 Ming-ming Ou1,2,†Chun-tao Sun3,4,†Li Zhang3,4Ling-li Kong3,4Lin-xin Zhang1,2Jun Zheng1,2Pei-ying Zhang3,4
Ming-ming Ou1,2,†Chun-tao Sun3,4,†Li Zhang3,4Ling-li Kong3,4Lin-xin Zhang1,2Jun Zheng1,2Pei-ying Zhang3,4 Yu-mei Wang1,2*Xiao-fei Lin5,6*Xin Yang1,2*
Yu-mei Wang1,2*Xiao-fei Lin5,6*Xin Yang1,2*
- 1Neonatal Disease Screening Center, Huai'an Maternal and Child Health Care Hospital Affiliated to Yangzhou University, Huai'an, China
- 2Neonatal Disease Screening Center, The Huai'an Maternity and Child Clinical College of Xuzhou Medical University, Huai'an, China
- 3Department of Child Health, Huai'an Maternal and Child Health Care Hospital Affiliated to Yangzhou University, Huai'an, China
- 4Department of Child Health, The Huai'an Maternity and Child Clinical College of Xuzhou Medical University, Huai'an, China
- 5Department of Pediatrics, Huai'an Maternal and Child Health Care Hospital Affiliated to Yangzhou University, Huai'an, China
- 6Department of Pediatrics, The Huai'an Maternity and Child Clinical College of Xuzhou Medical University, Huai'an, China
Objectives: This study aimed to determine the incidence and disease spectrum of inherited metabolic diseases (IMDs) among newborns in Huai'an City, China.
Methods: Expanded newborn screening for IMDs using tandem mass spectrometry (MS/MS) enables the simultaneous analysis of more than 40 metabolites and the identification of approximately 50 types of IMDs. Next-generation sequencing (NGS), targeting hundreds of IMD-associated genes, was subsequently performed for genetic analysis of patients identified through screening. Between June 2018 and December 2024, in total, 161,966 newborns in Huai'an were screened using MS/MS. Ultimately, 57 patients were diagnosed with IMDs based on plasma amino acid and acylcarnitine profiling, urinary organic acid analysis, and molecular genetic testing, performed via NGS. Data were analyzed using descriptive statistics.
Results: Fifty-seven cases of IMDs were diagnosed, corresponding to an overall incidence rate of 1 in 2,842. Among these, 28 cases involved amino acid metabolism disorders (1 in 5,785), 17 cases of organic acid metabolism disorders (1 in 9,527), and 12 cases of fatty acid oxidation disorders (1 in 13,497). The three most common IMDs were phenylalanine hydroxylase deficiency (1 in 8,098), primary carnitine deficiency (1 in 23,138), and methylmalonic acidemia (1 in 32,393). Genetic testing revealed variants in all 57 patients, with 75 variants identified across 17 IMD-associated genes. Recurrent variants were observed in five IMDs, including PAH gene variants c.728G>A, c.611A>G, and c.721C>T for phenylketonuria, PAH c.158G>A, c.721C>T, and c.728G>A for mild hyperphenylalaninemia, SLC22A5 c.1400C>G for primary carnitine deficiency, MMACHC c.609G>A, c.567dup and c.482G>A for methylmalonic acidemia, ACADS c.1055C>T, and c.1130C>T for short-chain acyl-CoA dehydrogenase deficiency, and ACADSB c.923G>A for 2-methylbutyrylglycinuria. All these recurrent variants were reported as pathogenic or likely pathogenic, except PAH c.158G>A, which was classified as a variant of uncertain significance.
Conclusion: The majority of IMD patients in Huai'an carried pathogenic or potentially pathogenic variants identified through expanded newborn screening. Although MS/MS newborn screening followed by NGS confirmation cannot prevent the occurrence of IMDs, timely diagnosis combined with appropriate treatment and management can effectively prevent morbidity, reduce mortality, and support long-term symptom control. Overall, this study demonstrates that MS/MS-based newborn screening combined with molecular diagnosis was highly effective for the study early detection and management of IMDs in the Huai'an population.
1 Introduction
Inherited metabolic diseases (IMDs) constitute a large group of disorders in which a single gene defect results in a metabolic block, leading to biochemical abnormalities that may manifest at birth and/or later in life. IMDs are classified as single-gene genetic diseases, most of which follow an autosomal recessive pattern of inheritance. The majority of children with IMDs present with non-specific clinical manifestations at birth and may remain undiagnosed unless neonatal screening is conducted. Without prompt diagnosis and treatment, IMDs can impair the function of multiple organs and systems, cause progressive and irreversible damage to the nervous system, and lead to developmental delay or even death. These outcomes impose a considerable burden on affected families and society. Early diagnosis and timely intervention for neonates identified with IMDs through screening can prevent severe clinical consequences, such as mild to severe irreversible intellectual disability, lifelong disability, physical handicaps, coma, and early death (1).
Newborn screening (NBS) for IMDs represents a highly successful public-health initiative aimed at detecting life-threatening or long-term health conditions to reduce morbidity and mortality (2). Tandem mass spectrometry (MS/MS) serves as a cornerstone technology in NBS programs, enabling the rapid detection of numerous metabolites in dried blood spots (DBSs). The simultaneous quantification of amino acids and acylcarnitines allows identification of approximately 40–50 IMDs within a few days after birth (3). Expanded NBS using MS/MS has been widely adopted worldwide due to its advantages, including rapid and convenient analysis, significantly increased detection rates of IMDs (4), early diagnosis, prevention of premature death, and cost-effectiveness (5, 6).
With the advent of target capture and next-generation sequencing (NGS), it has become possible to sequence large panels of disease-related genes simultaneously, making NGS the preferred approach for identifying the genetic etiology in newborns with abnormal MS/MS screening results for IMDs. Since 2018, Huai'an City in China has implemented newborn screening for IMDs using tandem mass spectrometry, achieving a screening rate exceeding 98%. This study aimed to determine the incidence, disease spectrum, and gene spectrum of IMDs among newborns in Huai'an City, China.
2 Materials and methods
2.1 Subjects
In total, 210,395 newborns were born in Huai'an between June 2018 and December 2024. Informed and written consent was obtained from the parents of 161,966 newborns, who were subsequently enrolled in the MS/MS-based NBS program. In total, 48,429 parents declined participation in the MS/MS NBS. Ultimately, 57 patients were diagnosed with IMDs through NGS. The screening protocol used in this study was consistent with that used by other newborn screening centers in China (Figure 1). The protocol was reviewed and approved by the Ethics Committee of Huai'an Maternal and Child Health Care Hospital, affiliated with Yangzhou University (Review No. 2022077).
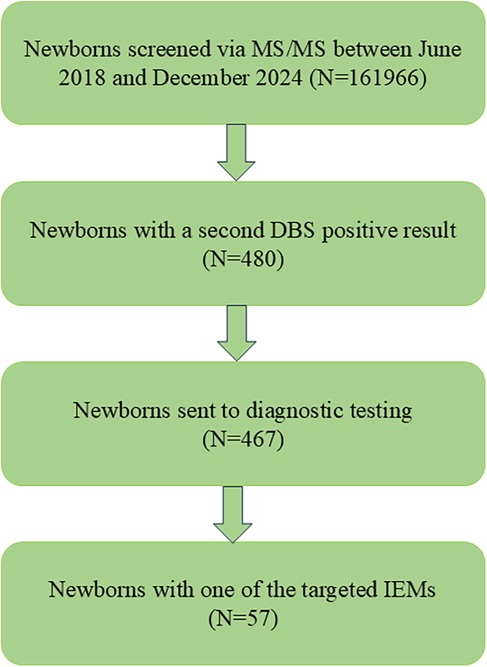
Figure 1. Flowchart of expanded newborn screening for IMDs and diagnosis of patients.
2.2 Expanded newborn screening
Heel-prick blood samples were collected from the newborns, and 11 amino acids, 30 acylcarnitines, free carnitine, and succinylacetone were analyzed as biomarkers for 27 IMDs using a non-derivatized MS/MS method at the Huai'an Newborn Disease Screening Center. Samples were collected at between 48 h and 20 days after birth (median: 4 days). Screening assays were performed using a commercial kit (PerkinElmer, USA) and a Waters HPLC-MS/MS system (TQD, Waters, USA). For each patient, a 3.2-mm DBS punch was taken using a Puncher 9 automatic drilling instrument (PerkinElmer, Finland) and placed in a 96-well U-bottom plate. Then, 100 µl of extraction solution containing internal standards was added to each well. After incubation at 45 °C for 45 min, 90 µl of the extract was transferred to a V-bottom plate and left at room temperature for 2 h. Subsequently, 25 µl of the extract was injected into the MS/MS instrument for metabolite analysis. Three levels of internal quality control—blank, low, and high were included in each batch to ensure analytical reliability.
2.3 Positive results for IMDs
The screening panel encompassed 27 types of IMDs. Each IMD was defined by two or more diagnostic indicators, including metabolites and metabolite ratios, with specific cut-off values. DBS results that met the positive criteria for IMDs were classified as suspected positive cases.
2.4 Genetic analysis
2.4.1 DNA extraction
In total, 5 ml of peripheral whole blood was collected from each subject in EDTA-containing anticoagulant tubes. Genomic DNA was extracted from 2 ml of peripheral blood using a Qiagen Blood DNA Mini Kit (Qiagen®, Hilden, Germany). DNA concentration was measured, and the extracted samples were stored at −20 °C. The remaining whole blood samples were stored at −80 °C for future use.
2.4.2 High throughput sequencing
High-throughput sequencing was conducted for all patients clinically diagnosed with IMDs using an expanded IMD gene panel (Genuine Diagnostic, Hangzhou, China) that included 346 genes related to IMDs. Target regions were captured using multiple probe hybridization, and the resulting capture products were purified with Agencourt AMPure XP beads (Beckman Coulter). The purified DNA was processed using a TruePrep™ DNA Library Prep Kit V2 for Illumina (Vazyme) and indexed with a TruePrep™ Index Kit V2 for Illumina (Vazyme). Library quality was assessed using a Qubit fluorometer and an Agilent 2100 bioanalyzer (High Sensitivity DNA Kit, Agilent Technologies). Sequencing libraries were quantified using an Illumina DNA Standards and Prime Premix Kit (Kapa Biosystems) and then subjected to massively parallel sequencing on the DNBSEQ-T7 platform. Paired-end reads were quality-trimmed using the Trimmomaticprogram and aligned to the human reference genome (UCSC Genome). Single-nucleotide polymorphisms and insertions/deletions were identified using the SAMtoolsprogram.
2.4.3 Sanger sequencing
All variants identified through high-throughput sequencing were confirmed by Sanger sequencing using specific primers. Polymerase chain reaction (PCR) amplification was performed using aTaKaRa LA PCR™ Kit ver. 2.1 (TaKaRa). PCR products were purified from agarose gels using aNucleoSpin® Gel and PCR Clean-up Kit (MACHEREY-NAGEL). Purified PCR products were diluted to a final concentration of 10 ng/µl and subjected to sequencing PCR using aBigDye® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems). Each reaction well received 10 µl of Hi-Di™ Formamide (Applied Biosystems). The samples were denatured at 95 °C for 5 min, cooled, and transferred to 96-well plates. Sequencing was performed using an ABI 3500XL Genetic Analyzer (Applied Biosystems).
2.5 Statistical analysis
Statistical analyses were conducted using SPSS software version 26.0 (IBM Corp., Armonk, NY, USA). Descriptive statistics were applied for count data.
3 Results
3.1 Screening of IMDs with MS/MS
In total, 161,966 newborns were screened using the expanded NBS program in Huai'an (Figure 1). Following the initial screening and subsequent repeat testing, 480 newborns with a second positive result were classified as “suspected positive”. Of these, 467 underwent NGS diagnostic testing, while the remaining 13 did not proceed with NGS. The biochemical indicators in these 13 newborns were only mildly abnormal, and their parents declined further genetic testing. During follow-up, the previously abnormal biochemical indicators in these cases returned to normal. Ultimately, 57 patients were diagnosed with IMDs based on MS/MS testing, urinary organic acid analysis, and molecular genetic testing using NGS.
3.2 Distribution of disease spectrum in children with IMDs
In total, 57 cases of IMDs were confirmed, corresponding to an overall incidence rate of 1 in 2,842 live births. Among these, 28 cases were amino acid metabolism disorders (1 in 5,785), 17 were organic acid metabolism disorders (1 in 9,527), and 12 were fatty acid oxidation disorders (1 in 13,497). The three most common IMDs by incidence were phenylalanine hydroxylase deficiency (1 in 8,098), primary carnitine deficiency (PCD; 1 in 23,138), and methylmalonic acidemia (MMA; 1 in 32,393) (Table 1, Figures 2A–D).
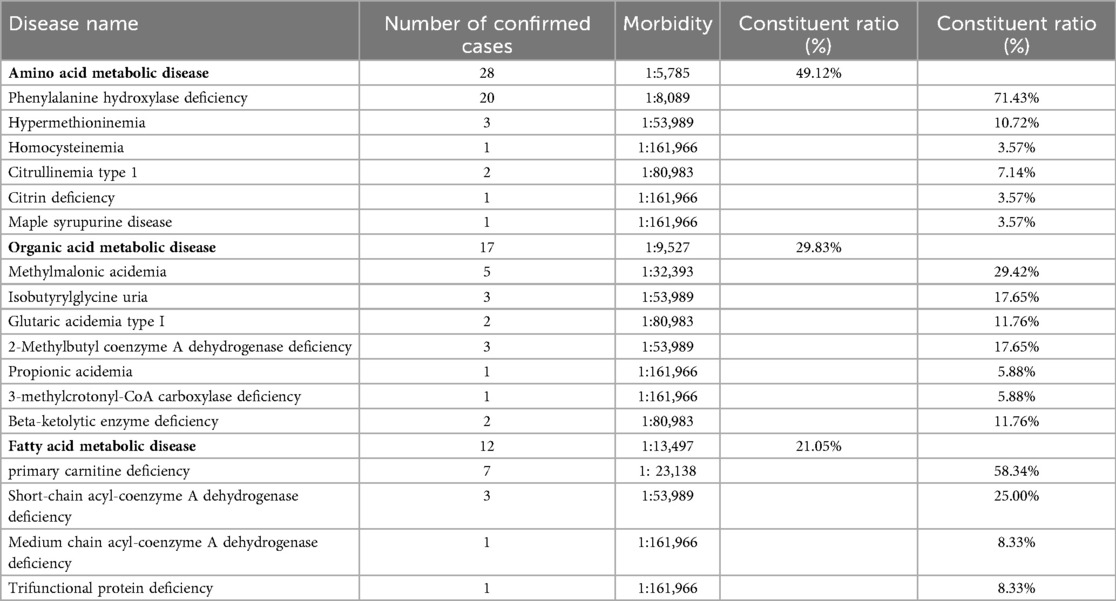
Table 1. Disease spectrum of 57 children with IMDs in 161,966 newborns.
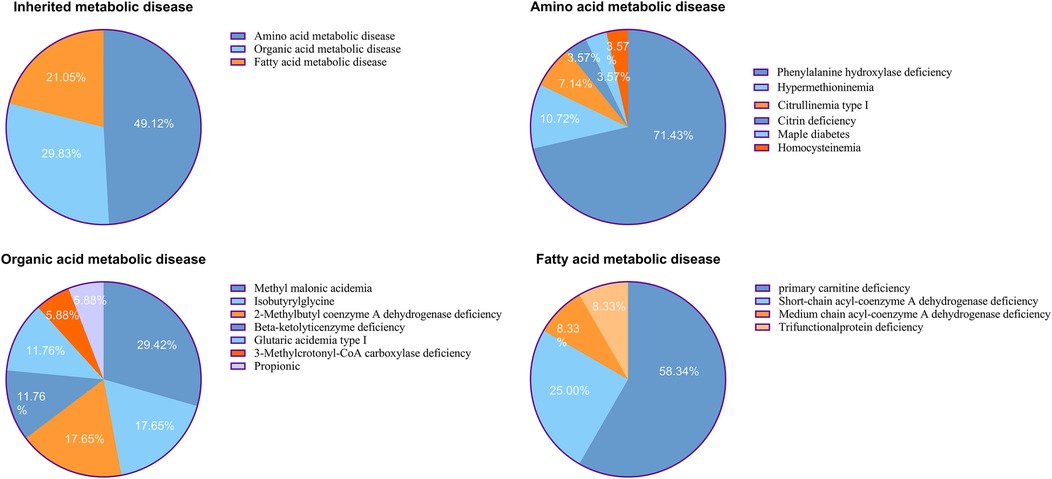
Figure 2. (A) Distributions of inherited metabolic disease. (B) Distributions of amino acid metabolic disease. (C) Distributions of organic acid metabolic disease. (D) Distributions of fatty acid metabolic disease.
3.3 Results of MS/MS in 57 children with IMDs
The results from the primary MS/MS screening and secondary recall testing in 57 children with IMDs revealed characteristic abnormalities in disease-related indices. The corresponding metabolite ratios and indicative values are presented in Table 2.
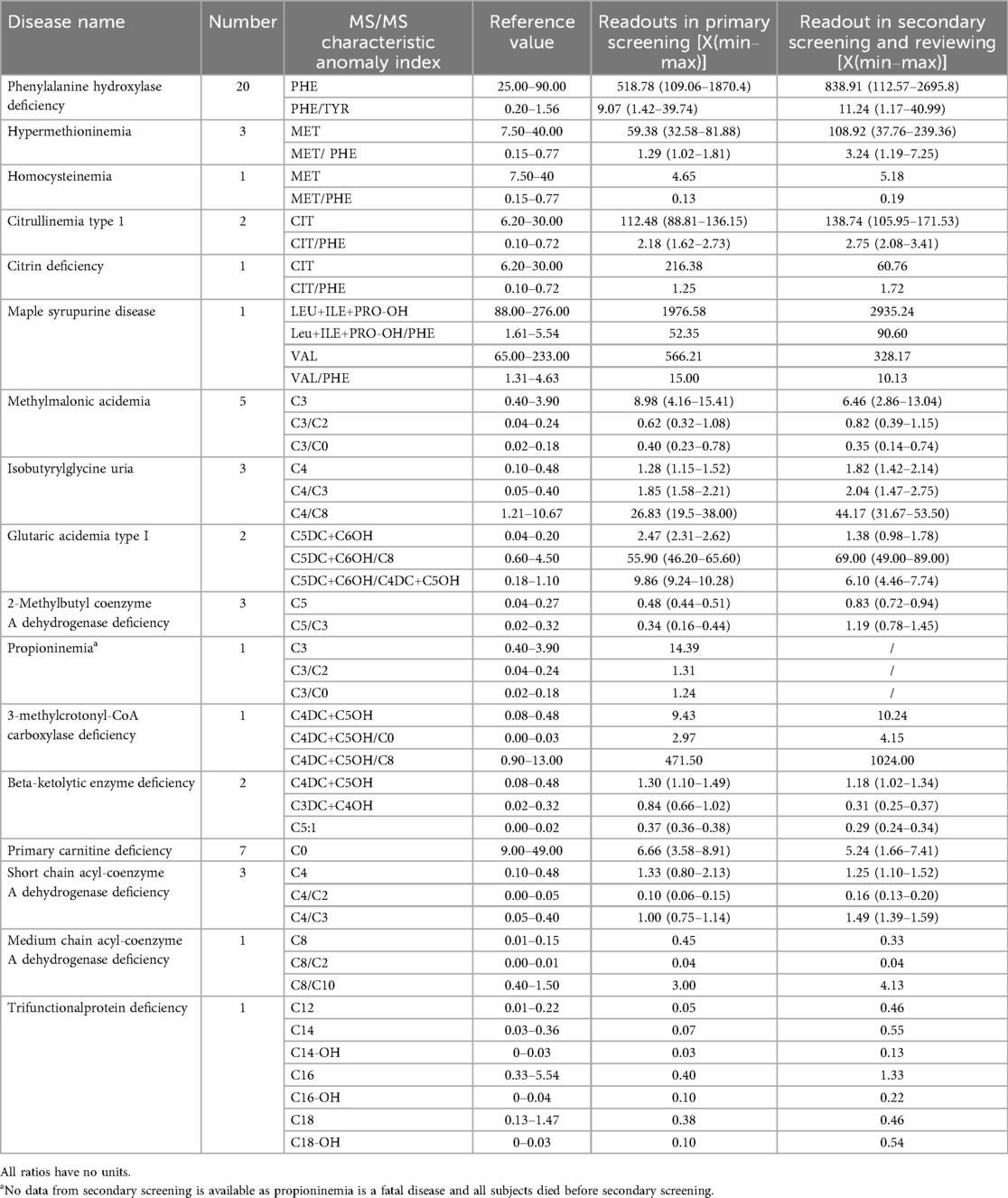
Table 2. Results of MS/MS in children with IMDs (μmol/L).
3.4 Variants in IMD patients identified by expanded newborn screening
Genetic analysis identified pathogenic or likely pathogenic variants in all 57 patients diagnosed with IMDs. In total, 75 variants were detected across 17 IMD-associated genes. Several recurrent variants were observed among 5 IMD types, including PAH gene variants c.728G>A, c.611A>G, and c.721C>T in phenylketonuria; PAH gene variants c.158G>A, c.721C>T, and c.728G>A in mild hyperphenylalaninemia; SLC22A5 gene variant c.1400C>Gin PCD; MMACHC gene variants c.609G>A, c.567dup, and c.482G>A in MMA; ACADS gene variants c.1055C>T and c.1130C>Tin short-chain acyl-CoA dehydrogenase deficiency (SCADD); and ACADSB gene variant c.923G>A in 2-methylbutyryl-CoA dehydrogenase deficiency (SBCADD). All of these recurrent variants have previously been reported as pathogenic or likely pathogenic, except for the PAH gene variant c.158G>A, which remains of uncertain significance (Table 3).
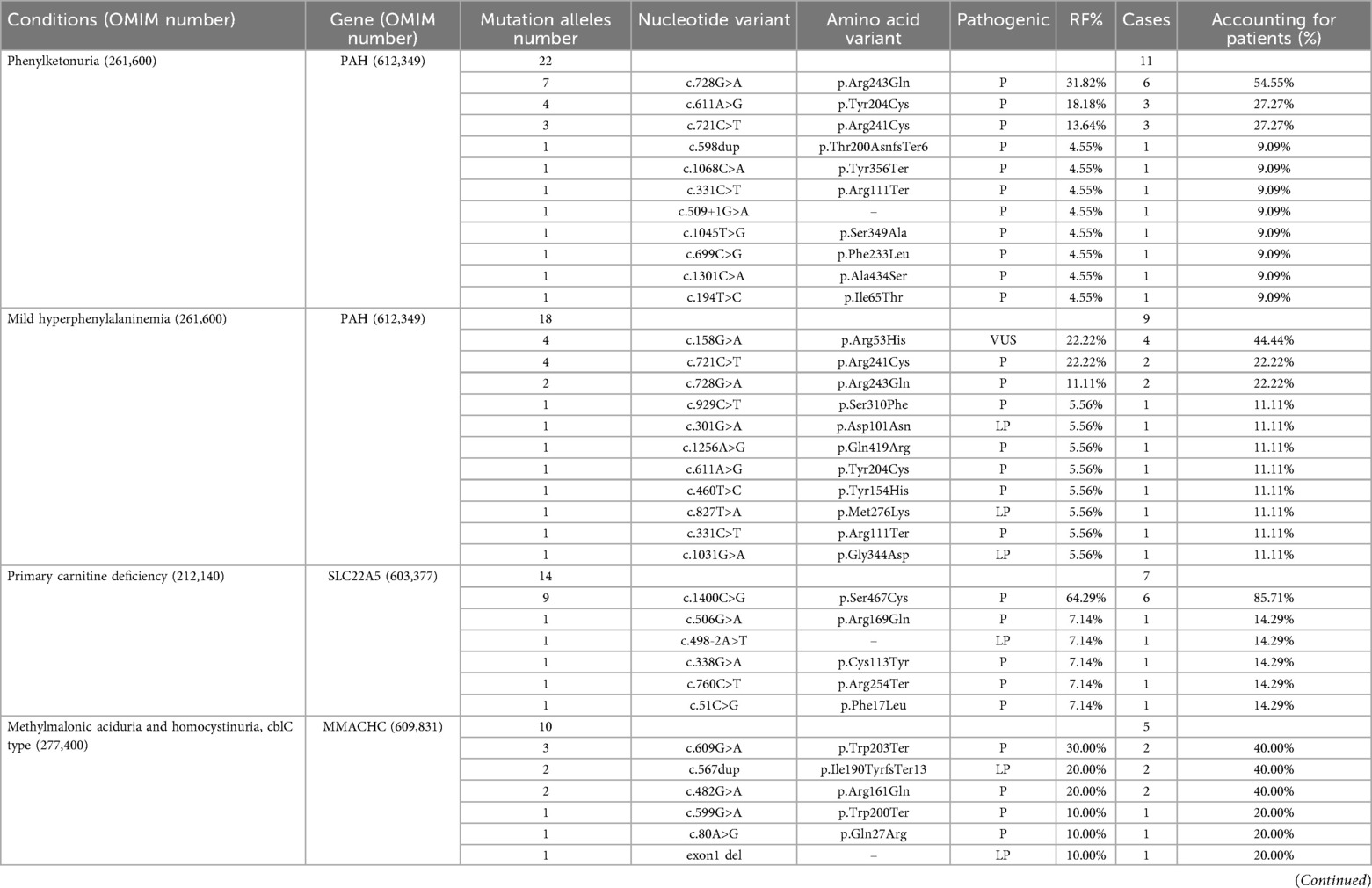
Table 3. Mutations in 57 patients with IEMs identified by expanded newborn screening.
4 Discussion
There are many types of IMDs, and the total number of identified disorders is estimated to exceed several thousand. Although the incidence of any single IMD is generally low, ranging from several per 10,000 to several per 100,000 live births, the cumulative incidence of all IMDs is considerably higher. Since the early 1990s, MS/MS has been widely implemented in neonatal screening programs for IMDs due to its relatively low cost, high sensitivity (99%), and high specificity (>99.8%) (7). The majority of IMDs detected through MS/MS are disorders of organic acid, amino acid, or fatty acid metabolism. Reported IMD types and their incidences vary substantially among countries and regions, reflecting differences in screening panels, population genetics, and healthcare systems.
As summarized in Table 4, expanded newborn screening programs worldwide demonstrate diverse detection rates. For example, approximately 40 types of IMDs are screened in Italy, with an incidence of about 1 in 6,041 (4). In Spain, 13 IMDs are screened with an incidence of approximately 1 in 2,577 (8). Germany, Japan, and Korea screen for 24, 18, and 24 IMDs, respectively, with corresponding incidences of 1/2,200, 1/8,557, and 1/13,205 (9). In China, 38 IMDs are screened by MS/MS across multiple neonatal screening centers, with an overall incidence of about 1 in 1,410 (10). In the present study, in total 161,966 newborns were screened in Huai'an, and 57 IMD cases were confirmed by NGS, corresponding to an incidence of 1/2,842. The three most common IMDs were phenylalanine hydroxylase deficiency (1/8,098), PCD (1/23,138), and MMA (1/32,393), consistent with previously reported data from other regions of China (10). All 57 affected children exhibited biochemical abnormalities of varying degrees that were successfully detected by MS/MS, confirming its reliability and critical role in early IMD screening and diagnosis.
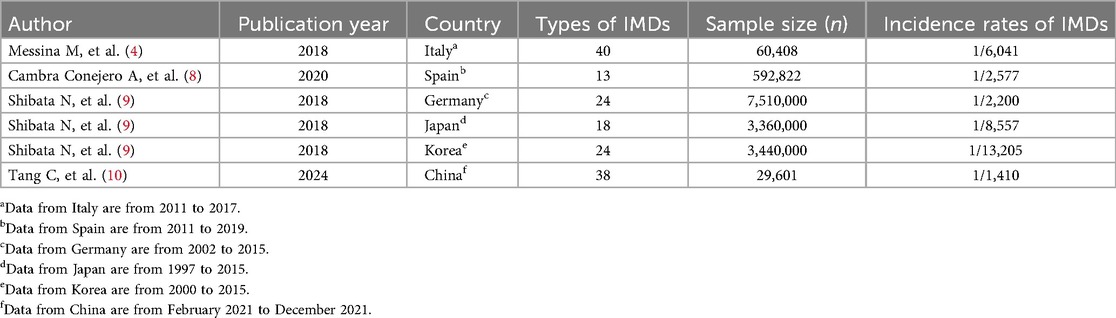
Table 4. Comparison of expanded newborn screening detection incidences of inherited metabolic diseases per country.
Hyperphenylalaninemia is the most prevalent amino acid metabolism disorder, resulting from a deficiency of either phenylalanine hydroxylase (PAH) or its cofactor, tetrahydrobiopterin (BH4). The deficiency leads to elevated phenylalanine concentrations in the blood, which can cause neurotoxicity and irreversible brain damage if left untreated. To date, more than 800 PAH gene variants have been identified in patients with PAH deficiency (11), with several recurrent variants reported in distinct populations. The most frequent variants include c.168+5G>C in Western Iranians (12), IVS10-11G>A in Iranians (13, 14) and Spaniards (15), c.1238G>C in Japanese (16), c.1162G>A in Brazilians (17), c.728G>A in Chinese (11, 18, 19), c.1068C>A and c.728G>A in South Koreans (20), c.1222C>T in Australians (21), and c.782G>A in Syrians (22).
In the Huai'an cohort, all 20 patients diagnosed with PAH deficiency harbored variants in the PAH gene. The detected variant included c.728G>A, c.611A>G, c.721C>T, c.598dup, c.1068C>A, c.331C>T, c.509+1G>A, c.1045T>G, c.699C>G, c.1301C>A, c.194T>C, c.158G>A, c.929C>T, c.301G>A, c.1256A>G, c.460T>C, c.827T>A, and c.1031G>A. Consistent with findings from other Chinese populations, the most frequent variant in this study was c.728G>A, accounting for 22.5% of alleles, followed by c.721C>T (17.5%) and c.611A>G (12.5%).
PCD was the second most common IMD detected in this study. PCD is caused by defects in the carnitine transporter protein encoded by the SLC22A5 gene, leading to impaired fatty acid β-oxidation within cells. The reported incidence of PCD in China varies regionally, ranging from 1/26,777 to 1/8,938 (18, 23, 24), while the incidence in Huai'an was 1/23,138. More than 110 SLC22A5variants have been identified to date, with distinct recurrent variants observed across different ethnic groups (25). For example, c.844T>C is common among Caucasian patients (26–28), while c.1400C>Gis the predominant hotspot variant in Southeast Asian populations (29–31). In Chinese populations, recurrent variants are relatively consistent across regions. The c.1400C>G variant is the most frequent, with a relative frequency of 50%, accounting for 80% of PCD cases in Suzhou (22). Chen et al. reported c.760C>T (32.9%) and c.1400C>G (21.1%) as the most common variants (32). Similarly, in the Huai'an cohort, c.1400C>G was the predominant SLC22A5 variant, with a relative frequency of 64.29%, aligning with findings from most other studies.
MMA was identified as the third most prevalent IMD and the most common organic acid metabolic disorder among the Huai'an population. MMA can be classified according to the specific defect into two categories: cobalamin (cbl) metabolic defects and methylmalonyl-CoA mutase deficiency (MUT type). It is a genetically and phenotypically heterogeneous disorder characterized by diverse clinical manifestations and significant morbidity. Currently, isolated MMA is attributed to variants in five genes—MMADHC, MUT, MCEE, MMAA, and MMAB. Among Chinese populations, MUT gene variants are the most frequently observed pathogenic type, with more than 400 related variant sites cataloged in ClinVar. The cblC type of MMA is the most common form associated with concurrent homocysteinemia. Reported incidences of MMA are approximately 1/50,000 in Japan (33) and 1/250,000 in Germany (34). However, in mainland China, the incidence ranges from 1/3,920 to 1/26,000 (18, 35). The incidence of MMA in Huai'an, approximately 1/32,393, is evidently higher than that in Japan and Germany but lower than in Shandong, Henan, Beijing, Shanghai, and Suzhou.
In this study, five patients were diagnosed with MMA, and all were classified as the cblC type. These patients carried six MMACHC gene variants: c.609G>A, c.567dup, c.482G>A, c.599G>A, c.80A>G, and an exon 1 deletion. These variants accounted for 28.6% of the total detected genetic sites, a distribution consistent with findings from most Chinese studies (36–38). All children diagnosed with MMA in the present cohort exclusively exhibited the combined type, and no variants in other related genes were identified. Given the limited sample size, the precision of variant frequency representation in the affected population may be constrained. Nevertheless, the identification of recurrent MMACHC gene variants—c.609G>A, c.567dup, and c.482G>A—highlights their importance as potential hotspots for subsequent MMA gene screening, prenatal diagnosis, and genetic counseling.
SCADD is an autosomal recessive disorder of fatty acid oxidation caused by defects in the ACADS gene, leading to the accumulation of butyrylcarnitine (C4) and ethylmalonic acid in urine. The ACADS gene is located on chromosome 12q24.31, spans approximately 13 kb, contains 10 exons, and encodes a 412-amino-acid enzyme. In this study, three cases of SCADD were identified with ACADS gene variants at four sites: c.1055C>T, c.881C>T, c.1130C>T, and c.164C>T. Among these, c.1055C>T and c.1130C>T exhibited the highest variant frequencies, each accounting for 33.33% of the detected sites. According to the American College of Medical Genetics and Genomics (ACMG) variant classification standards, c.881C>T (p.Ala294Val) was deemed clinically insignificant. Variants c.511C>T and c.625G>A are prevalent in Euramerican and Jewish populations (39), whereas c.1031A>G, c.164C>T, and c.323G>A have been reported in Japan (40). Long-term follow-up studies of SCADD cases detected through newborn screening have shown most patients remain asymptomatic, underlying a rationale that SCADD should not be routinely screened in newborns (41, 42). SCADD has been labeled a “biochemical phenotype” rather than a clinically significant monogenic disorder (43). This remains a controversial issue regarding the clinical significance of SCADD and whether it should be included in newborn screening panels.
Isobutyryl-CoA dehydrogenase deficiency (IBDD) is a rare autosomal recessive metabolic disorder that results from defects in the ACAD8gene, which encodes an enzyme responsible for the catabolism of the essential amino acid valine. Disruption of this metabolic pathway leads to the accumulation of isobutyrylglycine, which is excreted in urine and detectable through organic acid analysis. Accordingly, the condition is often referred to as isobutyrylglycinuria. The ACAD8gene is located on chromosome 11q25, comprises 11 exons, and encodes a member of the acyl-CoA dehydrogenase family. In this study, three cases of isobutyrylglycinuria were diagnosed, all associated with ACAD8 gene variants. Five variants— c.1000C>T, c.286G>A, c.526G>A, c.236G>A, and c.568-3C>G—as well as one structural variant (g.UNK1134123466 deletion) were identified. Based on ACMG classification criteria, c.526G>A, c.236G>A, and c.568-3C>G were categorized as variants of uncertain clinical significance. Reports from both Chinese and international studies imply that most IBDD patients are asymptomatic, making it difficult to establish clear genotype-phenotype correlations.
The etiology of hyperhomocysteinemia (HHCY) is multifactorial and can be categorized into congenital and acquired causes. Congenital factors primarily involve genetic variants in key enzymes such as thioether-β-synthase, methionine synthase reductase, methylenetetrahydrofolate reductase (MTHFR), and methionine synthase. These genetic defects lead to deficiencies in cofactors required for homocysteine (HCY) metabolism, resulting in HCY accumulation and subsequent development of HHCY. In the present study, one case of homocysteinemia was identified, in which a homozygous variant of the MTHFR gene (c.1316T>C) was detected. This case was initially identified through MS/MS screening, which revealed methionine levels below the diagnostic threshold, and was subsequently confirmed through molecular diagnosis using NGS. These findings indicate that HHCY can be detected by MS/MS-based newborn screening, acknowledging that screening for low methionine without screening for elevated HCY wouldn't effectively screen for all genetic etiologies of HHCY.
Citrullinemia type I (CTLN1), also referred to as argininosuccinate synthase (ASS) deficiency, is caused by pathogenic variants in the ASS1gene, a key enzyme in the urea cycle. The disorder is characterized by hyperammonemia and elevated citrulline levels and follows an autosomal recessive inheritance pattern (44). The ASS1 gene is located on chromosome 9q34.11, spans approximately 56 kb, comprises 16 exons, and encodes a 412-amino-acid protein. Reported common variants include c.1085G>T and c.970G>A in Turkish patients, and c.1088G>A in German patients (45, 46). In East Asian populations, c.421-2A>G is the most frequently observed variant, whereas c.970G>A predominates in Korean patients (47, 48). Additionally, c.794G>A is the most common variant reported among Japanese CTLN1 patients (49). In the present study, two cases of CTLN1 were diagnosed, and four heterozygous ASS1 gene variants were detected: c.349G>A, c.805G>A, c.919C>T, and c.1069C>G.
Maple syrup urine disease (MSUD) is an autosomal recessive disorder affecting the catabolism of branched-chain amino acids. The condition derives its name from the characteristic sweet, “maple syrup–like” odor of the urine. MSUD results from a functional defect in the branched-chain α-keto acid dehydrogenase complex. The primary pathogenic genes implicated in this disorder include BCKDHA (34%), BCKDHB (29%), DLD, and DBT. In this study, we identified an MSUD case in which two heterozygous variants of the BCKDHB gene were detected: c.93_103dup and c.673_675del. According to the ACMG variant classification guidelines, the c.673_675del (p.Leu226del) variant is classified as having uncertain clinical significance, warranting further functional validation. It shows how NGS as a standalone first tier screen would be insufficient as VUS clinical correlation is only feasible because of the MS/MS NBS result.
3-Methylcrotonyl-CoA carboxylase deficiency (3-MCCD) is an autosomal recessive inborn error of leucine metabolism that exhibits considerable phenotypic variability. Most affected individuals remain asymptomatic unless secondary systemic carnitine deficiency develops. However, severe cases may present with leukodystrophy, developmental delay, hypoglycemia, metabolic acidosis, failure to thrive, lactic acidosis, and hyperammonemia (50). Deficiency of 3-MCCC, caused by pathogenic variants in either MCCC1 or MCCC2, leads to elevated levels of 3-hydroxyisovalerylcarnitine in the blood. The enzyme comprises α and β subunits encoded by the MCCC1 and MCCC2 genes, respectively. Previous studies have shown that MCCC2variants are more frequently reported worldwide, whereas MCCC1variants are predominant in Chinese populations (51). In the present study, one patient with 3-MCCD was diagnosed, carrying a homozygous MCCC2 gene variant (c.728G>A).
5 Conclusions
This study reports, for the first time, the results of newborn screening for IMDs and their epidemiological characteristics in Huai'an over the past 7 years. Among 161,966 newborns screened using MS/MS, 57 children were diagnosed with 17 different types of IMDs, yielding an overall incidence rate of 1 in 2,842. The three most prevalent disorders were phenylalanine hydroxylase deficiency, PCD, and MMA, findings that were consistent with the reported incidence patterns of these diseases in other regions of China.
Of the total 77 genetic variants identified, 49.35% (38/77) were classified as pathogenic, 32.47% (25/77) as likely pathogenic, and 18.18% (14/77) as variants of uncertain clinical significance, the latter requiring further functional validation. Several recurrent variants were identified across 10 IMDs, including PAH gene variants (c.728G>A, c.611A>G, and c.721C>T) for phenylketonuria; PAH gene variants (c.158G>A, c.721C>T, and c.728G>A) for mild hyperphenylalaninemia; SLC22A5gene variant (c.1400C>G) for PCD; MMACHC gene variants (c.609G>A, c.567dup, and c.482G>A) for MMA; ACADS gene variants (c.1055C>T and c.1130C>T) for SCADD; and ACADSB gene variant (c.923G>A) for SBCADD. All these recurrent variants have been previously reported as pathogenic or likely pathogenic, except for the PAH gene variant c.158G>A.
In conclusion, the majority of the IMD patients identified through expanded newborn screening in Huai'an carried pathogenic or potentially pathogenic variants. These findings underscore the clinical value of combining MS/MS-based screening with NGS for the accurate diagnosis and genetic confirmation of IMDs in newborns.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving humans were approved by Ethics Committee of Huai'an Maternal and Child Health Hospital, Jiangsu Province, China. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
M-mO: Investigation, Data curation, Resources, Writing – review & editing. C-tS: Resources, Writing – original draft, Supervision, Investigation. LZ: Resources, Writing – review & editing. L-lK: Resources, Writing – original draft. L-xZ: Writing – review & editing, Data curation, Resources. JZ: Resources, Data curation, Writing – original draft. P-yZ: Software, Resources, Writing – original draft, Investigation. Y-mW: Methodology, Project administration, Writing – review & editing, Writing – original draft, Funding acquisition. X-fL: Software, Writing – review & editing, Project administration, Methodology, Formal analysis. XY: Software, Methodology, Writing – original draft, Data curation, Resources.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. Health Research Project of the Jiangsu Provincial Health Commission (F202220), the Natural Science Research Project of the Huai’an Municipal Science and Technology Bureau (HAB2024040), and the author's home institution. The aforementioned three funding sources provided all project research funding.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Therrell BL, Padilla CD, Loeber JG, Kneisser I, Saadallah A, Borrajo GJ, et al. Current status of newborn screening worldwide: 2015. Semin Perinatol. (2015) 39(3):171–87. doi: 10.1053/j.semperi.2015.03.002
2. Loeber JG, Platis D, Zetterstrom RH, Almashanu S, Boemer F, Bonham JR, et al. Neonatal screening in Europe revisited: an ISNS perspective on the current state and developments since 2010. Int J Neonatal Screen. (2021) 7(1):15. doi: 10.3390/ijns7010015
3. Watson MS, Mann MY, Lloyd-Puryear MA, Rinaldo P, Rodney Howell R, editors. Newborn screening: toward a uniform screening panel and system. Genet Med. (2006) 8 Suppl 1(Suppl 1):1S–252S. doi: 10.1097/01.gim.0000223891.82390.ad
4. Messina M, Meli C, Raudino F, Pittala A, Arena A, Barone R, et al. Expanded newborn screening using tandem mass spectrometry: seven years of experience in Eastern Sicily. Int J Neonatal Screen. (2018) 4(2):12. doi: 10.3390/ijns4020012
5. Tang C, Liu S, Wu M, Lin S, Lin Y, Su L, et al. Clinical and molecular characteristics of carnitine-acylcarnitine translocase deficiency: experience with six patients in Guangdong China. Clin Chim Acta. (2019) 495:476–80. doi: 10.1016/j.cca.2019.05.018
6. Zhao Z, Chen C, Sun X, Zhou D, Huang X, Dong H. Newborn screening for inherited metabolic diseases using tandem mass spectrometry in China: outcome and cost-utility analysis. J Med Screen. (2022) 29(1):12–20. doi: 10.1177/09691413211021621
7. Adhikari AN, Gallagher RC, Wang Y, Currier RJ, Amatuni G, Bassaganyas L, et al. The role of exome sequencing in newborn screening for inborn errors of metabolism. Nat Med. (2020) 26(9):1392–7. doi: 10.1038/s41591-020-0966-5
8. Cambra Conejero A, Martinez Figueras L, Ortiz Temprado A, Blanco Soto P, Martin Rivada A, Palomino Perez L, et al. Newborn screening program in the community of Madrid: evaluation of positive cases. Rev Esp Salud Publica. (2020) 94:e202012185.33372917
9. Shibata N, Hasegawa Y, Yamada K, Kobayashi H, Purevsuren J, Yang Y, et al. Diversity in the incidence and spectrum of organic acidemias, fatty acid oxidation disorders, and amino acid disorders in Asian countries: selective screening vs. expanded newborn screening. Mol Genet Metab Rep. (2018) 16:5–10. doi: 10.1016/j.ymgmr.2018.05.003
10. Tang C, Li L, Chen T, Li Y, Zhu B, Zhang Y, et al., Newborn screening for inborn errors of metabolism by next-generation sequencing combined with tandem mass spectrometry. Int J Neonatal Screen. (2024) 10(2):28. doi: 10.3390/ijns10020028
11. Zhang Z, Gao JJ, Feng Y, Zhu LL, Yan H, Shi XF, et al. Mutational spectrum of the phenylalanine hydroxylase gene in patients with phenylketonuria in the central region of China. Scand J Clin Lab Invest. (2018) 78(3):211–8. doi: 10.1080/00365513.2018.1434898
12. Alibakhshi R, Moradi K, Mohebbi Z, Ghadiri K. Mutation analysis of PAH gene in patients with PKU in western Iran and its association with polymorphisms: identification of four novel mutations. Metab Brain Dis. (2014) 29(1):131–8. doi: 10.1007/s11011-013-9432-0
13. Esfahani MS, Vallian S. A comprehensive study of phenylalanine hydroxylase gene mutations in the Iranian phenylketonuria patients. Eur J Med Genet. (2019) 62(9):103559. doi: 10.1016/j.ejmg.2018.10.011
14. Rastegar Moghadam M, Shojaei A, Babaei V, Rohani F, Ghazi F. Mutation analysis of phenylalanine hydroxylase gene in Iranian patients with phenylketonuria. Med J Islam Repub Iran. (2018) 32:21. doi: 10.14196/mjiri.32.21
15. Aldamiz-Echevarria L, Llarena M, Bueno MA, Dalmau J, Vitoria I, Fernandez-Marmiesse A, et al. Molecular epidemiology, genotype-phenotype correlation and Bh4 responsiveness in Spanish patients with phenylketonuria. J Hum Genet. (2016) 61(8):731–44. doi: 10.1038/jhg.2016.38
16. Dateki S, Watanabe S, Nakatomi A, Kinoshita E, Matsumoto T, Yoshiura K, et al. Genetic background of hyperphenylalaninemia in Nagasaki, Japan. Pediatr Int. (2016) 58(5):431–3. doi: 10.1111/ped.12924
17. Vieira Neto E, Laranjeira F, Quelhas D, Ribeiro I, Seabra A, Mineiro N, et al. Mutation analysis of the PAH gene in phenylketonuria patients from Rio De Janeiro, southeast Brazil. Mol Genet Genomic Med. (2018) 6(4):575–91. doi: 10.1002/mgg3.408
18. Wang T, Ma J, Zhang Q, Gao A, Wang Q, Li H, et al. Expanded newborn screening for inborn errors of metabolism by tandem mass spectrometry in Suzhou, China: disease spectrum, prevalence, genetic characteristics in a Chinese population. Front Genet. (2019) 10:1052. doi: 10.3389/fgene.2019.01052
19. Zhou YA, Ma YX, Zhang QB, Gao WH, Liu JP, Yang JP, et al. Mutations of the phenylalanine hydroxylase gene in patients with phenylketonuria in Shanxi, China. Genet Mol Biol. (2012) 35(4):709–13. doi: 10.1590/S1415-47572012005000069
20. Lee YW, Lee DH, Kim ND, Lee ST, Ahn JY, Choi TY, et al. Mutation analysis of PAH gene and characterization of a recurrent deletion mutation in Korean patients with phenylketonuria. Exp Mol Med. (2008) 40(5):533–40. doi: 10.3858/emm.2008.40.5.533
21. Ho G, Alexander I, Bhattacharya K, Dennison B, Ellaway C, Thompson S, et al. The molecular bases of phenylketonuria (PKU) in New South Wales, Australia: mutation profile and correlation with tetrahydrobiopterin (Bh4) responsiveness. JIMD Rep. (2014) 14:55–65. doi: 10.1007/8904_2013_284
22. Murad H, Dabboul A, Moassas F, Alasmar D, Al-Achkar W. Mutation spectrum of phenylketonuria in Syrian population: genotype-phenotype correlation. Gene. (2013) 528(2):241–7. doi: 10.1016/j.gene.2013.07.001
23. Guo K, Zhou X, Chen X, Wu Y, Liu C, Kong Q. Expanded newborn screening for inborn errors of metabolism and genetic characteristics in a Chinese population. Front Genet. (2018) 9:122. doi: 10.3389/fgene.2018.00122
24. Sun Y, Wang YY, Jiang T. Clinical features and genotyping of patients with primary carnitine deficiency identified by newborn screening. J Pediatr Endocrinol Metab. (2017) 30(8):879–83. doi: 10.1515/jpem-2017-0002
25. Han L, Wang F, Wang Y, Ye J, Qiu W, Zhang H, et al. Analysis of genetic mutations in Chinese patients with systemic primary carnitine deficiency. Eur J Med Genet. (2014) 57(10):571–5. doi: 10.1016/j.ejmg.2014.08.001
26. Burwinkel B, Kreuder J, Schweitzer S, Vorgerd M, Gempel K, Gerbitz KD, et al. Carnitine transporter Octn2 mutations in systemic primary carnitine deficiency: a novel Arg169gln mutation and a recurrent Arg282ter mutation associated with an unconventional splicing abnormality. Biochem Biophys Res Commun. (1999) 261(2):484–7. doi: 10.1006/bbrc.1999.1060
27. Vaz FM, Scholte HR, Ruiter J, Hussaarts-Odijk LM, Pereira RR, Schweitzer S, et al. Identification of two novel mutations in Octn2 of three patients with systemic carnitine deficiency. Hum Genet. (1999) 105(1-2):157–61. doi: 10.1007/s004399900105
28. Wang Y, Ye J, Ganapathy V, Longo N. Mutations in the organic cation/carnitine transporter Octn2 in primary carnitine deficiency. Proc Natl Acad Sci U S A. (1999) 96(5):2356–60. doi: 10.1073/pnas.96.5.2356
29. Koizumi A, Nozaki J, Ohura T, Kayo T, Wada Y, Nezu J, et al. Genetic epidemiology of the carnitine transporter Octn2 gene in a Japanese population and phenotypic characterization in Japanese pedigrees with primary systemic carnitine deficiency. Hum Mol Genet. (1999) 8(12):2247–54. doi: 10.1093/hmg/8.12.2247
30. Nezu J, Tamai I, Oku A, Ohashi R, Yabuuchi H, Hashimoto N, et al. Primary systemic carnitine deficiency is caused by mutations in a gene encoding sodium Ion-dependent carnitine transporter. Nat Genet. (1999) 21(1):91–4. doi: 10.1038/5030
31. Tang NL, Ganapathy V, Wu X, Hui J, Seth P, Yuen PM, et al. Mutations of Octn2, an organic cation/carnitine transporter, lead to deficient cellular carnitine uptake in primary carnitine deficiency. Hum Mol Genet. (1999) 8(4):655–60. doi: 10.1093/hmg/8.4.655
32. Chen YC, Chien YH, Chen PW, Tang L-S, Chiu N, Hwu PC, et al. Carnitine uptake defect (primary carnitine deficiency): risk in genotype-phenotype correlation. Hum Mutat. (2013) 34(4):655. doi: 10.1002/humu.22286
33. Shigematsu Y, Hirano S, Hata I, Tanaka Y, Sudo M, Sakura N, et al. Newborn mass screening and selective screening using electrospray tandem mass spectrometry in Japan. J Chromatogr B Analyt Technol Biomed Life Sci. (2002) 776(1):39–48. doi: 10.1016/s1570-0232(02)00077-6
34. Schulze A, Lindner M, Kohlmuller D, Olgemoller K, Mayatepek E, Hoffmann GF. Expanded newborn screening for inborn errors of metabolism by electrospray ionization-tandem mass spectrometry: results, outcome, and implications. Pediatrics. (2003) 111(6 Pt 1):1399–406. doi: 10.1542/peds.111.6.1399
35. Han B, Cao Z, Tian L, Zou H, Yang L, Zhu W, et al. Clinical presentation, gene analysis and outcomes in young patients with early-treated combined methylmalonic acidemia and homocysteinemia (cblc type) in Shandong province, China. Brain Dev. (2016) 38(5):491–7. doi: 10.1016/j.braindev.2015.10.016
36. Hu S, Mei S, Liu N, Kong X. Molecular genetic characterization of cblc defects in 126 pedigrees and prenatal genetic diagnosis of pedigrees with combined methylmalonic aciduria and homocystinuria. BMC Med Genet. (2018) 19(1):154. doi: 10.1186/s12881-018-0666-x
37. Wang C, Li D, Cai F, Zhang X, Xu X, Liu X, et al. Mutation spectrum of mmachc in Chinese pediatric patients with cobalamin C disease: a case series and literature review. Eur J Med Genet. (2019) 62(10):103713. doi: 10.1016/j.ejmg.2019.103713
38. Zhou W, Li H, Wang C, Wang X, Gu M. Newborn screening for methylmalonic acidemia in a Chinese population: molecular genetic confirmation and genotype phenotype correlations. Front Genet. (2018) 9:726. doi: 10.3389/fgene.2018.00726
39. Jethva R, Bennett MJ, Vockley J. Short-chain acyl-coenzyme a dehydrogenase deficiency. Mol Genet Metab. (2008) 95(4):195–200. doi: 10.1016/j.ymgme.2008.09.007
40. Shirao K, Okada S, Tajima G, Tsumura M, Hara K, Yasunaga S, et al. Molecular pathogenesis of a novel mutation, G108d, in short-chain acyl-Coa dehydrogenase identified in subjects with short-chain acyl-Coa dehydrogenase deficiency. Hum Genet. (2010) 127(6):619–28. doi: 10.1007/s00439-010-0822-7
41. Kelly CL, Rhead WJ, Kutschke WK, Brix AE, Hamm DA, Pinkert CA, et al. Functional correction of short-chain acyl-Coa dehydrogenase deficiency in transgenic mice: implications for gene therapy of human mitochondrial enzyme deficiencies. Hum Mol Genet. (1997) 6(9):1451–5. doi: 10.1093/hmg/6.9.1451
42. Maguolo A, Rodella G, Dianin A, Nurti R, Monge I, Rigotti E, et al. Diagnosis, genetic characterization and clinical follow up of mitochondrial fatty acid oxidation disorders in the new era of expanded newborn screening: a single centre experience. Mol Genet Metab Rep. (2020) 24:100632. doi: 10.1016/j.ymgmr.2020.100632
43. Wolfe L, Jethva R, Oglesbee D, Vockley J. Short-chain acyl-CoA dehydrogenase deficiency. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle (1993–2025).
44. Zielonka M, Kolker S, Gleich F, Stutzenberger N, Nagamani SCS, Gropman AL, et al. Early prediction of phenotypic severity in citrullinemia type 1. Ann Clin Transl Neurol. (2019) 6(9):1858–71. doi: 10.1002/acn3.50886
45. Kose E, Unal O, Bulbul S, Gunduz M, Haberle J, Arslan N. Identification of three novel mutations in fourteen patients with citrullinemia type 1. Clin Biochem. (2017) 50(12):686–9. doi: 10.1016/j.clinbiochem.2017.01.011
46. Woo HI, Park HD, Lee YW. Molecular genetics of citrullinemia types I and II. Clin Chim Acta. (2014) 431:1–8. doi: 10.1016/j.cca.2014.01.032
47. Lee BH, Kim YM, Heo SH, Kim GH, Choi IH, Lee BS, et al. High prevalence of neonatal presentation in Korean patients with citrullinemia type 1, and their shared mutations. Mol Genet Metab. (2013) 108(1):18–24. doi: 10.1016/j.ymgme.2012.11.011
48. Woo HI, Ki CS, Lee SY, Kim JW, Song J, Jin DK, et al. Mutation spectrum of the Ass1 gene in Korean patients with citrullinemia type I. Clin Biochem. (2013) 46(3):209–13. doi: 10.1016/j.clinbiochem.2012.10.008
49. Gao HZ, Kobayashi K, Tabata A, Tsuge H, Iijima M, Yasuda T, et al. Identification of 16 novel mutations in the argininosuccinate synthetase gene and genotype-phenotype correlation in 38 classical citrullinemia patients. Hum Mutat. (2003) 22(1):24–34. doi: 10.1002/humu.10230
50. Forsyth R, Vockley CW, Edick MJ, Cameron CA, Hiner SJ, Berry SA, et al. Outcomes of cases with 3-methylcrotonyl-Coa carboxylase (3-mcc) deficiency—report from the inborn errors of metabolism information system. Mol Genet Metab. (2016) 118(1):15–20. doi: 10.1016/j.ymgme.2016.02.002
Keywords: tandem mass spectrometry, inherited metabolic disorders, next generation sequencing, NGS, pathogenic or likely pathogenic variant, genetic variant
Citation: Ou M-m, Sun C-t, Zhang L, Kong L-l, Zhang L-x, Zheng J, Zhang P-y, Wang Y-m, Lin X-f and Yang X (2025) Incidence and disease spectrum of inherited metabolic diseases screened by tandem mass spectrometry in Huai'an from 2018 to 2024. Front. Pediatr. 13:1629840. doi: 10.3389/fped.2025.1629840
Received: 26 May 2025; Accepted: 31 October 2025;
Published: 21 November 2025.
Edited by:
Amy Brower, Creighton University, United StatesReviewed by:
Joanne Adelberg, Howard University, United StatesCraig Baker, University of Nebraska Medical Center, United States
Copyright: © 2025 Ou, Sun, Zhang, Kong, Zhang, Zheng, Zhang, Wang, Lin and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yu-mei WangMTM3NzAzNTc4ODJAeXp1LmVkdS5jbg==;Xiao-fei Lin, MTM4NjE1NjEzMTBAeXp1LmVkdS5jbg==; Xin Yang, eXg4MDEwMDVAMTI2LmNvbQ==
†These authors share first authorship