 Shirui Jiang1
Shirui Jiang1 Wei Wang
Wei Wang Liqin Zhu
Liqin Zhu Wei Liu
Wei Liu- 1First Central Hospital of Tianjin Medical University, Tianjin, China
- 2Tianjin Children’s Hospital, Children’s Hospital, Tianjin University, Tianjin, China
- 3Precision Medicine Laboratory, Precision Medicine Center, Tianjin Children’s Hospital, Children’s Hospital, Tianjin University, Tianjin, China
- 4Tianjin University of Traditional Chinese Medicine, Tianjin, China
- 5Department of Pharmacy, Tianjin First Central Hospital, Tianjin, China
Background: Pediatric primary cardiomyopathies (PCMs) are rare diseases with complex causes and nonspecific treatment. The influence of electrolytes and amino acids (AAs) on cardiomyopathies has not been extensively studied. This study aimed to explore clinical characteristics and the usage of electrolytes and AAs in children with PCMs.
Methods: Children diagnosed with PCMs who had genetic test reports were included. Relevant information was collected and processed, and clinical characteristics and mutated genes were clarified. Gene databases were searched to explore related electrolytes and AAs in the treatment of PCMs. The effect of calcium was explored in children with DCM. Paired samples T tests and nonparametric Wilcoxon signed-rank tests were performed for comparison between before and after using calcium.
Results: In this study, 27 children with gene test results were enrolled to perform gene-related analysis. The median age was 2.5 years old. Mutated genes were collected, including pathogenic, likely pathogenic, uncertain significance, and other mutations. The most frequently mutated genes related to dilated cardiomyopathy (DCM) were TTN, MYH7, NEXN, TNNI3, and SCN5A. In hypertrophic cardiomyopathy (HCM), MYBPC3, MYH7, PRKAG2, RAF1, and RBM20 were prevalent. Calcium and AAs (serine, cysteine, arginine, tyrosine, and alanine) were related to the mutated genes detected in children with PCMs. In addition, 17 children treated with calcium showed significant improvement in heart function.
Conclusions: For children with DCM, calcium supplements may be beneficial. AAs, including serine, cysteine, and arginine, could be used for supplementary treatment in children with DCM and HCM.
1 Introduction
In the statement of the American Heart Association, cardiomyopathies (CMs) are rare in children, with high mortality (1). Causes are heterogeneous in pediatric CMs, ranging from genetic mutations to systemic diseases. The classifications of CMs are complicated and varied. According to the World Health Organization, CMs are classified into primary and secondary. Primary cardiomyopathies (PCMs) are heart muscle diseases in the absence of secondary pathogenic causes. Based on morphologic abnormalities and pathophysiology, PCMs were grouped into dilated cardiomyopathy (DCM), hypertrophic cardiomyopathy (HCM), restrictive cardiomyopathy (RCM), and arrhythmogenic right ventricular cardiomyopathy (ARVC) (2). According to the European Society of Cardiology in 2023, left ventricular noncompaction cardiomyopathy (LVNC) was also categorized as a type of CMs characterized by unique changes in morphology and function of the left ventricle (3).
DCM is defined as left ventricular (LV) dilatation and systolic dysfunction in the absence of abnormal loading conditions and coronary artery disease (4). In DCM, common direct causes are gene mutations, infections, and autoimmunity (5). Many genes are associated with pediatric DCM, such as TTN, MYH7, RBM20, TNNT2, and DSP (6). HCM is a primary inherited cardiac disease characterized by LV hypertrophy without the existence of abnormal loading factors (e.g., congenital heart diseases, hypertension) (8, 9). TTN, MYH7, MYH6, PRKAG2, and MYBPC3 are related to pediatric HCM (6). RCM, ARVC, and LVNC are rare in CMs. RCM is described as a myocardial disorder characterized by structural and functional abnormalities, with the characteristics of restrictive ventricular filling (9, 10). LVNC, also called LV noncompaction, is characterized by excessive trabeculation in the left ventricle (11, 12). ARVC is a right ventricular (RV) inherited CM. Mutations of the gene encoding sarcomeric or cytoskeletal proteins are the common causes of RCM and LVNC (9, 11). Mutations of genes encoding desmosome proteins play a key role in the pathogenesis of ARVC (13).
Due to the rarity and high mortality, the research related to PCMs in children was limited. In addition, clinical manifestations, disease progression, and drug reactions between children and adults are different. Therefore, it is essential to focus on the research of PCMs in children. At present, the treatment of pediatric CMs is empirical and symptomatic. For instance, medicines used for DCM are diuretics, cardiotonic drugs, vasodilators, and cardiovascular protective drugs (6). Despite the reliance on empirical approaches, little attention has been paid to investigating nutrients, such as electrolytes and amino acids (AAs). A few articles described the effect of electrolytes or AAs on pediatric CMs. For instance, children with hypocalcemic dilated cardiomyopathy could benefit from calcium supplementation (14). It was reported that serine and cysteine could exert cardioprotective effects (15, 16). This study aimed to explore the clinical characteristics of PCMs in children and the influence of appropriate supplementation of electrolytes and AAs to provide more references in the treatment of pediatric PCMs in clinical practice.
2 Methods
2.1 Study population
Children who had been diagnosed with PCMs and had gene test results in Tianjin Children's Hospital from 2019 to 2023 were enrolled. The detailed inclusion and exclusion criteria were provided in Supplementary Material S1.
The inclusion criteria were as follows: DCM was described as age and body surface area corrected left ventricular end-diastolic diameter (LVDD) higher than 112%, left ventricular ejection fraction (LVEF) lower than 45%, and/or left ventricular fractional shortening (LVFS) lower than 25% (1, 17). HCM was described as an increased LV wall thickness, represented by a higher mean ± two standard deviations (SD) (18). RCM was described as biatrial enlargement, normal ventricular diameter, and LV diastolic dysfunction (1). ARVC was mainly diagnosed by the revised task force criteria (19). LVNC was described as the ratio of non-compaction to compaction higher than two in the end-diastolic stage (20).
In addition, some secondary pathogenic characteristics were excluded. Cardiovascular factors, including hypertension, congenital heart disease, arrhythmia, and aortic stenosis, were excluded. Extracardiac factors such as thyroid or parathyroid diseases, infections, and maternal diabetes during pregnancy were also excluded (1, 17).
2.2 Data collection and statistical analysis
Demographic and PCMs characteristics such as age, gender, height, weight, type of PCMs, gene test results, laboratory examination, and imaging examination were collected. Normality tests were used for continuous variables. Normal continuous variables were presented by mean ± SD, and non-normal data were presented by the median and interquartile range (IQR).
2.3 Gene test, data processing and analysis
Peripheral blood genomic DNA was extracted using the QIAamp DNA Blood Mini Kit (Qiagen, Hidden, Germany). DNA quantification and purity testing were performed by Qubit DNA Assay Kit in the NanoDrop 2000 spectrophotometer (Thermo Scientific, USA). Genomic DNA was fragmented by Tagment DNA Enzyme 1 and then subjected to PCR amplification to construct a sequencing library. The Qubit 3.0 Fluorometer was used to detect library concentration. High-throughput sequencing was performed on the Illumina Novaseq 6000 sequencing platform, and the target for average sequencing depth was 200×. All data were compared to the reference sequence using the BWA algorithm. The ANNOVAR tool was used to annotate data. The Genome Analysis Toolkit was used to analyze gene variations. The 1,000genomes, ESP6500, HGMD, and other databases were used for screening and annotation. Tools such as SIFT and Polyphen-2 were used for bioinformatics analysis of missense mutations. Based on the frequency, function, and inheritance mode of various gene variations, literature, and other relevant information, the classification of mutated genes was conducted in accordance with the guidelines established by the American College of Medical Genetics.
Genecards and Drugbank are large public databases that integrate information from various databases and literature, which could list genes and their related electrolytes and AAs. PubMed and Web of Science were used to search for articles to explore the influence of calcium and AAs on CMs. The search keywords were “cardiomyopathy, dilated cardiomyopathy, hypertrophic cardiomyopathy, restrictive cardiomyopathy, arrhythmogenic right ventricular cardiomyopathy, left ventricular noncompaction cardiomyopathy, calcium, amino acids, serine, cysteine, arginine, alanine, and tyrosine”.
2.4 Preliminary exploration of calcium supplements
To evaluate the therapeutic effects of calcium, children with primary DCM admitted from 2019 to 2023 were included. All participants received calcium, and underwent echocardiography both before and after treatment. Three patients used oral calcium carbonate D3 (150–300 mg once a day), and 14 patients used oral calcium lactate (250 mg twice or three times a day, or 500 mg once or twice a day). Normality tests were performed to assess the distribution of data. Paired samples T tests and nonparametric Wilcoxon signed-rank tests were performed for comparison between before and after medication.
3 Results
3.1 Demographic and clinical characteristics
A total of 27 children were enrolled in this study. The demographic and clinical characteristics of children with PCMs were summarized in Tables 1, 2. The demographic and clinical details of each child were provided in Supplementary Table S1. Supplementary Table S2 was involved in the echocardiography indicators of DCM, HCM, and LVNC. Sixteen children were diagnosed with DCM, seven with HCM, and another four with RCM, ARVC, or LVNC.
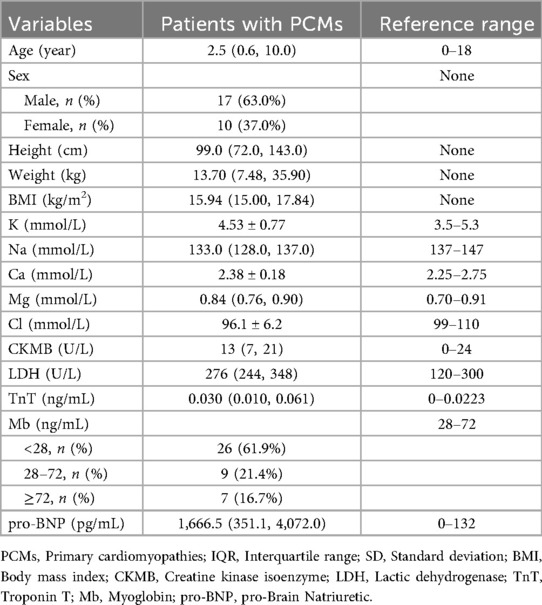
Table 1. Demographic and clinical characteristics of patients with PCMs.
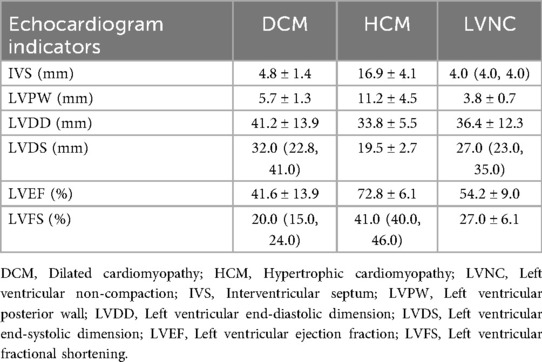
Table 2. The characteristics of echocardiogram indicators for patients with DCM, HCM, and LVNC.
The medians and IQRs of age were 2.5 (0.6, 10.0) years old. There were 17 males (63.0%) and 10 females (37.0%). Body mass index values were lower in these children, with a median and IQR of 15.94 (15.00, 17.84). Among the electrolytes, the median of Na+ (133.0 mmol/L) and the mean of Cl− (96.1 mmol/L) were lower than the normal ranges. Compared with the normal range of 0–132 pg/mL, the pro-brain natriuretic peptide in these children was significantly elevated, with a value of 1,666.5 (351.1, 4,072.0) pg/mL. In children with DCM, the values of LVDD and left ventricular end-systolic dimension (LVDS) increased to 41.2 ± 13.9 mm and 32.0 (22.8, 41.0) mm, respectively. The LVEF was lower than the normal range, with a value of 41.6% ± 13.9%. In children with HCM, the interventricular septum and LV posterior wall exceeded the normal ranges, with values of 16.9 ± 4.1 mm and 11.2 ± 4.5 mm, respectively.
3.2 Mutated gene characteristics
In this study, mutated genes were collected and classified into four categories: pathogenic, likely pathogenic, uncertain significance, and other mutations. The mutated genes found in DCM, HCM, and LVNC are available in Supplementary Table S3. Among them, 28 types of genes were related to calcium and AAs.
In children with DCM, seven genes were identified as pathogenic, such as TTN, MYH7, and TNNT2 (Figure 1a). Sixteen genes were defined as likely pathogenic and of uncertain significance, such as NEXN, SCN5A, TNNI3, MYH6, DSP, and VCL (Figure 1b). There were also some other mutated genes, such as TTN and JPH2.
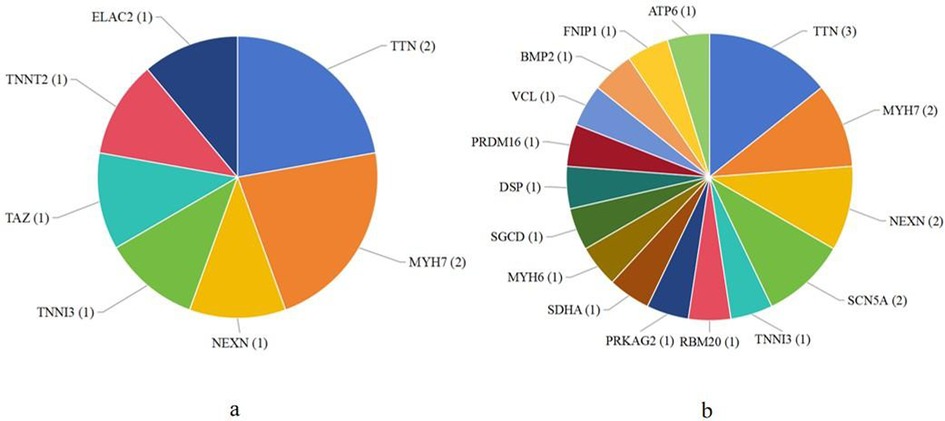
Figure 1. The number of patients related to mutated genes found in dilated cardiomyopathy. (a) The number of patients with pathogenic mutated genes; (b) The number of patients with likely pathogenic and uncertain significance mutated genes.
In children with HCM, three pathogenic mutated genes were detected, such as MYH7, PRKAG2, and RAF1. Fourteen likely pathogenic and uncertain significance mutated genes were identified, such as MYBPC3, RBM20, MYH6, and SLC22A5. TTN was also found as other mutated gene in HCM.
In children with RCM, DES was detected as a pathogenic mutated gene. In the likely pathogenic and/or uncertain significance mutated genes, two genes were found in children with ARVC, and 10 genes were found in children with LVNC.
3.3 Relationship between calcium and AAs and PCMs
Based on gene databases, calcium and 13 types of AAs were found to be associated with mutated genes in this study. Detailed information was provided in Table 3. A total of 21 genes were related to calcium. Among them, eight mutated genes (e.g., TNNI3, TNNT2) were found in DCM, whereas three mutated genes were found in HCM. MYH7 and MYH6 were found in both DCM and HCM. Additionally, DES was associated with RCM. Thirteen types of AAs were found, related to 22 genes. In genes related to DCM, the most frequent AAs were serine (e.g., SCN5A, MYH7), alanine (e.g., SCN5A, TNNT2), tyrosine (e.g., DSP, VCL), arginine (e.g., SCN5A, MYH7), and cysteine (e.g., MYH7, VCL). In genes related to HCM, arginine (e.g., MYBPC3, MYH7), tyrosine (e.g., RAF1, SLC22A5), serine (e.g., MYH7, RAF1), cysteine (e.g., MYH7, RAF1), and alanine (RAF1) were also found. Serine, cysteine, and tyrosine were found to be related to mutated genes in RCM, ARVC, and LVNC. TTN, a gene found in DCM, HCM, RCM, ARVC, and LVNC, was also related to serine and cysteine.
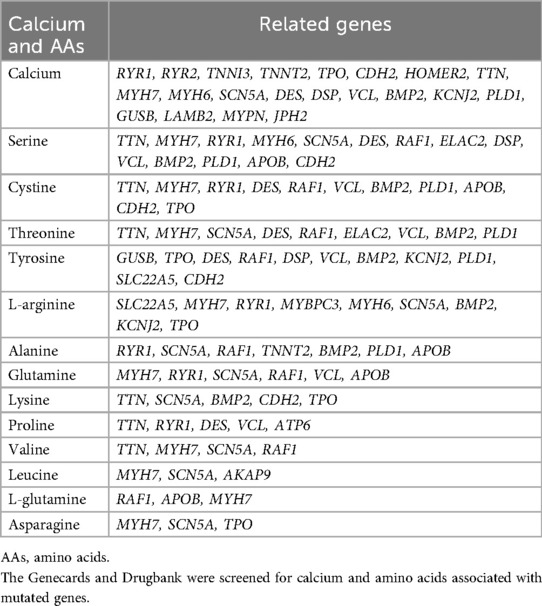
Table 3. Calcium, amino acids, and their related genes.
3.4 Influence of calcium on DCM
The details of children treated with calcium were listed in Supplementary Table S4. The comparison of echocardiographic parameters of DCM before and after using calcium was listed in Table 4. There were significant improvements in LVDD (P = 0.013), LVDS (P = 0.028), LVEF (P = 0.004), and LVFS (P = 0.013) after using calcium. The level of LVDD and LVDS decreased from 39.7 to 33.5 and from 33.5 to 29.0, respectively. The concentration level of LVEF and LVFS increased from 32.7 to 39.6 and from 15.3 to 18.5, respectively.
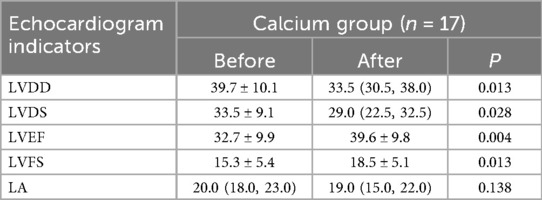
Table 4. Echocardiographic parameters for children with DCM treated with calcium.
4 Discussion
The low incidence and poor progression of CMs result in challenging treatment. Although the medication used for pediatric CMs is similar to adults, such as beta-blockers and calcium channel blockers, the differences in causes, complications, and outcomes are significant between adults and children with CMs (6). In addition, due to the small sample size of pediatric PCMs and limited medication experience, reliable medication for pediatric PCMs is more difficult. Thus, it is significant to explore clinical manifestations and appropriate treatment methods for pediatric PCMs. This is the first study exploring the influence of electrolytes and AAs on pediatric PCMs.
In this study, most of the mutated genes were sarcomere protein genes. Some mutated genes were only found in one phenotype. For example, TNNI3 and TNNT2 were only found in DCM. MYBPC3 and RAF1 were only found in HCM. In addition, one mutated gene could also be associated with distinct phenotypes. For instance, MYH7 and PRKAG2 were found in DCM and HCM. NEXN was found in DCM and LVNC. TTN was found in DCM, HCM, RCM, ARVC, and LVNC.
The mechanisms of CMs induced by various gene mutations are as follows. TTN encodes titin, a giant sarcomere protein that functions as a significant component of the Z-disk (21, 22). In HCM, functional changes in myosin resulting from TTN mutation could increase the binding to α-actinin, whereas there is a decrease in DCM (21). Regarding Z-disk element mutations, several hypotheses indicated that HCM is characterized by a “stiff sarcomere”, whereas DCM is characterized by a “loose sarcomere”. Therefore, the binding among Z-disk elements in HCM and DCM will be tight and loose, respectively (21). MYH7 encodes β-myosin heavy chain, which is an essential part of the sarcomere. In HCM, MYH7 mutations are critical pathogenic causes. A hypothesis indicated that HCM is a sarcomere disease. It arises as a compensatory mechanism for reduced power generation caused by MYH7 mutations (21). In two cases with DCM linked to MYH7 mutations, damaged sarcomere functions were observed (21, 23). Mutations of TNNI3, TNNT2, RAF1, and NEXN could be found in both DCM and HCM. TNNI3 and TNNT2 are common genes encoding sarcomere components, playing a critical role in Ca2+ regulation of muscle contraction and relaxation (24). RAF1 encodes the component of the RAS-mitogen-activated protein kinases pathway, which plays crucial roles in myocardial biology (25). NEXN encodes nexilin, which is a protein of the Z-disk and contributes to its stability (26).
Most of our patients were detected with more than one mutation. It was reported that genetic causes of DCM could be divided into monogenic, polygenic, and multifactorial (e.g., environmental exposures) (5). There is an increasing recognition of polygenic mutations as the cause of HCM rather than monogenic.
Based on previous studies, calcium and some AAs were related to CMs (27–30). This study also detected that calcium and some types of AAs were associated with several genes of enrolled children. Therefore, supplements of calcium and these AAs may be potential therapeutic strategies for pediatric PCMs.
Calcium plays a key role in cardiac contraction coupling and electrophysiological signal conducting. Thus, defective intracellular calcium handling could result in contractile dysfunction, arrhythmia, and cellular hypertrophy (31). Mutated genes associated with sarcomere components (e.g., MYH7, TNNT2, and TNNI3) and Z-disk components (e.g., TTN) in HCM may result in increased Ca2+ sensitivity. Whereas, the situation may be contrary in DCM (21, 32). The mutations of TNNI3 and TNNT2 in DCM could reduce Ca2+ sensitivity and prolong reuptake time, resulting in lower Ca2+ concentration in the systolic stage and reduced myocardial contractility (32). Therefore, for children with DCM, calcium-based agents may serve as a potential therapeutic option. In our study, children given calcium expressed significant improvements in heart function. The average age of these children was one and a half years old. Older children treated with calcium showed relatively poor therapeutic effects from our limited information. Calcium supplements were suitable for younger children (less than or equal to one year old) with DCM. Of course, the effects of the base therapeutics regimen could not be ignored.
Additionally, the binding of Ca2+ to troponin C triggers actomyosin complex activation, contributing to sarcomere shortening and myocardium contraction (33). Increased Ca2+ sensitivity in HCM could increase sarcomere tension and excessive myocardial contraction (21). As a first-in-class selective cardiac myosin inhibitor for HCM, mavacamten reversibly combines with myosin ATPase and decreases activated myosin heads to increase actomyosin dissociation and make myocardium relaxation (34). Aficamten, a next-generation cardiac myosin inhibitor characterized by its shorter half-life and reduced drug-drug interaction, is currently undergoing clinical trials to investigate its potential role in HCM treatment (35).
This study also identified 13 AAs, which were associated with 22 genes detected in children with PCMs. Serine, cysteine, arginine, alanine, and tyrosine were related to genes found in both DCM and HCM. Serine and cysteine were also related to genes found in RCM, ARVC, and LVNC.
Some AAs have been reported to be beneficial to CMs. A study showed that the activated ATF4-dependent serine synthesis pathway could attenuate the DCM phenotype and improve systolic dysfunction by improving mitochondrial respiration and increasing levels of tricarboxylic acid cycle metabolites and ATP (16, 27, 36). In addition, the activated serine and one-carbon metabolism could reduce cardiac hypertrophy and improve ventricular function (37). It was reported that the activated serine and one-carbon pathways could reduce protein oxidation in mitochondria, retain ATP production, and prevent adverse ventricular remodeling in the context of myocardial hypertrophy (16). Additionally, some serine/threonine protein kinases may be beneficial for CMs. For instance, glycogen synthase kinase-3β could inhibit excessive myocardial contraction and suppress the expression of hypertrophy-related genes. Protein kinase G could reduce oxidative stress (38). Cysteine is involved in the synthesis of glutathione (GSH), which is integral to the endogenous protective system of the myocardium. It was reported that cysteine could stimulate the activity and expression of GSH peroxidase to protect against oxidative stress in myocardial cells (15). Furthermore, N-acetylcysteine, the metabolite of cysteine and precursor of GSH, could reverse hypertrophy and diastolic dysfunction in familial HCM (39). Arginine could produce NO, which plays a major role in protecting the myocardium by reducing inflammation in reperfusion and myocardial ischemia and maintaining myocardial contraction function (40, 41). In addition, Arginine could also eliminate oxygen radicals to protect the myocardium (41). Arginine has been suggested as a potential therapeutic option for mitochondrial CM and diabetic CM (42, 43). For this reason, arginine may be beneficial to pediatric PCMs, but the efficacy of arginine needs to be verified in clinical practice. Thus, children with PCMs may benefit from appropriate AA supplementation.
However, the link between cardiac function and two AAs, namely alanine and tyrosine, remains controversial. For example, alanine could exert a dual impact on heart function. On one hand, it was reported that levels of alanine were higher in patients with microvascular disease and lower in those with atherosclerosis (44, 45). On the other hand, it may be due to the inability to infer precise causality in observational studies. The increased level of alanine in CMs may be related to ischemic myocardium rather than being a risk factor for disorders (45). As for tyrosine, its oxidation products may lead to myocardial injury (46). However, another study also indicated that tyrosine could increase contractility in isolated atrial myocardium from patients with heart failure (47). Therefore, the influence of alanine and tyrosine in pediatric PCMs needs to be further studied.
This study has some limitations. Firstly, the current sample size is limited due to the rarity of pediatric PCMs. Larger samples are essential in the future to further explore the distribution of mutated genes in pediatric PCMs. Secondly, this is a single-center retrospective study, and bias in sample selection is unavoidable. Lastly, the influence of calcium and AAs in pediatric PCMs was inferred from genetic characteristics. The present study demonstrated that children who received calcium supplementation exhibited significant improvement in cardiac function. Of course, the efficacy of other potential drugs remains unexplored. Extensive preclinical and clinical validation studies are required to substantiate our findings.
5 Conclusion
Among the mutated genes detected in children with PCMs, 23 genes were found in DCM, while 19 genes were found in HCM. TNNI3 and TNNT2 were only found in DCM, whereas MYBPC3 and RAF1 were only found in HCM. Six types of mutated genes were observed in both DCM and HCM, such as MYH7, RBM20, and PRKAG2. Children with DCM, particularly those with a family history, may benefit from appropriate supplementation of calcium, serine, cysteine, and arginine to decrease morbidity, slow down the progression, and improve prognosis. Serine, cysteine, and arginine may also be beneficial to children with HCM. Additionally, serine and cysteine could also be applied as supplementary medication for children with RCM, LVNC, or ARVC.
Data availability statement
The original data included in this study are presented in Supplementary Material Data Sheet 1. Further inquiries can be directed to Liqin Zhu,MzA4MTkzMjdAbmFua2FpLmVkdS5jbg==.
Ethics statement
The studies involving humans were approved by Ethic Committee of the Tianjin Children's Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required from the participants or the participants' legal guardians/next of kin in accordance with the national legislation and institutional requirements.
Author contributions
SJ: Writing – original draft. AZ: Writing – review & editing. JD: Writing – review & editing. WW: Writing – review & editing. JW: Writing – review & editing. HC: Writing – review & editing. LZ: Writing – review & editing. WL: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. The support of Tianjin Outstanding Health Professional Selection and Training Program (TJSJMYXYC-D2-031).
Acknowledgments
The authors wish to express their gratitude for the clinical data from Tianjin Children's Hospital.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2025.1631632/full#supplementary-material
References
1. Lipshultz SE, Law YM, Asante-Korang A, Austin Eric D, Dipchand Anne I, Everitt Melanie D, et al. Cardiomyopathy in children: classification and diagnosis: a scientific statement from the American Heart Association. Circulation. (2019) 140(1):e9–e68. doi: 10.1161/CIR.0000000000000682
2. Soler R, Rodríguez E, Remuiñán C, Bello MJ, Díaz A. Magnetic resonance imaging of primary cardiomyopathies. J Comput Assist Tomogr. (2003) 27(5):724–34. doi: 10.1097/00004728-200309000-00009
3. Arbelo E, Protonotarios A, Gimeno JR, Gimeno JR, Arbustini E, Barriales-Villa R, et al. 2023 ESC guidelines for the management of cardiomyopathies. Eur Heart J. (2023) 44(37):3503–626. doi: 10.1093/eurheartj/ehad194
4. Japp AG, Gulati A, Cook SA, Cowie MR, Prasad SK. The diagnosis and evaluation of dilated cardiomyopathy. J Am Coll Cardiol. (2016) 67(25):2996–3010. doi: 10.1016/j.jacc.2016.03.590
5. Heymans S, Lakdawala NK, Tschöpe C, Klingel K. Dilated cardiomyopathy: causes, mechanisms, and current and future treatment approaches. Lancet (London, England). (2023) 402(10406):998–1011. doi: 10.1016/S0140-6736(23)01241-2
6. Lee TM, Hsu DT, Kantor P, Towbin JA, Ware SM, Colan SD, et al.. pediatric cardiomyopathies. Circ Res. (2017) 121(7):855–73. doi: 10.1161/CIRCRESAHA.116.309386
7. Curran L, De Marvao A, Inglese P, McGurk KA, Schiratti P-R, Clement A, et al. Genotype-phenotype taxonomy of hypertrophic cardiomyopathy. Circ Genom Precis Med. (2023) 16(6):e004200. doi: 10.1161/CIRCGEN.123.004200
8. Moak JP, Kaski JP. Hypertrophic cardiomyopathy in children. Heart (Br Cardiac Soc). (2012) 98(14):1044–54. doi: 10.1136/heartjnl-2011-300531
9. Ditaranto R, Caponetti AG, Ferrara V, Parisi V, Minnucci M, Chiti C, et al. Pediatric restrictive cardiomyopathies. Front Pediatr. (2021) 9:745365. doi: 10.3389/fped.2021.745365
10. Habib G, Bucciarelli-Ducci C, Caforio AL, Cardim N, Charron P, Cosyns B, et al. Multimodality imaging in restrictive cardiomyopathies: an EACVI expert consensus document in collaboration with the “working group on myocardial and pericardial diseases” of the European society of cardiology endorsed by the Indian academy of echocardiography. Eur Heart J Cardiovasc Imaging. (2017) 18(10):1090–121. doi: 10.1093/ehjci/jex034
11. Towbin JA, Lorts A, Jefferies JL. Left ventricular non-compaction cardiomyopathy. Lancet (London, England). (2015) 386(9995):813–25. doi: 10.1016/S0140-6736(14)61282-4
12. Van Waning JI, Moesker J, Heijsman D, Boersma E, Majoor-Krakauer D. Systematic review of genotype-phenotype correlations in noncompaction cardiomyopathy. J Am Heart Assoc. (2019) 8(23):e012993. doi: 10.1161/JAHA.119.012993
13. Corrado D, Link MS, Calkins H. Arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. (2017) 376(1):61–72. doi: 10.1056/NEJMra1509267
14. Garg A, Azad S, Kumar K, Bhatia M, Radhakrishnan S. Role of cardiac magnetic resonance imaging in hypocalcemia-induced dilated cardiomyopathy in pediatric population. Indian J Radiol Imaging. (2021) 31(4):837–43. doi: 10.1055/s-0041-1740541
15. King N, Lin H, Suleiman MS. Cysteine protects freshly isolated cardiomyocytes against oxidative stress by stimulating glutathione peroxidase. Mol Cell Biochem. (2010) 343(1–2):125–32. doi: 10.1007/s11010-010-0506-6
16. Perea-Gil I, Seeger T, Bruyneel AAN, Termglinchan V, Monte E, Lim EW, et al. Serine biosynthesis as a novel therapeutic target for dilated cardiomyopathy. Eur Heart J. (2022) 43(36):3477–89. doi: 10.1093/eurheartj/ehac305
17. Subspecialty Group of Cardiology, the Society of Pediatrics, Chinese Medical Association; Pediatric Cardiovascular Disease Committee, College of Cardiovascular Physicians, Chinese Medical Doctor Association; Editorial Board, Chinese Journal of Pediatrics. Expert consensus on diagnosis and treatment of dilated cardiomyopathy in children (2024). Zhonghua er ke za zhi = Chin J Ped. (2024) 62(9):811–25. doi: 10.3760/cma.j.cn112140-20240410-00253
18. Elliott PM, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, et al. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy. Kardiol Pol. (2014) 72(11):1054–126. doi: 10.5603/KP.2014.0212
19. Graziano F, Zorzi A, Ungaro S, Bauce B, Rigato I, Cipriani A, et al. The 2023 European task force criteria for diagnosis of arrhythmogenic cardiomyopathy: historical background and review of main changes. Rev Cardiovasc Med. (2024) 25(9):348. doi: 10.31083/j.rcm2509348
20. Jenni R, Oechslin E, Schneider J, Jost CA, Kaufmann P. Echocardiographic and pathoanatomical characteristics of isolated left ventricular non-compaction: a step towards classification as a distinct cardiomyopathy. Heart (Br Cardiac Soc). (2001) 86(6):666–71. doi: 10.1136/heart.86.6.666
21. Kimura A. Molecular etiology and pathogenesis of hereditary cardiomyopathy. Circ J. (2008) 72(Suppl A):A38–48. doi: 10.1253/circj.CJ-08-0050
22. Loescher CM, Hobbach AJ, Linke WA. Titin (TTN): from molecule to modifications, mechanics, and medical significance. Cardiovasc Res. (2022) 118(14):2903–18. doi: 10.1093/cvr/cvab328
23. Fatkin D, Graham RM. Molecular mechanisms of inherited cardiomyopathies. Physiol Rev. (2002) 82(4):945–80. doi: 10.1152/physrev.00012.2002
24. Sheng JJ, Jin JP. TNNI1, TNNI2 and TNNI3: evolution, regulation, and protein structure-function relationships. Gene. (2016) 576(1 Pt 3):385–94. doi: 10.1016/j.gene.2015.10.052
25. Dhandapany PS, Razzaque MA, Muthusami U, Kunnoth S, Edwards JJ, Mulero-Navarro S, et al. RAF1 mutations in childhood-onset dilated cardiomyopathy. Nat Genet. (2014) 46(6):635–9. doi: 10.1038/ng.2963
26. Hassel D, Dahme T, Erdmann J, Meder B, Huge A, Stoll M, et al. Nexilin mutations destabilize cardiac Z-disks and lead to dilated cardiomyopathy. Nat Med. (2009) 15(11):1281–8. doi: 10.1038/nm.2037
27. Chen S, Zou Y, Song C, Cao K, Cai K, Wu Y, et al. The role of glycolytic metabolic pathways in cardiovascular disease and potential therapeutic approaches. Basic Res Cardiol. (2023) 118(1):48. doi: 10.1007/s00395-023-01018-w
28. Li X, He W, Song Q, Ding Q, Zhang X, Zeng Z, et al. The prognostic value of serum calcium levels in elderly dilated cardiomyopathy patients. Glob Heart. (2024) 19(1):25. doi: 10.5334/gh.1304
29. Princen F, Bard E, Sheikh F, Zhang SS, Wang J, Zago WM, et al. Deletion of Shp2 tyrosine phosphatase in muscle leads to dilated cardiomyopathy, insulin resistance, and premature death. Mol Cell Biol. (2009) 29(2):378–88. doi: 10.1128/MCB.01661-08
30. Xu M, Bermea KC, Ayati M, Kim HB, Yang X, Medina A, et al. Alteration in tyrosine phosphorylation of cardiac proteome and EGFR pathway contribute to hypertrophic cardiomyopathy. Commun Biol. (2022) 5(1):1251. doi: 10.1038/s42003-022-04021-4
31. Zaffran S, Kraoua L, Jaouadi H. Calcium handling in inherited cardiac diseases: a focus on catecholaminergic polymorphic ventricular tachycardia and hypertrophic cardiomyopathy. Int J Mol Sci. (2023) 24(4):3365. doi: 10.3390/ijms24043365
32. Robinson P, Sparrow AJ, Patel S, Malinowska M, Reilly SN, Zhang Y-H, et al. Dilated cardiomyopathy mutations in thin-filament regulatory proteins reduce contractility, suppress systolic Ca(2+), and activate NFAT and Akt signaling. Am J Physiol Heart Circ Physiol. (2020) 319(2):H306–h19. doi: 10.1152/ajpheart.00272.2020
33. Alves ML, Gaffin RD, Wolska BM. Rescue of familial cardiomyopathies by modifications at the level of sarcomere and Ca2+ fluxes. J Mol Cell Cardiol. (2010) 48(5):834–42. doi: 10.1016/j.yjmcc.2010.01.003
34. Braunwald E, Saberi S, Abraham TP, Elliott PM, Olivotto I. Mavacamten: a first-in-class myosin inhibitor for obstructive hypertrophic cardiomyopathy. Eur Heart J. (2023) 44(44):4622–33. doi: 10.1093/eurheartj/ehad637
35. Sykuta A, Yoon CH, Baldwin S, Rine NI, Young M, Smith A. Cardiac myosin inhibitors: expanding the horizon for hypertrophic cardiomyopathy management. Ann Pharmacother. (2024) 58(3):273–85. doi: 10.1177/10600280231180000
36. Fernández-Ruiz I. Modulating serine biosynthesis for the treatment of dilated cardiomyopathy. Nat Rev Cardiol. (2022) 19(9):575. doi: 10.1038/s41569-022-00751-4
37. Padrón-barthe L, Villalba-orero M, Gómez-Salinero JM, Acín-Pérez R, Cogliati S, López-Olañeta M, et al. Activation of serine one-carbon metabolism by calcineurin Aβ1 reduces myocardial hypertrophy and improves ventricular function. J Am Coll Cardiol. (2018) 71(6):654–67. doi: 10.1016/j.jacc.2017.11.067
38. Wu Y, Zou Y, Song C, Cao K, Cai K, Chen S, et al. The role of serine/threonine protein kinases in cardiovascular disease and potential therapeutic methods. Biomed Pharmacother. (2024) 177:117093. doi: 10.1016/j.biopha.2024.117093
39. Wilder T, Ryba DM, Wieczorek DF, Wolska BM, Solaro RJ. N-acetylcysteine reverses diastolic dysfunction and hypertrophy in familial hypertrophic cardiomyopathy. Am J Physiol Heart Circ Physiol. (2015) 309(10):H1720–30. doi: 10.1152/ajpheart.00339.2015
40. Sato H, Zhao ZQ, McGee DS, Williams MW, Hammon JW Jr, Vinten-Johansen J. Supplemental L-arginine during cardioplegic arrest and reperfusion avoids regional postischemic injury. J Thorac Cardiovasc Surg. (1995) 110(2):302–14. doi: 10.1016/S0022-5223(95)70226-1
41. Lass A, Suessenbacher A, Wölkart G, Mayer B, Brunner F. Functional and analytical evidence for scavenging of oxygen radicals by L-arginine. Mol Pharmacol. (2002) 61(5):1081–8. doi: 10.1016/S0026-895X(24)12201-8
42. Fiordelisi A, Cerasuolo FA, Avvisato R, Buonaiuto A, Maisto M, Bianco A, et al. L-Arginine supplementation as mitochondrial therapy in diabetic cardiomyopathy. Cardiovasc Diabetol. (2024) 23(1):450. doi: 10.1186/s12933-024-02490-x
43. Ikawa M, Yoneda M, Tanaka M. Energy states in mitochondrial cardiomyopathy. in vivo functional imaging and L-arginine therapy. Circ J. (2010) 74(12):2560–1. doi: 10.1253/circj.CJ-10-1062
44. Deidda M, Piras C, Cadeddu Dessalvi C, Congia D, Locci E, Ascedu F, et al. Blood metabolomic fingerprint is distinct in healthy coronary and in stenosing or microvascular ischemic heart disease. J Transl Med. (2017) 15(1):112. doi: 10.1186/s12967-017-1215-7
45. Hu S, Lin Z, Hu MJ, Tan J-S, Guo T-T, Huang X, et al. Causal relationships of circulating amino acids with cardiovascular disease: a trans-ancestry Mendelian randomization analysis. J Transl Med. (2023) 21(1):699. doi: 10.1186/s12967-023-04580-y
46. Zhang H, Yang Y, Yan B, Tang X, Cheng X, Le G, et al. Dityrosine administration induces myocardium injury and inflammatory response in mice. Wei Sheng Yan Jiu. (2018) 47(3):345–51. doi: 10.19813/j.cnki.weishengyanjiu.2018.03.001
Keywords: pediatric primary cardiomyopathies, mutated genes, calcium, amino acids, exploration
Citation: Jiang S, Zhang A, Deng J, Wang W, Wang J, Chen H, Zhu L and Liu W (2025) Exploration of calcium and amino acids for children with primary cardiomyopathies based on genetic characteristics. Front. Pediatr. 13:1631632. doi: 10.3389/fped.2025.1631632
Received: 20 May 2025; Accepted: 20 October 2025;
Published: 18 November 2025.
Edited by:
Emanuele Bobbio, Sahlgrenska University Hospital, SwedenReviewed by:
Pankaj Kumar Chauhan, University of Texas MD Anderson Cancer Center, United StatesFederica Conte, University of Twente, Netherlands
Copyright: © 2025 Jiang, Zhang, Deng, Wang, Wang, Chen, Zhu and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liqin Zhu, MzA4MTkzMjdAbmFua2FpLmVkdS5jbg==; Wei Liu, bGFuY2UxOTcxQDE2My5jb20=