 Hoon Seok Kim
Hoon Seok Kim Myungshin Kim
Myungshin Kim Jin-Soon Suh3
Jin-Soon Suh3- 1Department of Laboratory Medicine, Seoul St. Mary’s Hospital, College of Medicine, The Catholic University of Korea, Seoul, Republic of Korea
- 2Catholic Genetic Laboratory Center, Seoul St. Mary’s Hospital, College of Medicine, The Catholic University of Korea, Seoul, Republic of Korea
- 3Department of Pediatrics, Bucheon St. Mary’s Hospital, College of Medicine, The Catholic University of Korea, Seoul, Republic of Korea
- 4Department of Pediatrics, Seoul St. Mary’s Hospital, College of Medicine, The Catholic University of Korea, Seoul, Republic of Korea
Diagnosing Alport syndrome can be particularly challenging when targeted sequencing methods, such as panel-based next-generation sequencing (NGS), fail to identify pathogenic variants, especially deep intronic mutations. The syndrome is caused by mutations in type IV collagen genes (COL4A3, COL4A4, or COL4A5), with X-linked Alport syndrome (XLAS) accounting for approximately 80% of cases. Here, we report the case of a 4-year-old boy who presented with persistent microscopic hematuria detected during routine urinalysis. Although renal ultrasonography showed mild bilateral medullary nephrocalcinosis, no proteinuria was observed. His mother had been previously diagnosed with Alport syndrome by renal biopsy, but prior targeted sequencing failed to identify any disease-causing variants. To avoid an invasive renal biopsy in this pediatric patient, we directly performed whole genome sequencing (WGS), identifying a novel deep intronic hemizygous variant in the COL4A5 gene (c.2395 + 2723T > G). This variant, classified as a variant of uncertain significance (VUS) according to ACMG-AMP guidelines, was confirmed by Sanger sequencing to be hemizygous in the patient and heterozygous in his mother. The patient currently maintains normal renal function, vision, and hearing, with only microscopic hematuria persisting. This case highlights the diagnostic challenges posed by deep intronic variants in XLAS and demonstrates the clinical utility of WGS in cases where conventional genetic testing is inconclusive. Early genetic diagnosis facilitated timely intervention without requiring invasive procedures, emphasizing the growing role of comprehensive genomic sequencing in uncovering elusive genetic variants in clinically suspected Alport syndrome.
Introduction
Alport syndrome is an inherited kidney disorder characterized by progressive glomerular disease, often accompanied by sensorineural hearing loss and ocular abnormalities (1). This rare genetic condition affects approximately 1 in 50,000 individuals across all ethnicities and populations, representing the second most common monogenic kidney disease and accounting for 0.5% of adult and 1.7% of pediatric end-stage kidney disease cases in the United States (2).
First reported by Arthur Cecil Alport in 1927 as “Hereditary Familial Congenital Hemorrhagic Nephritis,” the diagnostic criteria for Alport syndrome have evolved over time (3). Historically, strict clinical criteria were required for diagnosis, including characteristic renal biopsy findings, sensorineural hearing loss, ocular abnormalities, and persistent hematuria and renal dysfunction, often accompanied by a relevant family history (4, 5). However, recent expert consensus guidelines recommend genetic confirmation as essential, defining Alport syndrome more broadly to include all individuals harboring pathogenic variants in COL4A3, COL4A4, or COL4A5, regardless of their clinical presentation (6–8). Although molecular technologies, such as direct sequencing and targeted panel-based next-generation sequencing (NGS), have become increasingly utilized due to their diagnostic reliability and non-invasiveness, these methods have limitations. Notably, they typically do not detect variants located in deep intronic or regulatory regions, potentially resulting in missed diagnoses (9–12).
Genetically, Alport syndrome is caused by pathogenic variants in type IV collagen genes (COL4A3, COL4A4, and COL4A5), which lead to abnormalities in the glomerular basement membrane resulting in kidney dysfunction and progressive renal failure (13, 14). The condition follows four primary inheritance patterns, with X-linked Alport syndrome (XLAS) being the most common, accounting for approximately 80% of cases due to mutations in the COL4A5 gene (15). XLAS primarily affects males, while female carriers are at risk of developing hematuria and renal failure. The remaining cases consist of autosomal recessive Alport syndrome (ARAS, 15%), autosomal dominant Alport syndrome (ADAS, 5%), and, very rarely, digenic inheritance (16, 17). While molecular genetic testing is recommended when Alport syndrome is clinically suspected, cost and accessibility considerations necessitate careful selection of testing methods and timing.
Importantly, emerging evidence indicates that deep intronic variants, which create cryptic splice sites or cause pseudo-exon inclusion, may account for cases undiagnosed by conventional exome sequencing (12). This suggests the inherent limitations of conventional NGS panel sequencing or exome sequencing approaches, underscoring the increasing importance of whole genome sequencing (WGS) in clinical diagnostics (9, 18).
In this study, we present a pediatric case of XLAS, where a novel deep intronic variant in the COL4A5 gene was successfully identified using WGS, underscoring the diagnostic complexities and clinical implications of such elusive genetic alterations.
Case description
A 4-year-old boy presented to our outpatient clinic with persistent microscopic hematuria initially detected during routine health screening urinalysis. At first, the hematuria was presumed to be due to cystitis, leading to an initial antibiotic treatment. However, microscopic hematuria persisted despite the absence of pyuria or bacteriuria, prompting referral to the pediatric nephrology clinic for further evaluation. Clinical examination revealed no proteinuria, hearing impairment, or other noticeable symptoms. Laboratory evaluation revealed a hemoglobin level of 11.8 g/dl, white blood cell count of 11,350/μl, platelet count of 532,000/μl, blood urea nitrogen of 11.2 mg/dl, serum creatinine of 0.41 mg/dl (estimated GFR 106 ml/min/1.73 m²), serum albumin of 4.1 g/dl, and total cholesterol of 135 mg/dl. Urinalysis confirmed significant microscopic hematuria (RBC >100/HPF) with a urine calcium/creatinine ratio of 0.022 mg/mg. Renal ultrasound identified grade I medullary nephrocalcinosis bilaterally (Figure 1), though kidney size and structure remained normal.
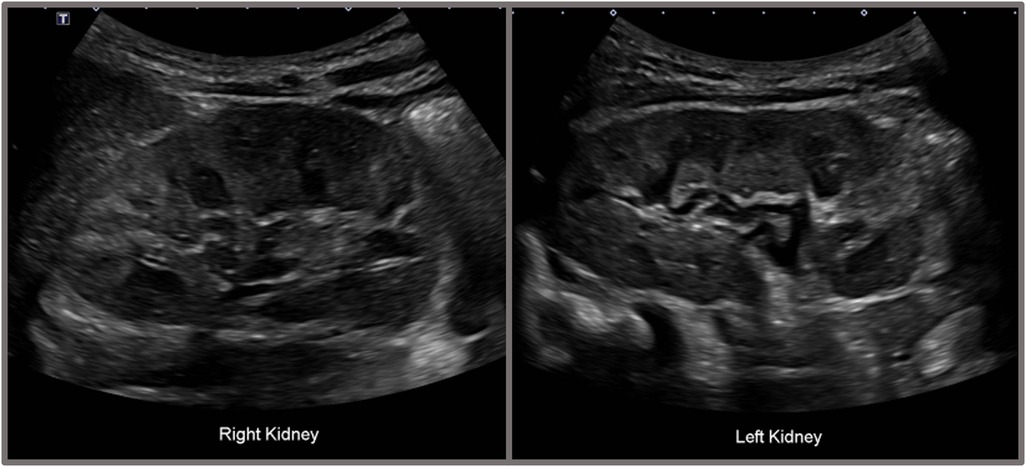
Figure 1. Renal ultrasonography of the patient showing bilateral grade I medullary nephrocalcinosis. Renal ultrasonography of the patient demonstrated bilateral grade I medullary nephrocalcinosis. The right kidney measured 7.8 × 3.7 cm, and the left kidney measured 7.3 × 3.8 cm, both within normal size limits. The cortical echogenicity of both kidneys was normal. Multiple hyperechogenic foci were observed in the medullary regions of both kidneys, and some medullary pyramids exhibited partial hyperechoic rims.
The patient's mother had been diagnosed with Alport syndrome approximately ten years earlier at the age of 25, following incidental detection of proteinuria and hematuria during routine screening. Her urinalysis at the time revealed red blood cells (RBCs) of 30–49/HPF, an albumin-to-creatinine ratio (ACR) of 332.77 mg/g, a protein-to-creatinine ratio (PCR) of 0.481 mg/g, and serum blood urea nitrogen (BUN)/creatinine levels of 15/0.69 mg/dl, corresponding to an estimated glomerular filtration rate (eGFR) of 103 ml/min/1.73 m². A renal biopsy demonstrated characteristic ultrastructural abnormalities on electron microscopy (Figure 2A), showing glomerular basement membranes (GBMs) with alternating thinning and thickening (150–1045 nm; mean 385 nm), compared to institutional reference values (296–481 nm). Immunofluorescence staining for type IV collagen α chains (α1, α3, and α5) showed diffuse linear expression of α1 and α3 chains, while the α5 chain was absent (Figures 2B–D). These findings established a definitive diagnosis of Alport syndrome in the patient's mother. However, her clinical exome sequencing including COL4A3, COL4A4, and COL4A5 failed to identify pathogenic variants. The patient's father and paternal uncles exhibited no abnormalities on urinalysis, and their renal function was within normal limits.
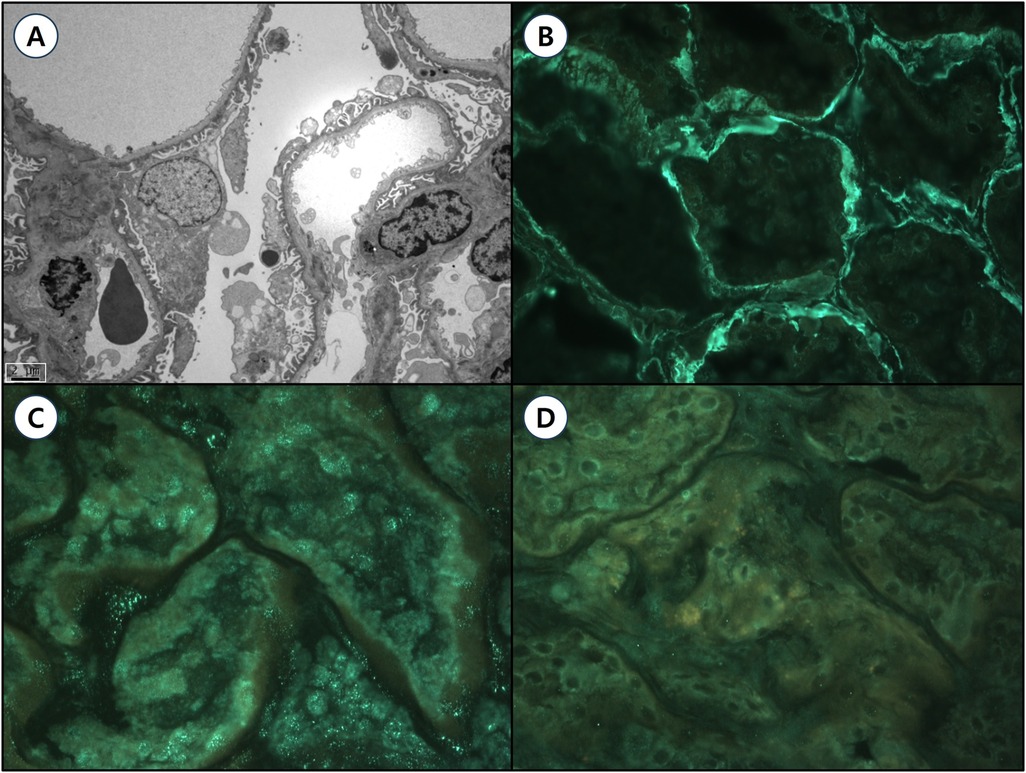
Figure 2. Renal pathological findings of the patient's mother with Alport syndrome. (A) Transmission electron microscopy (TEM, magnification ×4,000, scale bar = 2 μm) reveals ultrastructural abnormalities of the glomerular basement membranes (GBMs), characterized by alternating thinning and thickening (ranging from 150 to 1,045 nm; mean thickness 385 nm) compared to institutional reference values (296–481 nm). Moderate effacement of epithelial cell foot processes and focal rarefaction, degeneration, and granular material deposition were observed without definite lamellation. Mesangial basement membranes appear normal without significant deposits. (B–D) Immunofluorescence staining for type IV collagen α chains (magnification ×400): (B) α1 chain: Diffuse, strong linear positivity in both glomerular and tubular basement membranes (GBM 3+, TBM 3+). (C) α3 chain: Diffuse, weakly positive linear staining along the GBM (GBM 2+). (D) α5 chain: Negative staining in the GBM, consistent with X-linked Alport syndrome.
Given the persistent microscopic hematuria and strong family history, and in accordance with recent expert guidelines recommending genetic testing for persistent hematuria when targeted sequencing methods such as panel-based NGS fail to identify pathogenic variants, we opted for WGS instead of performing an invasive renal biopsy (6–8). WGS was conducted using genomic DNA extracted from peripheral blood, sequenced at an average depth of coverage of 32.45× on the Illumina NovaSeq 6000 platform, and processed using the EVIDENCE v4.1 pipeline (3billion, Seoul, Korea) based on Genome Reference Consortium Human Build 38 (GRCh38). A novel deep intronic hemizygous variant (NM_033380.3:c.2395 + 2723T > G) located at genomic position X-108609615-T-G (GRCh38), within intron 29 of the COL4A5 gene, was identified. This variant was absent in public population databases, including gnomAD v3.1.2, ClinVar, and HGMD (meeting ACMG criterion PM2), and in silico splice-site prediction analysis using SpliceAI indicated a moderate probability of altering splicing (SpliceAI score: 0.47, meeting ACMG criterion PP3). Subsequent Sanger sequencing confirmed hemizygosity in the patient and heterozygosity in his mother, supporting its pathogenic potential related to XLAS (Figure 3) (meeting ACMG criterion PP1). Accordingly, this variant was classified as a variant of uncertain significance (VUS) based on ACMG-AMP criteria PM2, PP3, and PP1, incorporating the Alport syndrome-specific modifications of the ACMG standards and guidelines for the interpretation of sequence variants (19, 20). Ophthalmological and otolaryngological examinations confirmed normal vision and hearing at the current stage. Identifying this elusive genetic variant through comprehensive genomic analysis allowed for an early genetic diagnosis, thereby avoiding invasive procedures and facilitating timely clinical management and counseling.
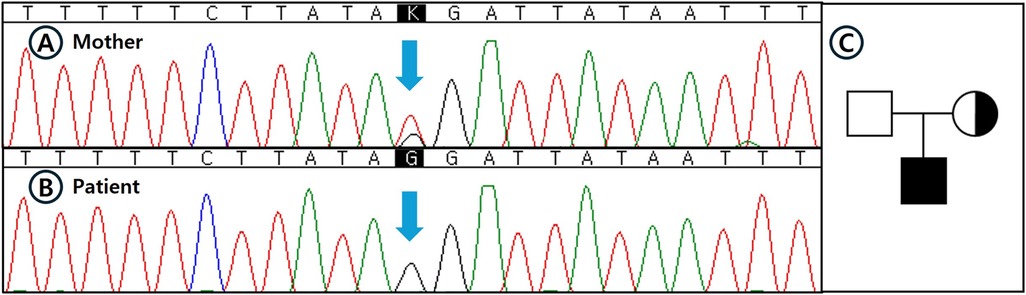
Figure 3. Sanger sequencing confirmation and pedigree analysis of the identified deep intronic COL4A5 variant. (A–B) Sanger sequencing validation of the deep intronic COL4A5 variant (NM_033380.3:c.2395 + 2723T > G) that was initially identified by whole genome sequencing (WGS): (A) The patient's mother showing a heterozygous state for the variant. (B) The patient demonstrating a hemizygous state for the variant. Blue arrows indicate the nucleotide substitution site. (C) Pedigree of the family affected by X-linked Alport syndrome (XLAS). Filled symbol indicates the patient (hemizygous), dotted circle represents the heterozygous mother, and open symbol indicates the unaffected father. The pedigree reflects confirmed genotypes based on sequencing results.
Discussion
This report describes a novel deep intronic variant (c.2395 + 2723T > G) in the COL4A5 gene, identified in a pediatric patient presenting with persistent microscopic hematuria and a family history of kidney disease. This variant was detected through WGS and confirmed by Sanger sequencing, highlighting diagnostic challenges associated with deep intronic variants. Indeed, conventional genetic testing methods such as targeted panel sequencing or exome sequencing typically fail to detect these intronic variants due to their inherent limitations in capturing non-coding regions. Previous studies have reported that approximately 15% of X-linked Alport syndrome (XLAS) cases involve splicing mutations, including deep intronic variants that cause cryptic splice site activation or pseudo-exon inclusion, disrupting the coding sequence and impairing normal protein function (9–12, 18). Such deep intronic variants are increasingly recognized as a significant cause of genetically unresolved Alport syndrome, emphasizing the importance of genetic testing approaches capable of identifying these elusive mutations.
Alport syndrome arises from structural abnormalities in the type IV collagen network of the glomerular basement membrane (GBM) (21). Type IV collagen, composed of six distinct alpha chains (α1–α6) (22–24), is the primary structural component of basement membranes. Pathogenic variants in the COL4A3, COL4A4, or COL4A5 genes disrupt the proper assembly and structure of collagen type IV, leading to impaired GBM integrity and progressive renal dysfunction, and may also affect ocular and cochlear structures (1, 24). Microscopic hematuria is the most common early symptom of XLAS, although some individuals remain asymptomatic. Importantly, males with XLAS typically progress to end-stage kidney disease within the first three decades of life, highlighting the necessity of timely diagnosis and intervention (25). Early therapeutic interventions, particularly renin-angiotensin-aldosterone system (RAAS) blockade, have demonstrated efficacy in slowing disease progression, underscoring the importance of early genetic diagnosis to facilitate prompt initiation of treatment (26). Nonetheless, the heterogeneity of clinical presentations and the limitations of standard genetic methods frequently contribute to delayed or missed diagnoses.
In this case, comprehensive genomic analysis using WGS uniquely facilitated the identification of a deep intronic COL4A5 variant that conventional exome and targeted sequencing had previously failed to detect. The identified variant (c.2395 + 2723T > G) was classified as a VUS according to ACMG-AMP guidelines with disease-specific modifications, based on its absence from population databases (PM2), computational predictions indicating a potential splicing impact (PP3), and co-segregation evidence within the family (PP1) (19, 20). Similar deep intronic variants causing cryptic splice site activation and pseudo-exon inclusion have been previously reported in COL4A5, reinforcing the potential clinical significance of these alterations (9–12, 18). However, this study has an important limitation: functional validation studies, such as RNA sequencing or minigene assays, were not performed. Such analyses are essential to definitively confirm the splicing impact and the pathogenicity of deep intronic variants, and future functional investigations will be crucial for clarifying the clinical relevance of the identified variant.
Clinically, classifying variants as uncertain significance creates considerable challenges in medical decision-making, as uncertain pathogenicity necessitates careful clinical correlation and cautious genetic counseling. Regular monitoring of renal function and clinical evaluation of affected and at-risk family members are advisable to detect potential disease progression or to obtain additional evidence that could clarify the variant's clinical significance. Confirming the pathogenicity of this variant through subsequent functional studies may provide critical prognostic insights, enabling more precise, personalized management and genetic counseling. Furthermore, early genetic diagnosis has broader clinical implications beyond merely confirming the diagnosis. It facilitates personalized patient management, enabling timely implementation of interventions, such as RAAS blockade therapy, that can significantly delay kidney disease progression. Identifying the genetic cause also provides essential information for genetic counseling, allowing at-risk family members to undergo targeted genetic evaluation and receive appropriate monitoring and early therapeutic interventions, thereby potentially reducing the necessity for invasive diagnostic procedures such as renal biopsy. Consequently, comprehensive genomic sequencing approaches, particularly WGS, represent an essential tool in diagnosing genetically elusive cases of Alport syndrome and improving patient outcomes through early and accurate genetic diagnosis.
Data availability statement
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Institutional Review Board of Seoul St. Mary's Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
HK: Data curation, Formal analysis, Funding acquisition, Methodology, Visualization, Writing – original draft, Writing – review & editing. MK: Data curation, Methodology, Writing – review & editing. JS: Data curation, Investigation, Writing – review & editing. YL: Conceptualization, Data curation, Investigation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by Research Fund of Seoul St. Mary’s Hospital, The Catholic University of Korea.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript. The author(s) verify and take full responsibility for the use of generative AI in the preparation of this manuscript. Generative AI was used minimally for English language editing and manuscript proofreading.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Gubler M, Levy M, Broyer M, Naizot C, Gonzales G, Perrin D, et al. Alport’s syndrome. A report of 58 cases and a review of the literature. Am J Med. (1981) 70(3):493–505. doi: 10.1016/0002-9343(81)90571-4
2. Saran R, Li Y, Robinson B, Abbott KC, Agodoa LY, Ayanian J, et al. US renal data system 2015 annual data report: epidemiology of kidney disease in the United States. Am J Kidney Dis. (2016) 67(3 Suppl 1):S1–305. doi: 10.1053/j.ajkd.2015.12.014
3. Alport AC. Hereditary familial congenital haemorrhagic nephritis. Br Med J. (1927) 1(3454):504–6. doi: 10.1136/bmj.1.3454.504
4. Rambausek M, Hartz G, Waldherr R, Andrassy K, Ritz E. Familial glomerulonephritis. Pediatr Nephrol. (1987) 1(3):416–8. doi: 10.1007/bf00849246
5. Grünfeld JP, Grateau G, Noel LH, Charbonneau R, Gubler MC, Savage CO, et al. Variants of Alport’s syndrome. Pediatr Nephrol. (1987) 1(3):419–21. doi: 10.1007/bf00849247
6. Savige J, Ariani F, Mari F, Bruttini M, Renieri A, Gross O, et al. Expert consensus guidelines for the genetic diagnosis of Alport syndrome. Pediatr Nephrol. (2019) 34(7):1175–89. doi: 10.1007/s00467-018-3985-4
7. Savige J, Lipska-Zietkiewicz BS, Watson E, Hertz JM, Deltas C, Mari F, et al. Guidelines for genetic testing and management of Alport syndrome. Clin J Am Soc Nephrol. (2022) 17(1):143–54. doi: 10.2215/cjn.04230321
8. Torra R, Lipska-Zietkiewicz B, Acke F, Antignac C, Becker JU, Cornec-Le Gall E, et al. Diagnosis, management and treatment of the Alport syndrome - 2024 guideline on behalf of ERKNet, ERA and ESPN. Nephrol Dial Transplant. (2025) 40(6):1091–106. doi: 10.1093/ndt/gfae265
9. Nozu K, Vorechovsky I, Kaito H, Fu XJ, Nakanishi K, Hashimura Y, et al. X-linked Alport syndrome caused by splicing mutations in COL4A5. Clin J Am Soc Nephrol. (2014) 9(11):1958–64. doi: 10.2215/cjn.04140414
10. Wang X, Zhang Y, Ding J, Wang F. mRNA analysis identifies deep intronic variants causing Alport syndrome and overcomes the problem of negative results of exome sequencing. Sci Rep. (2021) 11(1):18097. doi: 10.1038/s41598-021-97414-0
11. Qian P, Bao Y, Huang HM, Suo L, Han Y, Li ZJ, et al. A deep intronic splice variant of the COL4A5 gene in a Chinese family with X-linked Alport syndrome. Front Pediatr. (2022) 10:1009188. doi: 10.3389/fped.2022.1009188
12. Boisson M, Arrondel C, Cagnard N, Morinière V, Arkoub ZA, Saei H, et al. A wave of deep intronic mutations in X-linked Alport syndrome. Kidney Int. (2023) 104(2):367–77. doi: 10.1016/j.kint.2023.05.006
13. Miner JH. Pathology vs. Molecular genetics: (re)defining the spectrum of Alport syndrome. Kidney Int. (2014) 86(6):1081–3. doi: 10.1038/ki.2014.326
14. Gillion V, Dahan K, Cosyns JP, Hilbert P, Jadoul M, Goffin E, et al. Genotype and outcome after kidney transplantation in Alport syndrome. Kidney Int Rep. (2018) 3(3):652–60. doi: 10.1016/j.ekir.2018.01.008
15. Feingold J, Bois E, Chompret A, Broyer M, Gubler MC, Grünfeld JP. Genetic heterogeneity of Alport syndrome. Kidney Int. (1985) 27(4):672–7. doi: 10.1038/ki.1985.63
16. Rheault MN, Kashtan CE, editors. Inherited glomerular diseases. In: Pediatric Nephrology. Seventh edn. Berlin, Heidelberg: Springer (2015). p. 777–803. doi: 10.1007/978-3-662-43596-0_79
17. Mencarelli MA, Heidet L, Storey H, van Geel M, Knebelmann B, Fallerini C, et al. Evidence of digenic inheritance in Alport syndrome. J Med Genet. (2015) 52(3):163–74. doi: 10.1136/jmedgenet-2014-102822
18. King K, Flinter FA, Nihalani V, Green PM. Unusual deep intronic mutations in the COL4A5 gene cause X linked Alport syndrome. Hum Genet. (2002) 111(6):548–54. doi: 10.1007/s00439-002-0830-3
19. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
20. Savige J, Storey H, Watson E, Hertz JM, Deltas C, Renieri A, et al. Consensus statement on standards and guidelines for the molecular diagnostics of Alport syndrome: refining the ACMG criteria. Eur J Hum Genet. (2021) 29(8):1186–97. doi: 10.1038/s41431-021-00858-1
21. Brazel D, Oberbäumer I, Dieringer H, Babel W, Glanville RW, Deutzmann R, et al. Completion of the amino acid sequence of the alpha 1 chain of human basement membrane collagen (type IV) reveals 21 non-triplet interruptions located within the collagenous domain. Eur J Biochem. (1987) 168(3):529–36. doi: 10.1111/j.1432-1033.1987.tb13450.x
22. Barker DF, Hostikka SL, Zhou J, Chow LT, Oliphant AR, Gerken SC, et al. Identification of mutations in the COL4A5 collagen gene in Alport syndrome. Science. (1990) 248(4960):1224–7. doi: 10.1126/science.2349482
23. Kalluri R. Basement membranes: structure, assembly and role in tumour angiogenesis. Nat Rev Cancer. (2003) 3(6):422–33. doi: 10.1038/nrc1094
24. Zehnder AF, Adams JC, Santi PA, Kristiansen AG, Wacharasindhu C, Mann S, et al. Distribution of type IV collagen in the cochlea in Alport syndrome. Arch Otolaryngol Head Neck Surg. (2005) 131(11):1007–13. doi: 10.1001/archotol.131.11.1007
25. Temme J, Kramer A, Jager KJ, Lange K, Peters F, Müller GA, et al. Outcomes of male patients with Alport syndrome undergoing renal replacement therapy. Clin J Am Soc Nephrol. (2012) 7(12):1969–76. doi: 10.2215/cjn.02190312
Keywords: alport syndrome, COL4A5, deep intronic variant, whole genome sequencing, pediatric nephrology
Citation: Kim HS, Kim M, Suh J-S and Lee Y (2025) Case Report: Whole genome sequencing identifies a novel deep intronic COL4A5 variant of uncertain significance in X-linked Alport syndrome. Front. Pediatr. 13:1639471. doi: 10.3389/fped.2025.1639471
Received: 2 June 2025; Accepted: 17 July 2025;
Published: 7 August 2025.
Edited by:
Giulia Pascolini, Institute of Immaculate Dermatology (IRCCS), ItalyReviewed by:
Ammar Husami, Cincinnati Children’s Hospital Medical Center, United StatesMartina Lipari, Local Health Authority Rome 2, Italy
Copyright: © 2025 Kim, Kim, Suh and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yeonhee Lee, c3RhcnBoaWxlQGNtY251Lm9yLmty
†ORCID:
Hoon Seok Kim
orcid.org/0000-0001-8980-8177
Myungshin Kim
orcid.org/0000-0001-8632-0168
Yeonhee Lee
orcid.org/0000-0002-5816-6375