 Lina Almohammadi
Lina Almohammadi Lama Alsayel
Lama Alsayel Mohammad Aljumaa
Mohammad Aljumaa Raghad Alhuthil
Raghad Alhuthil Afaf Alsagheir
Afaf Alsagheir- 1College of Medicine, Alfaisal University, Riyadh, Saudi Arabia
- 2Section of Pediatric Endocrinology, Department of Pediatrics, King Faisal Specialist Hospital and Research Centre, Riyadh, Saudi Arabia
Selenocysteine insertion sequence-binding protein 2 (SECISBP2) is crucial for the biosynthesis of selenoproteins, including iodothyronine deiodinases, which play a vital role in thyroid hormone metabolism. Mutations in SECISBP2 can disrupt thyroid function, leading to various clinical manifestations across multiple systems. We present the case of a 3-year-old Saudi female who was referred for genetic testing due to poor growth, developmental abnormalities, and notable facial dysmorphism. Laboratory tests indicated elevated FT4 levels, low T3 levels, and modestly elevated TSH values. Whole-exome sequencing revealed a homozygous pathogenic variant in the SECISBP2 gene (c.358C>T; p.Arg120Ter), correlating with the laboratory findings and the patient's clinical presentation. Additional variants of uncertain significance (VUS) in the ARCN1 and DNA2 genes were also identified but were considered clinically insignificant due to inheritance patterns and the absence of corresponding phenotypes in heterozygous family members. Treatment with liothyronine (L-T3) led to significant clinical improvement in growth and energy levels over a two-year follow-up period. This case highlights the importance of identifying the specific biochemical profile associated with SECISBP2 deficiency and advocates for the inclusion of SECISBP2 in genetic testing panels for endocrine and neurodevelopmental disorders to prevent diagnostic delays. The therapeutic efficacy of liothyronine in such cases is further supported.
1 Introduction
The human body expresses 25 selenoproteins, three of which—iodothyronine deiodinases—are essential for thyroid hormone metabolism and the conversion of thyroxine (T4) to triiodothyronine (T3) (1). Selenocysteine insertion sequence-binding protein 2 (SECISBP2) is crucial for facilitating the incorporation of selenocysteine into these proteins by recoding the UGA stop codon within the 3′-untranslated region of selenoprotein mRNAs (2–4). Mutations in SECISBP2 disrupt this process, leading to abnormal selenoprotein production and a range of multisystem manifestations, including growth delay, developmental disabilities, and thyroid hormone abnormalities (2).
Biochemically, SECISBP2 deficiency is characterized by a distinct thyroid profile: elevated or normal TSH levels, high serum T4, low serum T3, and elevated reverse T3, which result from impaired activity of the three deiodinases (4, 5). The clinical phenotype can be complex, demonstrating variable neurological, skeletal, and endocrine involvement.
This report presents the clinical and molecular findings of a pediatric patient with a homozygous pathogenic SECISBP2 mutation, emphasizing the diagnostic challenges and therapeutic responses observed. The clinical course highlights the necessity of focusing on SECISBP2-related thyroid dysfunction while considering the potential—though not definitive—contributions of coexisting variants.
2 Case description
from a local hospital in the Al-Hasa region (eastern Saudi Arabia) in December 2019 for further genetic evaluation due to unexplained growth retardation, developmental delay, and dysmorphological features.
She was born full-term via lower-segment cesarean section following an uneventful pregnancy. The neonatal period was unremarkable, and she was discharged on the same day as her mother. However, developmental concerns became apparent during infancy. She was able to sit independently by 10 months, walked at 1 year and 11 months, and exhibited delays in speech along with mild intellectual disability. Additional concerns included poor weight gain, limited oral intake, and diffuse hair loss.
On physical examination, the patient's weight was 8.4 kg [−5.81 standard deviation scores (SDS)], height was 78 cm (−4.54 SDS), BMI was 13.8 (below the 4th percentile), and head circumference was 38 cm (below the 3rd percentile) (Figure 1). Dysmorphic features included microcephaly, bilateral ptosis, refractive error (for which she wore glasses), large protruding ears, and a small nose, hands, and feet. The examination revealed no gross skeletal anomalies. She was the first child of consanguineous parents (first-degree cousins), both of whom are healthy and medically free. She has a healthy younger brother aged 7 months. The mid-parental height was 162 cm, and the patient's projected adult height fell significantly below this target, further indicating potential pathological growth failure.
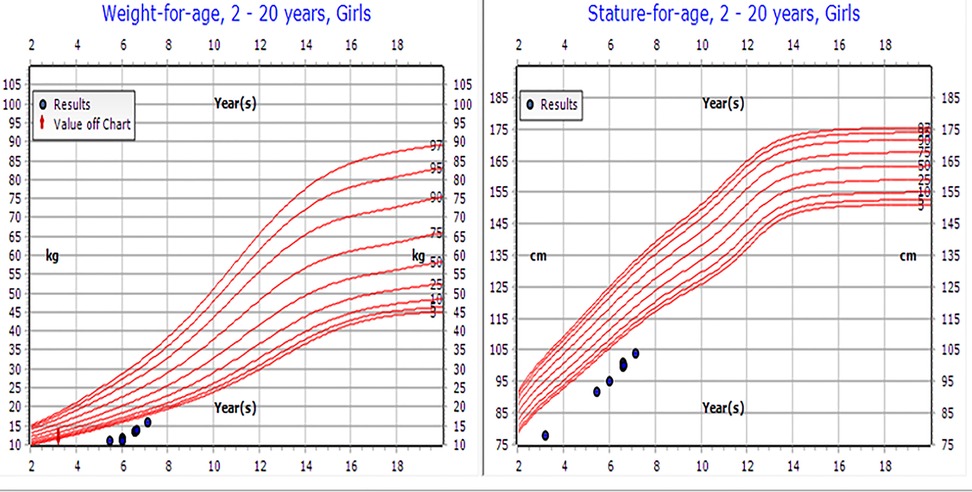
Figure 1. Patient's growth chart.
2.1 Investigation
Initial laboratory testing revealed thyroid function abnormalities, including elevated FT4 levels (33.5 pmol/L; reference range: 12–22), low T3 levels (1.1 nmol/L; reference range: 1.3–3.1), and mildly elevated TSH levels (5.32 mU/L; reference range: 0.27–4.2) (Table 1). These findings were indicative of impaired peripheral conversion of T4 to T3.
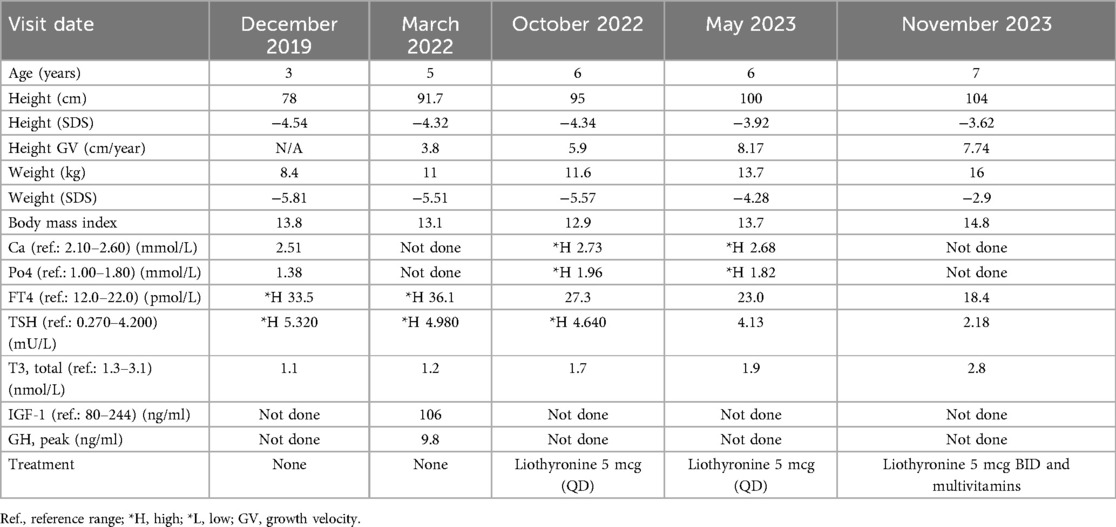
Table 1. Patient information during follow-up visits.
Whole-exome sequencing and deletion/duplication analysis were performed on DNA extracted from peripheral blood. A homozygous pathogenic variant in the SECISBP2 gene (NM_024077.5): c.358C>T; p.(Arg120Ter) was identified. Additionally, a homozygous VUS in the ARCN1 gene (NM_001655.5): c.1004A>G; p.(Lys335Arg) was detected, along with a heterozygous VUS in the DNA2 gene (NM_001080449.3): c.3084G>T; p.(Lys1028Asn) (Figure 2 and Table 2).

Figure 2. Family pedigrees of the affected patient with identified mutations: (a) homozygous SECISBP2 (NM_024077.5):c.358C>T; p.(Arg120Ter). (b) homozygous ARCN1 (NM_001655.5):c.1004A>G; p.(Lys335Arg). (c) heterozygous DNA2 (NM_001080449.3):c.3084G>T; p.(Lys1028Asn).
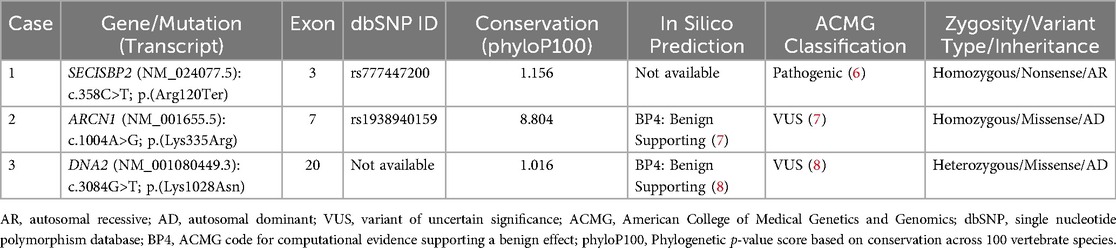
Table 2. Summary of reported genetic variants and predicted impact.
Segregation analysis indicated that the 7-month-old brother was homozygous for the ARCN1 variant but exhibited no clinical symptoms. The father was heterozygous for both the SECISBP2 and ARCN1 variants, while the mother carried heterozygous variants in SECISBP2, ARCN1, and DNA2. The DNA2 variant was also identified in the mother in a heterozygous state, and she showed no phenotype consistent with mitochondrial or myopathic disease.
Given the biochemical and clinical phenotype, along with the inheritance pattern, the SECISBP2 mutation was considered causative. The ARCN1 and DNA2 variants did not align with the patient's clinical presentation and are more likely to be benign or only contributory in an oligogenic context.
The patient was lost to in-person follow-up during the COVID-19 pandemic in 2020 and 2021 but continued to be monitored through virtual consultations. At age 5 (March 2022), she presented with fatigue, hypersomnia (sleeping up to 14–16 h), constipation, and recurrent hair loss. Biochemical testing indicated persistent low T3 levels, elevated FT4 and TSH levels, and mildly elevated serum calcium and phosphate levels (Table 1). Her dietary intake included daily milk with cornflakes, which may have affected her mineral levels.
A neck ultrasound revealed a non-visualized left thyroid lobe (Figure 3). A pituitary MRI was conducted to exclude central causes of growth failure and thyroid dysfunction; the imaging results were unremarkable. A dysmorphic skeletal survey showed anterolisthesis of S2 over S3 (Figure 4), but no other significant abnormalities were noted. Bone age, assessed through a hand radiograph, was estimated at 28 months, despite her chronological age of 5 years, indicating delayed skeletal maturation.
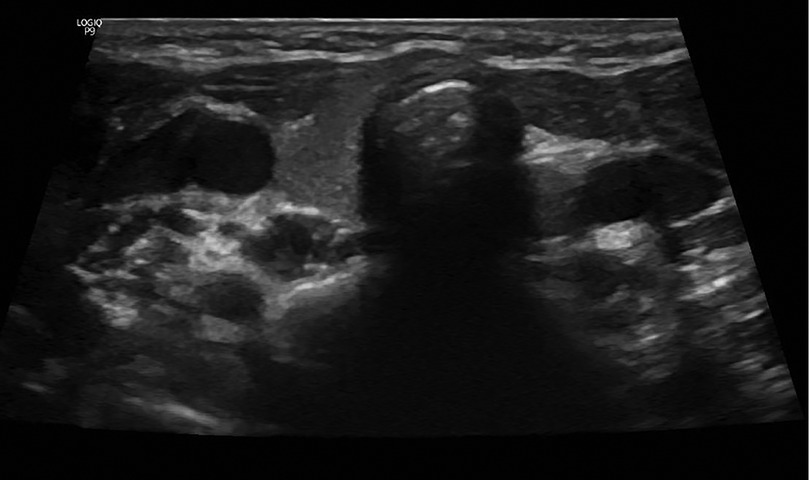
Figure 3. Neck ultrasound showing a non-visualized left thyroid lobe.
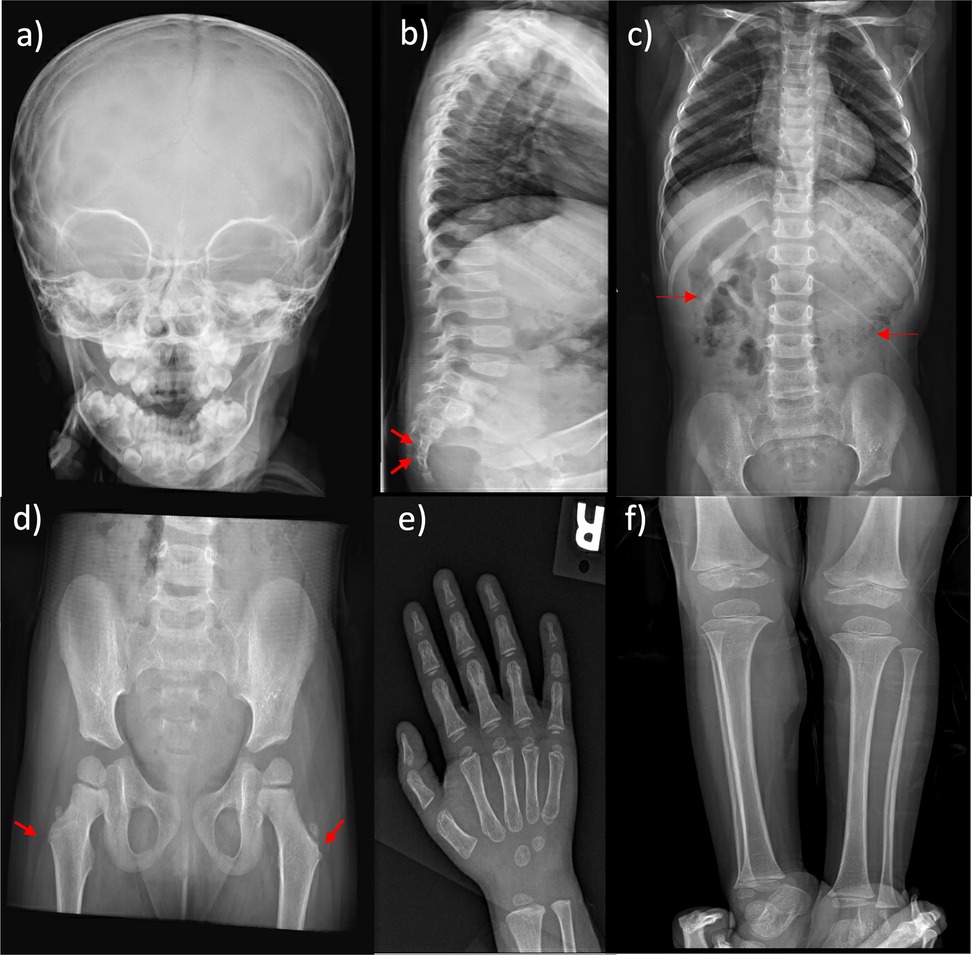
Figure 4. Skeletal survey at 4 years of age. (a) The skull exhibits patent sutures and an unremarkable skull base. (b) The spine shows more than 50% anterolisthesis of S2 over S3 (arrows). Vertebral alignment, height, and disc spaces in the cervical, thoracic, and lumbar regions are within normal limits, with no segmentation or formation anomalies. (c) The abdomen reveals the presence of fecal matter throughout the colon (arrows) and a rounded calcific density over the right 11th rib on the anteroposterior view. (d) The pelvis exhibits mild bilateral coxa valga (arrows), whereas the rest of the bony pelvis is unremarkable. (e,f) Upper and lower extremities show no skeletal dysmorphic features.
2.2 Treatment
The patient was started on liothyronine (Cytomel) at a dosage of 5 mcg once daily (QD) to address her persistent low T3 levels and normalize thyroid function. Over the following months, there was significant improvement in clinical symptoms, including constipation, fatigue, and hair loss. The dosage was subsequently increased to 5 mcg twice daily (BID) for maintenance. Additionally, a multivitamin supplement was prescribed to help prevent potential micronutrient deficiencies.
2.3 Outcome and follow-up
At the most recent follow-up in November 2023, the patient, now 7 years old, demonstrated significant clinical improvement. Her weight increased from −5.81 to −2.9 SDS, and her height improved from −4.54 to −3.62 SDS. Her annual growth velocity reached 8 cm per year. Although her bone age remained delayed, the enhancements in height and energy levels indicate a positive response to liothyronine therapy (see Table 1 and Figure 1). Moreover, her cognitive and speech development has remained stable.
3 Discussion
This report presents a complex genetic case involving a homozygous pathogenic mutation in the SECISBP2 gene (6), along with two additional VUS in the ARCN1 (7) and DNA2 genes (8). Among these, the SECISBP2 variant (c.358C>T; p.Arg120Ter) provides the most compelling molecular explanation for the patient's biochemical and clinical phenotype. SECISBP2 is essential for selenoprotein synthesis, and its disruption impairs the function of deiodinases necessary for thyroid hormone metabolism. This results in a characteristic biochemical profile that includes elevated or normal TSH, elevated T4, low T3, and elevated reverse T3 (1–3). Clinical manifestations can include growth failure, delayed bone maturation, neurodevelopmental delays, and dysmorphic features.
Although the prevalence of SECISBP2 mutations is currently unknown, they have been reported across various populations (2, 5, 9–14). The homozygous nonsense mutation identified in our patient, c.358C>T (p.Arg120Ter), is currently considered a pathogenic variant (14). This same mutation was observed in a Brazilian girl with delayed bone age, congenital myopathy, and neurological impairment who carried compound heterozygous SECISBP2 mutations (11). Another case described a girl of Guinean descent who presented with hypotonia and feeding difficulties while carrying the same homozygous variant (12). In both instances, the biochemical profiles were similar, and while L-T3 therapy improved some systemic manifestations, neurocognitive deficits persisted.
Other reported genotypes, such as a homozygous c.382C>T (p.Arg128Ter) mutation in an African boy, presented only with short stature and delayed bone age, despite being clinically euthyroid. This suggests phenotypic variability that may be linked to alternative start codons or residual protein activity (13). Moreover, Stoupa et al. emphasized the heterogeneity of SECISBP2 presentations, which can include absence of speech, seizures, and features of autism spectrum disorder across six unrelated families. Some patients experienced misdiagnosis or inappropriate treatment due to misinterpretation of thyroid function tests and the exclusion of SECISBP2 from neurodevelopmental gene panels (12). These findings underscore the critical importance of early recognition and comprehensive genetic screening in children who present with multisystem symptoms and atypical thyroid function profiles.
In addition to the pathogenic SECISBP2 mutation, our patient carried a homozygous missense variant in ARCN1 (c.1004A>G; p.Lys335Arg) and a heterozygous missense variant in DNA2 (c.3084G>T; p.Lys1028Asn). While mutations in ARCN1 have been associated with craniofacial dysmorphism and developmental delay (15), and DNA2 variants with mitochondrial myopathy and external ophthalmoplegia (16, 17), neither variant was deemed causative in this case due to several reasons. The ARCN1 variant, for instance, was found to be homozygous in the patient's phenotypically normal 7-month-old brother, while the heterozygous DNA2 variant was inherited from the asymptomatic mother. Additionally, both variants are missense mutations that had not been previously reported as pathogenic in population or clinical databases, and in silico prediction tools classified them as benign (6, 7).
Taken together with their autosomal dominant inheritance patterns, these findings suggest limited clinical relevance of the ARCN1 and DNA2 variants in this case. However, the potential for an oligogenic contribution cannot be entirely ruled out. Interactions between genes may influence the severity or range of SECISBP2-related phenotypes, which is a possibility that warrants further investigation.
Regarding management, there are currently no standardized treatment guidelines for SECISBP2 deficiency. Our patient demonstrated significant clinical improvement in both growth and energy levels following treatment with liothyronine (L-T3). While bone age remained delayed, there was evidence of catch-up growth, and thyroid hormone levels gradually returned to normal. These findings support previous research indicating that L-T3 can alleviate symptoms, although neurodevelopmental progress may be limited if treatment is started late. It is essential to include SECISBP2 in gene panels for thyroid dysfunction and developmental delay to avoid diagnostic delays and inappropriate treatment.
In conclusion, this case illustrates the value of considering SECISBP2 deficiency in pediatric patients who present with growth retardation and abnormal thyroid function tests. While additional genetic variants may be identified through whole-exome sequencing, their interpretation should take into account the clinical context, family segregation, population frequency, and in silico predictions. Timely diagnosis and personalized management can lead to significantly improved outcomes for affected individuals.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by King Faisal Specialist Hospital and Research Centre. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants' legal guardians/next of kin. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
LA: Writing – original draft, Data curation, Investigation. LA: Data curation, Investigation, Writing – original draft. MA: Investigation, Writing – original draft, Data curation. RA: Investigation, Data curation, Writing – original draft. AA: Supervision, Conceptualization, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Fu J, Korwutthikulrangsri M, Gönç EN, Sillers L, Liao XH, Alikaşifoğlu A, et al. Clinical and molecular analysis in 2 families with novel compound heterozygous SBP2 (SECISBP2) mutations. J Clin Endocrinol Metab. (2020) 105:e6–11. doi: 10.1210/clinem/dgz169
2. Çatli G, Fujisawa H, Kirbiyik Ö, Mimoto MS, Gençpinar P, Özdemir TR, et al. A novel homozygous selenocysteine insertion sequence binding protein 2 (SECISBP2, SBP2) gene mutation in a turkish boy. Thyroid. (2018) 28:1221–3. doi: 10.1089/thy.2018.0015
3. Lee KW, Shin Y, Lee S, Lee S. Inherited disorders of thyroid hormone metabolism defect caused by the dysregulation of selenoprotein expression. Front Endocrinol. (2022) 12:803024. doi: 10.3389/fendo.2021.803024
4. Seeher S, Atassi T, Mahdi Y, Carlson BA, Braun D, Wirth EK, et al. SECISBP2 is essential for embryonic development and enhances selenoprotein expression. Antioxid Redox Signal. (2014) 21:835–49. doi: 10.1089/ars.2013.5358
5. Dumitrescu AM, Liao XH, Abdullah MS, Lado-Abeal J, Majed FA, Moeller LC, et al. Mutations in SECISBP2 result in abnormal thyroid hormone metabolism. Nat Genet. (2005) 37:1247–52. doi: 10.1038/ng1654
6. National Center for Biotechnology Information. ClinVar; [VCV0001323572.3] NM_024077.5(SECISBP2):c.358C>T (p.Arg120Ter). Bethesda (MD): National Library of Medicine (US) (2021). Available online at: https://www.ncbi.nlm.nih.gov/clinvar/variation/1323572/ (Accessed August 3, 2025)
7. VarSome. Variant NM_001655.5:c.1004A>G [Internet]. Lausanne (Switzerland): Saphetor SA (2023). c2018–2025. Available online at: https://varsome.com/variant/hg38/NM_001655.5%3Ac.1004A%3EG?annotation-mode=germline (Accessed August 3, 2025)
8. VarSome. Variant NM_001080449.3:c.3084G>T [Internet]. Lausanne (Switzerland): Saphetor SA (2023). c2018–2025. Available online at: https://varsome.com/variant/hg38/DNA2(NM_001080449.3)%3Ac.3084G%3ET?annotation-mode=germline (Accessed August 3, 2025)
9. Dumitrescu AM, Cosmo CD, Liao XH, Weiss RE, Refetoff S. The syndrome of inherited partial SBP2 deficiency in humans. Antioxid Redox Signal. (2010) 12:905–20. doi: 10.1089/ars.2009.2892
10. Schoenmakers E, Agostini M, Mitchell C, Schoenmakers N, Papp L, Rajanayagam O, et al. Mutations in the selenocysteine insertion sequence–binding protein 2 gene lead to a multisystem selenoprotein deficiency disorder in humans. J Clin Invest. (2010) 120(12):4220–35. doi: 10.1172/JCI43653
11. Hamajima T, Mushimoto Y, Kobayashi H, Saito Y, Onigata K. Novel compound heterozygous mutations in the SBP2 gene: characteristic clinical manifestations and the implications of GH and triiodothyronine in longitudinal bone growth and maturation. Eur J Endocrinol. (2012) 166(4):757–64. doi: 10.1530/EJE-11-0812
12. Azevedo MF, Barra GB, Naves LA, Ribeiro Velasco LF, Godoy Garcia Castro P, de Castro LC, et al. Selenoprotein-related disease in a young girl caused by nonsense mutations in the SBP2 gene. J Clin Endocrinol Metab. (2010) 95:4066–71. doi: 10.1210/jc.2009-2611
13. Stoupa A, Franca MM, Abdulhadi-Atwan M, Fujisawa H, Korwutthikulrangsri M, Marchand I, et al. Severe neurodevelopmental phenotype, diagnostic, and treatment challenges in patients with SECISBP2 deficiency. Genet Med. (2024) 26(12):101280. doi: 10.1016/j.gim.2024.101280
14. Di Cosmo C, McLellan N, Liao XH, Khanna KK, Weiss RE, Papp L, et al. Clinical and molecular characterization of a novel selenocysteine insertion sequence-binding protein 2 (SBP2) gene mutation (R128X). J Clin Endocrinol Metab. (2009) 94:4003–9. doi: 10.1210/jc.2009-0686
15. Izumi K, Brett M, Nishi E, Drunat S, Tan ES, Fujiki K, et al. ARCN1 mutations cause a recognizable craniofacial syndrome due to COPI-mediated transport defects. Am J Hum Genet. (2016) 99:451–9. doi: 10.1016/j.ajhg.2016.06.011
16. Ronchi D, Di Fonzo A, Lin W, Bordoni A, Liu C, Fassone E, et al. Mutations in DNA2 link progressive myopathy to mitochondrial DNA instability. Am J Hum Genet. (2013) 92:293–300. doi: 10.1016/j.ajhg.2012.12.014
Keywords: SECISBP2 gene, thyroid function, dysmorphic features, short stature, Saudi Arabia
Citation: Almohammadi L, Alsayel L, Aljumaa M, Alhuthil R and Alsagheir A (2025) Case Report: A homozygous selenocysteine insertion sequence-binding protein 2 (SECISBP2) gene mutation in a pediatric patient. Front. Pediatr. 13:1637116. doi: 10.3389/fped.2025.1637116
Received: 28 May 2025; Accepted: 11 August 2025;
Published: 22 August 2025.
Edited by:
Ronald Cohen, The University of Chicago, United StatesReviewed by:
Alexandra Dumitrescu, University of Chicago Medical Center, United StatesErdal Kurnaz, Ministry of Health, Türkiye
Copyright: © 2025 Almohammadi, Alsayel, Aljumaa, Alhuthil and Alsagheir. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Afaf Alsagheir, QVNhZ2hlaXJAa2ZzaHJjLmVkdS5zYQ==