 Wanting Xu1,2,3,4
Wanting Xu1,2,3,4 Yan Yang1,2,3Lan Kang1,2
Yan Yang1,2,3Lan Kang1,2 Ling Guo3Jing Liu3Yan Zeng3Lei Li4Ai Chen4Rong Zhang1,2*
Ling Guo3Jing Liu3Yan Zeng3Lei Li4Ai Chen4Rong Zhang1,2* Wenbin Dong1,2,3*
Wenbin Dong1,2,3*
- 1Division of Neonatology, Department of Pediatrics, The Affiliated Hospital of Southwest Medical University, Luzhou, Sichuan, China
- 2Department of Perinatology, The Affiliated Hospital of Southwest Medical University, Luzhou, Sichuan, China
- 3Pediatrics, Sichuan Clinical Research Center for Birth Defects, Luzhou, Sichuan, China
- 4Pediatrics, Chengdu Second People’s Hospital, Chengdu, Sichuan, China
Primary ciliary dyskinesia (PCD) is a rare genetically heterogeneous disorder characterized by dysfunctional motile cilia, with or without detectable ultrastructural abnormalities. This study focuses on a homozygous mutation in the rare radial spoke head component 4A (RSPH4A) gene in a Chinese adolescent girl with PCD. The patient, an 11-year and 3-month-old girl, developed neonatal pneumonia after birth and gradually presented with persistent perennial rhinitis and recurrent productive cough. Lung CT scan indicated bronchiectasis, and whole-exome sequencing (WES) exhibited a novel pathogenic homozygous c.351dup (p. Pro118Serfs*2) frameshift mutation in the RSPH4A gene. A literature review reported that 21 pathogenic variants in RSPH4A have been discovered. WES recognized disease-causing mutations in PCD, and c.351dup (p. Pro118Serfs*2) frameshift mutation in RSPH4A may become a hotspot in Chinese patients.
Introduction
Primary ciliary dyskinesia (PCD) is a rare, congenital disease induced by mutations in genes that encode ciliary components, primarily manifesting as recurrent respiratory infections, chronic sinusitis, and secretory otitis media (1). PCD is diagnosed based on a combination of genetic sequence testing, high-speed video microscopy tests for cilia beat frequency and pattern, transmission electron microscopy (TEM), and nasal nitric oxide (2). Its prevalence varies between 1/15,000 and 1/30,000 live births, with >50 genes and over 2,000 causative mutations (3). The proteins that make up the motile cilia structure in the upper and lower respiratory tracts are impacted by genetic mutations (4).
The RSPH4A gene (c.921+3_921+6delAAGT) was first identified as a pathogenic variant in Puerto Rico and linked to the ancestor's haplotype in 2013 (5), and there were only a few reports of variants in China. Prior research verified the presence of RSPH4A in the radial spoke heads of cilia's microtubules. Its variations mainly damage the ciliary ultrastructure and result in the absence of radial spoke heads, which impairs mucus clearance (6). In this study, we reported a novel homozygous frameshift mutation in RSPH4A (GRCh37/hg19:chr6:116938137, Exon:1/6, NM_001010892.3 c.351dup p.Pro118Serfs*2), which has never been documented before in the literature on patients with RSPH4A-related disease. Additionally, we conducted a literature review to clarify the clinical characteristics and provide compound variations for early identification of this PCD genotype.
Case report
A teenage girl, aged 11 years and 3 months, was admitted to the respiratory department of our hospital with a 1-week history of cough and 1-day history of fever. She came from a non-consanguineous family, according to the family history, and her parents showed no symptoms (Figure 1A). Born at term, the patient was admitted to a neonatal unit and diagnosed as neonatal pneumonia at 24 h after birth. She had no history of prolonged oxygen dependency and displayed situs solitus. She had persistent perennial rhinitis and a recurrent productive cough since she was 7 years old. She takes montelukast orally every day and continuously inhales powdered budesonide and formoterol fumarate. Additionally, the patient was diagnosed with sinusitis and secretory otitis media by the otolaryngologist based on nasal obstruction, purulent nasal discharge for over 4 years, and the results of the otoscopy examination. Her motor, cognitive, and language development was normal, with no hydrocephalus. At 11 years old, she was admitted to our respiratory department for community-acquired pneumonia. Chest radiograph indicated bilateral pleural thickening and inflammation of lower fields of both lungs (Figure 1B). Bronchoscopy revealed a lot of white and viscous secretions in the bronchial cavities bilaterally. A bacterial culture of bronchoalveolar lavage fluid (BALF) exhibited Pseudomonas aeruginosa. Transmission electron microscope (TEM) showed microvilli with few cilia on the epithelial surface (Figure 1C). Electrocardiogram, echocardiography, myocardial enzymes, liver and kidney function tests, plasma electrolytes, serum mycoplasma and chlamydia antibodies, and immunoglobulin levels were unremarkable.
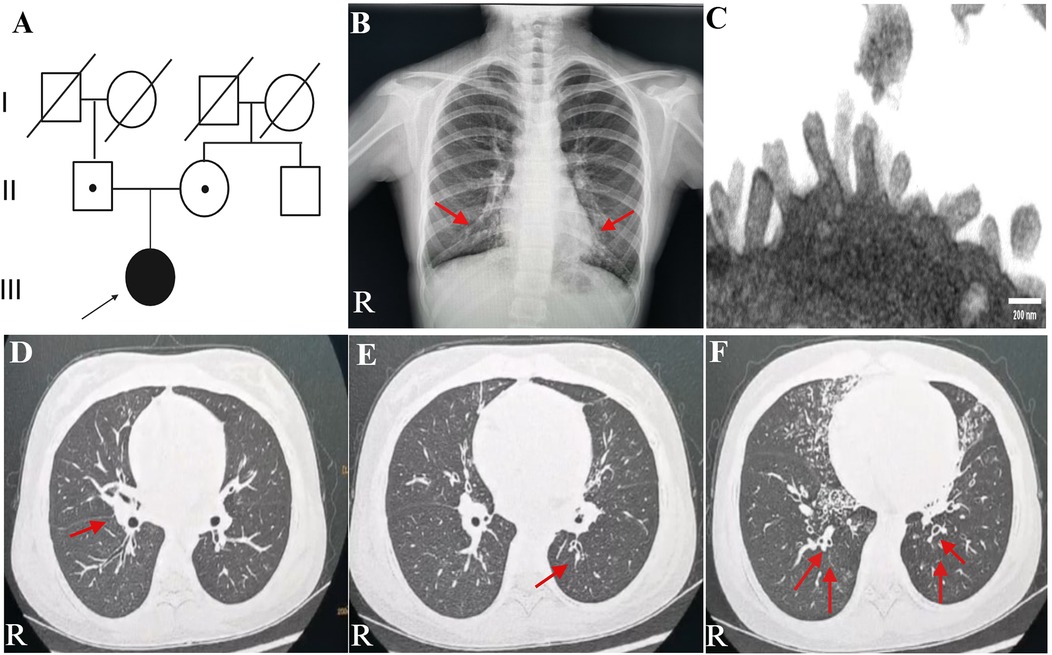
Figure 1. Family history and clinical characteristics of the patient. (A) This PCD patient came from a non-consanguineous family, and her parents showed no clinical symptoms. The teenage female proband inherited a homozygous mutation from both heterozygous carrier parents. The proband is identified by the arrow. (B) Chest radiograph indicated bilateral pleural thickening, inflammation of lower fields of both lungs, and no ectopic heart. Inflammation infiltration images are shown by the red arrows. (C) TEM indicated microvilli on the epithelial surface. (D–F) Axial view: lung CT scan indicated infections in the middle lobe of the right lung and the lower lingual segment of the upper lobe of the left lung and inflammation of lower lobes of both lungs, with slightly dilated bronchi in the lower lobes of both lungs. As is shown by the red arrows. PCD, primary ciliary dyskinesia; R, right; TEM, transmission electron microscope.
On physical examination, the adolescent patient indicated no shortness of breath. Auscultation exhibited coarse to moderate dampness and phlegm rales.
Auxiliary examination
A lung CT scan indicated infections in the middle lobe of the right lung and the lower lingual segment of the upper lobe of the left lung and inflammation of the lower lobes of both lungs, with slightly dilated bronchi in the lower lobes of both lungs (Figures 1D–F). The first-time pulmonary function (PF) test showed increased airway resistance and decreased ventilation function of small airways, with forced vital capacity (FVC) at 82.9% of the predicted value, forced expiratory capacity in 1 s (FEV1) at 78.5% of the predicted value, peak expiratory flow rate (PEF) at 58.0% of the predicted value, and forced expiratory flow at 75% of forced vital capacity with 50.2% of the predicted value. Multiplex combined detection of respiratory pathogens testing of BALF was positive for P. aeruginosa DNA and Streptococcus pneumoniae DNA and negative for Mycobacterium tuberculosis DNA. Her fractional exhaled nitric oxide (FeNO) level was 5.8 ppb. The nasal nitric oxide (nNO) was measured at 16.0 ppb (9.6 nl/min). The patient no longer had a fever following 1 week of intravenous ceftriaxone infusion, 3 days of oral azithromycin anti-infection treatment, and 5 days of glucocorticoid inhalation; however, the patient continued to experience recurrent wet coughing. The second-time PF test exhibited that the flow velocity of medium and small airways decreased, and the function of small airways was impaired. Re-examination showed FVC at 94.9% of the predicted value, FEV1 at 86.1% of the predicted value, PEF at 82.2% of the predicted value, and forced expiratory flow at 75% of forced vital capacity with 34.8% of the predicted value. The second time of FeNO measurement was 8.0 ppb, and nNO was 7.0 ppb (4.2 nl/min). PFs and fractional nitric oxide examination results are illustrated in Figure 2.
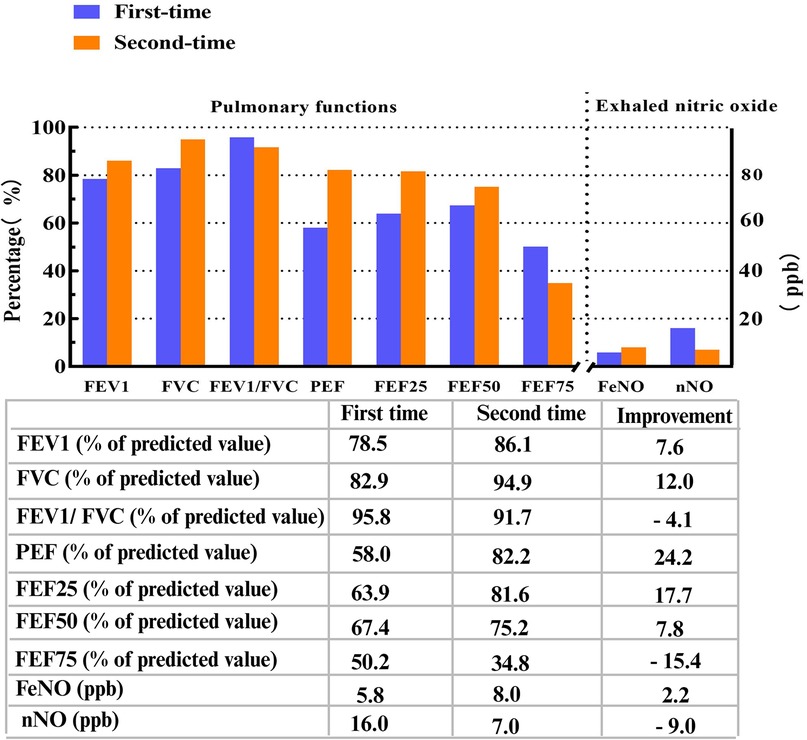
Figure 2. Pulmonary functions and fractional nitric oxide examination detections. FEV1, FVC, FEV1/FVC, PEF, FEF25, FEF50, and FEF75 were all identified as percentages of predicted values. FEV1/FVC ratio, percent of predicted value of FEV1 to FVC ratio; FEV1, forced expiratory volume in 1 s; FVC, forced vital capacity; PEF, peak expiratory flow rate; FEF25, forced expiratory flow at 25% of forced vital capacity; FEF50, forced expiratory flow at 50% of forced vital capacity; FEF75, forced expiratory flow at 75% of forced vital capacity; FeNO, fractional exhaled nitric oxide; FnNO, fractional nasal nitric oxide. FeNO and FnNO were indicated as ppb.
Diagnosis
Given the previous experience of full-term born, neonatal pneumonia, prolonged nasal obstruction, purulent nasal discharge, secretory otitis media, bronchiectasis, and extremely low nNO results, the suspected diagnosis is PCD. In addition, Primary Ciliary Dyskinesia Rule (PICADAR) was applied to the patient who has a recurrent purulent cough starting in early childhood. It was used to predict the likelihood of having PCD based on seven questions (7). The total score of this adolescent patient was 6, whose detailed scoring items were summarized (Supplementary Table S1). Based on four general clinical features, PCD was evaluated using an additional scoring system created by the American Thoracic Society (ATS) score (8). Two criteria for this patient were positive, and the specificity of PCD was 72% (Supplementary Table S2).
Follow-up
Whole-exome sequencing (WES, KingMed Diagnostics, Sichuan, China) verified a homozygous c.351dup (p. Pro118Serfs*2) in the RSPH4A gene, approving a definitive diagnosis of PCD (Figure 3). After bronchoscopy, alveolar lavage to clear the respiratory tract, and low-dose oral administration of azithromycin and expectorants, her recurrent wet cough improved. A follow-up examination lung CT scan 2 weeks after discharge revealed multiple bronchiectases with infection in both lungs, which had considerably improved of infections from the initial findings. However, compared with the previous images, there was minimal change in the small patchy ground-glass density shadow in the posterior segment of the right lung's upper lobe. At the 3-month follow-up after discharge, the patient had improved in nasal obstruction, with no recurrent fever or wheezing. Body mass index was appropriate for age and gender. As a measure of inflammation, C-reactive protein was normalized in the blood test to 2.1 mg/L. The patient adhered to the prescribed inhaled corticosteroids and oral montelukast taken daily. No liver function, emotional, or behavioral changes were reported.
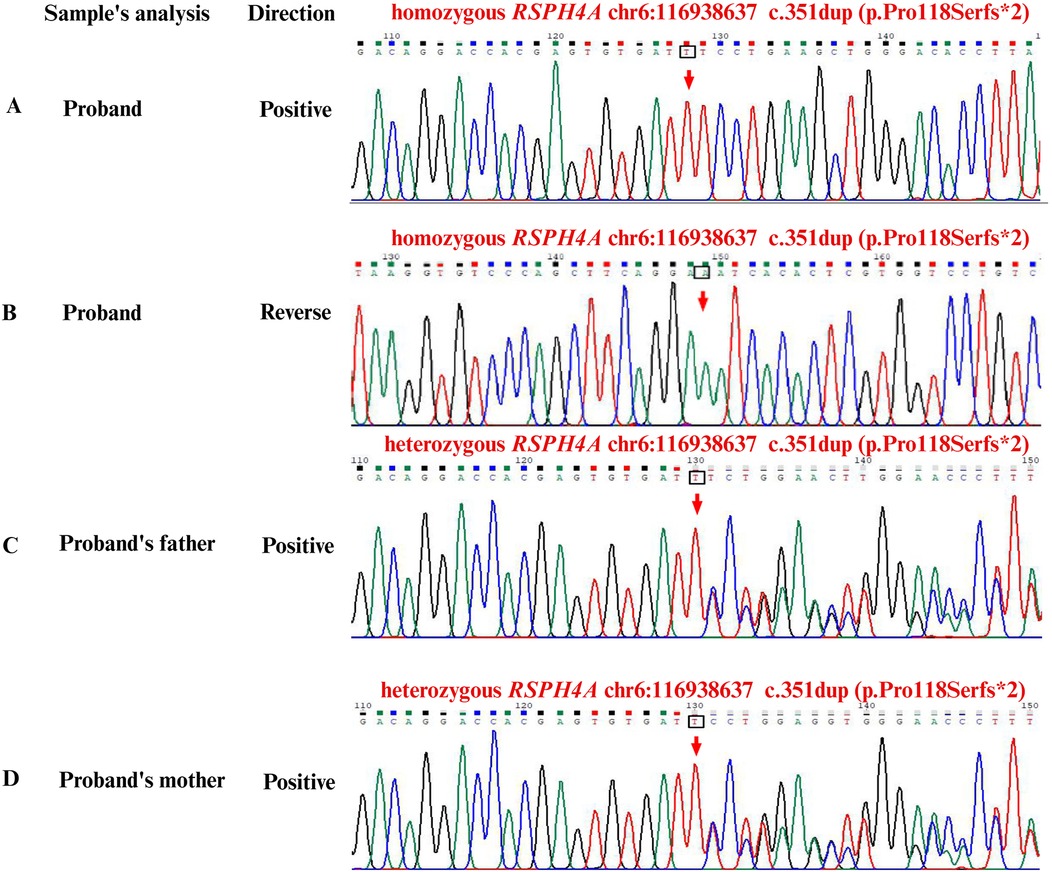
Figure 3. Sanger second-generation sequencing for capturing the whole-exome and gene sequencing. (A) Positive sequence of homozygous RSPH4A: c.351dup (p.Pro118Serfs*2) frameshift variant in the patient. (B) Reverse sequence of homozygous RSPH4A: c.351dup (p.Pro118Serfs*2) frameshift variant in the patient. (C) Positive sequence of heterozygous RSPH4A: c.351dup (p.Pro118Serfs*2) carrier in the father. (D) Positive sequence of heterozygous RSPH4A: c.351dup (p.Pro118Serfs*2) carrier in the mother. Green was represented as adenine. Black was represented as guanine. Red was represented as thymine. Blue was represented as cytosine. Hom, homozygous; Het, heterozygous.
Literature review
Using the keywords “primary ciliary dyskinesia,” “immotile-cilia syndrome,” and “RSPH4A gene,” a PubMed literature search was conducted to locate articles published between 2012 and 2025. A total of 12 articles were searched, and clinical characteristics and functional roles of RSPH4A mutations in PCD are summarized in Table 1.
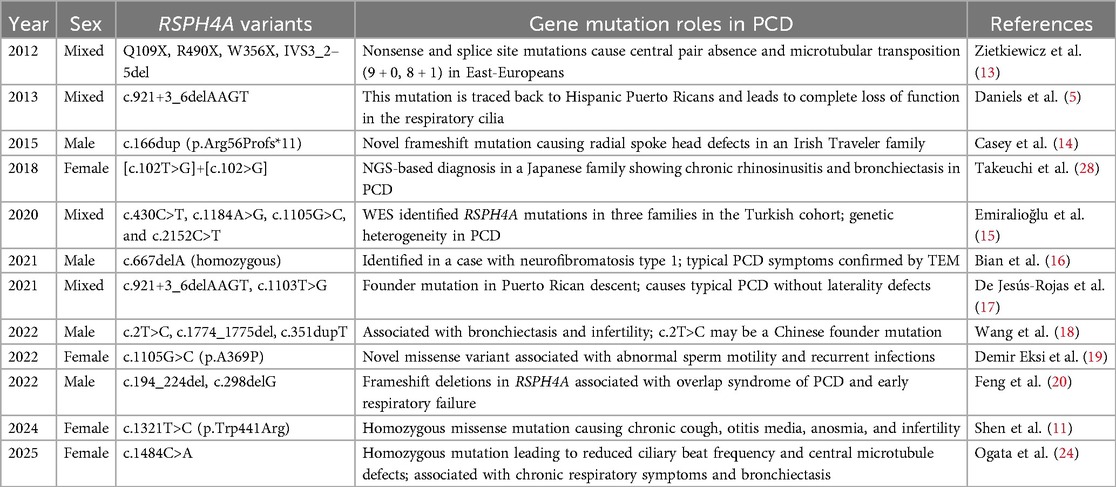
Table 1. Roles of RSPH4A mutations in PCD patients.
Discussion
PCD is a rare genetic disease characterized by progressive lung disorder, and 54 pathogenic genes have been discovered as causing variants to date (9). In this case, TEM was non-diagnostic, and the final diagnosis was made based on clinical characteristics and WES sequence confirmation, which was in line with diagnostic paradigm for PCD in the DAW1 gene mutation (10). In a ciliary cross section, nine sets of outer microtubule doublets are usually grouped around a central pair, forming a 9 + 2 ultrastructure. The T-shaped radial spokes join the central pair of microtubules to the outer doublets. RSPH4A mutant is an essential structure, as it serves as the central part of the radial spoke head (11). RSPH4A variants primarily damage the ciliary ultrastructure and result in the loss of radial spoke heads, which impairs mucus clearance (6).
Previous research revealed that RSPH4A mutations are rare in East Asia, accounting for 1.5% of Chinese PCD patients (12). Zietkiewicz et al. (13) firstly reported RSPH4A mutations in/around exons 1 and 3, such as Q109X, R490X, W356X, and IVS3_2–5del, demonstrating that microtubule transposition phenotype was present in nearly half of the ciliary cross sections examined in patients with RSPH4A mutations. Daniels et al. (5) described a novel splice site mutation (c.921+3_6delAAGT) in RSPH4A, which could be traced back to Hispanic Puerto Ricans and led to complete loss of function in the respiratory cilia. Casey et al. (14) identified a new mutation in RSPH4A (c.166dup; p.Arg 56Pro*11) gene, which caused the loss of all annotated domains, including the radial spoke domain, by introducing an early stop codon at residue 66. Takeuchi et al. conducted a targeted next-generation sequencing panel and discovered a novel disease-causing mutation in exon 1 of RSPH4A: [c.102T>G]+[c.102T>G] in nucleotide change. WES identified RSPH4A mutations, such as c.430C>T, c.1184A>G, c.1105G>C, and c.2152C>T in three families in the Turkish cohort, and proved genetic heterogeneity in PCD by Emiralioğlu et al. (15). A homozygous mutation c.667delA, p.S223Afs*15 in the RSPH4A gene was identified in a case with neurofibromatosis type 1, and typical PCD symptoms were confirmed by TEM (16). De Jesús-Rojas et al. (17) demonstrated a likely pathogenic variant c.1103T>G (p.Val368Gly) in the RSPH4A gene, which caused typical PCD without laterality defects. Wang et al. (18) identified heterozygous variants c.2T>C, p.(Met1Thr) and c.1774_1775del, p.(Leu592Aspfs*5) in the RSPH4A gene, which were associated with bronchiectasis and infertility, and c.2T>C, p.(Met1Thr) may be a hotspot variant in Chinese. Another novel missense variant c.1105G>C (p.A369P) in the RSPH4A gene was also associated with abnormal sperm motility and recurrent infections in PCD, and the t-NGS panel was helpful for efficient and exact identification (19). As for deletion frameshift mutations, Feng et al. (20) revealed c.194_224del and c.298delG in the RSPH4A gene, which were also associated with PCD overlap syndrome, as well as early respiratory failure. A novel homozygous mutation c.1321T>C (p.Trp441Arg) in exon 3 of RSPH4A was reported by Shen et al. (11), causing symptoms with chronic cough, otitis media, anosmia, and infertility. Prior research indicated that distinct RSPH4A mutations consistently led to PCD with specific phenotypic patterns, which were characterized by respiratory symptoms without laterality defects. De Jesús-Rojas et al. discovered that RSPH4A (c.921+3_6delAAGT) mutation, as the Puerto Rican founder mutation, demonstrated a very consistent clinical characteristic in several patients, confirming the absence of laterality defects and the presence of typical bronchiectasis and measurable olfactory impairment (21, 22). As Cryoelectron tomography shown by Yoke et al., RSPH4A KO mice exhibited a complete absence of all three radial spoke heads (RS1, RS2, RS3) in tracheal cilia. They verified that RSPH4A assisted in generating the planar beating of motile cilia by constructing the distal architecture of radial spokes in the trachea, as well as the ependymal tissues (23). The latest homozygous variant c.1484C>A in the RSPH4A gene was discovered in Japan, resulting in a reduction in ciliary beat frequency and central microtubule defects, which was also associated with chronic respiratory symptoms and bronchiectasis (24). These indicated that different RSPH4A genes led to similar basic clinical symptoms, mainly affecting respiratory epithelia.
In the present research, we analyzed the genetic sequence of an adolescent PCD patient combined with recurrent productive cough, purulent nasal discharge, secretory otitis media, bronchiectasis, and extremely low nNO results. WES results indicated a novel frameshift mutation c.351dup (p. Pro118Serfs*2) in the RSPH4A gene. This mutation carried by the proband was inherited from her parents, who carried a heterozygous mutant gene respectively and generated homozygosity in the patient. This frameshift variant c.351dup (p. Pro118Serfs*2) in RSPH4A discovered in this adolescent patient has not been reported in the databases or literature yet, including ClinVar (https://www.ncbi.nlm.nih.gov/clinvar), LOVD3 (https://databases.lovd.nl/shared/genes/RSPH4A), and gnomAD (https://gnomad.broadinstitute.org/). All information was searched on 16 May 2025. It is suggested that c.351dup (p. Pro118Serfs*2) in RSPH4A is a rare, novel variant, which is first reported in a PCD patient. Additionally, in silico analysis verified that the variant also has an influence on the protein's function. MutationTaster (https://www.genecascade.org/MutationTaster2021/) analyzed the c.351dup (p. Pro118Serfs*2) in RSPH4A and identified it as “disease causing” with a high probability score of 1.0. It was suggested that the pathogenic frameshift mutation was pathogenic and could potentially result in nonsense-mediated mRNA decay (NMD). VarSome (https://varsome.com) classified the variant as met PVS1, PM2, and likely PP3, indicating it as likely pathogenic (Supplementary Files 3, 4). Additionally, the timeline of the diagnostic process of the PCD patient is summarized in Figure 4. According to the Genome Aggregation Database (gnomAD, v2.1.1), the RSPH4A c.351dup (p.Pro118Serfs*2) variant has not been identified in individuals from diverse global populations, indicating it is a novel frameshift variant with no reported cases to date.
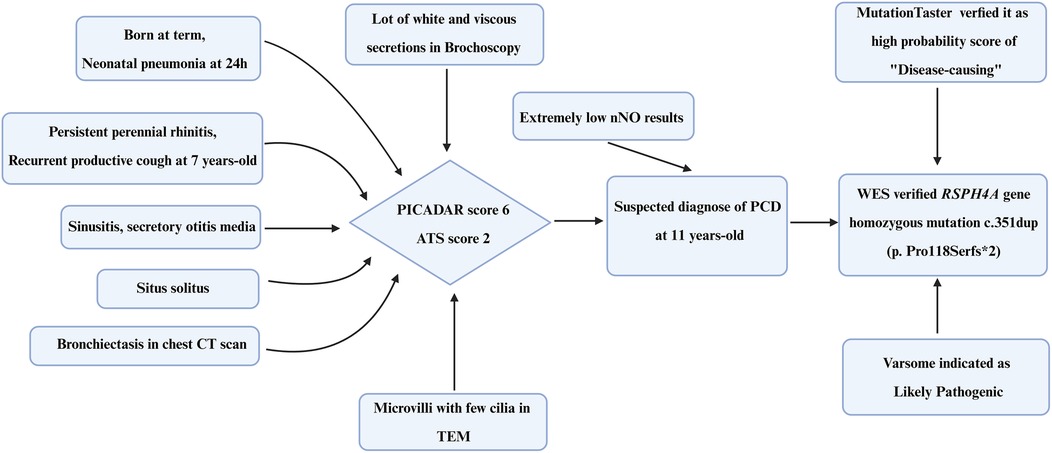
Figure 4. Timeline of the diagnostic process of the PCD patient. PICADAR, Primary Ciliary Dyskinesia Rule; ATS, American Thoracic Society; TEM, transmission electron microscopy; WES, whole-exome sequencing.
As for pathogens and lung functions in PCD patients, Rubbo et al. (25) indicated that P. aeruginosa was isolated from airway samples in adults and induced obviously lower FEV1, FVC, and FEF25%–75% z-scores, while the results were not very prominent in children. Marthin et al. (26) reported an international consensus on prevention and administration of PCD patients infected with P. aeruginosa, which recommended eradication even in asymptomatic situations. Impulse Oscillometry (IOS) was used to assess preschoolers with PCD, and a notable variation in airway resistance and reactance was reported, compared with healthy children (27). In our research, bacterial culture of BALF from this PCD patient exhibited P. aeruginosa. She received 1 week of intravenous ceftriaxone infusion and 3 days of oral azithromycin administration so as to eradicate this bacterial infection. Two weeks after discharge, a follow-up lung CT scan revealed that the bilateral lung infections had considerably improved, but multiple bronchiectases still existed.
Apart from these, several limitations in this case deserve to be taken into account. For example, high-speed video microscopy and immunofluorescence staining are recommended in analyzing ciliary ultrastructure for the next step. Combined with the second-generation sequencing results, ciliary functions caused by disease-inducing gene mutation were further identified. Furthermore, long-term monitoring of lung function, exercise evaluation, hearing evaluation, rehabilitation training, and psychotherapy feedback are all necessary to enhance the quality of life for PCD patients.
Conclusion
We report a case of a PCD patient with a novel RSPH4A mutation. Although PCD caused by gene mutation of RSPH4A has been reported several times in China, the first case of a pathogenic homozygous frameshift mutation c.351dup (p. Pro118Serfs*2) in the RSPH4A gene is reported worldwide. Reporting recently discovered gene mutations is essential for PCD research.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Ethics statement
Written informed consent was obtained from the individual(s), and the minor(s)' legal guardians/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
WX: Investigation, Conceptualization, Writing – original draft, Writing – review & editing, Data curation, Methodology, Formal analysis. YY: Methodology, Writing – review & editing, Data curation. LK: Formal analysis, Writing – review & editing. LG: Methodology, Writing – review & editing, Supervision. JL: Writing – review & editing, Data curation. YZ: Writing – review & editing, Data curation, Validation. LL: Writing – review & editing. AC: Writing – review & editing. RZ: Data curation, Writing – review & editing, Supervision. WD: Methodology, Writing – review & editing, Supervision.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. The Wu Jieping Medical Foundation Clinical Research (No. 320.6750. 2023-24-10) and Chinese National Natural Science Foundation (No. 82371710) provided funding for this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2025.1630730/full#supplementary-material
References
1. Horton K, Wing PAC, Jackson CL, McCormick CJ, Carroll MP, Lucas JS. Interplay between respiratory viruses and cilia in the airways. Eur Respir Rev. (2025) 34(175):1–16. doi: 10.1183/16000617.0224-2024
2. Muhonen EG, Zhu A, Sempson S, Bothwell S, Sagel SD, Chan KH. Management of middle ear disease in pediatric primary ciliary dyskinesia. Int J Pediatr Otorhinolaryngol. (2025) 192:112297. doi: 10.1016/j.ijporl.2025.112297
3. Collison R, Hyatali SA, Kamenova A, Rashed A, Riley D, Kumar K, et al. Primary ciliary dyskinesia: aetiology, diagnosis and clinical management. Clin Med (Lond). (2025) 25(3):100319. doi: 10.1016/j.clinme.2025.100319
4. Zhou W, Li Y, Zheng H, He M, Zhang M, Chen Q, et al. Whole exome sequencing enhances diagnosis of hereditary bronchiectasis. Orphanet J Rare Dis. (2025) 20(1):142. doi: 10.1186/s13023-025-03661-z
5. Daniels ML, Leigh MW, Davis SD, Armstrong MC, Carson JL, Hazucha M, et al. Founder mutation in RSPH4A identified in patients of Hispanic descent with primary ciliary dyskinesia. Hum Mutat. (2013) 34(10):1352–6. doi: 10.1002/humu.22371
6. Zhao Y, Pinskey J, Lin J, Yin W, Sears PR, Daniels LA, et al. Structural insights into the cause of human RSPH4A primary ciliary dyskinesia. Mol Biol Cell. (2021) 32(12):1202–09. doi: 10.1091/mbc.E20-12-0806
7. Behan L, Dimitrov BD, Kuehni CE, Hogg C, Carroll M, Evans HJ, et al. PICADAR: a diagnostic predictive tool for primary ciliary dyskinesia. Eur Respir J. (2016) 47(4):1103–12. doi: 10.1183/13993003.01551-2015
8. Leigh MW, Ferkol TW, Davis SD, Lee HS, Rosenfeld M, Dell SD, et al. Clinical features and associated likelihood of primary ciliary dyskinesia in children and adolescents. Ann Am Thorac Soc. (2016) 13(8):1305–13. doi: 10.1513/AnnalsATS.201511-748OC
9. Barber AT, Davis SD, Ferkol TW, Shapiro AJ, Atkinson J, Sagel SD, et al. The association of neonatal respiratory distress with ciliary ultrastructure and genotype in primary ciliary dyskinesia. Pediatr Pulmonol. (2025) 60(5):e71091. doi: 10.1002/ppul.71091
10. Leslie JS, Hjeij R, Vivante A, Bearce EA, Dyer L, Wang J, et al. Biallelic DAW1 variants cause a motile ciliopathy characterized by laterality defects and subtle ciliary beating abnormalities. Genet Med. (2022) 24(11):2249–61. doi: 10.1016/j.gim.2022.07.019
11. Shen C, Shen Y, Huang W, Zhang A, Zou T, Guo D, et al. A novel homozygous RSPH4A variant in a family with primary ciliary dyskinesia and literature review. Front Genet. (2024) 15:1364476. doi: 10.3389/fgene.2024.1364476
12. Guan Y, Yang H, Yao X, Xu H, Liu H, Tang X, et al. Clinical and genetic spectrum of children with primary ciliary dyskinesia in China. Chest. (2021) 159(5):1768–81. doi: 10.1016/j.chest.2021.02.006
13. Zietkiewicz E, Bukowy-Bieryllo Z, Voelkel K, Klimek B, Dmenska H, Pogorzelski A, et al. Mutations in radial spoke head genes and ultrastructural cilia defects in East-European cohort of primary ciliary dyskinesia patients. PLoS One. (2012) 7(3):e33667. doi: 10.1371/journal.pone.0033667
14. Casey JP, McGettigan PA, Healy F, Hogg C, Reynolds A, Kennedy BN, et al. Unexpected genetic heterogeneity for primary ciliary dyskinesia in the Irish traveller population. Eur J Hum Genet. (2015) 23(2):210–7. doi: 10.1038/ejhg.2014.79
15. Emiralioğlu N, Taskiran EZ, Kosukcu C, Bilgic E, Atilla P, Kaya B, et al. Genotype and phenotype evaluation of patients with primary ciliary dyskinesia: first results from Turkey. Pediatr Pulmonol. (2020) 55(2):383–93. doi: 10.1002/ppul.24583
16. Bian C, Zhao X, Liu Y, Chen M, Zheng S, Tian X, et al. Case report of neurofibromatosis type 1 combined with primary ciliary dyskinesia. Front Med. (2021) 15(6):933–37. doi: 10.1007/s11684-021-0860-7
17. De Jesús-Rojas W, Reyes De Jesus D, Nieves AM, Mosquera RA, Martinez-Cruzado JC. Primary ciliary dyskinesia: ancestral haplotypes analysis of the RSPH4A founder mutation in Puerto Rico. Cureus. (2021) 13(9):e17673. doi: 10.7759/cureus.17673
18. Wang L, Wang R, Yang D, Lu C, Xu Y, Liu Y, et al. Novel RSPH4A variants associated with primary ciliary dyskinesia-related infertility in three Chinese families. Front Genet. (2022) 13:922287. doi: 10.3389/fgene.2022.922287
19. Demir Eksi D, Yilmaz E, Basaran AE, Erduran G, Nur B, Mihci E, et al. Novel gene variants associated with primary ciliary dyskinesia. Indian J Pediatr. (2022) 89(7):682–91. doi: 10.1007/s12098-022-04098-z
20. Feng M, Yu X, Yue Y, Zhong J, Wang L. Novel mutations in RSPH4A and TTN genes lead to primary ciliary dyskinesia-hereditary myopathy with early respiratory failure overlap syndrome. Genes Dis. (2023) 10(3):743–45. doi: 10.1016/j.gendis.2022.10.013
21. De Jesús-Rojas W, Reyes-De Jesus D, Mosquera RA. Primary ciliary dyskinesia diagnostic challenges: understanding the clinical phenotype of the Puerto Rican RSPH4A founder mutation. Diagnostics (Basel). (2021) 11(2):281. doi: 10.3390/diagnostics11020281
22. De Jesus MA, De Jesús-Rojas W. Assessing olfactory acuity in primary ciliary dyskinesia with the RSPH4A founder mutation. J Clin Med. (2025) 14(10):3612. doi: 10.3390/jcm14103612
23. Yoke H, Ueno H, Narita A, Sakai T, Horiuchi K, Shingyoji C, et al. Rsph4a is essential for the triplet radial spoke head assembly of the mouse motile cilia. PLoS Genet. (2020) 16(3):e1008664. doi: 10.1371/journal.pgen.1008664
24. Ogata R, Kido T, Sakamoto N, Murakami R, Tokito T, Yura H, et al. Primary ciliary dyskinesia in a Japanese woman caused by a novel RSPH4A variant. Respir Investig. (2025) 63(4):507–09. doi: 10.1016/j.resinv.2025.04.007
25. Rubbo B, Kant A, Zhang K, Allegorico A, Basilicata S, Boon M, et al. Associations between respiratory pathogens and lung function in primary ciliary dyskinesia: cross-sectional analysis from the PROVALF-PCD cohort. ERJ Open Res. (2024) 10(5):00253-2024. doi: 10.1183/23120541.00253-2024
26. Marthin JK, Lucas JS, Boon M, Casaulta C, Crowley S, Destouches DMS, et al. International BEAT-PCD consensus statement for infection prevention and control for primary ciliary dyskinesia in collaboration with ERN-LUNG PCD Core Network and patient representatives. ERJ Open Res. (2021) 7(3):00301-2021. doi: 10.1183/23120541.00301-2021
27. Sunman B, Yalcin E, Alboga D, Capraz Yavuz B, Altay Tanyer E, Emiralioğlu N, et al. Impulse oscillometry is useful in detecting lung function abnormalities in preschoolers with primary ciliary dyskinesia but not cystic fibrosis: a cross-sectional study results. Pediatr Allergy Immunol Pulmonol. (2025) 38(2):71–7. doi: 10.1089/ped.2024.0142
Keywords: adolescent patient, primary ciliary dyskinesia, homozygous mutations, RSPH4A, case report
Citation: Xu W, Yang Y, Kang L, Guo L, Liu J, Zeng Y, Li L, Chen A, Zhang R and Dong W (2025) Analysis of clinical and genetic features in an adolescent patient with primary ciliary dyskinesia induced by homozygous mutation in the RSPH4A gene: a case report. Front. Pediatr. 13:1630730. doi: 10.3389/fped.2025.1630730
Received: 22 May 2025; Accepted: 15 July 2025;
Published: 7 August 2025.
Edited by:
Seyed Abbas Rafat, University of Tabriz, IranReviewed by:
Jingjing Feng, Fudan University, ChinaRyotaro Hashizume, Mie University, Japan
Kawther Mohammed Nasser Al Adawi, Sultan Qaboos University Hospital, Oman
Copyright: © 2025 Xu, Yang, Kang, Guo, Liu, Zeng, Li, Chen, Zhang and Dong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rong Zhang, emhhbmdyb25nOTYwNEAxNjMuY29t; Wenbin Dong, ZG9uZ3dlbmJpbjIwMDBAMTYzLmNvbQ==