 Nazi Tabatadze1,2,3*
Nazi Tabatadze1,2,3* Otar Koniashvili2,3
Otar Koniashvili2,3 Gia Melikishvili2
Gia Melikishvili2 Thierry Bienvenu4,5Laurence Cuisset6
Thierry Bienvenu4,5Laurence Cuisset6 Flora Tassone7,8
Flora Tassone7,8 Dragana D. Protic9,10Nino Naneishvili3,11Maia Zarandia12Sopo Gverdtsiteli13,14Sophio Kakabadze15Tamar Gachechiladze1,2Mariam Melikishvili2Zurab Kuchukashvili16Tamaz Giunashvili16Ketevan Silagava16Giorgi Mamardashvili2,3Mariam Chipashvili17Ketevan Barabadze1,18
Dragana D. Protic9,10Nino Naneishvili3,11Maia Zarandia12Sopo Gverdtsiteli13,14Sophio Kakabadze15Tamar Gachechiladze1,2Mariam Melikishvili2Zurab Kuchukashvili16Tamaz Giunashvili16Ketevan Silagava16Giorgi Mamardashvili2,3Mariam Chipashvili17Ketevan Barabadze1,18 Leonard Abbeduto19,20
Leonard Abbeduto19,20 Randi J. Hagerman19,21
Randi J. Hagerman19,21
- 1Faculty of Medicine, Ivane Javakhishvili Tbilisi State University, Tbilisi, Georgia
- 2Department of Pediatrics, MediClubGeorgia Medical Center, Tbilisi, Georgia
- 3Faculty of Medicine, Tbilisi State Medical University, Tbilisi, Georgia
- 4Université Paris Cité, Institute of Psychiatry and Neuroscience of Paris (IPNP), INSERM U1266, “Genetic vulnerability to addictive and psychiatric disorders” Team, Paris, France
- 5Service de médecine génomique des maladies de système et d’organe, Hôpital Cochin, Assistance Publique, Centre Université de Paris Cité, Paris, France
- 6Assistance Publique – Hôpitaux de Paris, APHP, Centre Universitaire Paris, Hôpital Cochin, Laboratoire de Génétique et Biologie Moléculaires, Paris, France
- 7Medical Investigation of Neurodevelopmental Disorders Institute, University of California Davis, Sacramento, CA, United States
- 8Department of Biochemistry and Molecular Medicine, School of Medicine, University of California Davis, Sacramento, CA, United States
- 9Department of Pharmacology, Clinical Pharmacology and Toxicology, Faculty of Medicine, University of Belgrade, Belgrade, Serbia
- 10Fragile X Clinic, Special Hospital for Cerebral Palsy and Developmental Neurology, Belgrade, Serbia
- 11Center for Mental Health and Prevention of Addiction, Tbilisi, Georgia
- 12Department of Molecular and Medical Genetics, Tbilisi State Medical University, Tbilisi, Georgia
- 13Department of Epilepsy Genetics and Personalized Medicine, Danish Epilepsy Center, Dianalund, Denmark
- 14Department of Regional Health Research, University of Southern Denmark, Odense, Denmark
- 15Department of Internal Medicine, Ascension Saint Joseph Hospital, Chicago, IL, United States
- 16Faculty of Exact and Natural Science, Ivane Javakhishvili Tbilisi State University, Tbilisi, Georgia
- 17Department of Pharmacology, Tbilisi State University, Tbilisi, Georgia
- 18Tbilisi State Medical University and High Technology Medical Center University Clinic, Tbilisi, Georgia
- 19MIND Institute, University of California Davis Health System, Sacramento, CA, United States
- 20Department of Psychiatry and Behavioral Sciences, University of California Davis School of Medicine, Sacramento, CA, United States
- 21Department of Pediatrics, School of Medicine, University of California, Sacramento, CA, United States
Fragile X syndrome (FXS) is the most common inherited form of intellectual disability (ID) and autism spectrum disorder (ASD). Despite its clinical importance, data on FXS prevalence in Georgia remains limited. This study aims to assess the prevalence of FXS in individuals with ID and/or ASD in Georgia and to review current diagnostic and management approaches. A total of 441 patients (n = 332 males and n = 109 females) diagnosed with ID and/or ASD based on DSM-5 criteria underwent genetic testing for FXS using a PCR-based approach. The FXS full mutation was identified in 25 patients (5.7%), and four individuals were carriers of the premutation. One patient had a large FMR1 deletion, thus the prevalence of the full mutation (FM) was 5.9%, and the prevalence of a premutation was 0.9%. The FXS-positive cohort showed a significant male predominance (80.77%). Among patients with ASD, 1.9% tested positive for FXS, and these individuals displayed more severe behavioral problems, requiring more intensive intervention. Phenotypic features such as a long face (76.9%), joint hypermobility (61.5%), and flat feet (53.8%) were commonly observed. The study underscores a significant diagnostic delay, with the average age of clinical ID/ASD diagnosis at 8.42 years and a lag of 4.63 years before FXS is identified. Compared to the U.S., where FXS diagnosis occurs at 35–41 months, Georgia faces significant barriers, including low awareness, lack of early screening, and limited access to genetic testing. Efforts to address these challenges include public awareness campaigns and integration of early genetic testing protocols.
1 Introduction
Fragile X syndrome (FXS) is the most common inherited form of intellectual disability, with a prevalence of approximately 1 in 4,000 to 1 in 5,000 males and 1 in 6,000 to 1 in 8,000 females (1). The behavioral features of FXS are variable, but it is estimated to account for 1%–6% of autism spectrum disorders (ASD), making it the most common single-gene condition associated with ASD (2).
FXS is caused by a deficiency or absence of the Fragile X messenger ribonucleoprotein (FMRP), primarily due to the expansion of CGG trinucleotide repeats in the promoter region of the fragile X messenger ribonucleoprotein 1 (FMR1) gene, leading to hypermethylation and epigenetic silencing. Individuals with more than 200 CGG trinucleotide repeats have full mutation (FM) leading to FXS, whereas those with 55–200 repeats carry a premutation (PM) (3). In addition to CGG repeat expansions, other genetic changes such as point variations, complete or partial deletions, and splicing errors have also been reported to cause FXS (4, 5). Once diagnosed, tailored preventive care is warranted from infancy through early adulthood (6). This proactive approach helps to improve developmental outcomes and enhances the quality of life for individuals with FXS.
This report is the first to focus on the targeted population-based prevalence of FXS in Georgia. Population-based studies are essential to provide a comprehensive understanding of the true extent of the challenges associated with the condition, identify at-risk groups, and inform health policy and resource allocation.
2 Materials and methods
A total of 441 individual patients meeting the inclusion criteria for Intellectual and Developmental Disabilities (IDD) and/or ASD were selected from the two largest cities of Georgia, Tbilisi and Batumi. The diagnoses of IDD and/or ASD were established based on DSM-5 criteria (5th ed.; DSM-5; American Psychiatric Association, 2013) and further supported by ancillary assessments, including the Autism Diagnostic Observation Schedule (ADOS), Autism Diagnostic Interview-Revised (ADI-R), Wechsler Preschool and Primary Scale of Intelligence (WPPSI), Wechsler Intelligence Scale for Children (WISC), and Vineland Adaptive Behavior Scales, Third Edition (Vineland-3).
The first cohort of 116 patients was tested with the FastFraX™ FMR1 Identification Kit (The Biofactory, Singapore), a PCR-based assay that uses melt curve analysis to detect CGG repeat expansions in the fragile X messenger ribonucleoprotein 1 (FMR1) gene. As an external quality control, all positive results were retested with triplet repeat primed PCR using AmplideX® FMR1 PCR (Asuragen, Austin, TX) at the Genomic Medicine Laboratory, Hôpital Cochin, France. The second cohort of 325 patients was tested directly using the AmplideX® PCR/CE FMR1 Kit (Asuragen, Austin, TX).
CGG repeats were categorized according to ACMG guidelines: Normal: <44 repeats (ACMG); Intermediate: 45–54 repeats (ACMG); Premutation: 55–200 repeats; Full mutation: >200 repeats (7).
In one patient, high clinical suspicion and an abnormal PCR amplification curve suggested a possible deletion or copy number variation, prompting us to perform array-CGH. The analysis was performed at the UC Davis MIND Institute, California. Copy number variants (CNVs) were compared with public databases, including DGV and DECIPHER, and classified according to established guidelines Additional family testing was performed in mother, two sisters and his maternal uncle after obtaining informed consent.
Clinical information was collected retrospectively from medical records. This included demographic details, clinical history, neurological, behavioral and developmental assessments, laboratory findings, imaging studies, and treatment history.
3 Results
A total of 441 patients were genetically evaluated for FXS, of whom 75.28% (n = 332) were male and 24.72% (n = 109) female. Among them, 59.88% (n = 264) were diagnosed with ASD, 89.83% (n = 396) had ID, and 19.77% (n = 87) had both ASD and ID. The mean age at molecular diagnosis of FXS was 13.05 years, compared to a mean age of 8.42 years at clinical diagnosis of ID or ASD.
PCR analysis detected FM in (N = 25) patients, while an additional (N = 4) patients had PM. Additionally, (N = 1) patient had a large deletion identified through Array-CGH, bringing the FM rate in this cohort to approximately 5.9% and 6.8%, respectively, (30/441) with any fragile X mutation.
The cohort had a significant male predominance, with 80.77% of the patients being male and 19.23% female, resulting in a sex ratio of approximately 4.2:1. Among the patients with the full mutation, 21 were male and 5 were female. All male patients with the full mutation had moderate to severe ID, and 4 were additionally diagnosed with both ID and ASD. Among the female patients, 4 were diagnosed with mild ID, and 1 with ASD. All four patients with the premutation exhibited learning disabilities, a feature consistently documented in previous studies (8).
Among the 26 patients with identified fragile X mutations, several phenotypic features were documented (Table 1). The most common was a long and narrow face (76.9%, n = 20), followed by joint hypermobility (61.5%, n = 16) and flat feet (53.9%, n = 14). Other frequent findings included prominent ears (50.0%, n = 13), soft and velvety skin (50.0%, n = 13), and a prominent jaw (38.5%, n = 10). Less frequent but notable features included constipation (34.6%, n = 9), sleep disorders (23.1%, n = 6), and seizures (15.4%, n = 4). Macroorchidism was present in all postpubertal males (100%, n = 4). Obesity was observed in 11.5% (n = 3), while macrocephaly and strabismus were each present in 7.7% (n = 2).
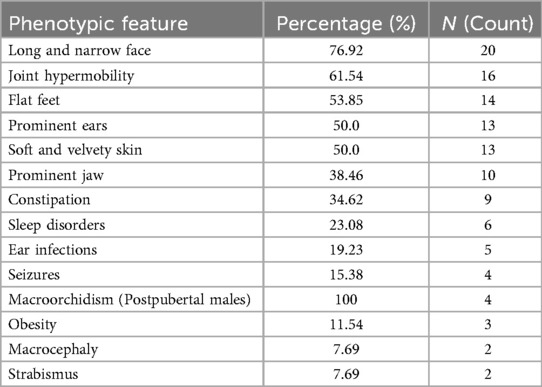
Table 1. Phenotypic features in patients with fragile X mutations.
Beyond these physical manifestations, diagnostic subgrouping further clarified the clinical picture. Of the 441 patients, 264 (59.9%) were diagnosed with ASD. Within this group, 5 patients (1.9%) also tested positive for FXS (FXS-ASD), while the remaining 259 (98.1%) had ASD alone. Importantly, patients with FXS-ASD exhibited more severe behavioral difficulties—including anxiety, aggression, and self-injurious behaviors—than those with idiopathic ASD or FXS alone.
4 Discussion
Developing countries continue to struggle with the challenges associated with early diagnosis and treatment of FXS. This struggle is further compounded by the fact that FXS remains poorly understood among senior medical students in developing countries. One study found that none of the cohorts achieved a 60% passing threshold in FXS knowledge, with most students reporting inadequate education on neurodevelopmental disorders (NDDs). Barriers such as stigma, limited access to testing, the financial burden of testing, and systemic health care deficiencies exacerbate delayed diagnoses and poor care (9).
In the US, boys with FXS are typically diagnosed with FXS at an average age of 35–37 months, while girls are diagnosed later, at around 41.6 months. There is an average delay of 24 months between the first concern (11.6 months for boys, 16 months for girls) and formal diagnosis of FXS (10). In contrast, based on our study in the Georgian cohort, the mean age at molecular diagnosis of FXS was 13.05 years, compared to a mean age of 8.42 years at clinical diagnosis of ID or ASD, reflecting an average diagnostic delay of 4.63 years.
To address the delay in diagnosis and the associated challenges, we have conducted numerous awareness campaigns and established active social platforms. These efforts have significantly increased Georgia's position on the global map for FXS awareness and advocacy. This proactive approach aims to improve developmental outcomes and enhance the quality of life for individuals with FXS. While the American Academy of Pediatrics (AAP) recommends testing patients with ASD and/or ID for FXS, limited resources and awareness in most developing countries result in a two-step diagnostic process. This involves first identifying physical stigmata associated with FXS, followed by genetic testing. Such an approach risks overemphasizing physical features, potentially delaying early interventions that are critical to improving psychosocial outcomes.
In addition, the lack of resources often makes multidisciplinary care inaccessible for patients with FXS, further limiting their access to comprehensive treatment.
The current practice in the management of FXS involves both non-pharmacologic and pharmacologic approaches. Although the evidence base for the efficacy of a specific treatment approach is quite limited. Non-pharmacological interventions, such as physiotherapy, occupational therapy, and speech and language therapy, are essential in addressing motor and communication delays. Applied Behavior Analysis (ABA) is particularly beneficial for children with FXS, especially those with comorbid ASD, to improve social and communication deficits. Cognitive-behavioral therapy (CBT) is effective in managing anxiety, attention-deficit hyperactivity disorder (ADHD), and social deficits in higher-functioning individuals (11, 12). Educational programs that emphasize sensory integration, social interaction, and life skills contribute significantly to improvements in adaptive behavior. Pharmacologically, targeted treatments aim to address the dysregulated pathways characteristic of FXS. Medications such as selective serotonin reuptake inhibitors (SSRIs), psychostimulants, antiepileptics such as lamotrigine, and antipsychotics are used to manage symptoms such as anxiety, ADHD, seizures, and behavioral problems. Novel treatments, including metformin and cannabidiol (CBD), have shown promise in modulating synaptic plasticity and improving behavioral outcomes by targeting molecular pathways such as mTOR and the endocannabinoid system (13–16).
Over the past decade, considerable research has been conducted to better understand the pathophysiology of FXS and to develop targeted treatments. While some approaches have shown promise, the results have been mixed. For example, metabotropic glutamate receptor 5 (mGluR5) antagonists, such as mavoglurant, failed to show significant improvements in behavioral symptoms compared to placebo (17). Similarly, GABAergic system modulators such as arbaclofen showed some improvement but lacked statistical significance (18). In contrast, Phosphodiesterase-4D (PDE4D) inhibitors, such as zatolmilast, have shown encouraging results, including improvements in cognitive function and behavior, highlighting their promise as a therapeutic option for FXS (19).
Advances in gene therapy have also shown potential. Recent studies have shown that targeting specific R-loops can induce the contraction of CGG repeats and reactivate the silenced FMR1 gene in FXS cells, offering a novel and promising approach (20).
5 Conclusion
In conclusion, FXS remains a significant yet underdiagnosed cause of ID and ASD, particularly in resource-limited settings. Georgia continues to face major challenges in autism and FXS care, particularly due to the lack of standardized referral pathways after a clinical diagnosis and the limited availability of resources for patient education and advocacy. Notably, in our cohort the male-to-female prevalence ratio of FXS was 4.2:1, compared with the approximately 2:1 ratio reported internationally. This disparity likely reflects the historical under-recognition of FXS in females, as the condition has often been perceived primarily as a disorder affecting boys with ID and ASD. Consequently, many affected girls may remain undiagnosed, underscoring the importance of raising awareness of the female phenotype and ensuring more inclusive diagnostic approaches. To address gaps, in close collaboration with NFXF we have established a Fragile X clinic and national patient registry, also launched an annual Fragile X Awareness campaign. As a next step, we are preparing a mixed-methods study that will integrate qualitative and quantitative approaches to evaluate current knowledge and practices among healthcare professionals. Together, these initiatives aim to enhance early identification, strengthen patient and family support systems, and promote nationwide advocacy for individuals affected by FXS and related conditions. Our study underscores the urgent need to integrate early genetic testing into routine pediatric and neurodevelopmental care pathways, as this would facilitate earlier diagnosis and enable tailored management strategies that ultimately improve patient outcomes. Increased public and professional education about FXS, combined with global collaboration, holds promise for reducing diagnostic delays, improving access to multidisciplinary care, and ultimately improving the quality of life for individuals affected by this condition.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
Ethical approval was not required for the studies involving humans as it involved a retrospective analysis of fully anonymized clinical and genetic data obtained during routine diagnostic procedures. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent to participate in this study was not required from the participants or the participants legal guardians/next of kin in accordance with the national legislation and the institutional requirements.
Author contributions
NT: Investigation, Software, Conceptualization, Funding acquisition, Supervision, Writing – review & editing, Visualization, Data curation, Resources, Project administration, Writing – original draft, Formal analysis, Validation, Methodology. OK: Funding acquisition, Formal analysis, Writing – original draft, Methodology, Visualization, Resources, Software, Supervision, Investigation, Conceptualization, Project administration, Validation, Data curation, Writing – review & editing. GMe: Data curation, Visualization, Software, Validation, Formal analysis, Resources, Conceptualization, Investigation, Writing – review & editing, Project administration, Supervision, Funding acquisition, Writing – original draft, Methodology. TB: Writing – review & editing, Formal analysis, Writing – original draft, Methodology. LC: Writing – review & editing, Writing – original draft, Formal analysis. FT: Writing – original draft, Investigation, Writing – review & editing, Methodology. DP: Writing – original draft, Writing – review & editing, Data curation, Validation. NN: Writing – original draft, Formal analysis, Data curation, Writing – review & editing, Investigation. MZ: Writing – original draft, Formal analysis, Data curation, Investigation, Writing – review & editing. SG: Investigation, Writing – review & editing, Writing – original draft. SK: Formal analysis, Writing – original draft, Writing – review & editing. TGa: Formal analysis, Writing – review & editing, Writing – original draft. MM: Formal analysis, Writing – original draft, Writing – review & editing. ZK: Software, Writing – review & editing, Methodology, Writing – original draft. TGi: Methodology, Writing – original draft, Writing – review & editing, Software. KS: Writing – original draft, Writing – review & editing, Methodology, Software. GMa: Writing – review & editing, Writing – original draft, Formal analysis, Data curation. MC: Project administration, Formal analysis, Writing – original draft, Validation, Resources, Data curation, Visualization, Supervision, Conceptualization, Writing – review & editing, Software, Methodology, Investigation, Funding acquisition. KB: Supervision, Writing – review & editing, Funding acquisition, Writing – original draft, Software, Formal analysis, Resources, Investigation, Data curation, Project administration, Validation, Visualization, Methodology, Conceptualization. LA: Writing – review & editing, Resources, Formal analysis, Software, Writing – original draft, Data curation, Methodology, Validation, Conceptualization, Investigation, Supervision, Project administration. RH: Formal analysis, Investigation, Validation, Resources, Data curation, Project administration, Supervision, Writing – review & editing, Methodology, Software, Conceptualization, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. APC was funded by the authors.
Acknowledgments
The authors would like to thank Asuragen, Inc. for the generous in-kind donation of 116 AmplideX® PCR/CE FMR1 Reagent kits used in the molecular analysis. We also acknowledge the administrative and technical support provided by MediClubGeorgia Medical Center and the Department of Molecular Genetics at Hôpital Cochin, Paris. Special thanks to the patients and families whose anonymized data made this research possible.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript. Polishing of the manuscript (minor grammatical mistakes).
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Hunter J, Rivero-Arias O, Angelov A, Kim E, Fotheringham I, Leal J. Epidemiology of fragile X syndrome: a systematic review and meta-analysis. Am J Med Genet A. (2014) 164A(7):1648–58. doi: 10.1002/ajmg.a.36511
2. Kaufmann WE, Kidd SA, Andrews HF, Budimirovic DB, Esler A, Haas-Givler B, et al. Autism spectrum disorder in fragile X syndrome: cooccurring conditions and current treatment. Pediatrics. (2017) 139(Suppl 3):S194–206. doi: 10.1542/peds.2016-1159F
3. Hagerman RJ, Berry-Kravis E, Hazlett HC, Bailey DB, Moine H, Kooy RF, et al. Fragile X syndrome. Nat Rev Dis Primers. (2017) 3(1):17065. doi: 10.1038/nrdp.2017.65
4. Jiraanont P, Manor E, Tabatadze N, Zafarullah M, Mendoza G, Melikishvili G, et al. De novo large deletion leading to fragile X syndrome. Front Genet. (2022) 13:884424. doi: 10.3389/fgene.2022.884424
5. Sitzmann AF, Hagelstrom RT, Tassone F, Hagerman RJ, Butler MG. Rare FMR1 gene mutations causing fragile X syndrome: a review. Am J Med Genet A. (2018) 176(1):11–8. doi: 10.1002/ajmg.a.38504
6. Hersh JH, Saul RA, Committee on Genetics. Health supervision for children with fragile X syndrome. Pediatrics. (2011) 127(5):994–1006. doi: 10.1542/peds.2010-3500
7. Monaghan KG, Lyon E, Spector EB, erican College of Medical Genetics and Genomics. ACMG standards and guidelines for fragile X testing: a revision to the disease-specific supplements to the standards and guidelines for clinical genetics laboratories of the American college of medical genetics and genomics. Genet Med. (2013) 15(7):575–86. doi: 10.1038/gim.2013.61
8. Bourgeois JA, Coffey SM, Rivera SM, Hessl D, Gane LW, Tassone F, et al. A review of fragile X premutation disorders: expanding the psychiatric perspective. J Clin Psychiatry. (2009) 70(6):852–62. doi: 10.4088/JCP.08m04476
9. Protic D, Salcedo-Arellano MJ, Stojkovic M, Saldarriaga W, Ávila Vidal LA, Miller RM, et al. Raising knowledge and awareness of fragile X syndrome in Serbia, Georgia, and Colombia: a model for other developing countries? Yale J Biol Med. (2021) 94(4):559–71.34970093
10. Bailey DB Jr, Raspa M, Bishop E, Holiday D. No change in the age of diagnosis for fragile x syndrome: findings from a national parent survey. Pediatrics. (2009) 124(2):527–33. doi: 10.1542/peds.2008-2992
11. Niu M, Han Y, Dy ABC, Du J, Jin H, Qin J, et al. Fragile X syndrome: prevalence, treatment, and prevention in China. Front Neurol. (2017) 8:254. doi: 10.3389/fneur.2017.00254
12. Protic DD, Aishworiya R, Salcedo-Arellano MJ, Tang SJ, Milisavljevic J, Mitrovic F, et al. Fragile X syndrome: from molecular aspect to clinical treatment. Int J Mol Sci. (2022) 23(4):1935. doi: 10.3390/ijms23041935
13. Aishworiya R, Valica T, Hagerman R, Restrepo B. An update on psychopharmacological treatment of autism spectrum disorder. Neurotherapeutics. (2022) 19(1):248–62. doi: 10.1007/s13311-022-01183-1
14. Gantois I, Popic J, Khoutorsky A, Sonenberg N. Metformin for treatment of fragile X syndrome and other neurological disorders. Annu Rev Med. (2019) 70(1):167–81. doi: 10.1146/annurev-med-081117-041238
15. Tartaglia N, Bonn-Miller M, Hagerman R. Treatment of fragile X syndrome with cannabidiol: a case series study and brief review of the literature. Cannabis Cannabinoid Res. (2019) 4(1):3–9. doi: 10.1089/can.2018.0053
16. Berry-Kravis E, Hagerman R, Budimirovic D, Erickson C, Heussler H, Tartaglia N, et al. A randomized, controlled trial of ZYN002 cannabidiol transdermal gel in children and adolescents with fragile X syndrome (CONNECT-FX). J Neurodev Disord. (2022) 14(1):56. doi: 10.1186/s11689-022-09466-6
17. Berry-Kravis E, Portes VD, Hagerman R, Jacquemont S, Charles P, Visootsak J, et al. Mavoglurant in fragile X syndrome: results of two randomized, double-blind, placebo-controlled trials. Sci Transl Med. (2016) 8(321):321ra5. doi: 10.1126/scitranslmed.aab4109
18. Berry-Kravis E, Hagerman R, Visootsak J, Budimirovic D, Kaufmann WE, Cherubini M, et al. Arbaclofen in fragile X syndrome: results of phase 3 trials. J Neurodevelop Disord. (2017) 9:3. doi: 10.1186/s11689-016-9181-6
19. Berry-Kravis EM, Harnett MD, Reines SA, Reese MA, Ethridge LE, Outterson AH, et al. Inhibition of phosphodiesterase-4D in adults with fragile X syndrome: a randomized, placebo-controlled, phase 2 clinical trial. Nat Med. (2021) 27(5):862–70. doi: 10.1038/s41591-021-01321-w
Keywords: Fragile X syndrome, FMR1, intellectual disability, autism spectrum disorder, prevalence, Georgia, genetic testing
Citation: Tabatadze N, Koniashvili O, Melikishvili G, Bienvenu T, Cuisset L, Tassone F, Protic DD, Naneishvili N, Zarandia M, Gverdtsiteli S, Kakabadze S, Gachechiladze T, Melikishvili M, Kuchukashvili Z, Giunashvili T, Silagava K, Mamardashvili G, Chipashvili M, Barabadze K, Abbeduto L and Hagerman RJ (2025) Prevalence of Fragile X syndrome in Georgian patients with autism spectrum disorder and/or intellectual disability: cross-sectional study and review of current approaches. Front. Pediatr. 13:1645386. doi: 10.3389/fped.2025.1645386
Received: 11 June 2025; Accepted: 15 September 2025;
Published: 26 September 2025.
Edited by:
Alfredo Brusco, University of Turin, ItalyReviewed by:
Dejan Budimirovic, Johns Hopkins University, United StatesFrancois Bolduc, University of Alberta, Canada
Copyright: © 2025 Tabatadze, Koniashvili, Melikishvili, Bienvenu, Cuisset, Tassone, Protic, Naneishvili, Zarandia, Gverdtsiteli, Kakabadze, Gachechiladze, Melikishvili, Kuchukashvili, Giunashvili, Silagava, Mamardashvili, Chipashvili, Barabadze, Abbeduto and Hagerman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nazi Tabatadze, bl90YWJhdGFkemVAaG90bWFpbC5jb20=