 Sofia Maria Carlotta Arnaboldi1,2*
Sofia Maria Carlotta Arnaboldi1,2* Martha Caterina Faraguna3,4
Martha Caterina Faraguna3,4 Antonella Colombini1Alessandra Sala1Veronica Leoni1Marco Spinelli1
Antonella Colombini1Alessandra Sala1Veronica Leoni1Marco Spinelli1 Giacomo Gotti1Laura Rachele Bettini1,4Viola Crescitelli3
Giacomo Gotti1Laura Rachele Bettini1,4Viola Crescitelli3 Anna Commone5Serena Gasperini3,†Carmelo Rizzari1,4,†
Anna Commone5Serena Gasperini3,†Carmelo Rizzari1,4,†
- 1Department of Pediatrics, Pediatric Hematology-Oncology Unit, Fondazione IRCCS San Gerardo dei Tintori, Monza, Italy
- 2Fondazione Matilde Tettamanti e Menotti De Marchi Onlus, Monza, Italy
- 3Department of Pediatrics, Fondazione IRCCS San Gerardo dei Tintori, Monza, Italy
- 4School of Medicine and Surgery, University of Milano-Bicocca, Monza, Italy
- 5Rare Disease Centre, Fondazione IRCCS San Gerado dei Tintori, Monza, Italy
Treating Acute Lymphoblastic Leukemia (ALL) in patients with genetic disorders poses significant challenges for onco-hematologists. Mucopolysaccharidosis type IVA (MPS-IVA) is a lysosomal storage disorder that clinically manifests with progressive and multi-systemic comorbidities, primarily affecting the bone, cartilage, spine, heart and lungs. We report a unique case of B-lineage ALL in a patient with MPS-IVA, who was successfully cured with a personalized chemo-radiotherapy approach. The treatment strategy aimed to balance a curative-intent chemotherapy attempt with the minimization of life-threatening complications. This case highlights the importance of individualized therapy in managing ALL in the context of complex comorbidities.
Introduction
Acute Lymphoblastic Leukemia (ALL) is the most common malignancy in childhood (1). Currently, 5-year overall survival rates for pediatric ALL are reported to exceed 90% across several treatment protocols (1). However, certain subgroups of patients continue to experience inferior outcomes. Among these, children with genetic disorders have usually poorer survival rates compared to other cohorts (2–4). The treatment of ALL in patients with concomitant genetic syndromes is associated with major challenges. In patients with preexisting organ dysfunction, a personalized treatment plan is often required and must be tailored on a case-by-case basis. In this regard, patients with mucopolysaccharidosis type IVA (MPS-IVA) present with a multi-systemic progressive disease, with the bone and cartilage, spine, heart and lungs being the most affected organs (5). MPS-IVA is a lysosomal storage disorder caused by mutations in the GALNS gene (NM_000512.5), which leads to a deficiency of the enzyme N-acetyl-galactosamine-6-sulfate sulfatase (GALNS) and the accumulation of glycosaminoglycans (GAG) keratan sulfate and chondroitin 6-sulfate. Phenotypically, MPS-IVA is prominently characterized by short stature and skeletal dysplasia. Further manifestations of the disease consist in restrictive lung disease, hepatomegaly, recurrent airway infections, hypoacusia, corneal clouding, valvular heart disease and dental abnormalities. The central nervous system (CNS) is not directly involved by GAG accumulation and subjects present normal intelligence; nevertheless, patients have a high risk of developing neurological complications due to atlanto-occipital instability and spinal cord compression secondary to extra-dural GAG accumulation, leading to cervical myelopathy and quadriparesis. No curative treatment is available for MPS-IVA. The benefit of hematopoietic stem cell transplantation—the gold standard treatment for mucopolysaccharidosis type I (Hurler syndrome)—is controversial in Morquio syndrome (6). In this context, Enzyme Replacement Therapy (ERT) with elosulfase alfa emerged as a therapeutic option and was approved by EMA and U.S. FDA in 2014. ERT was associated with a decrease in GAG accumulation, stability in motor performance and an improvement in forced vital capacity, but had no effect on skeletal disorders (7). Although mucopolysaccharidoses are not typically associated with an increased risk of malignancy, cancers have been reported in other inherited metabolic storage disorders, including multiple myeloma in Gaucher disease (8) and hepatocellular carcinoma in glycogen storage disease type I (9). Among MPS, only one previous case of Acute Myeloid Leukemia was reported in a 2.5-year-old girl with Hurler syndrome (10). However, that patient died one week after diagnosis. The case reported here represents the first patient with MPS-IVA and B-cell precursor ALL (B-ALL), who was successfully treated with a conventional chemotherapy-based regimen.
Case presentation
At a young age, our female patient underwent clinical investigations due to short stature, skeletal dysplasia and gait impairment. She was diagnosed with MPS-IVA at 4.5 years due to reduced lysosomal GALNS activity in leukocytes and increased urinary keratan sulphate excretion. A compound heterozygosity identified in the GALNS gene (c.239C > T; p.Ser80Leu and c.850TTC; p.Phe285del) further confirmed the diagnosis. Since the age of 6, the patient presented spastic paraparesis in the lower limbs due to stenosis of the craniocervical junction and of C7-T3 tract, associated with sensory deficits and hypotonia in the upper limbs. She underwent multiple spinal surgeries—including decompressive laminectomy from C1 to T3, fixation of the craniocervical junction and posterior spinal decompression with instrumentation from T9 to L1 (Figures 1A–C)—resulting in partial clinical improvement. The clinical phenotype was of a classic form of MPS-IVA, characterized by low stature (height 100 cm at age 18), bilateral corneal clouding, bilateral transmissive hypoacusia, severe restrictive pneumopathy, hip dysplasia and scoliosis. No heart valve involvement was documented. At the age of 17, she experienced an episode of acute respiratory insufficiency. A chest CT scan at that time revealed a trachea of reduced caliber (Figure 1B) associated with abnormal nocturnal oximetry findings, for which CPAP therapy was prescribed and used when clinically indicated. Due to paraparesis, the patient was highly dependent on carers. She required limited assistance in eating and drinking, self-hygiene, and dressing the upper body part, while required full help to go to the toilet, putting on/taking off clothes from the lower body part and moving around.
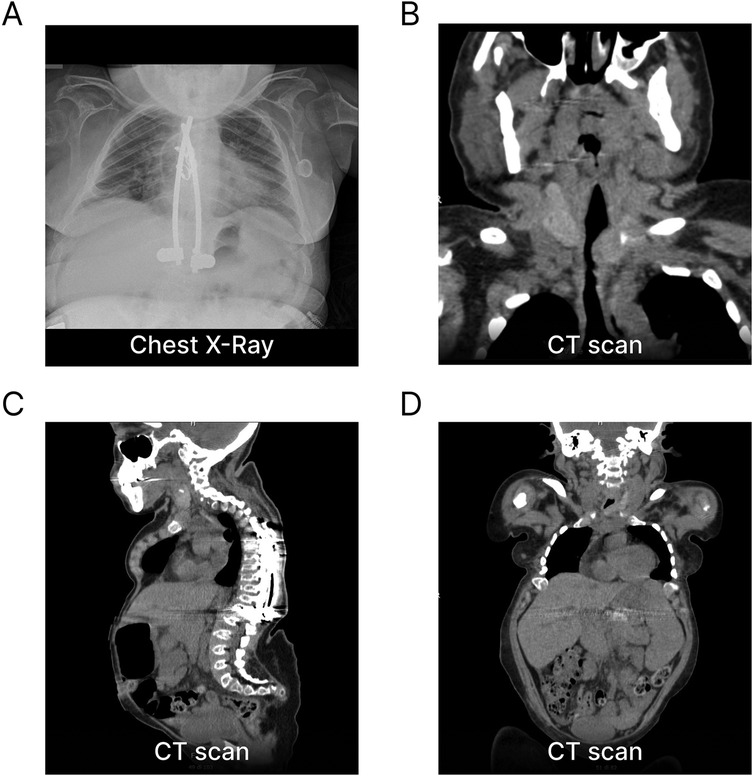
Figure 1. Imaging sections of phenotypic manifestations of our patient with Morquio syndrome. (A) Chest x-ray at B-ALL onset showing the absence of a mediastinal mass and evidence of spinal instrumentation with fixation from T9 to L1. (B) Frontal CT scan demonstrating a reduced-caliber trachea secondary to extra-luminal GAG accumulation. (C) Sagittal CT scan revealing skeletal dysplasia and severe anatomical abnormalities of the vertebral spine and thorax, including marked thoracic kyphoscoliosis and cervical and sacrolumbar hyperlordosis. (D) Total-body CT scan at B-ALL onset showing stable hepatosplenomegaly. GAG, glycosaminoglycans.
At 18 years, an urgent ophthalmologic evaluation was prompted by the acute onset of decreased vision in the right eye and revealed ipsilateral retinal hemorrhages. During the assessment, she reported a one-month history of pallor and asthenia. Blood tests revealed severe anemia, thrombocytopenia and neutropenia, with lymphoblasts in the peripheral blood smears (Hb 52 g/L, platelets 11.0 × 109/L, WBC 3.6 × 109/L, neutrophils 8%, blasts 25%). The patient was thus transferred to our Pediatric Hematology-Oncology Unit with the suspicion of an acute leukemia.
At the initial clinical examination, the patient presented with an upper respiratory tract infection and stable hepatosplenomegaly (Figure 1D). Bone marrow (BM) aspiration revealed a monomorphic infiltration of 50% lymphoblasts, confirming the diagnosis of ALL. The immunophenotype was consistent with the B-II subtype (EGIL criteria) (Figure 2). Molecular testing was negative for the translocations t(9;22), t(4;11), t(12;21) and t(1;19). Due to the poor quality of lymphoblast DNA at diagnosis, lymphoid clonality could not be determined for subsequent PCR-based minimal residual disease (MRD) assessment. A chest x-ray ruled out any mediastinal mass and laboratory tests were negative for tumor lysis syndrome. Due to the prior lumbar fixation surgery, lumbar puncture was contraindicated (Figures 1A–C), preventing both the evaluation of CNS involvement and the administration of intrathecal chemotherapy. However, neuroimaging exams were negative for focal leukemic involvement.
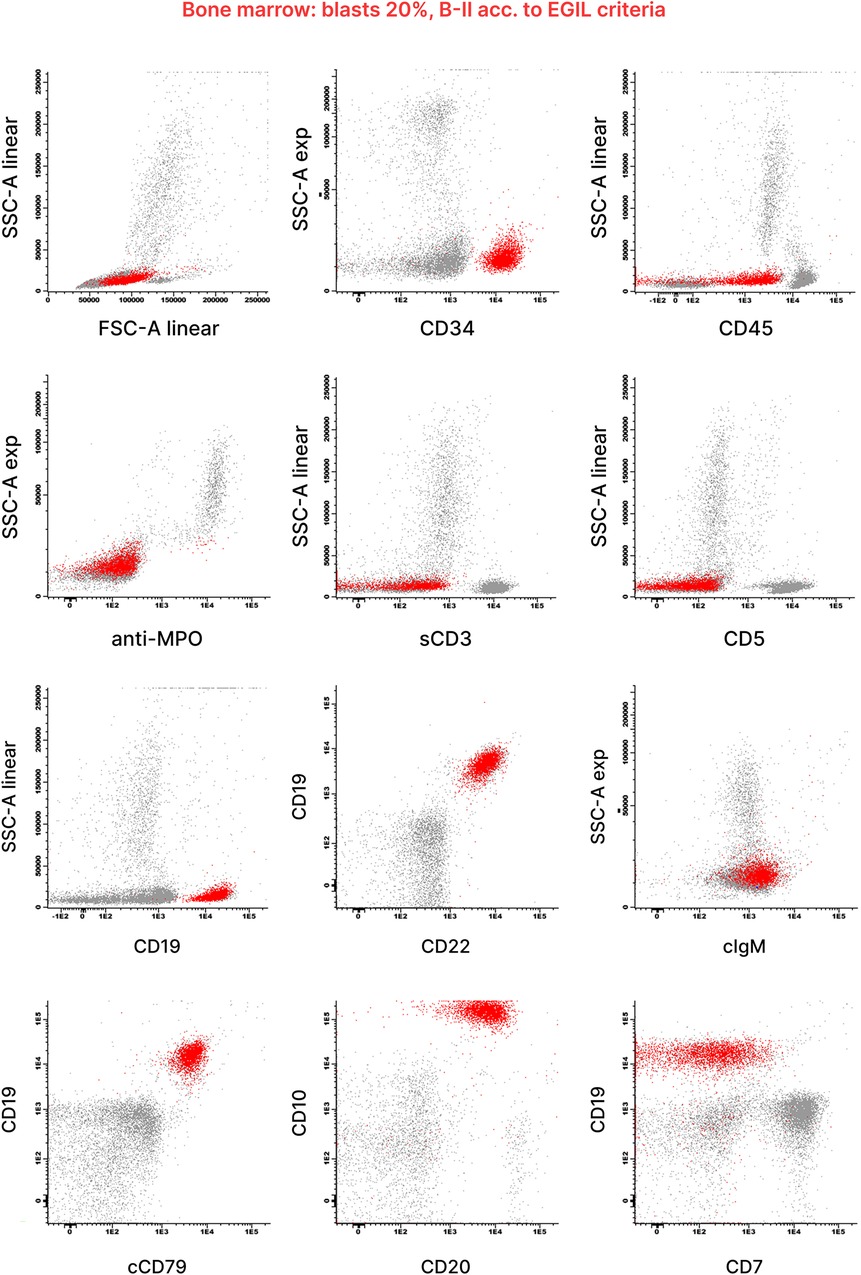
Figure 2. Diagnostic flow cytometry of bone marrow blood from this patient. Flow cytometric analysis of the bone marrow aspirate revealed a blast population (highlighted in red) with uniform expression of B-lymphoid markers CD19, CD20, CD22 and CD10. Antigen expression was analyzed within the blast gate, identified using light scatter parameters and CD45/CD19. The results were generated according to the diagnostic standards approved by the AIEOP-BFM-ALL FLOW-SG, based on the WHO 2008 classification, EGIL criteria and the Bethesda recommendations. Scatter plots were kindly provided by the Hematology Diagnostic Laboratory of Fondazione M. Tettamanti.
Considering the severe comorbidities of the patient (reduced body surface area, restrictive pneumopathy, prior lumbar fixation surgery, increased risk of cardio-toxicity), a personalized treatment plan was designed (Figures 3A,B). Induction chemotherapy was based on the AIEOP LLA 87 protocol (NCT00613457) (Figure 3B). A BM aspirate on day +15 showed a MRD of 0.823% by flow cytometry, indicating a good response to the initial treatment. During the ninth dose of native E. coli L-Asparaginase, the patient developed a grade 3 hypersensitivity reaction (CTCAE v3.0), requiring early discontinuation of the infusion. On day +32 of Induction, a dose of liposomal daunorubicin (DaunoXome®) was administered under metabolic and echocardiographic monitoring. Due to the development of grade 3 hypertransaminasemia (CTCAE v3.0), further doses of anthracyclines were omitted. The clinical course of this phase was otherwise uneventful. Given the high risk for this patient of developing a potentially life-threatening infection, systemic broad-spectrum antibiotic was administered prophylactically during the initial treatment. A BM aspirate performed at the end of Induction (timepoint 1—TP1) documented the morphological complete remission (CR). Because of the stable clinical conditions and good treatment tolerance, chemotherapy was continued according to the phase IB (Consolidation) of the AIEOP-BFM ALL 2009 protocol (NCT01117441). Due to the inability to perform any intrathecal chemotherapy, CNS prophylaxis was provided via cranial radiotherapy (CRT) upon discontinuation of systemic chemotherapy during this phase (Figure 3A). A BM aspirate performed at timepoint 2 (TP2) confirmed a continuous CR. Therefore, the Intensification phase was personalized on the AIEOP-BFM ALL 2009 protocol M (extracompartimental phase) and consisted of an increased dose of 6-mercaptopurine (60 mg/m2/day PO) and four courses of methotrexate (500 mg/m2 IV). Reinduction was also derived from the same protocol, and was complicated by an episode of Coronavirus-associated pneumonia, leading to respiratory insufficiency requiring non-invasive ventilatory support. In light of the episode of pneumonia, the two scheduled doses of cyclophosphamide were omitted to avoid myelotoxicity. The intensive phase of chemotherapy was completed approximately one year after diagnosis. Continuation was adapted from the Intermediate-Risk AIEOP ALL 95 protocol (NCT00613457) (Figure 3B). During the early phase of Continuation, the patient developed another episode of pneumonia complicated by acute respiratory insufficiency. Subsequently, considering also several episodes of grade 3 thrombocytopenia (CTCAE v3.0), no further monthly vincristine and five-day dexamethasone pulses were administered. For the view of the whole chemotherapy treatment plan, see Figure 3B. The chemotherapy treatment was discontinued 2.5 years after the diagnosis (Figures 3A,B).
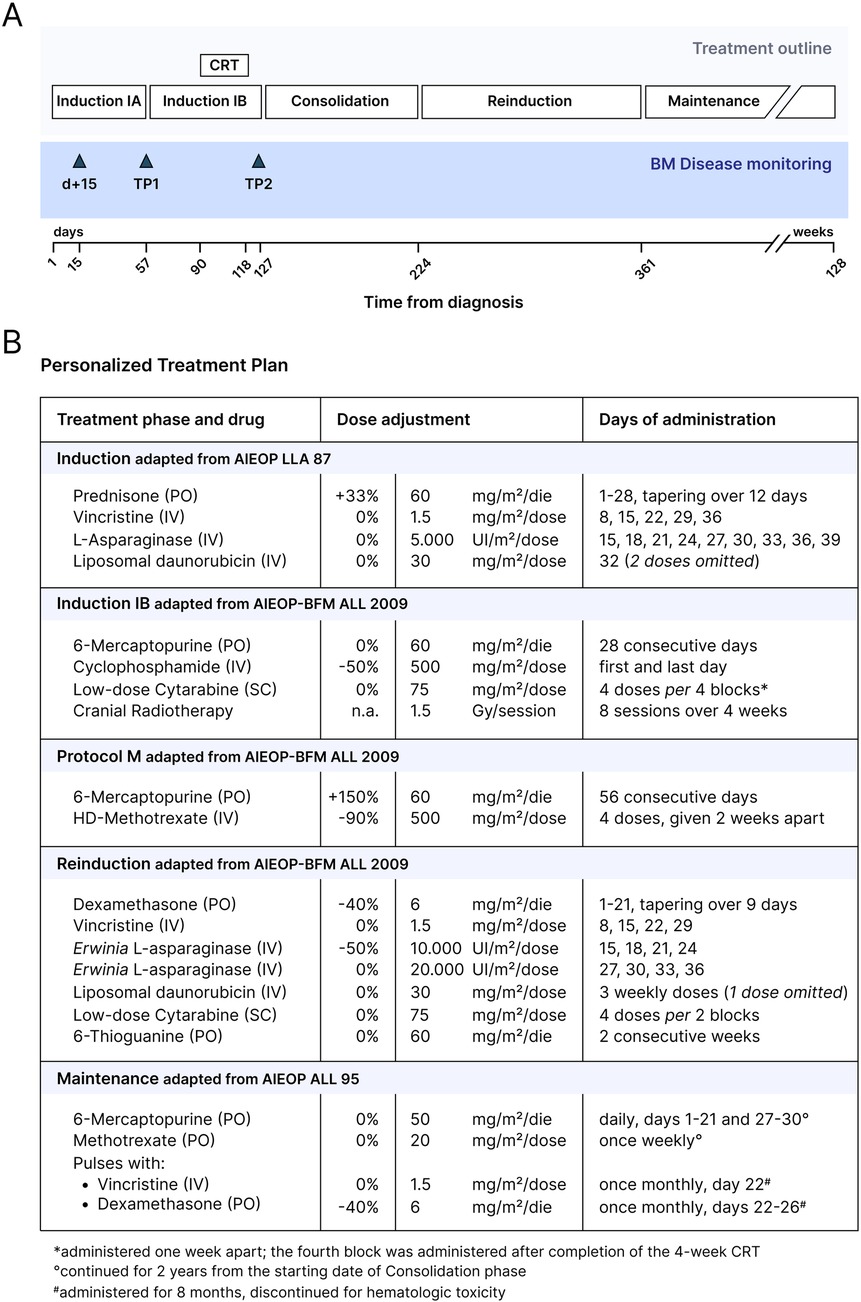
Figure 3. Personalized treatment plan for B-ALL in this patient with Morquio syndrome. (A) Overview of the personalized treatment plan and BM disease monitoring designed for the patient. (B) Table summarizing the chemotherapy regimen. Dose adjustments with respect to the protocol-based treatment phases are outlined. CRT, cranial radiotherapy; BM, bone marrow; TP1, timepoint 1; TP2, timepoint 2; PO, orally administered; IV, intravenously; SC, subcutaneously.
Upon EMA approval and after the end of chemotherapy, ERT with elosulfase alfa was started based on clinical stability and a joint decision between the patient, her family and physicians. ERT start was followed by an improvement in limb strength and sensitivity, a reduction in secretions of the upper airways, stability in cardiac and pulmonary function, and normalization of urinary GAG (Figure 4).
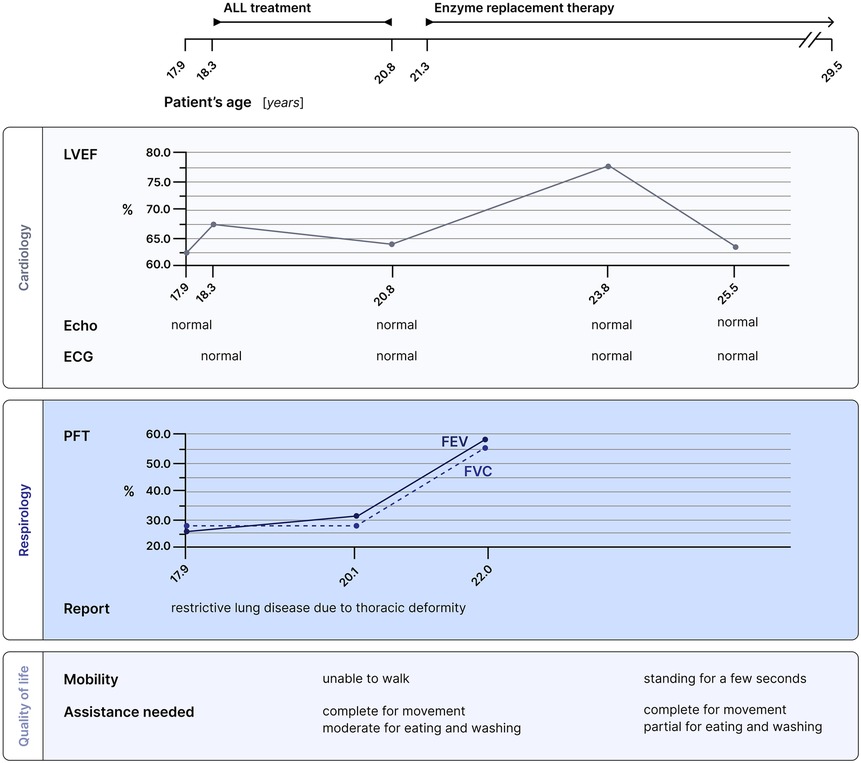
Figure 4. Multidisciplinary follow-up for this patient with Morquio syndrome. Cardiology, respirology and quality of life assessments are presented in relation to both ALL treatment and enzyme replacement therapy. Pulmonary function tests showed improvement, and the patient gained limb strength and the ability to stand for a few seconds under enzymatic therapy. LVEF, left ventricular ejection fraction; ECG, electrocardiogram; PFT, pulmonary function test; FEV, forced expiratory volume; FVC, forced vital capacity.
Currently, the patient remains in continuous CR ten years after the diagnosis of B-ALL. To date, no long term, treatment-related toxicities have been observed (Figure 4).
Discussion
We report a unique case of successful treatment of B-ALL in a patient with MPS-IVA. The definition of a treatment plan for this patient was particularly challenging due to the rarity of both the underlying metabolic disorder and ALL, the presence of severe comorbidities, the high risk of life-threatening chemotherapy-related complications and the need to deliver effective treatment. To ensure a comprehensive and multidisciplinary approach, the patient was managed at our center, where expertise in both pediatric Hematology-Oncology and Inborn Errors of Metabolism was available. Although our patient had already turned 18 at the time of diagnosis and would normally have been treated in an adult setting, it was agreed to administer a pediatric-oriented and personalized chemotherapy regimen. This decision was supported by encouraging survival outcomes reported over the past 2–3 decades in adolescents and young adults treated with pediatric-inspired regimens compared to adult protocols (11). In addition, our patient had a body surface area typically observed in a 9-year-old girl.
Our patient presented with severe comorbidities, requiring a stepwise approach to defining a personalized treatment plan, which was adjusted on the disease characteristics, the treatment response and the chemotherapy-related toxicities. Indeed, anthracyclines were administered with a reduced schedule and dosage under close metabolic and echocardiographic monitoring; in addition, the liposomal formulation was chosen for its reduced cardiotoxicity (12). To maintain treatment intensity, the native E. coli L-asparaginase was substituted with the Erwinia chrysanthemi L-asparaginase in Reinduction phase due to an allergic reaction observed at the end of Induction (13). Furthermore, monthly vincristine and dexamethasone pulses—administered during Continuation—were discontinued due to persistent hematological toxicity.
A major concern was the increased risk of pulmonary infection due to the patient's severe restrictive lung disease and thoracic deformity, so that antibacterial prophylaxis with systemic broad-spectrum antibiotics was administered during Induction. Additionally, any episodes of febrile neutropenia were cautiously managed with systemic antibacterial and antifungal therapy.
Another major challenge in this case was the anatomical contraindication to intrathecal chemotherapy due to severe spinal abnormalities and prior lumbar fixation surgery. Considering the high anesthesiologic and infection risks associated with the placement of an intraventricular reservoir, CRT was considered the only viable alternative for CNS prophylaxis. This option was extensively discussed by the treating medical team, given the potential long-term neurocognitive side effects associated with CRT (14), and was finally delivered during phase IB.
Our patient was successfully treated with a personalized, but conventional, chemo-radiotherapy regimen. Nowadays, new effective and less toxic treatment options are available, primarily represented by immunotherapeutic agents, such as Blinatumomab (15–17). The Children's Oncology Group COG AALL1731 clinical trial (NCT03914625) has recently reported that the addition of Blinatumomab to frontline chemotherapy for NCI standard-risk B-ALL with an average or higher risk of relapse significantly improved disease-free survival, leading to the study's early termination at the first interim analysis (17). Notably, also fragile patients, including infants and those with genetic disorders, have been recently reported to particularly benefit from Blinatumomab (15, 16).
To the best of our knowledge, this is the first report of B-ALL in a patient with MPS-IVA. This case emphasizes the importance of individualized treatment planning in patients with ALL and complex comorbidities, for whom a curative-intent approach should rely on multidisciplinary collaboration, tailored therapies, careful monitoring of treatment-related toxicities and response-adapted chemo-radiotherapy.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
Ethical approval was not required for the studies involving humans because Institutional regulations do not require Ethics Committee approval for the publication of case reports. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
SA: Investigation, Visualization, Writing – original draft, Conceptualization. MF: Conceptualization, Investigation, Writing – original draft. AC: Writing – review & editing. AS: Writing – review & editing. VL: Writing – review & editing. MS: Writing – review & editing. GG: Writing – review & editing. LB: Writing – review & editing. VC: Writing – review & editing. AC: Writing – review & editing. SG: Supervision, Conceptualization, Writing – review & editing. CR: Writing – review & editing, Conceptualization, Supervision.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This article has been partially funded by the Italian Ministry of Health, ‘Ricerca Corrente’ Program.
Acknowledgments
We sincerely acknowledge Fondazione Maria Letizia Verga and Fondazione Mariani for their valuable support. Several authors of this publication are members of the European Reference Network for Hereditary Metabolic Disorders (MetabERN) and the European Reference Network for Paediatric Cancer (ERN PaedCan). This article has been partially funded by the Italian Ministry of Health, ‘Ricerca Corrente’ Program.
Conflict of interest
CR has received support for participating (travel grants) and speaking (honoraria) at symposia sponsored by the pharmaceutical company AMGEN.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Pui CH, Yang JJ, Hunger SP, Pieters R, Schrappe M, Biondi A, et al. Childhood acute lymphoblastic leukemia: progress through collaboration. J Clin Oncol. (2015) 33(27):2938–48. doi: 10.1200/JCO.2014.59.1636
2. Buitenkamp TD, Izraeli S, Zimmermann M, Forestier E, Heerema NA, Van Den Heuvel-Eibrink MM, et al. Acute lymphoblastic leukemia in children with down syndrome: a retrospective analysis from the Ponte di Legno study group. Blood. (2014) 123(1):70–7. doi: 10.1182/blood-2013-06-509463
3. Qian M, Cao X, Devidas M, Yang W, Cheng C, Dai Y, et al. TP53 germline variations influence the predisposition and prognosis of B-cell acute lymphoblastic leukemia in children. J Clin Oncol. (2018) 36(6):591–9. doi: 10.1200/JCO.2017.75.5215
4. Rabin KR, Devidas M, Chen Z, Ji L, Kairalla J, Hitzler JK, et al. Outcomes in children, adolescents, and young adults with down syndrome and ALL: a report from the children’s oncology group. J Clin Oncol. (2024) 42(2):218–27. doi: 10.1200/JCO.23.00389
5. Tomatsu S, Montaño AM, Oikawa H, Smith M, Barrera L, Chinen Y, et al. Mucopolysaccharidosis type IVA (morquio A disease): clinical review and current treatment. Curr Pharm Biotechnol. (2011) 12(6):931–45. doi: 10.2174/138920111795542615
6. Yabe H, Tanaka A, Chinen Y, Kato S, Sawamoto K, Yasuda E, et al. Hematopoietic stem cell transplantation for morquio A syndrome. Mol Genet Metab. (2016) 117(2):84–94. doi: 10.1016/j.ymgme.2015.09.011
7. Mitchell JJ, Burton BK, Bober MB, Campeau PM, Cohen S, Dosenovic S, et al. Findings from the morquio A registry study (MARS) after 6 years: long-term outcomes of MPS IVA patients treated with elosulfase alfa. Mol Genet Metab. (2022) 137(1–2):164–72. doi: 10.1016/j.ymgme.2022.08.007
8. Mistry PK, Taddei T, Vom Dahl S, Rosenbloom BE. Gaucher disease and malignancy: a model for cancer pathogenesis in an inborn error of metabolism. Crit Rev Oncog. (2013) 18(3):235–46. doi: 10.1615/CritRevOncog.2013006145
9. Zangeneh F, Limbeck GA, Brown BI, Emch JR, Arcasoy MM, Goldenberg VE, et al. Hepatorenal glycogenosis (type I glycogenosis) and carcinoma of the liver. J Pediatr. (1969) 74(1):73–83. doi: 10.1016/S0022-3476(69)80010-7
10. Chen KT, McKenna RW, Desnick RJ. Acute myelogenous leukaemia in Hurler’s syndrome. J Med Genet. (1978) 15(3):239–42. doi: 10.1136/jmg.15.3.239
11. Testi AM, Valsecchi MG, Conter V, Vignetti M, Paoloni F, Giona F, et al. Difference in outcome of adolescents with acute lymphoblastic leukemia (ALL) enrolled in pediatric (AIEOP) and adult (GIMEMA) protocols. Blood. (2004) 104(11):1954–1954. doi: 10.1182/blood.V104.11.1954.1954
12. Jones RL, Berry GJ, Rubens RD, Miles DW. Clinical and pathological absence of cardiotoxicity after liposomal doxorubicin. Lancet Oncol. (2004) 5(9):575–7. doi: 10.1016/S1470-2045(04)01570-0
13. Rizzari C, Conter V, Starý J, Colombini A, Moericke A, Schrappe M. Optimizing asparaginase therapy for acute lymphoblastic leukemia. Curr Opin Oncol. (2013) 25(Supplement 1):S1–9. doi: 10.1097/CCO.0b013e32835d7d85
14. Piette C, Suciu S, Bertrand Y, Uyttebroeck A, Vandecruys E, Plat G, et al. Long-term outcome evaluation of medium/high risk acute lymphoblastic leukaemia children treated with or without cranial radiotherapy in the EORTC 58832 randomized study. Br J Haematol. (2020) 189(2):351–62. doi: 10.1111/bjh.16337
15. Van Der Sluis IM, De Lorenzo P, Kotecha RS, Attarbaschi A, Escherich G, Nysom K, et al. Blinatumomab added to chemotherapy in infant lymphoblastic leukemia. N Engl J Med. (2023) 388(17):1572–81. doi: 10.1056/NEJMoa2214171
16. Hodder A, Mishra AK, Enshaei A, Baird S, Bhuller K, Elbeshlawi I, et al. Blinatumomab for first-line treatment of children and young persons with B-ALL. J Clin Oncol. (2024) 42(8):907–14. doi: 10.1200/JCO.23.01392
Keywords: acute lymphoblastic leukemia, mucopolysaccaridosis, Morquio a syndrome, B-lineage acute lymphoblastic leukemia, enzyme replacement therapy (ERT), chemo-radiotherapy
Citation: Arnaboldi SMC, Faraguna MC, Colombini A, Sala A, Leoni V, Spinelli M, Gotti G, Bettini LR, Crescitelli V, Commone A, Gasperini S and Rizzari C (2025) Case report: Successful treatment of a patient presenting with a very rare association of acute lymphoblastic leukemia and mucopolysaccharidosis type IVA. Front. Pediatr. 13:1644720. doi: 10.3389/fped.2025.1644720
Received: 10 June 2025; Accepted: 11 August 2025;
Published: 4 September 2025.
Edited by:
Victor Aquino, University of Texas Southwestern Medical Center, United StatesReviewed by:
Tomasz Szczepanski, Medical University of Silesia, PolandTakeshi Isoda, Tokyo Medical and Dental University, Japan
Karolina M. Stepien, Salford Royal NHS Foundation Trust, United Kingdom
Jerzy Kowalczyk, Medical University of Lublin, Poland
Copyright: © 2025 Arnaboldi, Faraguna, Colombini, Sala, Leoni, Spinelli, Gotti, Bettini, Crescitelli, Commone, Gasperini and Rizzari. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sofia Maria Carlotta Arnaboldi, cy5hcm5hYm9sZGk1QGNhbXB1cy51bmltaWIuaXQ=
†These authors have contributed equally to this work and share senior authorship