 Y. Y. Tan1
Y. Y. Tan1 V. S. Rajadurai
V. S. Rajadurai S. Chandran
S. Chandran- 1Department of Neonatology, KK Women’s and Children’s Hospital, Singapore, Singapore
- 2Pediatric Obstetrics & Gynecology Clinical Program, Duke NUS Medical School, Singapore, Singapore
- 3Perinatal Palliative Care, KK Women’s and Children’s Hospital, Singapore, Singapore
- 4Pediatric Academic Clinical Program, Lee Kong Chian School of Medicine, Singapore, Singapore
- 5Pediatric Academic Clinical Program, Yong Loo Lin School of Medicine, Singapore, Singapore
- 6Pediatric Academic Clinical Program, Duke NUS Medical School, Singapore, Singapore
Carnitine palmitoyltransferase II (CPT II) deficiency is a rare inherited disorder of mitochondrial oxidation of long-chain fatty acids (LCFA). Carnitine is the sole carrier of LCFA, which is transferred to the cellular mitochondria for β-oxidation; CPT II plays a vital role in this process. Three phenotypic forms of CPT II deficiency exist: lethal neonatal (LNF), severe infantile hepato-cardio-muscular, and mild adult myopathic forms. The LNF is the most severe type of CPT II deficiency. Management should be guided by shared decision-making with parents, taking into account the severity of the disease, the goals of care, and the quality of life for both the patient and their family. We present a consanguineous couple with two siblings in consecutive pregnancies with LNF of CPT II deficiency who exhibited similar symptoms, both antenatally and postnatally. Antenatal assessments revealed cardiomegaly, ventriculomegaly, and polycystic kidneys. The first sibling received all supportive measures, including extracorporeal life support, but succumbed. Parents, counseled antenatally by the perinatal palliative care team, opted for comfort care for the second sibling, who passed away on day 3 of life. Cardiac, renal, and cerebral malformations were consistent in fetal and neonatal ultrasound scans of both siblings, who had biochemical and molecular diagnoses, confirming CPT II deficiency. The fetal imaging signs served as reliable indicators for early diagnosis, especially in the background of consanguinity. We present the cases of the siblings, including a literature review on fetal and neonatal characteristics, as well as the biochemical and molecular features of CPT II deficiency.
Introduction
Carnitine regulates the balance between free CoA and acyl-CoA both inside and outside the cell. A critical step in fatty acid oxidation is the transport of long-chain fatty acids (LCFAs) into the mitochondria via the carnitine shuttle facilitated by carnitine palmitoyltransferase (CPT) I and II and carnitine-acylcarnitine translocase (CACT), which play a crucial role in energy production. The carnitine shuttle regulates the entry of acyl-CoA esters into the mitochondria. The shuttle requires three major enzymes, namely, CPT I, CACT, and CPT II (1). There are two isoforms of CPT I: Liver (L-CPT I) and the muscle forms (M-CPT I). The lungs, pancreas, ovary, intestine, spleen, and brain also have L-CPT I. In the fasting state, the LCFA are activated in the cytosol of the cytoplasm to form fatty acyl CoA, which then enters the mitochondria. CPT I in the outer mitochondrial membrane facilitates the conversion of acyl-CoA and carnitine to acylcarnitine and free CoA, allowing its entry into the intermembranous space. CPT I is upregulated in the fasting state when low levels of intracellular malonyl-CoA are present. Acylcarnitine is then translocated through the inner mitochondrial membrane in exchange for free carnitine in the presence of transporter CACT. The CPT II enzyme is in the matrix side of the inner mitochondrial membrane, encoded by the CPT II gene (chromosome 1p32.3). In the last step, CPT II enzyme activity converts acylcarnitine back to acyl-CoA's, which undergoes β-oxidation, which involves dehydrogenation, hydration, another dehydrogenation, and thiolytic cleavage. Regenerated carnitine is pumped back to the cytosol by CACT to be recycled again in the carnitine shuttle (Figure 1) (1, 2).
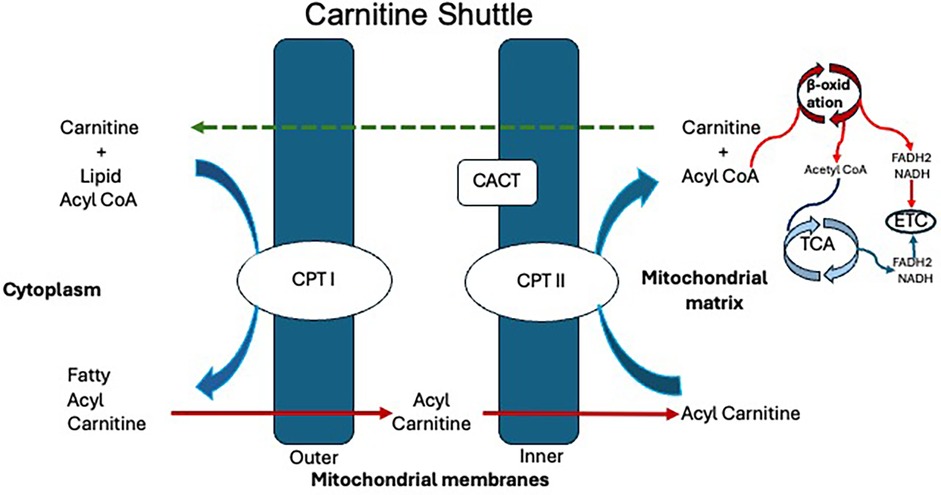
Figure 1. Carnitine shuttle. CACT, carnitine-acylcarnitine translocase; CoA, coenzyme A; CPT I, carnitine palmitoyltransferase I; CPT II, carnitine palmitoyltransferase II; ECT, electron transport chain; FADH2, flavin adenine dinucleotide; NADH, nicotinamide adenine dinucleotide.
CPT II deficiency has three main forms: (1) severe lethal neonatal form (LNF), inherited in an autosomal recessive manner. Affected infants are symptomatic at birth with hypoketotic hypoglycemia, seizures, cardiomyopathy/arrythmias, liver failure and death occurring within the first few weeks of life, (2) severe infantile (hepatocardiomuscular) form, with symptomatology appearing from 6 to 24 months of life in a similar presentation but less severe than LNF form and (3) myopathic form, which is the least severe type and they become symptomatic in infancy to adulthood with myalgia and weakness following exercise or fasting (3–5).
Lethal neonatal form of CPT II remains a challenge in early diagnosis and treatment. We present a consanguineous family who had two children in consecutive pregnancies with LNF presented in a similar way in the fetal and neonatal periods.
Case presentation
We describe a pair of siblings—an older female (patient A) and a younger male (patient B) with CPT II deficiency born by normal vaginal delivery to a first-degree consanguineous couple who have two healthy male living children. The mother had a history of an early pregnancy loss. The sibling's clinical presentations and investigations were consistent with the severe LNF of CPT II deficiency.
Antenatally noted fetal nephromegaly with renal cysts, cardiomegaly, and ventriculomegaly in both pregnancies. Siblings were born small for gestational age (SGA) and did not require active resuscitation. There was no family history of inborn errors of metabolism (IEM).
Patient A presented with hypothermia and hypoglycemia on day 1 of life while on observation in special care nursery for fetal anomalies. With in hours of stabilization of glucose profile and temperature, baby developed apnea, seizures, intractable arrhythmias, and circulatory collapse. She required full respiratory support. Hyperkalemia and acidosis worsened and was treated accordingly. Following persistent cardiorespiratory failure and intractable ventricular arrythmias she was placed on extracorporeal membrane oxygenation(ECMO) support and hemodialysis. ECMO support was withdrawn after documentation of severe intracranial bleed and succumbed on day 14 of life. The parents were given genetic counseling following the death of patient A.
The family declined prenatal diagnostic tests offered in subsequent pregnancy. Given the similar presentation antenatally to patient A, the parents chose to pursue comfort care for patient B and decided against full resuscitation and active medical intervention. Parents agreed to needful investigations and genetic testing at birth. Before the birth of patient B, advanced care planning, including milk bonding and memory making, was offered to parents by the perinatal palliative (PeriPal) care team. The patient was discharged home according to the antenatal plan on day 2, on a special milk feed. The baby passed away on day 3 of life at home with family members around. Figure 2 illustrates a timeline of the fetal and neonatal events of the siblings.
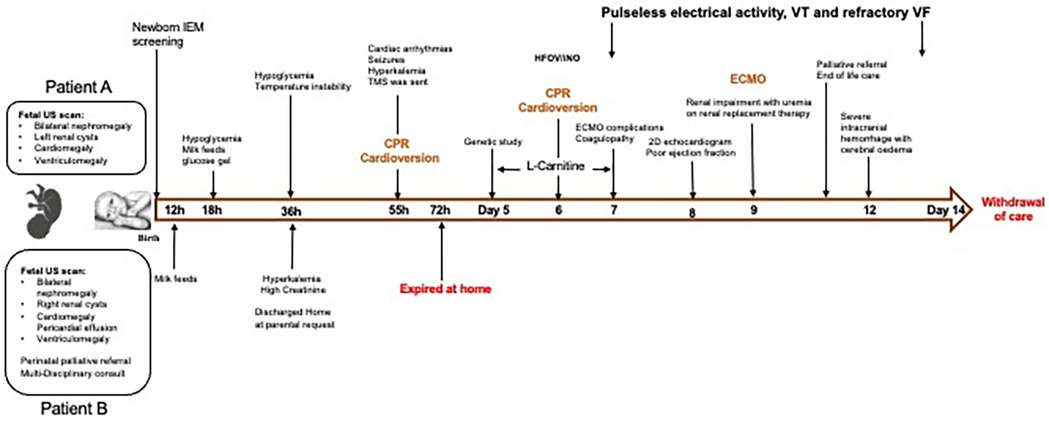
Figure 2. Timeline depicting the sequence of events from fetus to end of life of both siblings. CPR, cardiopulmonary resuscitation; ECMO, extracorporeal membrane oxygenation; HFOV, high frequency oscillatory ventilation; h, hour of age; IEM, inborn errors of metabolism; iNO, inhaled nitric oxide; TMS, tandem mass spectrometry; US, ultrasound; VF, ventricular fibrillation; VT, ventricular tachycardia.
A summary of the antenatal and postnatal findings and biochemical and genetic test results of patients A and B are given in Table 1. Table 2 shows the newborn screening results of the siblings. Figure 3 shows the genogram depicting the autosomal recessive inheritance pattern of CPT II deficiency in this family.
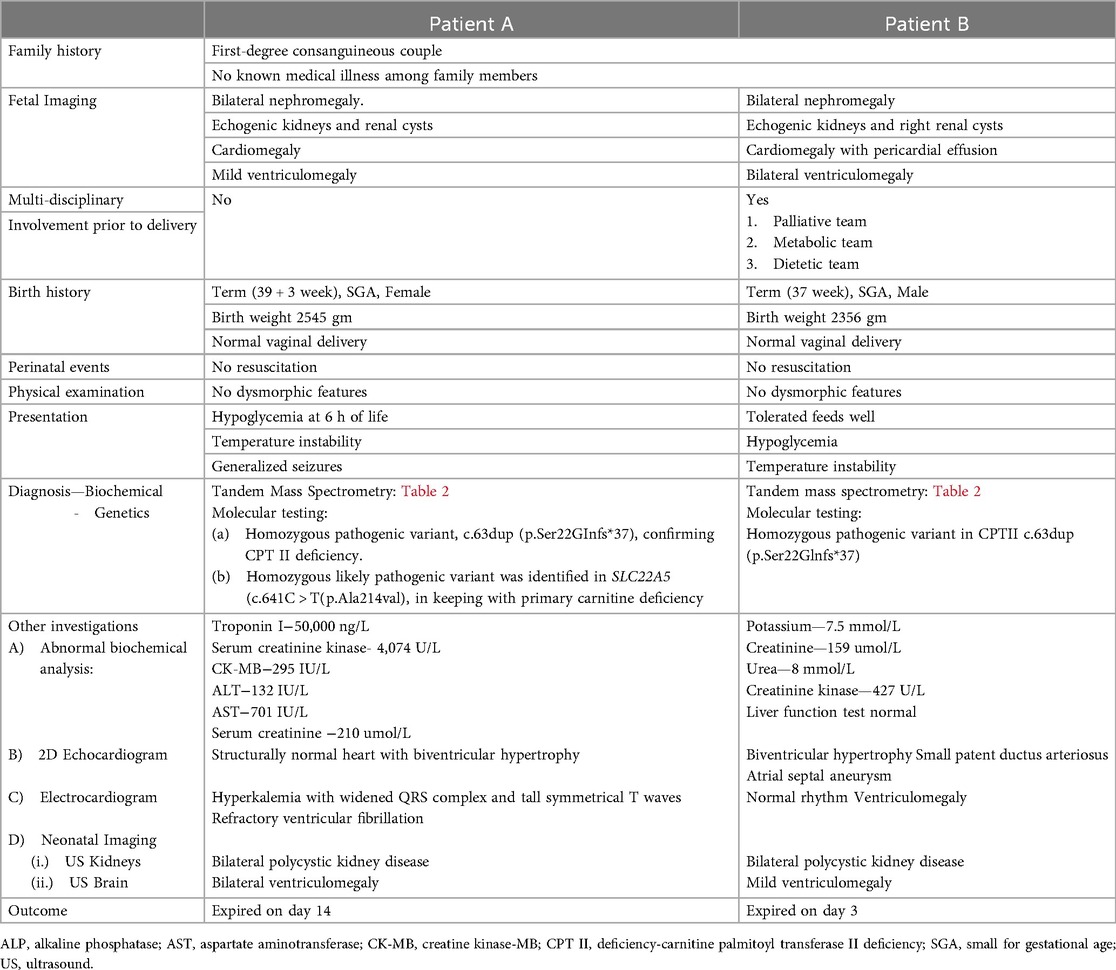
Table 1. Case description of patient A and patient B.
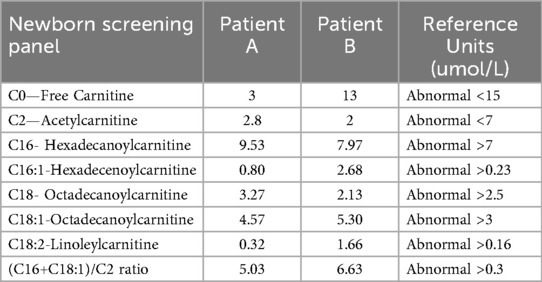
Table 2. Newborn screening panel.
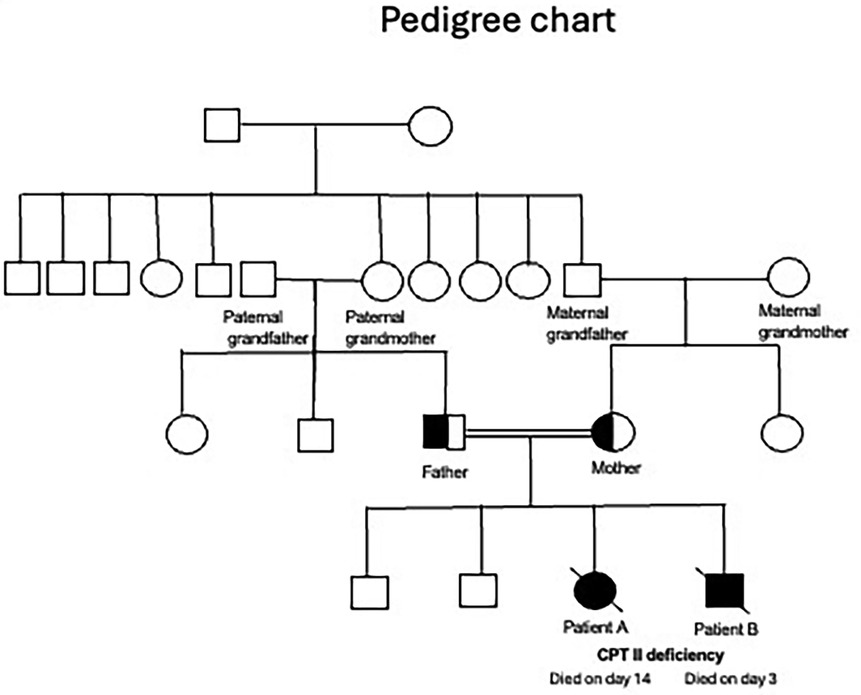
Figure 3. Genogram. CPT II, carnitine palmitoyltransferase II.
Patient A also had a likely pathogenic variant in SLC22A5 (p.Ala214Val) indicative of primary carnitine deficiency but needed additional data to prove that conclusively (Invitae, San Francisco, USA). But coexistence with pathogenic CPT II gene mutation may explain the more complicated neonatal period of patient A compared to patient B.
Pathophysiology of CPT II deficiency
The LNF is a multisystemic disease characterized by liver failure with hypoketotic hypo glycemia, cardiomyopathy, arrhythmias, and seizures. Among the CPT II deficiency cases reported, LNF constitutes 6%, infantile form 8%, and adult myopathic form 86% (6). Many CPT II gene variants can cause CPT II deficiency. However, only 16 variants were associated with LNF. The residual activity of the CPT II enzyme is very low in neonatal and infantile forms, resulting in abnormal LCFA profiles, notably with high levels of C16 and C18OH (5). This leads to energetic failure due to decreased β-oxidation, thus systemic lipid accumulation. Since the body is unable to use LCFA, it must rely on glucose to generate energy. While unable to metabolize fat, following the depletion of glucose stores, hypoketotic hypoglycemia sets in. Classically, IEM screen in neonates detect markedly abnormal acylcarnitine profile in LNF form but not always in infantile hepato-cardio-muscular form, which often presents later between 6 and 24 months of age (7, 8).
Clinical and metabolic features
In 1973, DiMauro and DiMauro reported the first case of an adult-onset form of CPT II deficiency (7). We described the presentation of LNF of CPT II deficiency in a family comprising two siblings born to a consanguineous couple. In the following discussion, we review the physiology of carnitine, the antenatal and neonatal presentations, and the diagnosis and management of CPT II deficiency.
Carnitine plays a crucial role in the entry of LCFA via carnitine shuttle into the mitochondrial matrix for β-oxidation and energy production in the form of adenosine triphosphate. Carnitine also provides acetyl-CoA for gluconeogenesis (1). Carnitine cycle defects include (a) primary carnitine deficiency, (b) CPT I deficiency, (c) CACT deficiency, and (d) CPT II deficiency (8). The reported patient A had two homozygous variants: one pathogenic CPT II gene, confirming CPT II deficiency and a likely pathogenic variant in SLC22A5 gene indicating primary carnitine deficiency (Table 1).
Antenatal phenotypes include facial dysmorphism, cerebral malformations, cystic dysplastic kidneys, cardiomegaly, and diffuse fatty infiltration (9). After reviewing 19 cases, Boemer et al. found that the malformations associated with CPT II deficiency include renal cysts/nephromegaly in 57%, cardiomegaly in 15%, and severe cerebral dysgenesis in 74%. Cerebral malformations observed included neuronal migration disorders, such as polymicrogyria and pachygyria, cystic dysplasia of the brain, cerebellar vermian hypoplasia, hydrocephalus, and agenesis of the corpus callosum (10). Maternal liberal fetal supply of glucose as a substrate leads to high concentrations of malonyl-CoA, which inhibits CPT I. The entry of LCFA via carnitine shuttle into the mitochondria is inhibited with low CPT I activity, impairing fatty acid oxidation (11). Oey et al. demonstrated high CPT II enzyme activity in the heart, liver, and brain in a 6-week-old embryo, explaining multiorgan involvement in CPT II deficiency (12). This was reflected in our siblings who presented antenatally with polycystic kidneys, ventriculomegaly, and cardiomegaly, showing that these clinical features in fetal scans could be an indicator of CPT II deficiency, particularly in cases involving consanguineous couples. In cases with a known family history, parents may be offered pre-implantation genetic diagnosis for future pregnancies (13, 14). Unfortunately, most fetuses with major malformations-particularly involving the brain and kidneys, are terminated, resulting in missed opportunities for metabolic/genetic screening.
The LNF of CPT II deficiency is often symptomatic shortly after birth. The affected infant may exhibit dysmorphic features, including microcephaly, overfolded helices, a sloping forehead, long and tapered fingers and toes, contractures, and hypoplastic toenails (15); however, none were observed in our case studies. Other manifestations of this multisystemic disease include cardiac arrhythmias, cystic dysplastic kidney, cardiomyopathy, and hypoketotic hypoglycemia (10, 16). Structurally abnormal brain and neuronal migration defects can result from defective β-oxidation, leading to impaired phospholipid synthesis and accumulation of toxic metabolites (9, 17). Both of our patients developed hypoglycemia as the presenting feature. Patient A experienced intractable seizures, cardiovascular collapse following recurrent arrhythmias, and severe hyperkalemia in the immediate newborn period and died at 2 weeks of age despite maximum optimal therapy. For the younger child, on fetal detection of renal and cardiac defects, parents were counseled towards comfort care if confirmed CPT II deficiency postnatally with biochemical profiling. Due to the early discharge of patient B, other life-threatening symptoms were not observed.
Among the various phenotypic forms of CPT II deficiency, the severe infantile hepatocardiomuscular type typically presents with hypoketotic hypoglycemia, liver failure, and cardiomyopathy within the first year of life. It can be fatal, even with early intervention. The more common myopathic form is found in adults, and symptoms of muscle pain and weakness usually appear during periods of fasting or with intercurrent illness (8, 18).
Diagnosis
The use of tandem mass spectrometry (MS/MS) is common in neonatal metabolic screening. Significant elevations of C16 and C18-acylcarnitine with decreased free carnitine indicate CPT II deficiency. Total and free carnitine levels are often reduced with higher-than-normal levels of acylcarnitine so that the acylcarnitine to free carnitine ratio is increased (19, 20). For a definitive diagnosis, rapid molecular genetic testing is recommended. Pathogenic variants of the CPT II gene, which cause either truncation of the protein or degradation of its mRNA, are associated with LNF of CPT II deficiency (8, 21). Mutation analysis can be done on fibroblasts, blood, skeletal muscle, and lymphoblasts (22–24). In LNF and infantile forms, intrafamilial phenotypic homogeneity is a known feature. Still, the prediction of the phenotype from prenatal tests remains scarce, and the genotype-phenotype correlation remains imprecise (21, 25).
Alternatively, the diagnosis can be made by detecting decreased CPT II enzyme activity in cultured fibroblasts (10). In LNF, CPT II activity in cultured fibroblast cells is 7%, whereas the enzyme activity is 2.5%–10% in infantile and 20%–46% in the milder adult myopathic forms. Liver function is often deranged with low prothrombin activity. Raised serum creatine kinase and transaminitis of 20–400 times higher have been observed biochemically (26). Prenatal and pre-implantation genetic testing can be offered to parents who are carriers of CPT II deficiency for future pregnancies (13). In both infants, molecular testing identified a homozygous pathogenic variant in CPT II c.63dup (p.Ser 22Glnfs*37), inherited from both parents. This sequence change creates a premature translational stop signal in the CPT II gene and is expected to result in an absent or disrupted protein product. Loss-of-function variants in the CPT II gene are known to be pathogenic (8).
Management
Suspect IEM in infants born to consanguineous couple with bad obstetric history or fetal brain/cardiac and/or renal anomalies. Early symptomatology in a neonate like lethargy, vomiting, poor suck, and disorientation should raise concern of IEM. At birth, dextrose infusion is recommended to slow down catabolism, helping to prevent recurrent hypoglycemia, awaiting tandem mass spectrometry report. Once abnormal acylcarnitine profile is detected, treatment focuses on nutrition with carbohydrate-rich (70%) and low-fat (<20%) feeds, incorporating medium-chain triglycerides to support glycolysis. Triheptanoin, a medium-chain triglyceride, is used in the treatment of fatty acid oxidation disorders (27). It provides a source of calories and fats needed for energy production. Studies have shown that triheptanoin reduces the likelihood of patients developing hypoglycemia, cardiomyopathy, rhabdomyolysis, and hepatomegaly (28). Despite the initiation of triheptanoin supplementation and a high-carbohydrate diet, our patient B passed away shortly thereafter, being an LNF of CPT II deficiency. The lack of active resuscitative efforts due to comfort care measures may also have contributed to the outcome in patient B.
Carnitine supplementation is a common practice in IEM, especially in primary and secondary carnitine deficiency. Carnitine has been used in the treatment of CPT II deficiency; however, its use remains controversial. It is intended to convert potentially toxic long-chain acyl-CoA compounds into acylcarnitine, which can then be excreted in the urine, depleting free CoA in the mitochondria (16, 29). In mouse models, acylcarnitine has been shown to regulate the hERG (human ether-a-go-go related gene) channel, which is crucial for cardiac repolarization and may contribute to the development of arrhythmias (30, 31). In the case of patient A, initiation of carnitine supplementation was associated with refractory arrhythmias, leading to discontinuation of the treatment. However, a Cochrane review found insufficient evidence to support the efficacy and safety of carnitine supplementation in IEM (32).
It is also important to avoid known triggers, including extreme temperatures, illness, and medications such as sodium valproate, diazepam, and ibuprofen (9, 33, 34). Frequent feedings are advised to prevent fasting, along with the administration of carnitine supplements (3, 8). In patients with cystic dysplastic kidney disease, maintaining adequate hydration is crucial to slow the progression to renal failure (35).
Bezafibrate, a class of hypolipidemic drugs, increases CPT II mRNA and normalizes enzyme activity in mild forms of CPT II deficient myoblasts. In a trial of fibrates in adult myopathic forms, muscle biopsy specimens showed a marked increase in palmitoyl L-carnitine oxidation levels by 60%–284% and CPT II mRNA levels by 20%–93%. Clinically, reduced episodes of rhabdomyolysis with significant improvement in quality of life over 6 months of treatment (36).
Advancements in antenatal diagnostic techniques have enabled the detection of a range of fetal anomalies of varying severity in CPT II deficiency. In some cases, treatment options may be limited or unavailable, or the condition may be potentially fatal, leading to neonatal death. The establishment of a structured program that includes PeriPal care alongside neonatologists could offer comprehensive support for vulnerable babies with life-limiting conditions. If parents choose to continue with the pregnancy, early counseling by the PeriPal team is beneficial to help alleviate parental anxiety, ensure clear, consistent communication, and make advance care plans for the baby (35). The team will collaborate with community palliative care (Star PALS team) services to ensure the transition of care from hospital to home, provide a comprehensive plan through home visits, and offer psychosocial support. In the event of demise, the team will also provide bereavement support.
Genetic counselling
CPT II deficiency is an autosomal recessive disorder with a probability of 25% being affected and 50% being carriers in future pregnancies for a consanguineous couple who are carriers for this variant. Molecular genetic testing for the CPT II gene or CPT II enzyme activity assay can give a prenatal diagnosis in pregnancies at increased risk for mild to severe forms of CPT II deficiency. To date, 16 variants have been reported in 26 lethal neonatal cases, born to 11 different families (5). Our reported cases had homozygous pathogenic variants in CPT II—c.63dup (p.Ser22GInfs*37). Patient A also had a homozygous likely pathogenic variant- SLC22A5 (c.641C>T(p.Ala214val), indicative of a diagnosis of primary carnitine deficiency. El-Hattab et al. reported the variability in presentation of systemic primary carnitine deficiency from metabolic decompensation in infancy to an asymptomatic adult (37).
Conclusion
Antenatal detection of fetal anomalies are reliable indicators for suspecting CPT II deficiency in the background of parental consanguinity, more so if a previous baby was affected. In a suspected case of CPT II deficiency the following are considered: (1) dextrose infusion to prevent catabolism till special milk feeds can be arranged, (2) a high carbohydrate and low-fat diet to provide substrate for glycolysis, (3) avoidance of known triggers, e.g., fasting, illness etc., (4) urgent IEM screen/genetic testing/echocardiography/and ultrasound scan of the brain and kidneys, (5) MCT supplementation, (6) intensive care monitoring, and (7) a multidisciplinary team including metabolic specialists and palliative care doctors. As the neonatal form of CPT II deficiency is typically fatal, early confirmation of the diagnosis should prompt advance care planning discussions with the parents to support timely, appropriate care and informed decision-making in the best interest of the child.
Patient's perspective
“The family was in shock after seeing our daughters struggle to survive. For the son, we were strong enough to decide on comfort care with the timely support of the palliative team.”
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Central Institutional Review Board/Singhealth, Singapore. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants' legal guardians/next of kin. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article. Written informed consent was obtained from the minor(s)' legal guardian for the publication of any potentially identifiable data included in this case report.
Author contributions
YT: Writing – original draft. KT: Conceptualization, Resources, Supervision, Writing – review & editing. AA: Resources, Supervision, Writing – review & editing. CK: Resources, Writing – review & editing. VR: Resources, Supervision, Writing – review & editing. SC: Conceptualization, Resources, Supervision, Validation, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
The authors would like to thank Vidal E and Banas JL, Department of Neonatology, KK Women's and Children's Hospital, Singapore, for preparing the figures. We want to thank Dr. Krishna G R and Dr. Neha Garg, Department of Neonatology, and Dr. James Lim Soon Chuan, Chief Scientific Officer, Biochemical Genetics and National Expanded Newborn Screening, Department of Pathology, KK Women's and Children's Hospital, Singapore for their involvement in the diagnosis and treatment of this patient. We are grateful to the Children's ICU, Cardiology, Genetics, and the ECMO team of KK Women's and Children's Hospital, Singapore, for their expertise in managing this case.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Longo N, Frigeni M, Pasquali M. Carnitine transport and fatty acid oxidation. Biochim Biophys Acta. (2016) 1863: 2422–35. doi: 10.1016/j.bbamcr.2016.01.023
2. Joshi PR, Zierz S. Muscle carnitine palmitoyltransferase II (CPT II) deficiency: a conceptual approach. Molecules. (2020) 25:1784. doi: 10.3390/molecules25081784
3. Malik S, Paldiwal AA, Korday CS, Jadhav SS. Neonatal carnitine palmitoyltransferase II deficiency: a lethal entity. J Clin Diagn Res. (2015) 9:SD01–2. doi: 10.7860/JCDR/2015/13600.6560
4. Tan YY, Fong WYN, Chan JH, Chandran S. Do renal and cardiac malformations in the fetus signal carnitine palmitoyltransferase II deficiency? A rare lethal fatty acid oxidation defect. BMJ Case Rep. (2022) 19(12):e251321. doi: 10.1136/bcr-2022-251321
5. Tran TCM, Ta VT, Bui TB, Vu CD, Pham TN. A case study of lethal neonatal CPT II deficiency: novel insights from genetic analysis. Mol Genet Metab Rep. (2024) 41:101170. doi: 10.1016/j.ymgmr.2024.101170
6. Wieser T, Deschauer M, Olek K, Hermann T, Zierz S. Carnitine palmitoyltransferase II deficiency: molecular and biochemical analysis of 32 patients. Neurology. (2003) 60(8):1351–3. doi: 10.1212/01.WNL.0000055901.58642.48
7. DiMauro S, DiMauro PM. Muscle carnitine palmitoyltransferase deficiency and myoglobinuria. Science. (1973) 182(4115):929–31. doi: 10.1126/science.182.4115.929
8. Wieser T. Carnitine palmitoyltransferase II Deficiency. [Updated 2019 Jan 3]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025 (2004).
9. Elpeleg ON, Hammerman C, Saada A, Shaag A, Golazand E, Hochner-Celnikier D, et al. Antenatal presentation of carnitine palmitoyltransferase II deficiency. Am J Med Genet. (2001) 102:183–7. doi: 10.1002/ajmg.1457
10. Boemer F, Deberg M, Schoos R, Caberg JH, Gaillez S, Dugauguier C, et al. Diagnostic pitfall in antenatal manifestations of CPT II deficiency. Clin Genet. (2016) 89:193–7. doi: 10.1111/cge.12593
11. Prip-Buus C, Pegorier JP, Duee PH, Kohl C, Girard J. Evidence that the sensitivity of carnitine palmitoyl transferase I to inhibition by malonyl-CoA is an important site of regulation of hepatic fatty acid oxidation in the fetal and newborn rabbit. Perinatal development and effects of pancreatic hormones in cultured rabbit hepatocytes. Biochem J. (1990) 269(2):409–15. doi: 10.1042/bj2690409
12. Oey NA, den boer MEJ, Wijburg FA, Vekemans M, Auge J, Steiner C, et al. Long-chain fatty acid oxidation during early human development. Pediatr Res. (2005) 57:755–9. doi: 10.1203/01.PDR.0000161413.42874.74
13. Bonnefont JP, Demaugre F, Prip-Buus C, Saudubray JM, Brivet M, Abadi L, et al. Carnitine palmitoyltransferase deficiencies. Mol Genet Metab. (1999) 68:424–40. doi: 10.1006/mgme.1999.2938
14. Vekemans BC, Bonnefont J-P, Aupetit J, Royer G, Droin V, Attie-Bitach T, et al. Prenatal diagnosis of carnitine palmitoyltransferase 2 deficiency in chorionic villi: a novel approach. Prenat Diagn. (2003) 23:884–7. doi: 10.1002/pd.713
15. North KN, Hoppel CL, De Girolami U, Kozakewich HP, Korson MS. Lethal neonatal deficiency of carnitine palmitoyltransferase II associated with dysgenesis of the brain and kidneys. J Pediatr. (1995) 127:414–20. doi: 10.1016/S0022-3476(95)70073-0
16. Vladutiu GD, Bennett MJ, Fisher NM, Smail D, Boriack R, Leddy J, et al. Phenotypic variability among first-degree relatives with carnitine palmitoyltransferase II deficiency. Muscle Nerve. (2002) 26:492–8. doi: 10.1002/mus.10217
17. Bonnet D, Martin D, De Lonlay P, Villain E, Jouvet P, Rabier D, et al. Arrhythmias and conduction defects as presenting symptoms of fatty acid oxidation disorders in children. Circulation. (1999) 100:2248–53. doi: 10.1161/01.CIR.100.22.2248
18. Almannai M, Alfadhel M, El-Hattab AW. Carnitine inborn errors of metabolism. Molecules. (2019) 24:3251. doi: 10.3390/molecules24183251
19. Wang T, Ma J, Zhang Q, Gao A, Wang Q, Li H, et al. Expanded newborn screening for inborn errors of metabolism by tandem mass spectrometry in Suzhou, China: disease spectrum, prevalence, genetic characteristics in a Chinese population. Front Genet. (2019) 10:1052. doi: 10.3389/fgene.2019.01052
20. Gempel K, Praun CV, Baumkotter J, Lehnert W, Ensenauer R, Gerbitz KD, et al. “Adult” form of muscular carnitine palmitoyltransferase II deficiency: manifestation in a 2-year-old child. Eur J Pediatr. (2001) 160:548–51. doi: 10.1007/s004310100802
21. Thuillier L, Rostane H, Droin V, Demaugre F, Brivet M, Kadhom N, et al. Correlation between genotype, metabolic data, and clinical presentation in carnitine palmitoyltransferase 2 (CPT2) deficiency. Hum Mutat. (2003) 21:493–501. doi: 10.1002/humu.10201
22. Smeets RJ, Smeitink JA, Semmekrot BA, Scholte HR, Wanders RJ, van den Heuvel LP. A novel splice site mutation in neonatal carnitine palmitoyl transferase II deficiency. J Hum Genet. (2003) 48:8–13. doi: 10.1007/s100380300001
23. Sigauke E, Rakheja D, Kitson K, Bennett MJ. Carnitine palmitoyltransferase II deficiency: a clinical, biochemical, and molecular review. Lab Invest. (2003) 83:1543–54. doi: 10.1097/01.LAB.0000098428.51765.83
24. Demaugre F, Bonnefont JP, Colonna M, Cepanec C, Leroux JP, Saudubray JM. Infantile form of carnitine palmitoyltransferase II deficiency with hepatomuscular symptoms and sudden death. Physiopathological approach to carnitine palmitoyltransferase II deficiencies. J Clin Invest. (1991) 87(3):859–64. doi: 10.1172/JCI115090
25. Joshi PR, Deschauer M, Zierz S. Carnitine palmitoyltransferase II (CPT II) deficiency: genotype-phenotype analysis of 50 patients. J Neurol Sci. (2014) 338:107–11. doi: 10.1016/j.jns.2013.12.026
26. Bonnefont JP, Djouadi F, Prip-Buus C, Gobin S, Munnich A, Bastin J, et al. Carnitine palmitoyltransferases 1 and 2: biochemical, molecular and medical aspects. Mol Aspects Med. (2004) 25:495–520. doi: 10.1016/j.mam.2004.06.004
27. Wehbe Z, Tucci S. Therapeutic potential of triheptanoin in metabolic and neurogenerative disease. J Inheri Metab Dis. (2020) 43:385–91. doi: 10.1002/jimd.12199
28. Roe CR, Brunengraber H. Anaplerotic treatment of long-chain fat oxidation disorders with triheptanoin: review of 15 years experience. Mol Genet Metab. (2015) 116:260–8. doi: 10.1016/j.ymgme.2015.10.005
29. Yoshino M, Tokunaga Y, Watanabe Y, Yoshida I, Sakaguchi M, Hata I, et al. Effect of supplementation of L-carnitine at a small dose on acylcarnitine profiles in serum and urine and the renal handling of acylcarnitines in a patient with multiple acyl-coenzyme A dehydrogenation defect. J Chromatogr B Analyt Technol Biomed Life Sci. (2003) 792:73–82. doi: 10.1016/S1570-0232(03)00310-6
30. Ferro F, Ouillé A, Tran TA, Fontanaud P, Bois P, Babuty D, et al. Long-chain acylcarnitines regulate the HERG channel. PLoS One. (2012) 7:e41686. doi: 10.1371/journal.pone.0041686
31. Nasser M, Javaheri H, Fedorowicz Z, Noorani Z. Carnitine supplementation for inborn errors of metabolism. Cochrane Database Syst Rev. (2012) 2012:CD006659. doi: 10.1002/14651858.CD006659.pub3
32. Spiekerkoetter U, Lindner M, Santer R, Grotzke M, Baumgartner MR, Boehles H, et al. Treatment recommendations in long-chain fatty acid oxidation defects: consensus from a workshop. J Inherit Metab Dis. (2009) 32:498–505. doi: 10.1007/s10545-009-1126-8
33. Deschauer M, Wieser T, Zierz S. Muscle carnitine palmitoyltransferase II deficiency: clinical and molecular genetic features and diagnostic aspects. Arch Neurol. (2005) 62:37–41. doi: 10.1001/archneur.62.1.37
34. Chandran S, Rajadurai VS, Abdul Haium AA, Hussain K. Current perspectives on neonatal hypoglycemia, its management, and cerebral injury risk. Res Rep Neonatol. (2015) 5:17–30. doi: 10.2147/RRN.S55353
35. Tewani KG, Jayagobi PA, Chandran S, Anand AJ, Thia EWH, Bhatia A, et al. Perinatal palliative care service: developing a comprehensive care package for vulnerable babies with life limiting fetal conditions. J Palliat Care. (2022) 37:471. doi: 10.1177/08258597211046735
36. Bonnefont JP, Bastin J, Behin A, Djouadi F. Bezafibrate for an inborn mitochondrial beta-oxidation defect. N Engl J Med. (2009) 360:838–40. doi: 10.1056/NEJMc0806334
Keywords: CPT II deficiency, carnitine, fatty acid oxidation, polycystic kidneys, ventriculomegaly
Citation: Tan Y. Y., Tewani K. G., Anand A. J., Kong C. X., Rajadurai V. S. and Chandran S. (2025) Case Report: Lethal neonatal form of CPT II deficiency in consecutive pregnancies: fetal-neonatal characteristics, biochemical and molecular review. Front. Pediatr. 13:1648282. doi: 10.3389/fped.2025.1648282
Received: 17 June 2025; Accepted: 22 October 2025;
Published: 18 November 2025.
Edited by:
Ronan Lordan, University of Pennsylvania, United StatesReviewed by:
Alex Staffler, Central Teaching Hospital of Bolzano/Bozen, ItalyAnne Chiaramello, George Washington University, United States
Copyright: © 2025 Tan, Tewani, Anand, Kong, Rajadurai and Chandran. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: S. Chandran, cHJvZnNjaGFuZHJhbjIwMTlAZ21haWwuY29t