic counseling, necessitating future research into its genetic characteristics to enhance prenatal diagnostic accuracy.
1 Introduction
Treacher Collins syndrome (TCS, OMIM 154500) is a rare developmental disorder with an incidence of 1:50,000 (1). It arises from the aberrant development of the first and second pharyngeal arches during the 5th to 8th gestational weeks. The major clinical characteristics encompass micrognathia, retrognathia, lower eyelid coloboma (with loss of medial eyelashes), microtia or anotia, external auditory canal atresia or stenosis, downslanting palpebral fissure secondary to lateral orbital hypoplasia, a large or protruding nose, and zygomatic bone hypoplasia. Minor features include a cleft lip with or without a concomitant cleft palate, preauricular hair displacement, airway obstruction, and conductive hearing loss due to middle and external ear malformations (2). TCS is typically diagnosed clinically at birth, supported by radiographic imaging and confirmed by molecular genetic testing. Management necessitates a staged, multidisciplinary approach across the patient’s lifespan.
TCS syndrome is genetically heterogeneous. The predominant gene involved is TCOF1, with additional contributions from POLR1B, POLR1C, and POLR1D. These genes exhibit distinct inheritance patterns: TCOF1 and POLR1B variants are autosomal dominant, POLR1C variants are autosomal recessive, and POLR1D variants can be either autosomal dominant (AD) or recessive (3).
As the primary pathogenic locus for TCS, TCOF1 is located at chromosome 5q32-q33.1. This gene comprises 28 exons and can encode the nucleolar phosphorylated Treacle protein (1,488 amino acids, 152 kDa) (4). Truncating variants in this gene lead to Treacle haploinsufficiency through three primary mechanisms: premature translational termination, impaired nucleolar localization, and compromised ribosomal RNA biogenesis. This functional impairment of Treacle disrupts neural crest cell (NCC) development, leading to craniofacial hypoplasia. Notably, genotype-phenotype associations remain contentious, and incomplete penetrance is mechanistically unexplained, implicating genetic modifiers or epigenetic regulators. Herein, we report a novel truncating TCOF1 variant, TCOF1:NM_001135243.2:c.3559delG (p.Ala1187GlufsTer21), in a classical TCS proband. The variant exhibits incomplete penetrance, as it was inherited from an unaffected mother. This case adheres to the CARE reporting checklist.
2 Case description
2.1 Clinical characteristics
The proband was vaginally delivered at term (40 weeks) to non-dysmorphic consanguineous Chinese parents (21 March 2023), with no history of perinatal asphyxia. He was their second child. The first pregnancy was complicated by sudden intrauterine fetal demise at 32 weeks of gestation. The gross morphological assessment identified ear malformations and mandibular hypoplasia (no ancillary testing or autopsy was conducted). The proband's birth weight was 3.5 kg (within ±1 SD) and length was 50 cm (within ±1 SD). He was admitted to the neonatology unit due to severe choking episodes during oral feeding.
A physical examination revealed normal skin and hair pigmentation, alongside the following characteristic craniofacial dysmorphisms: downslanting palpebral fissures, depressed nasal bridge, marked malar hypoplasia, mandibular hypoplasia, right microtia, and left external auditory canal atresia with preserved pinna architecture. Additional findings included a grade I cleft palate and a continuous murmur at the left second intercostal space. Limb and abdominal examinations were unremarkable. Laboratory tests (complete blood count, biochemistry) were normal. Echocardiography confirmed patent ductus arteriosus (PDA). The infant received symptomatic and supportive care and the parents received feeding guidance before discharge at parental request.
At the 1-month visit (28 April 2023), his weight was 4.2 kg (within ±1 SD) and length 54.5 cm (within ±1 SD). At the 6-month visit (7 October 2023), the patient’s Gesell developmental quotient (DQ) scores indicated developmental delay (defined as DQ < 75): gross motor was 41.5 (moderate deficiency), fine motor was 37.7 (severe deficiency), adaptive behavior was 56.6 (mild deficiency), language was 50.9 (moderate deficiency), and personal-social was 50.9 (moderate deficiency). Auditory brainstem response (ABR) thresholds [right ear: 65 dB normal hearing level (nHL); left ear: 55 dB nHL] suggested asymmetric hearing loss.
Given the proband's clinical features (craniofacial dysmorphism, palatal cleft, developmental delay, congenital heart malformations, and concomitant hearing abnormalities) and a family history (Figure 1) of auricular deformities and hearing impairment in the maternal grandmother, TCS was strongly suggested. Hearing aids were fitted after an otolaryngology consultation. Rehabilitation therapy was initiated. Genetic testing was recommended, but was deferred by the family due to the infant's age.
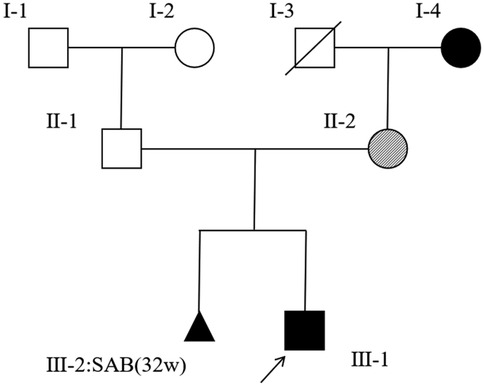
Figure 1. A pedigree of the family of the patient (Ⅲ-1). Squares indicate males, and circles indicate females. A black arrow represents affected subjects, clear symbols represent unaffected subjects, and crossed symbols represent deceased family members. Dashed circles indicate asymptomatic females carrying the causative variant. The aborted fetus is represented by a triangle.
At 1 year 8 months of age (24 December 2024), repeat echocardiography confirmed closure of the PDA (Figure 2). The patient’s Gesell DQ scores showed significant improvement with a gross motor score of 79.9 (borderline), fine motor score of 78.9 (borderline), adaptive behavior score of 77.7 (borderline), language score of 55.1 (persistent delay), and personal-social score of 80.5 (borderline). Following counseling regarding the infant's condition, the parents consented to genetic testing. The proband continues to receive rehabilitative therapy with regular auditory-verbal assessments. A staged surgical plan is proposed: palatoplasty beyond age 2, autologous microtia reconstruction in childhood, and orthognathic surgery in adolescence, supplemented by psychosocial support. The timeline is as shown in Figure 3.
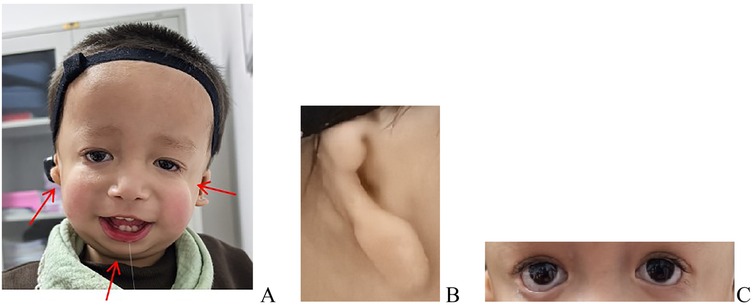
Figure 2. Dysmorphic features, including downslanting palpebral fissures, depressed nasal bridge, marked malar hypoplasia, mandibular hypoplasia, right microtia, and left external auditory canal atresia with preserved pinna architecture. (A) Overall appearance presentation. (B) Microtia. (C) Downslanting palpebral fissures and depressed nasal bridge.

Figure 3. The timeline of this case.
2.2 Variant sequencing
Genomic DNA was extracted from the proband's blood using the QIAamp DNA Blood Mini Kit (Qiagen) per the manufacturer's instructions. DNA quality was assessed using a Nanodrop 2000. After repairing the fragment ends and adding A-tails, DNA libraries were prepared by ligating adapters to the fragments. Whole-exome sequencing (WES) was performed on the proband's DNA, with exome enrichment conducted using the GenCap-WES Capture Kit. Labeled probes were hybridized in the liquid phase, followed by linear PCR amplification of the libraries. Library quality was assessed, and after passing QC, sequencing was carried out on the Illumina NextSeq500 platform (5, 6).
The WES reads were aligned to GRCh37.p10 using the Burrows–Wheeler Aligner (BWA). The BAM files underwent duplicate marking using GATK’s MarkDuplicates tool and base quality score recalibration using GATK’s Base Recalibrator tool. Finally, GATK’s HaplotypeCaller tool processed the Recal BAM to detect the single-nucleotide polymorphisms (SNPs) and deletions (indels).
A heterozygous frameshift variant in exon 22 of TCOF1 [chr5:g.149772312; TCOF1:NM_001135243.2:c.3559delG (p.Ala1187GlufsTer21)] was classified as “Likely Pathogenic” according to the American College of Medical Genetics and Genomics (ACMG) guidelines (PVS1 + PM2_Supporting). PVS1 indicates that this frameshift variant is predicted to cause loss of gene function, while PM2_Supporting indicates that it is absent from population databases. Searches of the Clinical Variant (ClinVar), Human Gene Mutation Database (HGMD), and Genome Aggregation Database (GnomAD) databases, combined with a comprehensive literature review, identified no prior reports of this variant, confirming it as a novel variant. The variant follows an AD inheritance pattern and was maternally inherited. Sanger sequencing confirmed the variant in the proband and his mother (Figure 4). Paternal and maternal grandmother samples were unavailable.

Figure 4. Heterozygous variant in exon 22 of the TCOF1 gene: TCOF1:NM_001135243.2:c.3559delG (p.Ala1187GlufsTer21). (A) The proband. (B) The proband's mother.
2.3 Sanger sequencing
Sanger sequencing was performed to validate the variants identified by whole-exome sequencing. The primers are as follows: F- 5′-GTGCAAGGAGTGTTGAAGCAG-3′, R- 5′-GAGGGATCGGGTAGACAGGAG-3′. The reference sequence NM_001135243.2 of TCOF1 was used.
3 Discussion
TCS is predominantly caused by TCOF1 variants (88.71% of cases) (7). Over 200 distinct pathogenic variants have been reported globally to date, distributed across exons 3, 5, 6, 10, 12, 13, 15, 16, 18, 20, 22, 23, and 24 (3, 8, 9). Exon 24 is a mutational hotspot region (17.75% frequency). A prototypical variant in this exon (c.4369_4373delAAGAA) induces a frameshift variant that disrupts the nuclear localization signal (NLS), compromising nucleolar localization of the Treacle protein, consequently leading to severe craniofacial malformations (reflected by higher Teber scores) (7, 10).
This study reports a pediatric TCS case harboring a novel TCOF1 variant, TCOF1:NM_001135243.2:c.3559delG (p.Ala1187GlufsTer21), located within exon 22. This deletion induces a frameshift at codon 1,187 (alanine), incorporating 21 aberrant amino acids and subsequent premature termination, resulting in a C-terminally truncated Treacle protein (Figure 5).
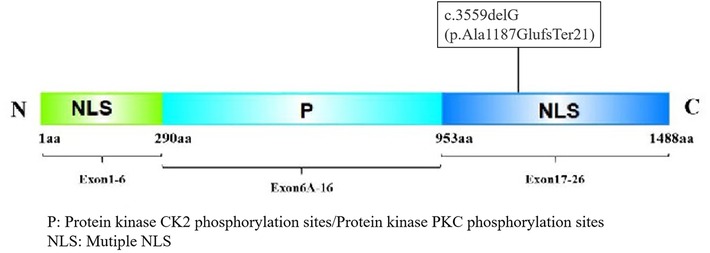
Figure 5. The Treacle protein consists of an N-terminal, C-terminal, and central repeat region. The N-terminal contains the LAQPVTLLDI sequence, which may serve as a nuclear export signal (NES) between 40 and 49 aa, and an NLS between 74 and 77 aa (11). The central structure contains many phosphorylation sites for casein kinase II (CK2) and protein kinase C (PKC). The C-terminus has an NLS, and the phosphorylation site of the Treacle protein by ATM/ATR kinase is located at the Serine-Glutamine/Threonine-Glutamine (SQ/TQ) site of the amino acid sequence (12). The C-terminus contains two SQ sites, at positions 1,199–1,200 and 1,216–1,217 of its amino acid sequence, of which positions 1,199–1,200 are necessary for nucleolar localization.
This variant exhibits a molecular mechanism analogous to the hotspot variant TCOF1:NM_001135243.2:c.4369_4373delAAGAA (p.Lys1457GlufsTer12). It disrupts the function of the critical C-terminal NLS within the Treacle protein. Specifically, the variant ablates the conserved serine residue at position 1199 (S1199), which serves as a phosphorylation target for ATM/ATR kinases (11, 12). Loss of S1199 phosphorylation prevents the treacle protein from localizing to nucleoli, impairing DNA damage response (DDR) activation and the repair of DNA double-strand breaks (DSBs) (13). These defects disrupt ribosome biogenesis and trigger apoptosis in cranial neural crest cells, manifesting as characteristic craniofacial dysmorphology, including malar hypoplasia, mandibular hypoplasia, cleft palate, and ossicular chain malformation (14).
The proband exhibited all typical TCS craniofacial features, plus cardiac malformation (11% incidence) and delayed motor development (1.7%–10% incidence) (3). This broader phenotypic spectrum suggests the following: (1) the premature truncation may impact multiple functional domains of the C-terminal within the Treacle protein; (2) the parents of this proband are consanguineous. Consanguinity elevates autozygosity, thereby increasing the probability that the affected child carries clinically undetected pathogenic variants in additional genes. These variants may engage in synergistic interactions with the TCOF1 mutation to enhance pathogenicity. This variant provides insight into the TCOF1 genotype–phenotype correlation and confirms the critical role of the C-terminal NLS domain in Treacle protein function.
Although individuals with TCS typically exhibit characteristic phenotypic features, incomplete penetrance is observed in carriers of pathogenic TCOF1 variants. The proband's mother, a heterozygous carrier of the c.3559delG variant, was asymptomatic, consistent with previous reports (15, 16). Variable expressivity arises from a multifactorial regulatory network involving the following: (1) protein functional compensation, where the residual N-terminal domain of the truncated Treacle protein may partially sustain ribosome biogenesis through interactions with ribosomal factors such as the DDX21 protein (17); (2) epigenetic modulation, such as DNA hypomethylation activating residual mutant allele transcription (18), while compensatory histone modifications (e.g., H3K27ac enrichment) may upregulate alternative gene expression pathways; (3) environmental stress interference, whereby embryonic oxidative stress may exacerbate phenotypic severity by inducing DNA damage in neural crest cells or degrading TCOF1-interacting regulators like CNBP (19). The dynamic interplay between genomic integrity, epigenetic landscapes, and environmental stressors poses significant challenges for genetic counseling and prenatal risk assessment.
TCS is a multisystem disorder that requires a multidisciplinary approach beyond surgery. We propose an integrated clinical protocol incorporating the following: (1) rigorous developmental surveillance incorporating serial anthropometry, Gesell Developmental Schedules, and age-stratified auditory brainstem response (ABR) testing to preempt auditory-speech delays; (2) staged craniofacial reconstruction with intervention timing determined by craniofacial 3D CT severity indices, feeding status, and neurodevelopmental milestones. Priority interventions include atresiaplasty for aural atresia, palatoplasty, and computer-aided design/computer-aided manufacturing (CAD/CAM)-guided zygomaticomaxillary complex (ZMC) reconstruction (20); (3) multimodal rehabilitation combining bone-anchored hearing aid (BAHA) devices with evidence-based auditory-verbal therapy (AVT) and orofacial myofunctional therapy (OMT); (4) targeted psychosocial support through structured body-image counseling and peer-mediated social-skills training. This framework simultaneously addresses craniofacial dysmorphology, functional deficits, and psychosocial vulnerability, thus facilitating adaptive self-identity development.
4 Conclusions
In conclusion, pathogenic TCOF1 gene variants are the primary genetic cause of TCS. These alter the encoded amino acid sequence of the Treacle protein, disrupting its phosphorylation and impairing critical functional domains. This dysfunction ultimately leads to aberrant craniofacial chondrogenesis, manifesting as the characteristic facial dysmorphology. The novel variant identified here affects a conserved phosphorylation site in Treacle's C-terminal domain, abolishing NLS function, providing in vivo evidence for TCOF1's role in TCS pathogenesis. Incomplete penetrance complicates genetic counseling. This variant expands the mutational spectrum of TCOF1, enriching databases critical for future diagnostic and population studies.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
This study involving humans was approved by Lanzhou University Second Hospital. This study was conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the legal guardian for the publication of this case report.
Author contributions
LZ: Data curation, Writing – review & editing, Writing – original draft, Conceptualization. FW: Writing – original draft, Data curation, Formal analysis. YZ: Methodology, Investigation, Writing – original draft. HZ: Investigation, Writing – original draft, Methodology. YL: Funding acquisition, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was funded by the Cuiying Scientific and Technological Innovation Program of Lanzhou University Second Hospital (grant number: CY2023-QN-A08).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Jiang Z, Mao K, Wang B, Zhu H, Liu J, Lang R, et al. Deciphering TCOF1 mutations in Chinese Treacher Collins syndrome patients: insights into pathogenesis and transcriptional disruption. Orphanet J Rare Dis. (2025) 20(1):57. doi: 10.1186/s13023-024-03508-z
2. Kadakia S, Samuel NH, Arvind KB, Masoud S, Yadranko D. Treacher Collins syndrome: the genetics of a craniofacial disease. Int J Pediatr Otorhinolaryngol. (2014) 78(6):893–8. doi: 10.1016/j.ijporl.2014.03.006
3. Marszałek-Kruk BA, Wójcicki P, Dowgierd K, Śmigiel R. Treacher Collins syndrome: genetics, clinical features and management. Genes (Basel). (2021) 12(9):1392. doi: 10.3390/genes12091392
4. Zhang X, Fan Y, Zhang Y, Xue H, Chen X. A novel mutation in the TCOF1 gene found in two Chinese cases of Treacher Collins syndrome. Int J Pediatr Otorhinolaryngol. (2013) 77(9):1410–5. doi: 10.1016/j.ijporl.2013.05.013
5. Feng L, Sun Y, Liu F, Wang C, Zhang C, Liu J, et al. Clinical features and functions of a novel LPL mutation C.986A>C (p.Y329S) in patient with hypertriglyceridemia. Curr Res Transl Med. (2022) 70(4):103337. doi: 10.1016/j.retram.2022.103337
6. Dai Y, Liang SR, Dong X, Zhao YH, Ren HT, Guan YZ, et al. Whole exome sequencing identified a novel DAG1 mutation in a patient with rare, mild and late age of onset muscular dystrophy-dystroglycanopathy. J Cell Mol Med. (2018) 23(2):811–8. doi: 10.1111/jcmm.13979
7. Ulhaq ZS, Nurputra DK, Soraya GV, Kurniawati S, Istifiani LA, Pamungkas SA, et al. A systematic review on Treacher Collins syndrome: correlation between molecular genetic findings and clinical severity. Clin Genet. (2022) 103(2):146–55. doi: 10.1111/cge.14243
8. Wang Y, Yin X, Han T, Peng W, Wu H, Liu X, et al. A novel silent deletion, an insertion mutation and a nonsense mutation in the TCOF1 gene found in two Chinese cases of Treacher Collins syndrome. Mol Genet Genomics. (2014) 289(6):1237–40. doi: 10.1007/s00438-014-0883-8
9. Conte C, D'apice MR, Rinaldi F, Gambardella S, Sangiuolo F, Novelli G. Novel mutations of TCOF1 gene in European patients with Treacher Collins syndrome. BMC Med Genet. (2011) 12:125. doi: 10.1186/1471-2350-12-125
10. Teber ÖA, Gillessen-kaesbach G, Fischer S, Böhringer S, Albrecht B, Albert A, et al. Genotyping in 46 patients with tentative diagnosis of Treacher Collins syndrome revealed unexpected phenotypic variation. Eur J Hum Genet. (2004) 12(11):879–90. doi: 10.1038/sj.ejhg.5201260
11. Grzanka M, Piekiełko-Witkowska A. The role of TCOF1 gene in health and disease: beyond Treacher Collins syndrome. Int J Mol Sci. (2021) 22:2482. doi: 10.3390/ijms22052482
12. Korsholm LM, Gál Z, Lin L, Quevedo O, Ahmad DA, Dulina E, et al. Double-strand breaks in ribosomal RNA genes activate a distinct signaling and chromatin response to facilitate nucleolar restructuring and repair. Nucleic Acids Res. (2019) 47(15):8019–35. doi: 10.1093/nar/gkz518
13. Ciccia A, Huang J, Izhar L, Sowa ME, Wade harper J, Elledge SJ. Treacher Collins syndrome TCOF1 protein cooperates with NBS1 in the DNA damage response. Proc Natl Acad Sci U S A. (2014) 111(52):18631–6. doi: 10.1073/pnas.1422488112
14. Kantaputra PN, Tripuwabhrut K, Intachai W, Carlson BM, Quarto N, Ngamphiw C, et al. Treacher Collins syndrome: a novel TCOF1 mutation and monopodial stapes. Clin Otolaryngol. (2020) 45(5):695–702. doi: 10.1111/coa.13560
15. Zsigmond A, Till Á, Pintér AL, Maász A, Szabó A, Hadzsiev K. Génvizsgálat Treacher Collins-Szindrómában. Orv Hetil. (2020) 161(52):2201–5. doi: 10.1556/650.2020.31945
16. Caluseriu O, Lowry BR, Mcleod R, Lamont R, Parboosingh JS, Bernier FP, et al. The Hutterite variant of Treacher Collins syndrome: a 28-year-old story solved. Am J Med Genet Part A. (2013) 161(11):2855–9. doi: 10.1002/ajmg.a.36172
17. Calo E, Gu B, Bowen ME, Aryan F, Zalc A, Liang J, et al. Tissue-selective effects of nucleolar stress and rDNA damage in developmental disorders. Nature. (2018) 554(7690):112–7. doi: 10.1038/nature25449
18. Kathryn HS, Shiang R. Regulation of the mouse Treacher Collins syndrome homolog (Tcof1) promoter through differential repression of constitutive expression. DNA Cell Biol. (2008) 27(11):589–600. doi: 10.1089/dna.2008.0766
19. Sakai D, Trainor PA. Face off against ROS: Tcof1/Treacle safeguards neuroepithelial cells and progenitor neural crest cells from oxidative stress during craniofacial development. Dev Growth Differ. (2016) 58(7):577–85. doi: 10.1111/dgd.12305
Keywords: Treacher Collins syndrome, TCOF1 gene, variant, treacle protein, case report
Citation: Zhang L, Wang F, Zhu Y, Zhang H and Liu Y (2025) Case Report: A novel heterozygous variant of the TCOF1 gene in Treacher Collins syndrome. Front. Pediatr. 13:1615309. doi: 10.3389/fped.2025.1615309
Received: 21 April 2025; Accepted: 28 August 2025;
Published: 16 September 2025.
Edited by:
Santasree Banerjee, Jilin University, ChinaReviewed by:
Asmaa Fawzy Abdel Aleem, National Research Centre, EgyptMatheus de Mello Copelli, University of Campinas (UNICAMP), Brazil
Copyright: © 2025 Zhang, Wang, Zhu, Zhang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yahong Liu, MjAwOGxpdXlhaG9uZ0AxNjMuY29t